

Abstract
Mesenchymal stem/stromal cells (MSC) are multipotent precursors endowed with the ability to home to primary and metastatic tumor sites, where they can integrate into the tumor-associated stroma. However, molecular mechanisms and outcome of their interaction with cancer cells have not been fully clarified. In this study, we investigated the effects mediated by bone marrow-derived MSC on human colorectal cancer (CRC) cells in vitro and in vivo. We found that MSC triggered epithelial-to-mesenchymal transition (EMT) in tumor cells in vitro, as indicated by upregulation of EMT-related genes, downregulation of E-cadherin and acquisition of mesenchymal morphology. These effects required cell-to-cell contact and were mediated by surface-bound TGF-β newly expressed on MSC upon coculture with tumor cells. In vivo tumor masses formed by MSC-conditioned CRC cells were larger and characterized by higher vessel density, decreased E-cadherin expression and increased expression of mesenchymal markers. Furthermore, MSC-conditioned tumor cells displayed increased invasiveness in vitro and enhanced capacity to invade peripheral tissues in vivo. Thus, by promoting EMT-related phenomena, MSC appear to favor the acquisition of an aggressive phenotype by CRC cells.
Keywords: colorectal cancer, MSC, EMT, surface-bound TGF-β
Mesenchymal stem/stromal cells (MSC) are multipotent precursors endowed with the ability to home to wounds and areas of chronic inflammation where they contribute to the regeneration of mesenchymal tissues, including bone, cartilage, muscle and adipose tissue.1–3 MSC have been shown to be actively recruited also to primary and metastatic tumors.2,4–6 This peculiar tropism of MSC together with the possibility to easily engineer them has prompted researchers to explore the potential use of MSC as cellular vehicles for targeted delivery of anti-cancer agents into the tumor microenvironment.4,7,8
However, the outcome of the interaction between MSC and tumor cells remains unclear. A number of studies have investigated the impact of exogenously administered MSC on tumor growth and metastasis formation. Depending on the tumor model used, MSC have been shown to either inhibit or promote the development and progression of different tumor types, including melanoma, glioma, breast, lung and colorectal cancer (CRC)9–17 as reviewed in Refs.6 and 18.
What’s new? —
Mesenchymal stem/stromal cells (MSCs) are recruited into tumor-associated stroma, but their interactions with cancer cells are not fully understood. In this study, the authors found that MSCs can trigger epithelial-to-mesenchymal transition (EMT) of human colorectal cancer (CRC) cells via cell-to-cell contact. This phenomenon required membrane-bound TGF-beta, which was newly expressed by MSCs upon cross-talk with tumor cells. These results reveal a novel mechanism by which MSCs can enhance the aggressiveness of tumor cells, and they suggest a potential new therapeutic target in CRC.
Various mechanisms have been implicated. On one hand, MSC have been shown to secrete factors directly impacting on proliferation and/or survival of tumor cells9,15–17 or promoting tumor vasculogenesis.17,19–21 On the other hand, MSC-tumor cell crosstalk has been demonstrated to result in de novo secretion of cytokine/chemokines increasing motility, tumorigenicity and metastatic capacity of tumor cells.11,22 In addition, upon differentiation into cancer-associated fibroblasts,23,24 MSC may create a stromal niche sustaining cancer progression.16,22,25 Furthermore, three recent studies have suggested that MSC can induce in tumor cells epithelial-to-mesenchymal transition (EMT),22,26,27 a complex process resulting in increased tumor cell motility, invasiveness and resistance to apoptosis.28 Molecular mechanisms mediating this particular phenomenon and impact on tumor progression in vivo remain to be thoroughly investigated.
CRC is a leading cause of cancer-related death worldwide.29 Progression and metastasis formation have been recognized to be linked to the occurrence of EMT possibly initiated by signals delivered by the stromal component within the tumor microenvironment.30,31
MSC have been shown to migrate to CRC and, through the secretion of soluble factors, to increase tumorigenicity of tumor cells.9,15,16,32 Very recently, CRC cells have been reported to prompt release of inflammatory cytokines by MSC which then, in a paracrine fashion, induce EMT in CRC cells in vitro.22 However, the precise molecular determinants of this bidirectional, reciprocal interaction and its consequence on CRC spreading in vivo remain to be addressed.
In this study, we examined the effects mediated by human bone marrow-derived MSC on CRC cells in vitro and in vivo, and, in particular, their ability to initiate EMT in tumor cells. We found that MSC strongly induce the expression of EMT-related genes in CRC cells in vitro in a cell-to-cell contact dependent manner. This phenomenon appears to be mediated by surface-bound TGF-β expressed on MSC upon cross-talk with tumor cells. Importantly, tumors developed by CRC cells exposed to MSC conditioning exhibit decreased E-cadherin expression, increased vessel density and increased invasive capacity.
Material and Methods
MSC isolation and characterization
MSC were derived from bone marrow cells of healthy donors, as previously described,33 and were subsequently expanded in α-MEM (GIBCO, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 1% HEPES, 1% sodium pyruvate, 1% kanamycin and 5 ng/mL FGF-2 (R&D Systems, Minneapolis, MN). Expanded cells were analyzed by flow cytometry for the expression of stromal markers, including CD105, CD73, CD90 and CD29 and the absence of hematopoietic and endothelial markers, such as CD45, CD34 and CD31 (Supporting Information Fig. S1). The capacity of MSC to differentiate into osteoblasts, adipocytes and chondroblasts was assessed as described in Ref.34 (data not shown).
Tumor cell lines
Established human CRC cell lines (HCT116, LS180, COLO205, HT29 and SW480) were purchased from European Collection of Cell Cultures (ECACC, Salisbury, UK). HCT116, LS180 and COLO205 were maintained in RPMI-1640 supplemented with 10% FBS, GlutaMAX-I, non-essential amino acids (NEAA), 100 mM sodium pyruvate, 10 mM HEPES (all from GIBCO) and 50 mM 2-mercaptoethanol (Sigma–Aldrich, St. Louis, MO). HT29 was maintained in McCoy’s 5A medium (Sigma) supplemented with 10% FBS and GlutaMAX-I. SW480 were cultured in L-15 Medium (Leibovitz) (Sigma–Aldrich) supplemented with 10% FBS and GlutaMAX-I. Kanamycin sulfate (GIBCO) was included with all media. Absence of mycoplasma contamination in cultured cells was verified by PCR testing prior to investigation.
Cocultures
CRC cells were cocultured with MSC, or normal skin fibroblasts as controls, at different ratios, for 5 days in tumor cell medium. In specific experiments, recombinant TGF-β (100 ng/mL, R&D Systems) or IL-6 (10 ng/mL, R&D Systems), the TGF-β inhibitors latency-associated peptide (LAP) (10 µg/mL, R&D Systems) or SB431542 (10 µg/mL, Sigma) or anti-IL-6 neutralizing antibodies (10 µg/mL, R&D Systems) were added to cultures as indicated. The lack of effect by the TGF-β inhibitors on basal E-cadherin expression was verified in preliminary experiments (data not shown). In experiments aimed at evaluating the role of cell-to-cell contact, MSC and tumor cells were plated in the upper and lower chambers, respectively, of transwell plates (0.4 µm pore size, Corning, Lowell, MA). Alternatively, tumor cells were cultured in the presence of MSC-conditioned medium harvested every 48 hr. Monocultures of MSC or tumor cells were used as controls. At the end of culture periods, supernatants were collected and cells were harvested and used for subsequent analyses.
Flow cytometric analysis and cell sorting
Phenotypes of expanded MSC were analyzed upon staining with the following antibodies: allophycocyanin (APC)-labeled anti-CD34 (clone 581), anti-CD90 (clone 5E10), phycoerythrin (PE)-labeled anti-CD31 (clone WM59), anti-CD73 (clone AD2), anti-CD44 (clone G44-26), anti-CD29 (clone MAR4), fluorescein-isothiocyanate (FITC)-labeled anti-CD45 (clone 2D1) (all from BD Biosciences, San Jose, CA) and anti-CD105 (clone SN6, AbDSerotec, Raleigh, NC). For the analysis of CRC cells in coculture with stromal cells, the following antibodies were used: APC-labeled anti-EpCAM (clone EBA-1), FITC-labeled anti-CD90 and PE-labeled anti-CD44, anti-CD166 (all from BD Biosciences) or anti-CD133 (Miltenyi Biotec, Auburn, CA) or PE-labeled anti-TGF-β (clone 9016, R&D Systems). Propidium iodide (PI, 0.5 µg/mL) was added to all samples prior to analysis. Samples were analyzed by a dual laser BD FACS Calibur flow cytometer (BD Biosciences), following exclusion of dead cells based on PI incorporation. Analysis was performed using FlowJo software (Tree Star, Ashland, OR). For sorting of tumor cells and MSC from cocultures, cells were stained with APC-labeled anti-EpCAM and FITC-labeled anti-CD90. Dead cells were excluded based on DAPI incorporation. Cell sorting was performed using a BD Influx cell sorter (BD Biosciences). Purity of sorted cells was ≥98%.
Chemoinvasion assay
LS180, HCT116 and HT29 cells growing alone or sorted following coculture were tested for invasiveness in a chemoinvasion assay as previously described.35,36 Briefly, tumor cells suspended in serum-free medium were seeded into upper chambers of transwell plates onto uncoated or matrigel-coated membranes (8 µm pore size, BD Biocoat Tumor invasion assay, BD Biosciences). Lower chambers contained medium supplemented with 5% FBS. Plates were incubated at 37°C. After 20 hr, inserts were removed and the numbers of cells that had migrated into the lower chambers were quantified by CyQUANT Cell Proliferation Assay Kit (Invitrogen, Carlsbad, CA). Percentages of invading cells were calculated according to the following formula: (mean of relative fluorescent units [RFU] of cells invading through matrigel-coated membranes/mean RFU of cells migrated through uncoated membranes) ×100.
Gene expression analysis
Total cellular RNA was extracted from individual cell populations sorted by flow cytometry using the RNeasy® Mini Kit (QIAGEN, Hombrechtikon, Switzerland) according to the manufacturer’s protocol. RNA concentration and purity were determined using NanoDrop1 ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA (1 µg) was reverse transcribed using M-MLV reverse transcriptase following manufacturer’s (Invitrogen) protocol, and cDNA samples were amplified and analyzed by quantitative real-time PCR (qRT-PCR) using ABI Prism 7300 (Applied Biosystems, Paisley, UK). Pre-developed Taqman® assays (Applied Biosystems) were used to evaluate the expression of TWIST, SNAIL1, SNAIL2, ZEB1, E-cadherin (ECAD), N-cadherin (NCAD) and TGF-β genes. The comparative CT method was used to quantify gene expression upon normalization using human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene as reference.
Western blot analysis
Cells lysates were obtained using 50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40 and 0.1% SDS. Protein concentrations were determined by the Pierce BCA protein concentration assay (Thermo Scientific, Waltham, MA). Western blotting procedures have been detailed previously.37 Blots were probed with mouse anti-E-cadherin (1:1,000, BD Biosciences), goat anti-β-actin (1:2,000, Santa Cruz, Santa Cruz, CA) and rabbit anti-GAPDH (1:5,000, Abcam, Cambridge, UK) antibodies. Secondary species-specific HRP-conjugated IgG (Santa Cruz) together with Pierce ECL (Thermo Fisher, Scientific Waltham, MA) were used for detection of immunoreactive proteins. Quantification of E-cadherin expression relative to β-actin or GAPDH was performed following densitometric analysis of immunoreactive signals using AIDA software (Raytest, Straubenhardt, Germany).
Cytokine/chemokine detection
Cytokine, chemokine and matrix metalloproteinase (MMP) contents in culture supernatants were assessed by the RayBio® Human Cytokine Antibody Array III and RayBio® Matrix Metalloproteinases Array (RayBio Tech, Norcross, GA), respectively, according to the manufacturer’s protocol. Following development, densitometric analysis was performed using Scion Image software for Windows (Scion Corporation, Frederick, MD). Semiquantitative assessment of protein content was obtained upon normalization of signals detected in each condition to corresponding positive controls. Specific ELISA kits were used to quantify IL-6 (BD Biosciences), IL-1β (eBioscience), MCP-1, Angiogenin, TGFβ and VEGF (all R&D Systems) release.
Analysis of tumorigenicity and metastatic potential in vivo
In vivo experiments were approved by the local veterinary officer. NOD/SCID mice, from Charles River Laboratories (Sulzfeld, Germany), were bred and maintained under specific pathogen-free conditions in our animal facility. Mice (8–10-week old) were injected subcutaneously (s.c.) in the flank with titrated numbers (102−106) of tumor cells alone, tumor cells mixed with equal numbers of MSC or tumor cells cocultured with MSC and sorted prior to injection. Matrigel© (BD Biosciences), diluted 1:1 in PBS, was used as cell suspension vehicle. Tumor formation was monitored twice weekly by palpation and caliper measurements. After 5 weeks, all mice were sacrificed and tumors, liver and lungs were harvested. Tumor volumes (in mm3) were determined according to the formula (length × width2)/2. Samples from all tissues were frozen for subsequent histological examination.
Immunofluorescence
For detection of pSMAD 2/3, tumor-MSC cocultures were performed in Chambers slide (BD Biosciences). At day 5, cells were fixed in methanol and stained with a rabbit monoclonal pSMAD 2/3-specific antibody (Cell Signaling) followed by a secondary species-specific Alexa Fluor 488-conjugated antibodies (Invitrogen). Nuclei were counterstained with DAPI.
Histological analysis of tumors was conducted in three tumors per group. Three to five cryosections (10 µm) were cut from each tumor and fixed in methanol. Sections were incubated with Cy3-conjugated mouse monoclonal anti-alpha smooth muscle actin (αSMA) (clone C6198, Sigma–Aldrich), rabbit monoclonal anti-E-cadherin (clone 24E10, Cell Signaling Technology), rabbit monoclonal anti-vimentin (clone D21H3, Cell Signaling Technology, Danvers, MA) or rat monoclonal anti-CD31 (clone MEC13.3, BD Biosciences) antibodies, followed by secondary species-specific Alexa Fluor 488-conjugated antibodies (Invitrogen). Nuclei were counterstained with DAPI. Sections were examined under an Olympus BX61 fluorescence microscope (Olympus Switzerland) and images captured with 10× and 20× magnification using a digital camera and AnalySIS software (Soft Imaging System GmbH). For quantification of microvessel density (MVD) images captured at 20× magnification were used. Numbers of individual CD31-positive cells within three to five randomly selected fields in each section were counted.
Analysis of tumor invasiveness in vivo
LS180 cells were transduced with lentiviral pGreenfire1-CMV reporter vector (Clontech Laboratories, Mountain View, CA) co-expressing green fluorescent protein (GFP) and firefly luciferase (GFP/Luc-LS180 cells). NOD/SCID mice were injected intravenously (i.v.), in the tail, with 105 GFP/Luc-LS180 cells cultured alone, or mixed with equal numbers of MSC, or cocultured with MSC and sorted, resuspended in 100 µL PBS. Tumor development in vivo was monitored weekly after injection using the LB983 NightOWL II imaging system (Berthold Technologies GmbH, Bad Wildbad, Germany) as described.38 Briefly, animals were injected intraperitoneally with 5 mg d-luciferin (Gold BioTechnology, St. Louis) in 300 µL sterile PBS, anesthetized with isoflurane and placed in the NightOWL imaging chamber. Images were acquired using IndiGo software (Berthold Technologies) with 2 min exposure under the maximum sensitivity settings, processed to colorize luminescence signal according to intensity and overlaid with the black and white photographic images. Following detection of positive signals in peripheral tissues, mice were sacrificed and tumor formation in organs of interest was confirmed by histological evaluation.
Statistical analysis
Statistical analyses were performed using two-tailed Student’s t-test, one-way ANOVA, or the Mann–Whitney-U test, as appropriate, using the SPSS version 19.0 software (IBM Corporation). p-Values ≤0.05 were considered significant.
Results
MSC promote proliferation of CRC cells in vitro
In initial studies, to validate the previously reported9 capacity of MSC to promote CRC cell survival and expansion in vitro, CRC cells from different established cell lines were cocultured with MSC from healthy donors at various MSC:CRC ratios. We confirmed a significant increase in CRC cell numbers after coculture with MSC, as compared to tumor cells cultured alone (Supporting Information Fig. S2a). For adherent cell lines (e.g., LS180, HCT116, HT29 and SW480), we noted that this effect was more prominent at low than at high MSC:CRC ratios, possibly due to the occurrence, in the latter case, of cell-to-cell contact inhibition. For the COLO205 cell line, which grows in suspension, numbers of tumor cells increased even at an MSC:CRC ratio of 10:1. Based on these results, we decided to perform subsequent coculture experiments using adherent cell lines in culture with MSC at a 1:1 ratio. MSC from different donors (n = 4) comparably enhanced CRC proliferation (data not shown). In contrast, normal skin fibroblasts did not increase, but rather decreased CRC cell numbers (Supporting Information Fig. S3a). Thus, the capacity to enhance CRC expansion appeared to be a MSC-specific feature.
Since MSC have been recently reported to specifically promote the expansion of CRC-derived CSC,16 the expression of putative CSC markers, including CD133, CD166 and CD44, was also evaluated (Supporting Information Fig. S2b). No major modifications of CD133 expression were observed in CRC cells cultured alone or in the presence of MSC. Expression of CD166 was slightly down-modulated in some cell lines including HCT116. In contrast, CD44 expression was decreased in all cell lines tested. Thus, the increase in CRC cell numbers observed following coculture with MSC was not accompanied by the expansion of cells expressing putative CSC phenotypes.
MSC induce EMT in CRC cells
The capacity of MSC to induce EMT in CRC cells was then investigated. LS180, HCT116 and HT29 cell lines were cocultured with MSC at 1:1 ratio and morphology, expression of EMT-markers and invasiveness were assessed in comparison to tumor cells cultured alone. Various morphological changes were observed depending on the cell line used. For example, HCT116 and HT29 cells acquired a more elongated shape, whereas LS180 cells formed enlarged spheroid-like structures (Fig. 1a).
Figure 1.

MSC induce EMT in CRC cells. LS180, HCT116 and HT29 cells were cultured alone (TC) or with MSC (TC + MSC) at 1:1 ratio for 5 days and were then sorted by flow cytometry, based on EpCAM expression. (a) Morphological evaluation by bright field microscopy of LS180, HCT116 and HT29 cells growing alone or in coculture. Magnifications: ×10 and ×20, scale bar 100 µm. (b) Expression levels of EMT-related genes were evaluated by quantitative PCR, using GAPDH as reference gene. Relative expression levels (mean ± SD) from a minimum of two independent experiments are shown. (c) E-cadherin and β-actin expression on LS180 cells cultured alone or sorted after coculture was evaluated by Western blot analysis. The mean fold change relative to β-actin from two independent experiment is indicated. (d) Sorted LS180, HCT116 and HT29 cells were tested in a chemoinvasion assay in comparison to tumor cells cultured alone. Percentages of invasion (mean ± SD) from three independent experiments are shown.*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.005.
All three cell lines, sorted after coculture with MSC (Supporting Information Fig. S4), exhibited a significant upregulation of TWIST, SNAIL1, SNAIL2, ZEB1 and N-cadherin gene expression (Fig. 1b). Concomitantly, a downregulation of E-cadherin was evident at both gene (Fig. 1b) and protein level (Fig. 1c). Furthermore, following coculture, CRC cells displayed a significantly higher invasive potential (Fig. 1d). In contrast, CRC cells cocultured with skin fibroblasts displayed increased expression of TWIST and N-cadherin genes only, albeit to a much lower extent than in CRC cultured with MSC (Supporting Information Fig. S3b). No upregulation of SNAIL1, SNAIL2 or ZEB1 gene expression was induced, and importantly, no downmodulation of E-cadherin was detected (Supporting Information Fig. S3b). Thus, MSC, but not fibroblasts, promote EMT induction in human CRC cells.
Soluble factors promoting EMT induction
EMT can be triggered by soluble factors released into the tumor microenvironment upon tumor–stroma interaction, including TGF-β, MMPs and, as shown more recently, inflammatory cytokines such as IL-1α, IL-1β, IFN-γ and TNF-α.22,26,39 To gain insight into mechanisms underlying MSC-mediated EMT induction in CRC cells, we analyzed the supernatants collected from CRC and MSC growing as monocultures or in coculture for the content of a panel of chemokines/cytokines (Supporting Information Table S1). Significant levels of IL-8, IL-10, GM-CSF, GRO, GROα, MIP1δ, MDC, Angiogenin, EGF and VEGF were detected in tumor cell supernatants. MSC released IL-6, MCP-1, MDC and VEGF. Notably, IL-6, MCP-1 and Angiogenin titers were significantly higher in coculture supernatants as compared to the cumulative amounts released by MSC and TC in monocultures (Figs. 2a and 2b). IL-1α, IL-1β, IFN-γ and TNF-α were not detectable under any of the experimental conditions tested (Fig. 2).
Figure 2.
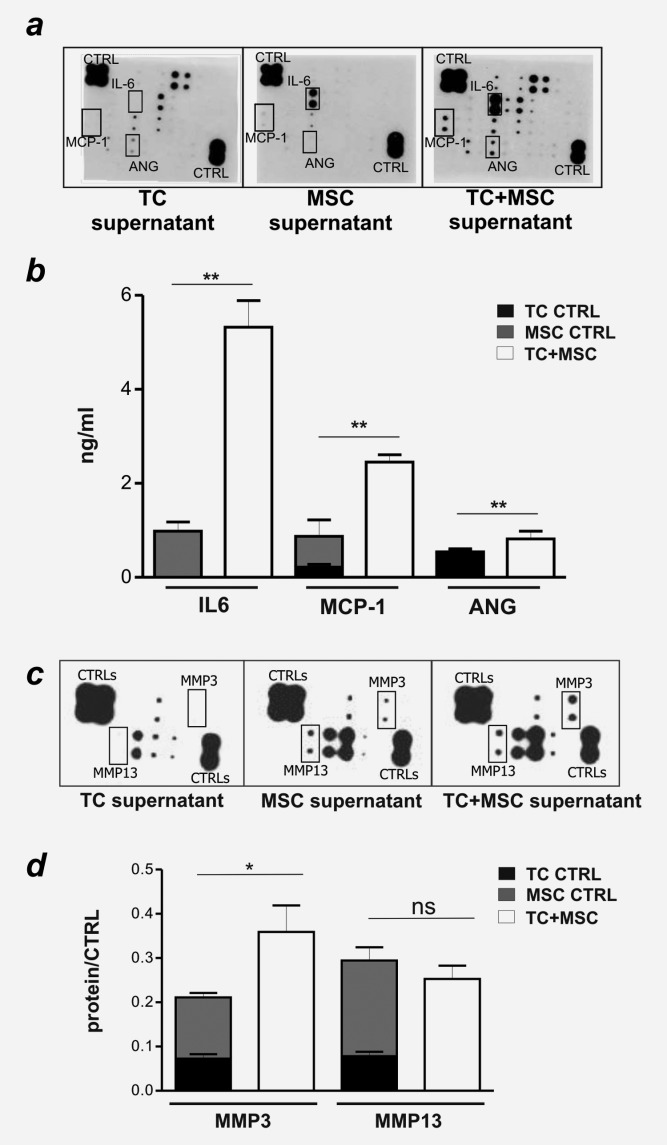
Soluble factors released during tumor cells-MSC coculture. (a, d) Supernatants of LS180 cells (TC) and MSC, cultured alone or in coculture (TC+MSC) were collected and assessed for cytokine/chemokine content by Chemokine/cytokine antibody array III® (a), ELISA (b) or for the presence of specific MMPs using RaybioMMP antibody array (c and d). (a) Duplicate dots in developed membranes mark the detection of specific chemokine/cytokines, or positive controls (upper left and lower right corners). (b) IL-6, MCP-1 and Angiogenin (ANG) contents quantified by ELISA. Mean ± SD of duplicate cultures are reported. (c) Duplicate dots in developed membranes mark the detection of specific MMPs, or positive controls (upper left and lower right corners). (d) Semi-quantitative analysis of MMP3 and MMP13 content (mean ± SD), obtained from the normalization of signals detected to the corresponding positive controls. *p ≤ 0.05, **p ≤ 0.01.
IL-6 is known to promote tumor cell proliferation,6 but does not per se promote EMT induction.22 Consistent with this notion, in our setting, blockade of IL-6 prevented MSC-dependent enhancement of tumor cell proliferation. Accordingly, addition of exogenous IL-6 to monocultures of CRC cells increased their numbers to an extent comparable to that induced by MSC (Supporting Information Fig. S5b).
Conditioned media collected from monocultures and cocultures of MSC and CRC were additionally assayed for the presence of ten MMPs/MMP-inhibitors (Supporting Information Table S2). MMP-1, -3, −8 and -13 were present in tumor cell and MSC monoculture supernatants, but only MMP-3 levels were significantly increased upon coculture (Figs. 2c and 2d).
Surface-bound TGF-β expressed by MSC induces EMT on CRC cells
We next investigated the potential role of TGF-β. Significant TGF-β expression was detected in CRC cells and MSC cultured alone or in coculture. Remarkably, upon coculture, TGF-β gene expression was strongly upregulated in MSC, but not in CRC cells (Fig. 3a). Intriguingly however, TGF-β protein amounts in coculture supernatants were not increased, as compared to the cumulative amounts released by MSC and TC in monocultures (Fig. 3b). As in hematopoietic cells TGF-β has been shown to be also expressed in membrane-bound form,40 we questioned whether this could also be the case for MSC. Strikingly, following coculture with LS180, HCT116 and HT29 cells MSC acquired surface TGF-β expression (Fig. 3c). Skin fibroblasts cultured with tumor cells did not display any significant surface TGF-β expression (Supporting Information Fig. S3c). These results suggest that the specific capacity of MSC to initiate EMT on CRC cells could be related to their expression of membrane-bound TGF-β.
Figure 3.
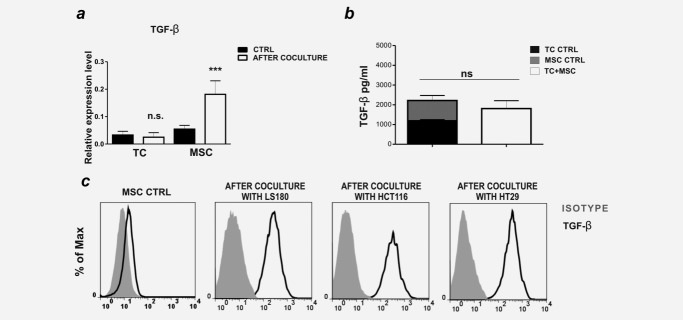
Surface-bound TGF-β expression is induced in MSC upon coculture with CRC cells. (a) LS180 cells (TC), and MSC, cultured alone or sorted following coculture, were tested for TGF-β gene expression levels by quantitative PCR. Means ± SD from three experiments are shown. (b) TGF-β content was measured in culture supernatants by ELISA. Mean ± SD of duplicate cultures are reported. (c) Surface TGF-β expression on MSC, cultured alone or in coculture with LS180, HCT116 or HT29 cells, was assessed by flow cytometry, upon gating on EpCAM-CD90+ cells. ***p ≤ 0.005.
Considering this possibility, we investigated the requirement for cell-to-cell contact in the induction of MSC-mediated EMT. CRC cells and MSC were indirectly cocultured in transwell plates, preventing direct contact between the two cell populations while allowing the exchange of soluble factors. Alternatively, tumor cells were cultured alone in the presence of MSC-conditioned medium. Strikingly, the indirect coculture of MSC and CRC completely abrogated the upregulation of EMT-related genes (Fig. 4a), thus suggesting a functional irrelevance of secreted cytokines. Indeed, exposure of CRC cells to MSC-derived conditioned medium also did not induce EMT (Fig. 4a). These findings strongly suggest that the phenomenon of EMT induction in CRC cells by MSC depends on cell-to-cell contact. Inclusion of the TGF-β inhibitors LAP or SB431542 during direct coculture protocols completely prevented the induction of EMT, at gene and protein level (Figs. 4b and 4c), thus demonstrating the role of TGF-β as a critical mediator. Furthermore, immunofluorescence analysis of cocultures revealed the phosphorylation of SMAD2/3 on tumor cells exposed to MSC (Fig. 4d).
Figure 4.
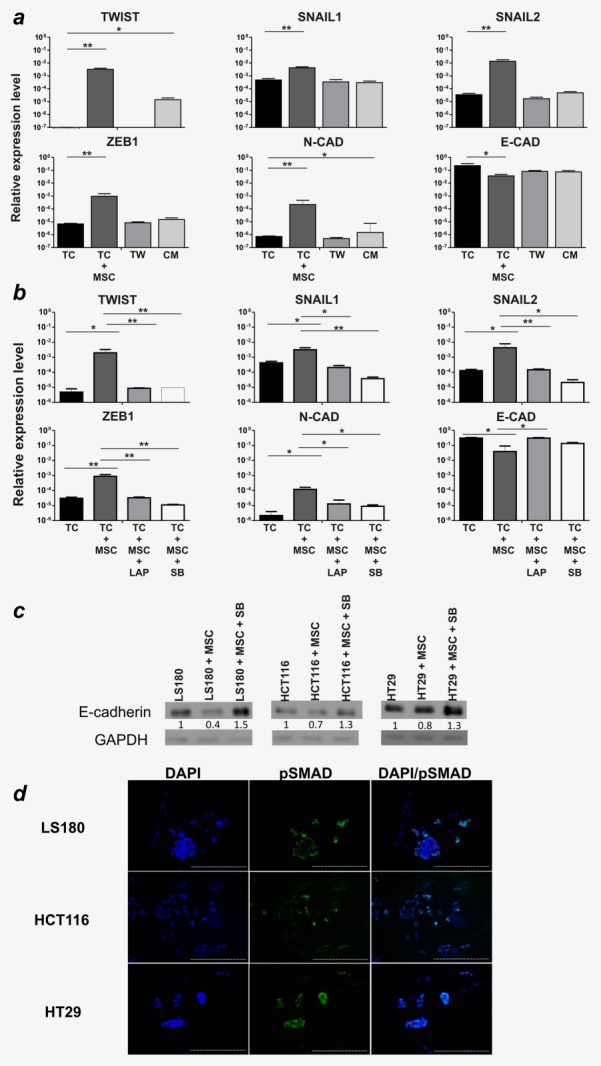
EMT induction in CRC cells is mediated by MSC-derived surface-bound TGF-β. (a) LS180 cells were cultured alone (TC), with MSC (TC+MSC) ratio 1:1, with MSC in transwell plates (TW), or with MSC-conditioned medium (CM) for 5 days. Following cell sorting, expression levels of EMT-related genes, relative to GAPDH, were assessed. Mean ± SD from two independent experiments are shown. (b) TC were cultured alone (TC) or with MSC (TC+MSC) in the absence, or presence of TGF-β-neutralizing LAP (TC+MSC+LAP) or SB431542 (TC+MSC+SB) (both at 10 µg/mL). After 5 days, tumor cells were sorted by flow cytometry and analyzed for EMT-related gene expression. Mean ± SD from two independent experiments are shown. (c) E-cadherin and GAPDH expression on LS180, HCT116 and HT29 cells cultured alone or after coculture with MSC, in the presence or absence of SB431542 (10 µg/mL) was evaluated by Western blot analysis. The fold changes relative to GAPDH are indicated. *p ≤ 0.05; **p ≤ 0.01. (d) Cocultures were stained with phospho-SMAD 2/3-specific antibody (green) and DAPI (blue) for nuclei counterstaining. Magnification ×10, scale bar 100 μm.
Taken together these data indicate that upon interaction with CRC cells MSC express high levels of membrane-bound TGF-β capable of initiating EMT in tumor cells.
Conditioning by MSC results in enhanced tumor volume and vessel density in vivo
We investigated the impact of MSC-mediated effects on tumor growth in vivo. NOD/SCID mice were injected s.c. with titrated numbers of LS180 cells alone, or admixed with an equal number of MSC, or with CRC cells cocultured with MSC and sorted prior to injection. Tumor development was monitored over time. All tumor cell preparations exhibited comparable tumorigenicity with tumor formation in almost all mice injected (Supporting Information Table S3). However, tumors formed from MSC-conditioned LS180 cells (either admixed with or pre-exposed to MSC) displayed slightly faster kinetics and significantly larger size as compared to those formed by untreated tumor cells (Figs. 5a and 5b). Importantly, upon immunohistochemical evaluation, tumors originating from MSC-conditioned LS180 cells showed weak and more heterogeneous E-cadherin expression (Fig. 5c). Furthermore, these tumors were characterized by a more abundant stromal component, as indicated by increased expression of mesenchymal markers, such as αSMA and vimentin, and by enhanced microvessel density (Figs. 5c and 5d). Thus, the conditioning of tumor cells by MSC appears to enhance tumor growth, possibly by promoting angiogenesis and stroma formation.
Figure 5.

MSC promote tumor development and invasion in vivo. (a–c): NOD/SCID mice (n = 5/group) were injected s.c. with 106 LS180 cells alone (TC), or mixed with equal numbers of MSC (TC+MSC), or with tumor cells cultured with MSC and sorted prior to injection (TC sorted). Tumor formation was monitored over time. (a) Tumor onset over time. (b) Tumor size measured after 5 weeks. Individual values plus mean (plotted as bar) from one out of two experiments performed with similar results are reported. (c) Tumor sections were stained with E-Cadherin- (green), αSMA- (red), Vimentin- (green) and CD31-specific (green) antibodies and with DAPI (blue) for nuclei counterstaining. Magnifications ×10 or ×20, scale bar 100 or 200 µm. (d) Microvessel density was quantified by counting numbers of individual CD31-positive cells within randomly selected fields in images captured at ×20 magnification. Seventeen fields from two different tumors per group were analyzed. *p ≤ 0.05, ***p ≤ 0.005. (e, f) NOD/SCID mice were injected i.v. with 105 GFP/Luc-LS180 (TC), TC+MSC or TC sorted. Mice were monitored for metastasis formation by non-invasive in vivo luciferase imaging. (e) Representative images of metastatic foci appearing in cervical lymph nodes (LN) and lungs are shown. Left panels: pseudocolor images of peak bioluminescence. The color bars indicate relative signal intensity. Right panels: histological evaluation of metastatic foci by IF. Tumor cells are detected based on GFP expression (green) counterstained by DAPI (blue). (f) Tumor incidence and location at end point (5 weeks) from two different experiments performed.
To assess whether MSC conditioning of CRC cells may favor metastasis formation at distant sites, livers and lungs of tumor-bearing mice were screened for the presence of tumor cells by histology. No LS180 cells in distant organs were detected in any of the tumor-bearing mice (data not shown). Therefore, as an alternative experimental modality to gain insights into MSC-mediated effects on the invasive potential of tumor cells in vivo, we compared the capacity of GFP-Luc LS180 cells, pre-cultured or admixed with MSC, and untreated GFP-Luc LS180 cells to engraft in peripheral tissues following i.v. injection. Starting from 3 weeks after injection, the presence of putative metastatic foci, as indicated by luciferase imaging, was detected in lungs (Fig. 5e, upper panel). After 5 weeks, palpable tumor masses, also positive for luciferase expression, appeared in the upper back of some mice at the position of deep cervical lymph nodes (Fig. 5e, lower panel). Histological evaluation confirmed the tumor origin of lung and lymph node metastatic foci (Fig. 5e, right panels). Importantly, mice injected with GFP-Luc LS180 cells admixed or pre-cultured with MSC showed a higher incidence of metastasis formation, as compared to mice injected with GFP-Luc LS180 cells alone (Fig. 5f), thus indicating that conditioning by MSC enhanced the invasive capacity of LS180 cells.
Discussion
Although the ability of MSC to home to tumor site is well documented, mechanisms and outcomes of their interaction with cancer cells remain to be fully clarified.
This study examined effects mediated by human bone marrow-derived MSC on CRC cells. In vitro investigations showed that MSC strongly induced the expression of a panel of EMT-related genes and reduced the expression of E-cadherin. Furthermore, MSC induced CRC cells to acquire mesenchymal morphology and increased their invasive potential. Induction of EMT in CRC cells by MSC required cell-to-cell contact and appeared to be mediated by surface-bound TGF-β expressed on MSC upon cross-talk with tumor cells. In vivo investigations showed that MSC-conditioned CRC cells formed larger tumor masses characterized by decreased E-cadherin expression, increased expression of mesenchymal markers and higher vessel density. Furthermore, MSC conditioning of CRC cells also enhanced their capacity to invade peripheral tissues.
Despite a large body of literature on the interaction between MSC and tumor cells, the role played by MSC in human CRC has only been addressed recently. A few papers have described a pro-tumorigenic effect of MSC on CRC cells, due to the secretion of soluble factors acting in a paracrine manner. In particular, MSC-derived IL-6 has been reported to increase tumor formation capacity of CRC cells, either by favoring preferential expansion and/or survival of CSC16 or by inducing in tumor cells the production of pro-angiogenic factors.32 MSC-derived neuroregulin 1 has been reported to enhance in vitro invasiveness as well as tumorigenicity of CRC cells.9 More recently tumor cell-derived IL-1α and β have been shown to elicit PGE2 production by MSC, resulting in triggering of EMT in tumor cells in vitro.22 MSC have also been shown to promote metastasis formation in vivo, but the mechanisms underlying this phenomenon remain unclear.
Our study confirms that MSC support CRC cell expansion in an IL-6-dependent manner in vitro.9 However, we could not detect a previously reported increased frequency of tumor cells expressing putative CSC markers, including CD133, CD166 and CD44.16 In vivo, tumors originated upon s.c. injection of CRC cells, either admixed or pre-cultured with MSC, showed larger volumes and increased microvessel density. However, MSC did not significantly affect tumor-initiating capacity of CRC cells, even upon injection of low tumor cell numbers, possibly due to the intrinsically high tumorigenicity of CRC cells from established cell lines. Thus, in our model MSC promoted CRC growth without apparently impacting on CSC, but rather by favoring tumor angiogenesis.
Importantly, we found that MSC act on CRC cells as EMT initiators. Tumor cells exposed to MSC respond with an upregulated expression of EMT-related genes in vitro, and displayed decreased E-cadherin expression both in vitro and in vivo. Moreover, upon exposure to MSC, some cell lines (e.g., HCT116 and HT29) acquired a mesenchymal morphology, also suggestive of EMT. However, at difference with previous reports,22,26,39 we found that neither tumor cells nor MSC released inflammatory cytokines possibly promoting EMT, including IL-1α, IL-1β, IFN-γ and TNF-α. Instead, significant amounts of TGF-β and MMP3 were detected in both mono- and cocultures. Nevertheless, exposure to culture supernatants was not sufficient to induce EMT gene signatures on CRC cells, suggesting a minor role played by secreted cytokines in this phenomenon. Conversely, there was no occurrence of EMT induction in CRC cells indirectly cocultured with MSC, thus indicating that in our system the induction of EMT gene signatures in CRC cells was not mediated by soluble factors, but rather required cell-to-cell contact.
Interestingly, we found that following interaction with tumor cells, MSC acquired surface TGF-β expression suggesting that membrane-bound TGF-β, rather than its corresponding soluble form, may act as critical EMT mediator. Membrane-bound TGF-β expression has been previously reported in some hematopoietic cell subsets,40 but has not yet been described on MSC. Notably, fibroblasts cocultured with CRC cells neither acquired surface TGF-β expression nor induced EMT in tumor cells. Thus, the capacity to express membrane-bound TGF-β may represent an essential feature of MSC that enables their initiation of EMT in CRC and possibly other types of carcinomas. Cell-to-cell contact-mediated upregulation of EMT-related genes has also been recently reported in breast cancer cells.27
The precise mechanisms leading to surface TGF-β expression and activation on MSC remain to be fully clarified. It is possible that some MMPs released in coculture, in particular MMP-3, contribute to cleavage of latent TGF-β into its active form.28 Conversely, protease-independent TGF-β activation by integrins binding to LAP has been reported.41 In preliminary experiments, we observed that blocking of αVβ6 integrin completely abrogated the MSC-mediated EMT (data not shown).
The signaling pathway activated by surface-bound TGF-β also deserves to be fully characterized. Unexpectedly, HCT116 cells were found to be susceptible to MSC-mediated EMT in a TGF-β-dependent manner, despite expressing a truncated, not functional form of TGF-β receptor II.42 This suggests that membrane-bound TGF-β may engage, on tumor cells, receptors other than TGF-βRII. For instance, expression of TGF-β R type III (TGF-β RIII) has recently been reported in primary colon cancers.43 Interestingly, TGF-β RIII has been found to mediate increased SMAD2 phosphorylation upon TGF-β exposure in cell lines carrying mutations in TGF-β RII gene.43 We indeed detected TGF-β RIII expression in several CRC cell lines, including HCT116 (data not shown). Further investigations are warranted to address the actual role of TGF-β RIII in MSC-mediated EMT in human CRC.
Increasing evidence supports the concept that EMT facilitates the acquisition of an invasive phenotype by cancer cells, thus favoring tumor spreading and metastasis formation.28,30 Indeed, loss of E-cadherin on primary CRC is associated with an infiltrating tumor margin and an increased metastasis occurrence.44–46 Consistent with this hypothesis, we found that MSC-conditioned CRC cells displayed increased invasiveness in vitro and formed tumor masses characterized by low E-cadherin expression. Following s.c. injection of tumor cells however, no spontaneous metastasis formation in distant organs was observed, possibly due to the relatively limited duration of the experiment, since for ethical reasons tumor-bearing mice were euthanized when tumor masses reached a 10 mm maximal diameter. In contrast, following systemic injection, CRC cells either admixed or pre-cultured with MSC, showed a higher capacity to invade lung tissues and lymph nodes as compared with non-conditioned CRC cells. Collectively, these findings suggest that MSC might enhance the invasiveness and metastatic potential of CRC cells in vivo as a consequence of EMT induction.
The cross-talk between MSC and tumor cells appears to be a major determinant of the phenomena observed. By mere interaction with tumor cells, MSC increased their capacity to release soluble pro-inflammatory and pro-angiogenic factors, including IL-6, MCP-1 and Angiogenin, and most importantly, to upregulate surface-bound TGF-β expression. Furthermore, MSC appeared to induce a long-lasting conditioning of tumor cells. MSC-mediated effects were detected in tumors originating from CRC cells administered together with MSC as well as in those developed by CRC cells pre-cultured with MSC. Thus, a transient exposure to MSC in vitro was sufficient to induce long-term modifications in tumor cells.
Although the conditions of interaction between MSC and tumor cells in vivo may differ substantially from those of our experimental settings, nevertheless the potential pro-tumorigenic effect of MSC should be taken into account when envisaging their possible clinical use in cancer patients.6,8,18
In conclusion, our data identify membrane-bound TGF-β as a novel mechanism enabling MSC to enhance aggressiveness of tumor cells and suggest a potential new therapeutic target in CRC.
Acknowledgments
The authors thank Prof. Alan Tyndall for helpful discussion and critical reviewing of the manuscript, and Dr. Chiara Giovenzana for helping in the initial set-up of the study. The authors have no potential conflicts of interest to disclose.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–9. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain G, Fox J, Ashton B, et al. Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25:2739–49. doi: 10.1634/stemcells.2007-0197. [DOI] [PubMed] [Google Scholar]
- Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–36. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- Hall B, Andreeff M, Marini F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. Handb Exp Pharmacol. 2007:263–83. doi: 10.1007/978-3-540-68976-8_12. [DOI] [PubMed] [Google Scholar]
- Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614–23. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazennec G, Jorgensen C. Concise review: adult multipotent stromal cells and cancer: risk or benefit? Stem Cells. 2008;26:1387–94. doi: 10.1634/stemcells.2007-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bexell D, Scheding S, Bengzon J. Toward brain tumor gene therapy using multipotent mesenchymal stromal cell vectors. Mol Ther. 2010;18:1067–75. doi: 10.1038/mt.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer RM, Kerin MJ. Mesenchymal stem cells and cancer: tumor-specific delivery vehicles or therapeutic targets? Hum Gene Ther. 2010;21:1506–12. doi: 10.1089/hum.2010.135. [DOI] [PubMed] [Google Scholar]
- De Boeck A, Pauwels P, Hensen K, et al. Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression through paracrine neuregulin 1/HER3 signalling. Gut. 2013;62:550–60. doi: 10.1136/gutjnl-2011-301393. [DOI] [PubMed] [Google Scholar]
- Djouad F, Plence P, Bony C, et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102:3837–44. doi: 10.1182/blood-2003-04-1193. [DOI] [PubMed] [Google Scholar]
- Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–63. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- Khakoo AY, Pati S, Anderson SA, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J Exp Med. 2006;203:1235–47. doi: 10.1084/jem.20051921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Otsu K, Das S, Houser SD, et al. Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood. 2009;113:4197–205. doi: 10.1182/blood-2008-09-176198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinagawa K, Kitadai Y, Tanaka M, et al. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int J Cancer. 2010;127:2323–33. doi: 10.1002/ijc.25440. [DOI] [PubMed] [Google Scholar]
- Tsai KS, Yang SH, Lei YP, et al. Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology. 2011;141:1046–56. doi: 10.1053/j.gastro.2011.05.045. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Sun Z, Han Q, et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia. 2009;23:925–33. doi: 10.1038/leu.2008.384. [DOI] [PubMed] [Google Scholar]
- Klopp AH, Gupta A, Spaeth E, et al. Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2010;29:11–9. doi: 10.1002/stem.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucerova L, Matuskova M, Hlubinova K, et al. Tumor cell behaviour modulation by mesenchymal stromal cells. Mol Cancer. 2010;9:129. doi: 10.1186/1476-4598-9-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Yang R, Banie L, et al. Effects of transplantation of adipose tissue-derived stem cells on prostate tumor. Prostate. 2010;70:1066–73. doi: 10.1002/pros.21140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Han ZP, Zhang SS, et al. Effects of inflammatory factors on mesenchymal stem cells and their role in the promotion of tumor angiogenesis in colon cancer. J Biol Chem. 2011;286:25007–15. doi: 10.1074/jbc.M110.213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HJ, Reinhardt F, Herschman HR, et al. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012;2:840–55. doi: 10.1158/2159-8290.CD-12-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra PJ, Mishra PJ, Humeniuk R, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–9. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaeth EL, Dembinski JL, Sasser AK, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4:e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quante M, Tu SP, Tomita H, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–72. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Y, Han Z, Liu Y, et al. Mesenchymal stem cells in inflammation microenvironment accelerates hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition. PLoS One. 2012;7:e43272. doi: 10.1371/journal.pone.0043272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin FT, Dwyer RM, Kelly J, et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT) Breast Cancer Res Treat. 2010;124:317–26. doi: 10.1007/s10549-010-0734-1. [DOI] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- Bastid J. EMT in carcinoma progression and dissemination: facts, unanswered questions, and clinical considerations. Cancer Metastasis Rev. 2012;31:277–83. doi: 10.1007/s10555-011-9344-6. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Hlubek F, Spaderna S, et al. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005;179:56–65. doi: 10.1159/000084509. [DOI] [PubMed] [Google Scholar]
- Huang WH, Chang MC, Tsai KS, et al. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene. 2013;32:4343–54. doi: 10.1038/onc.2012.458. [DOI] [PubMed] [Google Scholar]
- Bocelli-Tyndall C, Zajac P, Di Maggio N, et al. Fibroblast growth factor 2 and platelet-derived growth factor, but not platelet lysate, induce proliferation-dependent, functional class II major histocompatibility complex antigen in human mesenchymal stem cells. Arthritis Rheum. 2010;62:3815–25. doi: 10.1002/art.27736. [DOI] [PubMed] [Google Scholar]
- Di Maggio N, Mehrkens A, Papadimitropoulos A. Fibroblast growth factor-2 maintains a niche-dependent population of self-renewing highly potent non-adherent mesenchymal progenitors through FGFR2c. Stem Cells. 2012;30:1455–64. doi: 10.1002/stem.1106. [DOI] [PubMed] [Google Scholar]
- Albini A, Benelli R. The chemoinvasion assay: a method to assess tumor and endothelial cell invasion and its modulation. Nat Protoc. 2007;2:504–11. doi: 10.1038/nprot.2006.466. [DOI] [PubMed] [Google Scholar]
- Lugli A, Iezzi G, Hostettler I, et al. Prognostic impact of the expression of putative cancer stem cell markers CD133, CD166, CD44s, EpCAM, and ALDH1 in colorectal cancer. Br J Cancer. 2010;103:382–90. doi: 10.1038/sj.bjc.6605762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi MB, Philippova M, Ivanov D, et al. T-cadherin protects endothelial cells from oxidative stress-induced apoptosis. FASEB J. 2005;19:1737–9. doi: 10.1096/fj.05-3834fje. [DOI] [PubMed] [Google Scholar]
- Caceres G, Zhu XY, Jiao JA, et al. Imaging of luciferase and GFP-transfected human tumours in nude mice. Luminescence. 2003;18:218–23. doi: 10.1002/bio.729. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–7. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–44. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1—an intimate relationship. Eur J Cell Biol. 2008;87:601–15. doi: 10.1016/j.ejcb.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–8. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- Gatza CE, Holtzhausen A, Kirkbride KC, Morton A, et al. Type III TGF-beta receptor enhances colon cancer cell migration and anchorage-independent growth. Neoplasia. 2011;13:758–70. doi: 10.1593/neo.11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschos KA, Canovas D, Bird NC. The role of cell adhesion molecules in the progression of colorectal cancer and the development of liver metastasis. Cell Signal. 2009;21:665–74. doi: 10.1016/j.cellsig.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Minoo P, Baker K, Baumhoer D, et al. Urokinase-type plasminogen activator is a marker of aggressive phenotype and an independent prognostic factor in mismatch repair-proficient colorectal cancer. Hum Pathol. 2010;41:70–8. doi: 10.1016/j.humpath.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Karamitopoulou E, Zlobec I, Patsouris E, et al. Loss of E-cadherin independently predicts the lymph node status in colorectal cancer. Pathology. 2011;43:133–7. doi: 10.1097/PAT.0b013e3283425b7f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.