

Abstract
Background
Cardiac cell therapy has been proposed as one of the new strategies against myocardial infarction. Although several reports showed improvement of the function of ischemic heart, the effects of cell therapy vary among the studies and the mechanisms of the beneficial effects are still unknown. Previously, we reported that clonal stem cell antigen‐1–positive cardiac progenitor cells exerted a therapeutic effect when transplanted into the ischemic heart. Our aims were to identify the cardiac progenitor‐specific paracrine factor and to elucidate the mechanism of its beneficial effect.
Methods and Results
By using an antibody array, we found that soluble junctional adhesion molecule‐A (JAM‐A) was abundantly secreted from cardiac progenitor cells. Pretreatment of neutrophils with conditioned medium from cultured cardiac progenitor cells or soluble JAM‐A inhibited transendothelial migration and reduced motility of neutrophils. These inhibitory effects were attenuated by anti–JAM‐A neutralizing antibody. Injection of cardiac progenitor cells into infarct heart attenuated neutrophil infiltration and expression of inflammatory cytokines. Injection of soluble JAM‐A–expressing, but not of JAM‐A siRNA–expressing, cardiac progenitor cells into the infarct heart prevented cardiac remodeling and reduced fibrosis area.
Conclusions
Soluble JAM‐A secreted from cardiac progenitor cells reduces infiltration of neutrophils after myocardial infarction and ameliorates tissue damage through prevention of excess inflammation. Our finding may lead to a new therapy for cardiovascular disease by using the anti‐inflammatory effect of JAM‐A.
Keywords: cardiac progenitor cells, cell adhesion molecules, cell transplantation, inflammation, myocardial infarction
Introduction
Many randomized controlled clinical trials have demonstrated that cell therapy improves the function of ischemic heart. However, the results are inconsistent among the trials. One reason for improvement is said to be procedure‐related variables. Thus, the establishment of optimal protocol regarding to the cell source, timing of transplantation, and delivery methods is awaited.1–2 In addition to these technical issues, it has been recognized that an understanding of the mechanism of action is important to overwhelm the current limitation of cell therapy. Recently, the secretion of factors with paracrine effects by the transplanted cells has been considered as one of the mechanisms of the beneficial effects of cell therapy.3 Previously, we reported that among cardiac progenitor cells, bone marrow mononuclear cells (BMs), skeletal myoblasts (SMs), and adipose tissue–derived mesenchymal cells (AMCs), transplantation of cardiac progenitor cells (CPCs) most effectively prevents cardiac remodeling and dysfunction.4 The superior effect of CPC transplantation was based on both transdifferentiation of transplanted cells and paracrine mechanism. Recently, we have identified soluble vascular cell adhesion molecule 1 (VCAM‐1), which was secreted in a large amount from CPCs but not AMCs, attenuate cardiac remodeling after myocardial infarction (MI) through the promotion of angiogenesis and survival of cardiomyocytes.5 Various types of cells, such as BMs, SMs, AMCs, and CPCs, secrete many kinds of distinct proteins according to their organ‐specific profile. Therefore, it is conceivable that comprehensive analysis of secreted proteins from different cell sources may help us to find a new paracrine factor that can ameliorate cardiac function through cytoprotective and angiogenic potential or even some unidentified mechanisms.
We analyzed secreted proteins from CPCs, BMs, SMs, and AMCs by using a cytokine antibody array. Among 144 cytokines, we identified that junctional adhesion molecule‐A (JAM‐A), VCAM‐1, and granulocyte‐colony stimulating factor were exclusively released from CPCs. JAM‐A is a component of the junction of epithelial and endothelial cells, and recently it was reported that the soluble form of JAM‐A regulates transendothelial migration of leukocytes.6 We examined the potential mechanism of CPC transplantation from the facet of JAM‐A–mediated anti‐inflammatory effect by using a mouse MI model.
Methods
Animals
Wild‐type mice (C57Bl/6J, 10 to 12 weeks) were purchased from Japan SLC. All protocols were approved by the Institutional Animal Care and Use Committee of Chiba University Graduate School of Medicine and the Japanese Government Animal Protection and Management Law (No. 105).
Preparation of Cells
CPCs are clonal cells established from adult mouse heart stem cell antigen‐1–positive cells as described previously.4,7 CPCs were cultured in Iscove's modified Dulbecco's medium (Life Technologies) with 10% FBS and passaged before reaching confluence. AMCs were isolated from adult mice as described previously with a few modifications.8 Briefly, white adipose tissues were digested at 37°C in PBS with 2.5 mg/mL Dispase (Life Technologies) for 45 minutes. After filtration through 25‐μm filters and centrifugation, isolated AMCs were suspended in DMEM (Sigma‐Aldrich) supplemented with 10% FBS and penicillin‐streptomycin and cultured on 1% gelatin‐coated dishes. AMCs after passages 3 to 5 were used. SMs were isolated from the hindlimbs of adult mice as described previously.9 In brief, minced muscle tissues were digested in 0.05% trypsin‐EDTA and cultured in F‐10 medium (Life Technologies) with 20% horse serum and 2.5 ng/mL basic fibroblast growth factor (Promega) for 4 days. SMs were expanded in the medium with 20% FBS and used within 2 or 3 passages. BMs were harvested from adult mice. Mononuclear cells were subsequently separated by using Histopaque 1083 (Sigma‐Aldrich), suspended in Iscove's modified Dulbecco's medium.
Neonatal rat cardiomyocytes were isolated as previously described.10 Cardiomyocytes were plated at a field density of 1×106 cells/cm2 on 10‐cm culture dishes (BD Biosciences) and cultured in DMEM supplemented with 10% FBS. To obtain a chemotactic condition medium, cardiomyocytes were exposed to 12 hours of hypoxia (<0.1% O2 5% CO2–95% N2) in serum‐free DMEM without glucose.
Animal Surgery and Cell Transplantation
The mice were anesthetized via intraperitoneal injection of 50 mg/kg sodium pentobarbital and ventilated with use of a volume‐regulated respirator. MI was produced through ligation of the left anterior descending coronary artery with a 10‐0 Prolene suture. Transplantation of CPCs and self‐assembling nanopeptide (Puramatrix™ [PM] kindly provided from 3‐D Matrix, Ltd) was performed as previously described.4 Briefly, 10 μL of 0.1% PM‐CPC mixture containing 2×104 cells was directly injected into the border zone of infarct myocardium 2 times just after ligation of the coronary artery. Subsequently, 20 μL of 0.5% PM‐CPC mixture containing 2×104 cells was disseminated onto the surface of infarct area. To deliver soluble JAM‐A protein into the heart tissue, 0.25 μg/μL recombinant mouse JAM‐A Fc chimera (JAM‐A Fc; R&D Systems) was mixed with PM and transplanted in the same way as the CPCs. The final concentration of JAM‐A Fc in PM was 25 μg/mL. To detect the transplanted CPCs, they were infected with red fluorescent protein (RFP)‐expressing retroviral vector as described previously.4
Cytokine Antibody Array
CPCs, AMCs, and SMs (1.0×106) were seeded onto 10‐cm dishes. After incubation for 12 hours in the appropriate medium supplemented with 10% FBS, cells were thoroughly washed with PBS 3 times and medium was changed to serum‐depleted DMEM. Isolated BMs (1.0×106) were seeded onto 10‐cm dishes with serum‐depleted Iscove's modified Dulbecco's medium. After incubation for 24 hours in serum‐depleted medium, the supernatant was collected as conditioned medium (CM) and the contaminating cells were removed via centrifuge. The released cytokine was measured by using Mouse Cytokine Antibody Array (RayBiotech Inc and R&D Systems). The expression level of each cytokines was quantified by using Gene Pix 4000B (Molecular Devices, Inc).
Neutrophils Migration Assay
Human umbilical vein endothelial cells (HUVECs) (6×105) were seeded onto 1% gelatin–coated transwell culture inserts with 3‐μm pores (Corning) and grown for 2 days in 5% CO2 at 37°C. Human polymorphonuclear leukocytes were isolated as previously described.11 Briefly, whole blood was obtained from healthy laboratory volunteers. Leukocytes were separated from whole blood through dextran sedimentation and lysis of contaminating erythrocytes. Neutrophils were further purified by using Ficoll (Sigma‐Aldrich) density gradient centrifugation. Isolated neutrophils were preincubated with either 10 μg/mL JAM‐A Fc, 5 mL of CPC CM, or 5 mL of CPC CM with 1 μg/mL neutralizing anti‐mouse JAM‐A antibody (R&D Systems) for 30 minutes, washed, and added to the upper chamber (5×105 cells in RPMI medium with 0.2% BSA). A chemotactic gradient was created by the addition of 40 ng/mL tumor necrosis factor‐α (TNFα; R&D Systems) or hypoxic cardiomyocytes CM to the lower chamber. After 3 hours of incubation, transmigrated cells in the lower chamber were counted by using a hematocytometer.
For the assay of the length of neutrophil tails, neutrophils were purified from bone marrow by using EASYSEP (BD Pharmingen) according to the manufacture's instruction. After preincubation with either 10 μg/mL JAM‐A Fc, CPC CM, or CPC CM with 1 μg/mL neutralizing anti‐mouse JAM‐A antibody for 30 minutes, neutrophils (0.4×106 cells) were seeded onto fibronectin‐coated (20 μg/mL in PBS) coverslips for 10 minutes at 37°C followed by additional incubation with 5 nmol/L WKYMVm (Merck Millipore) for 20 minutes at 37°C. Cells were fixed with 4% paraformaldehyde for 20 minutes at room temperature and permeabilized with 1% NP‐40 for 5 minutes. Actin was stained with rhodamine phalloidin (Life Technologies). Images were taken made with fluorescent microscopy (Axioskop 40; Carl Zeiss), the length of neutrophil tails was measured by using Adobe Photoshop software version 12.0 for Macintosh (Adobe Systems), and the frequency of cells with different tail size was examined.
Morphological Examination and Immunohistochemistry
The transversal sections (4 μm) from paraffin embedded formalin fixed tissue were subjected to Masson's trichrome staining. Fibrous infarct area was measured as described previously.4 The paraffin sections were stained with anti–von Willebrand factor (vWF) (DAKO), and the detection of antibodies was performed by using VECSTATIN ABC kit (Vector Laboratories Inc) according to the manufacturer's instruction. Nuclei were stained with hematoxylin. For fluorescent immunohistochemistry, fresh frozen sections were fixed with 4% paraformaldehyde, treated with 0.1% NP‐40 and 2% BSA in PBS, and stained with anti–Ly‐6G (BD Pharmingen) or anti‐myeloproxidase (MPO) (Abcam) antibody. For the detection of JAM‐A, CD31, and Sca‐1, fresh frozen sections were fixed with chilled acetone, treated with 10% donkey serum in PBS and stained with anti–JAM‐A (Hycult Biotech), anti‐CD31 (BD Pharmingen), or anti–Sca‐1 (Ly6A/E, BD Pharmingen) antibody. For the detection of RFP‐positive cardiomyocytes, the isolated hearts were perfused and fixed with PLP (periodate lysine paraformaldehyde) Solution Set (Wako) before being frozen and then costained with anti‐RFP antibody (MBL) and anti–sarcomeric α‐actinin antibody (Sigma‐Aldrich). Antibodies were detected appropriate fluorescent conjugated secondary antibodies (Life Technologies or Jackson Immunoresearch Laboratory). Nuclei were stained with Hochst33342 (Sigma‐Aldrich). Images were taken with the use of bright field and fluorescent microscopy (Axioskop 40; Carl Zeiss). JAM‐A and CD31 immunofluorescence in the border and infarct areas were analyzed by using Adobe Photoshop software version 12.0.
Picrosirius Red Staining and Collagen Matrix Quality
Paraffin sections were deparaffinized and stained by using the Picrosirius Red Stain Kit (Polysciences, Inc) according to the manufacturer's instructions. The images of myocardial scar area were obtained through microscopy (Axioskop 40) with a polarizer filter to quantify thick, tightly packed collagen fibers as yellow‐red and thin, loosely assembled, immature fibers as green.12 The images were analyzed with the use of ImageJ software (National Institutes of Health) based on the hue component of the images, and the numbers of red, orange, yellow, and green pixels were calculated.13
Echocardiography
Echocardiography in spontaneous breathing mice was performed by using a Vevo770 (Visual Sonics) with a 25‐MHz imaging transducer. Two‐dimensional images and M‐mode tracing were recorded from the parasternal long‐axis view at midpapillary level to determine left ventricular internal diastole diameter (LVIDD) and left ventricular internal systolic diameter (LVISD). LV fractional shortening (FS) was calculated as %FS=[(LVIDD−LVISD)/LVIDD]×100.
Dihydroethidium Fluorescence Analysis
The frozen sections (4 μm thick) were prepared and stained with 10 μmol/L dihydroethidium (DHE) in Krebs‐HEPES buffer composed of (in mmol/L) 99.01 NaCl, 4.69 KCl, 1.87 CaCl2, 1.20 MgSO4, 1.03 K2HPO4, 25.0 NaHCO3, 20.0 Na‐HEPES, and 11.1 glucose (pH 7.4) at 37°C for 30 minutes in a dark. The intensities of fluorescence were quantitatively analyzed with the use of Adobe Photoshop software version 12.0 for Macintosh.
Quantitative Real‐Time PCR
Total RNA extraction and DNase treatment were performed by using SV total RNA Isolation Kit (Promega). RNA was DNase treated and reverse transcribed by using QuantiTect Reverse Transcription Kit (Qiagen). Real‐time quantitative PCR was performed by using Universal Probe Library (UPL) (Roche Molecular Diagnostics) and Light Cycler TaqMan Master kit (Roche). Relative levels of gene expression were normalized to the mouse 28S rRNA expression by using the delta delta CT method according to the manufacturer's instructions and shown as fold increase to sham‐operated mouse. Primer sequences and UPL number were listed in Table 1.
Table 1.
Primer Sequences and Universal Probe Library UPL Number
| Forward | Reverse | UPL No. | |
|---|---|---|---|
| Mouse 28S rRNA | agcgacactcggacttgc | cgaaggactccacaggtttt | 109 |
| Ly6G | ttgtggtcctactgtgtgcag | ggtgggaccccaatacaat | 19 |
| Myeroperoxidase | ggaaggagacctagaggttgg | tagcacaggaaggccaatg | 7 |
| Cxcl1 | agactccagccacactccaa | tgacagcgcagctcattg | 83 |
| Cxcl2 | cagaaaatcatccaaaagatactgaa | ctttggttcttccgttgagg | 26 |
| Cxcl3 | ccccaggcttcagataatca | gggatggatcgcttttctc | 69 |
| Ccl3 | caagtcttctcagcgccata | ggaatcttccggctgtagg | 40 |
| Interleukin‐1β | tgtaatgaaagacggcacacc | tcttctttgggtattgcttgg | 78 |
| Interleukin‐6 | gctaccaaactggatataatcagga | ccaggtagctatggtactccagaa | 6 |
| Interleukin‐10 | cagagccacatgctcctaga | tgtccagctggtcctttgtt | 41 |
| TNFα | tcttctcattcctgcttgtgg | ggtctgggccatagaactga | 49 |
TNF indicates tumor necrosis factor; UPL, Universal Probe Library.
Knockdown of JAM‐A Gene
Stealth small interfering RNA (siRNA) against mouse JAM‐A was designed and purchased from Invitrogen. The sequence for siRNA was agugaaugaagaauucaugaaggcc. Negative controls for siRNA were purchased from Life Technologies. Transfection of siRNA was performed by using Lipofectamine RNAiMax (Life Technologies). Knockdown of JAM‐A gene and reduction of JAM‐A secretion were validated by using quantitative real‐time PCR and ELISA (R&D Systems), respectively (Figure 1).
Figure 1.
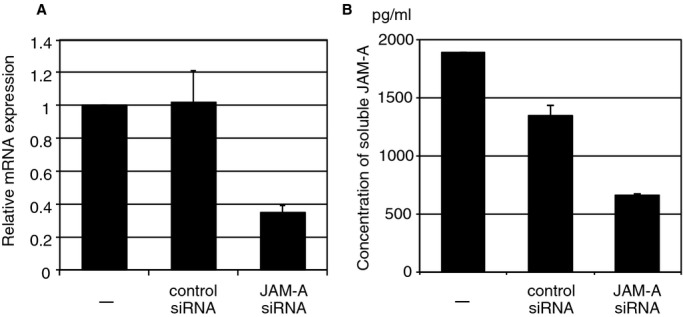
Validation of siRNA‐mediated knockdown of JAM‐A in CPC. A, Relative JAM‐A mRNA expression in JAM‐A siRNA–transfected CPC. RNA was collected from the cells 3 days after JAM‐A siRNA transfection (means±SE of duplicate samples). The results shown are representative of those from 3 independent experiments. B, Concentration of soluble JAM‐A in CPC CM. CM was collected from the cells 3 days after JAM‐A siRNA transfection (mean±SEM of duplicate samples). Nontransfected CPCs (−) and CPCs transfected with RNAi negative control were shown as control. CM indicates conditioned medium; CPC, cardiac progenitor cells; JAM‐A, junctional adhesion molecule‐A; siRNA, small interfering RNA.
Flow Cytometry
Cells were isolated from individual hearts as previously described. Briefly, the heart was rapidly removed, minced, and then digested for 20 minutes with prewarmed buffer containing 1% collagenase type 2 (Worthigton), 4.6 U/mL Dispase II (Life Technologies), and 2.4 mmol/L CaCl2. After the enzymatic reaction was stopped by adding 5 mL of cold PBS with 3% FBS, the cells were suspended in cold PBS with 3% FBS and then filtered through a 70‐μm cell strainer. Cells were stained at 4°C for 30 minutes simultaneously with 2 colors using the following antibodies: fluorescein PE‐Cy5–labeled rat anti‐mouse CD45, FITC‐labeled rat anti‐mouse Ly‐6G, and control rat IgG2b. All antibodies were purchased from eBioscience. Cell fluorescence was measured with EPICS ALTRA flow cytometer using EXPO32 software, version 1.2 (Beckman Coulter, Inc). Heart cell isolation and flow cytometry were performed on 3 mice per PM‐, CPC+PM–, or JAM‐A+PM–treated group.
Detection of Proliferative Endogenous CPCs and Cardiomyocytes
To label the proliferating cells, 50 mg/kg BrdU (Sigma‐Aldrich) was intraperitoneally injected into the mouse shortly after creating MI. The mice were treated with PM or JAM‐A+PM. Administration of BrdU was continued every 12 hours for 9 days. After 2 weeks, the hearts were excised, and paraformaldehyde‐fixed frozen sections were subjected to immunohistochemical analysis as described here earlier. For BrdU detection, 12% HCl was applied for 10 minutes before anti‐BrdU antibody (eBiosciences) was applied. For Nkx2.5 (Abcam) detection, Tyramide Signal Amplification Kits (Molecular Probe) were used according the manufacturer's instruction. The images were obtained with laser confocal microscopy (TCS‐SP5 Ver2.0; Leica Microsystems).
Statistical Analysis
The normality and variance of data were analyzed adjusted with a P value of 0.05 as the rejection criterion. Results are presented as mean±SEM. Statistical significance was calculated by using the unpaired Student t test for comparison between 2 groups or by 1‐way ANOVA–Tukey–Kramer post hoc test for multiple comparisons. To assess expression‐level changes in CD31 and JAM‐A over time, 2‐factor factorial ANOVA was used. In cases where the data were not normally distributed and/or the variances were not homogeneous, the significance was calculated by using Mann–Whitney U test for comparison between 2 groups or by the Kruskal–Wallis test, followed by the Steel–Dwass test for multiple comparisons. Statistical analysis was performed with the Microsoft Excel software program with the add‐in software Statcel3 (OMS, Japan). P<0.05 was considered statistically significant.
Results
JAM‐A Is Dominantly Secreted From CPCs
Previously, we have shown that the injection of CPCs reduces infarct size and restores cardiac function in comparison with the injection of BMs, SMs, and AMCs and that the beneficial effects are mediated not only by cardiomyogenesis but also by angiogenesis and antiapoptosis effects.4 This suggests that some of the humoral factors released from CPCs, but not from BMs, SMs, and AMCs, play an important role. When the quantity of secreted proteins from CPCs, BMs, SMs, and AMCs was measured by using a cytokine antibody array, CPCs secreted JAM‐A, VCAM‐1, and granulocyte‐colony stimulating factor more than twice as much as did BMs, SMs, and AMCs (Table 2; total dataset presented in Table 3).
Table 2.
Results of Cytokine Antibody Array
| CPCs to BMs | CPCs to AMCs | CPCs to SMs | ||||||
|---|---|---|---|---|---|---|---|---|
| Cytokine | Ratio of Net Intensity | Cytokine | Ratio of Net Intensity | Cytokine | Ratio of Net Intensity | |||
| Decorin | 289.94 | 243.61 | VCAM‐1 | 53.72 | 2.48 | JAM‐A | 16.06 | 25.04 |
| MCP‐1 | 32.02 | 18.98 | JAM‐A | 20.64 | 3.01 | VCAM‐1 | 10.37 | 3.40 |
| TWEAK R | 24.61 | 8.25 | GCSF | 2.61 | 7.49 | Pentraxin 3 | 8.34 | 3.04 |
| Growth arrest specific 1 | 16.68 | 20.56 | GCSF | 5.86 | 3.36 | |||
| MFG‐E8 | 13.57 | 27.83 | GITR ligand | 3.96 | 3.79 | |||
| TIMP‐1 | 11.63 | 9.96 | IL‐6R | 2.01 | 3.06 | |||
| VEGF | 11.43 | 13.87 | ||||||
| VCAM‐1 | 9.13 | 9.64 | ||||||
| JAM‐A | 6.62 | 10.99 | ||||||
| Galectin‐1 | 6.09 | 4.20 | ||||||
| GITR Ligand | 5.59 | 8.92 | ||||||
| GCSF | 4.94 | 2.59 | ||||||
| sTNF RI | 3.87 | 5.09 | ||||||
| Axl | 3.50 | 4.91 | ||||||
| RANTES | 2.55 | 3.62 | ||||||
| TACI | 2.47 | 9.46 | ||||||
| IGFBP‐2 | 2.15 | 2.25 | ||||||
| M‐CSF | 2.06 | 8.09 | ||||||
Each number indicates the ratio of normalized net fluorescent intensity of CPCs to BMs, AMCs, and SMs. The cytokines showing the ratio of normalized net fluorescent intensity more than 2.0 were presented. The results of 2 different samples were shown. AMC indicates adipose tissue‐derived mesenchymal cell; BM, bone marrow mononuclear cell; CPC indicates cardiac progenitor cell; GCSF, granulocyte‐colony stimulating factor; GITR, glucocorticoid‐induced tumor necrosis factor receptor family‐related gene; IGFBP‐2, insulin‐like growth factor binding protein 2; JAM‐A, junctional adhesion molecule‐A; MCP‐1, monocyte chemotactic protein‐1; M‐CSF, macrophage colony‐stimulating factor; MFG‐E8, milk fat globule–EGF factor 8 protein; RANTES, regulated upon activation normal T cell expressed and secreted; SMs, skeletal myoblasts; sTNF RI, soluble tumor necrosis factor receptor I; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor; TIMP1, tissue inhibitor of matrix metalloproteinase 1; TWEAK R, TNF‐related weak inducer of apoptosis receptor; VCAM‐1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor.
Table 3.
Total Dataset of Net Intensity Ratio of Mouse Cytokine Array
| CPCs vs BMs | CPCs vs BMs | ||
|---|---|---|---|
| Description | Net Intensity (Ratio) | Description | Net Intensity (Ratio) |
| Decorin | 243.6053593 | Decorin | 289.9391128 |
| MFG‐E8 | 27.83344467 | MCP‐1 | 32.01844262 |
| Growth arrest specific 1 | 20.55963322 | TWEAK R | 24.60629921 |
| MCP‐1 | 18.98397752 | Growth arrest specific 1 | 16.68112364 |
| ACE/CD143 | 14.92782397 | MFG‐E8 | 13.56520795 |
| VEGF | 13.87443635 | TIMP‐1 | 11.63453596 |
| JAM‐A | 10.98973559 | VEGF | 11.43053095 |
| Pentraxin 3 | 10.79074586 | VCAM‐1 | 9.132837116 |
| CD27 | 10.04580889 | JAM‐A | 6.618177486 |
| TIMP‐1 | 9.956688405 | Galectin‐1 | 6.091580826 |
| VCAM‐1 | 9.644875678 | GITR ligand | 5.588652799 |
| CD36 | 9.487486006 | GCSF | 4.944326879 |
| TACI | 9.458321905 | sTNF RI | 3.870478314 |
| RAGE | 8.942304253 | Axl | 3.504762973 |
| GITR ligand | 8.923870461 | RANTES | 2.551918788 |
| TWEAK R | 8.254911672 | TACI | 2.465841925 |
| M‐CSF | 8.089109632 | IGFBP‐2 | 2.150204699 |
| Prolactin | 7.795143626 | M‐CSF | 2.061830163 |
| CD40 ligand | 6.167091168 | ||
| IL‐17F | 5.970149254 | ||
| EGF | 5.686212073 | ||
| Cardiotrophin‐1 | 5.644615037 | ||
| IL‐20 | 5.562199294 | ||
| E‐cadherin | 5.515689378 | ||
| sTNF RI | 5.085538762 | ||
| Axl | 4.914922688 | ||
| IGFBP‐6 | 4.842310169 | ||
| Epiregulin | 4.580852038 | ||
| Neprilysin | 4.348620183 | ||
| IL‐17B | 4.34436948 | ||
| Amphiregulin | 4.212441868 | ||
| Galectin‐1 | 4.202969818 | ||
| Granzyme B | 3.777176787 | ||
| RANTES | 3.61856032 | ||
| IL‐17E | 3.389876473 | ||
| DKK‐1 | 3.306167656 | ||
| HGF | 3.228295363 | ||
| CXCL16 | 3.208676261 | ||
| IL‐1R4/ST2L | 3.172357347 | ||
| IL‐21 | 3.132733937 | ||
| IL‐28 | 3.116993224 | ||
| VEGF R1 | 2.977360153 | ||
| Endoglin | 2.888804151 | ||
| MAdCAM‐1 | 2.745585784 | ||
| 4‐1BB | 2.589278316 | ||
| GCSF | 2.585622902 | ||
| HAI‐1 | 2.500606397 | ||
| MIP‐3α | 2.461647531 | ||
| IL‐11 | 2.437425202 | ||
| IGFBP‐2 | 2.24696379 | ||
| Epigen | 2.129707719 | ||
| CPCs vs AMCs | CPCs vs AMCs | ||
|---|---|---|---|
| Description | Net Intensity (Ratio) | Description | Net Intensity (Ratio) |
| VCAM‐1 | 53.72011818 | GCSF | 7.488729462 |
| MIP‐1γ | 31.50797152 | JAM‐A | 3.012311316 |
| Growth arrest specific 1 | 23.48354977 | VCAM‐1 | 2.479888107 |
| MFG‐E8 | 20.97623393 | ||
| JAM‐A | 20.63855695 | ||
| sTNF RI | 16.48886177 | ||
| MCP1 | 15.4038109 | ||
| IL‐1ra/IL‐1F3 | 13.60100103 | ||
| ACE/CD143 | 12.95588521 | ||
| sTNF RII | 10.13880017 | ||
| CD36 | 10.1361282 | ||
| RAGE | 9.864462288 | ||
| Decorin | 9.685230024 | ||
| TACI | 9.212514279 | ||
| CD27 | 8.337224038 | ||
| Galectin‐1 | 7.900391859 | ||
| IL‐6 R | 7.464636286 | ||
| RANTES | 7.417572229 | ||
| GITR ligand | 7.180967564 | ||
| CD40 ligand | 7.065490027 | ||
| EGF | 7.050843633 | ||
| Prolactin | 6.247813265 | ||
| E‐cadherin | 6.179018525 | ||
| IL‐17F | 5.921714929 | ||
| IL‐20 | 5.60950025 | ||
| M‐CSF | 5.414243793 | ||
| Amphiregulin | 5.270897792 | ||
| CXCL16 | 5.184086927 | ||
| Granzyme B | 5.071045345 | ||
| Pentraxin 3 | 5.070505377 | ||
| Epiregulin | 4.833533119 | ||
| Neprilysin | 4.570634587 | ||
| IGFBP‐6 | 4.431956177 | ||
| VEGF | 4.394773735 | ||
| HGF | 4.286271501 | ||
| Axl | 4.18075931 | ||
| Cardiotrophin‐1 | 4.141815772 | ||
| 4‐1BB | 3.949291102 | ||
| IL‐21 | 3.563207742 | ||
| IL‐28 | 3.32499867 | ||
| VEGF R1 | 3.242331885 | ||
| TWEAK R | 3.223508402 | ||
| IL‐17B | 3.208120394 | ||
| MAdCAM‐1 | 3.103286691 | ||
| MIP‐3α | 2.828550255 | ||
| DKK‐1 | 2.756529529 | ||
| HAI‐1 | 2.685493768 | ||
| Osteopontin | 2.617437895 | ||
| GCSF | 2.610679769 | ||
| Pro‐MMP‐9 | 2.448897629 | ||
| Endoglin | 2.430818894 | ||
| IL‐11 | 2.400147849 | ||
| Epigen | 2.191636714 | ||
| IL‐17E | 2.168468302 | ||
| CPCs vs SMs | CPCs vs SMs | ||
|---|---|---|---|
| Description | Net Intensity (Ratio) | Description | Net Intensity (Ratio) |
| JAM‐A | 25.03567584 | JAM‐A | 16.05961328 |
| ACE/CD143 | 5.537006584 | VCAM‐1 | 10.36924896 |
| TACI | 4.99797582 | Pentraxin 3 | 8.343484573 |
| RAGE | 4.813246053 | GCSF | 5.857819017 |
| Endoglin | 4.256206613 | GITR Ligand | 3.964431124 |
| EGF | 3.917390078 | Growth arrest specific 1 | 2.759427584 |
| Epiregulin | 3.789572612 | Axl | 2.324024839 |
| GITR ligand | 3.788553265 | sTNF RII | 2.221955588 |
| Prolactin | 3.669563175 | IL‐6 R | 2.006175007 |
| Cardiotrophin‐1 | 3.573892004 | ||
| IL‐17F | 3.565214911 | ||
| IL‐20 | 3.46275975 | ||
| VCAM‐1 | 3.402772579 | ||
| HAI‐1 | 3.358104954 | ||
| GCSF | 3.357811781 | ||
| 4‐1BB | 3.324335465 | ||
| Granzyme B | 3.309526804 | ||
| CD36 | 3.282045896 | ||
| E‐cadherin | 3.237545164 | ||
| IL‐1ra/IL‐1F3 | 3.171703332 | ||
| IL‐6R | 3.060780989 | ||
| Neprilysin | 3.05722514 | ||
| Pentraxin 3 | 3.038839406 | ||
| CD27 | 3.02893542 | ||
| CD40 ligand | 3.004311187 | ||
| DKK‐1 | 2.978885658 | ||
| Amphiregulin | 2.925139822 | ||
| IL‐17B | 2.848207623 | ||
| MIP‐3α | 2.716623563 | ||
| IL‐17E | 2.588226159 | ||
| TWEAK | 2.344572432 | ||
| IL‐28 | 2.235206287 | ||
| IL‐21 | 2.211239732 | ||
| MAdCAM‐1 | 2.194450235 | ||
| IL‐11 | 2.159332853 | ||
| IL‐1R4/ST2L | 2.040633086 | ||
| Epigen | 2.003514164 | ||
ACE indicates angiotensin converting enzyme; BM, bone marrow mononuclear cell; CPC; cardiac progenitor cell; DKK‐1, Dickkopf‐related protein 1; EGF, epidermal growth factor; GCSF, granulocyte‐colony stimulating factor; GITR, glucocorticoid‐induced tumor necrosis factor receptor family‐related gene; HAI‐1, hepatocyte growth factor activator inhibitor type1; HGF, hepatocyte growth factor; IGFBP‐2, insulin‐like growth factor binding protein 2; IL, interleukin; MCP‐1, monocyte chemotactic protein‐1; M‐CSF, macrophage colony‐stimulating factor; MFG‐E8, milk fat globule–EGF factor 8 protein; MIP, Macrophage inflammatory protein; RAGE, receptor for advanced glycation end products; RANTES, regulated upon activation normal T cell expressed and secreted; sTNF RI, soluble tumor necrosis factor receptor I; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor; TIMP1, tissue inhibitor of matrix metalloproteinase 1; TWEAK R, TNF‐related weak inducer of apoptosis receptor; VCAM‐1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor. The results of two different samples were shown.
Soluble JAM‐A Protein and CPC Conditioned Medium Prevent Transendothelial Migration of Neutrophils
Recently, it was reported that JAM‐A is constitutively released from endothelial cells and epithelial cells via proteolytic shedding and inhibits neutorophil migration.6 Therefore, we examined whether CPC CM inhibits migration of neutrophils through HUVEC‐coated transwell filters toward the chemoattractants. TNFα induced a significant increase in the number of transmigrated neutrophils in comparison with control (control, 6.7±0.33; TNFα, 15±1.8: Figure 2A). Preincubation of neutrophils with JAM‐A Fc (4.7±0.67) or CPC CM (3.3±0.88) significantly inhibited transendothelial migration toward TNFα (Figure 2A). It has been reported that cardiomyocytes release neutrophil chemoattractant such as interleukin (IL)‐8 under hypoxia.14 Hypoxia‐exposed cardiomyocytes CM induced a significant increase in the number of transmigrated neutrophils in comparison with control (control, 7.7±0.88; hypoxia CM, 27±1.2: Figure 2B). Preincubation of neutrophils with JAM‐A Fc (6.0±0.58) or CPC CM (7.0±1.2) significantly inhibited transendothelial migration stimulated by hypoxia‐exposed cardiomyocyte CM (Figure 2B). When CPC CM was pretreated with neutralizing anti–JAM‐A antibody, the inhibitory effect of CPC CM on neutrophil migration under the stimulation with TNFα and hypoxia‐exposed cardiomyocyte CM was significantly attenuated (TNFα, 8.7±0.33; Figure 2A: hypoxia CM, 13±0.33; Figure 2B). This suggests that soluble form of JAM‐A released from CPCs may play an important role in prevention of inflammatory response in ischemic myocardium through the inhibition of the extravasation of neutrophils.
Figure 2.
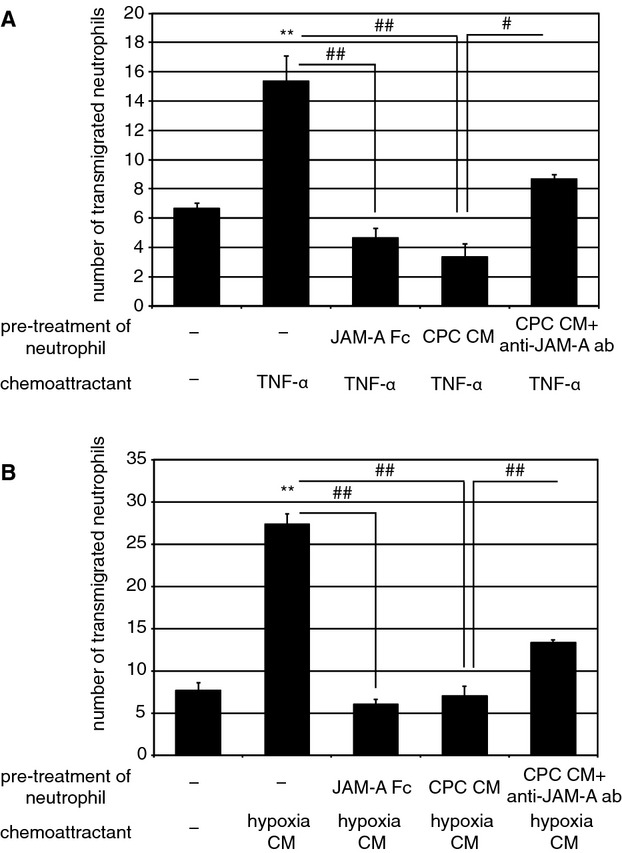
Soluble JAM‐A and CPC CM inhibit transendotherial migration of neutrophils. Neutrophils were pretreated at 37°C for 10 minutes with JAM‐A Fc (10 μg/mL), CPC CM, or CPC CM with anti–JAM‐A antibody (1 μg/mL). Neutrophils were allowed to migrate across HUVECs in response to TNFα (A) or hypoxia‐exposed cardiomyocytes CM (B) for 3 hours. Mean±SEM of 3 experiments. For statistical analysis, 1‐way ANOVA–Tukey–Kramer post hoc test was performed. Significant differences between control and stimulated groups are shown by asterisks (**P<0.01). Additional statistical comparisons are indicated by lines and number signs (##P<0.01). CM indicates conditioned medium; CPC, cardiac progenitor cell; HUVEC, human umbilical vein endothelial cell; JAM‐A, junctional adhesion molecule‐A; TNF, tumor necrosis factor.
Injection of CPCs Prevents Neutrophil Accumulation Through Secreted JAM‐A
Next, we examined whether the injection of CPCs reduces accumulation of neutrophils after MI. Immunohistochemical images of the injured myocardium 1 day after MI revealed that fewer Ly6G‐positive neutrophils were observed in CPC+PM–treated mice in comparison with PM‐treated mice (Figure 3A). The number of Ly6G‐positive neutrophils in the infarct area of CPC+PM–treated mice was significantly lower than that of PM‐treated mice (PM: 378±29/mm2; CPC+PM: 202±16/mm2, P<0.01. Figure 3B). Flow cytometric analysis revealed that percentages of CD45+Ly6G+ neutrophils of total live cells were decreased in CPC+PM–treated mice (23.0%) in comparison with PM‐treated mice (42.6%) at 1 day after MI (Figure 3C).
Figure 3.
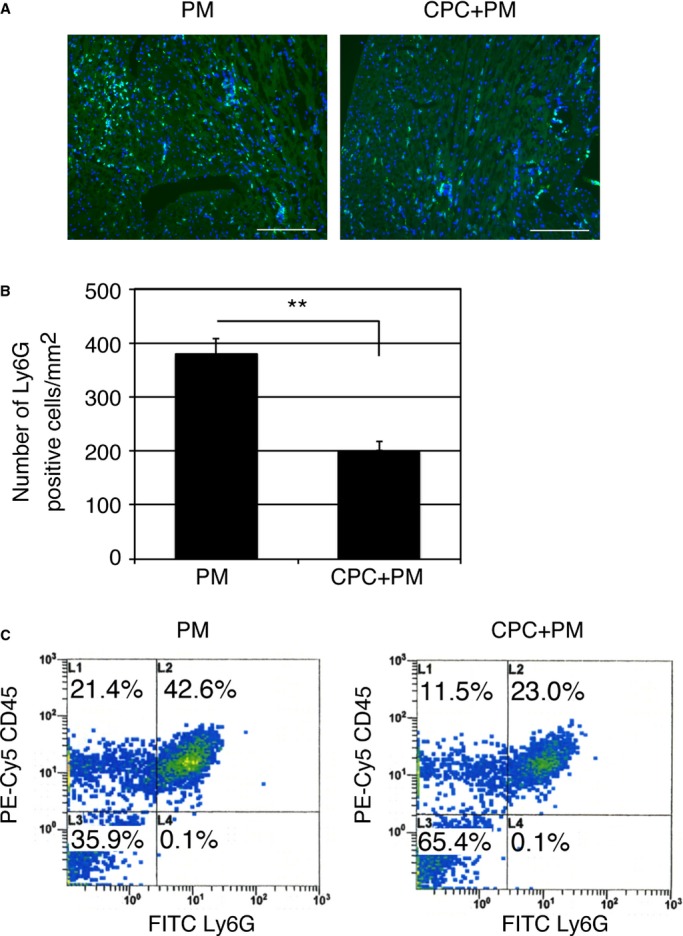
Injection of CPCs prevents neutrophil accumulation in ischemic myocardium. A, Immunohistochemical images of infarct area stained with anti‐Ly6G antibody on 1 day after MI. Mouse was treated with PM or CPC+PM. Ly6G and nuclei are shown in green and blue, respectively. Morphology of tissue is shown in nonspecific green fluorescence. Bars were 0.2 mm. B, Quantification of neutrophil accumulation in the infarct area of PM‐ and CPC+PM–treated mice. To obtain the number of Ly6G‐positive neutrophils/mm2 in infarct area, 2 heart sections at papillary muscle level were examined per mouse. An average of values obtained from 3 mice for each group was presented. Asterisks denote statistical significant differences between PM‐ and CPC+PM–treated groups after Student t test (**P<0.01). C, Representative dot plots from PM‐ or CPC+PM–treated MI hearts. Cell suspensions from PM‐ or CPC+PM–treated MI hearts were stained with anti–Ly‐6G and anti‐CD45 antibodies. Dot plots from a typical experiment of 2 performed are shown. CPC indicates cardiac progenitor cell; FITC, fluorescein isothiocyanate; MI, myocardial infarction.
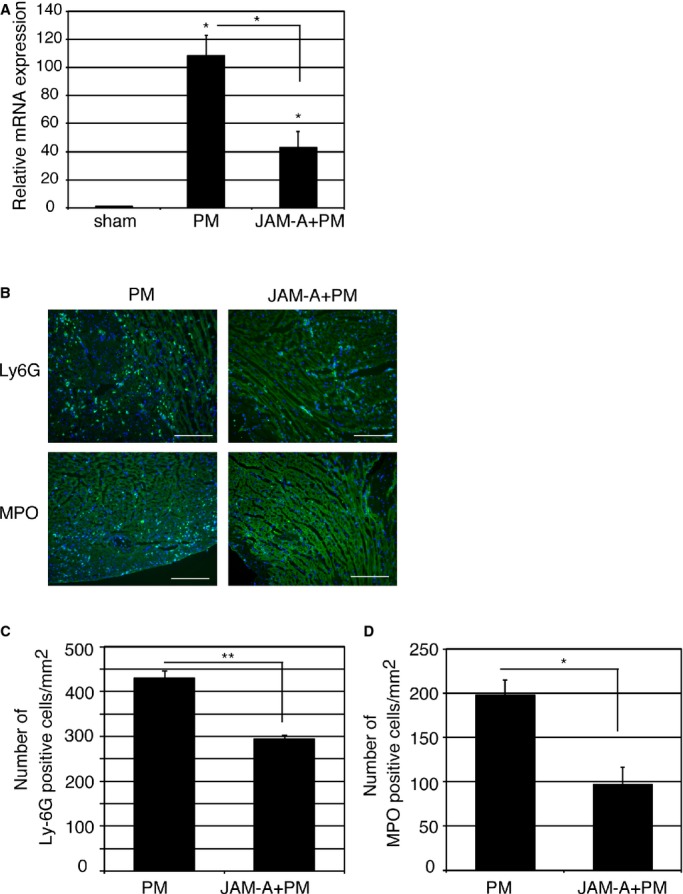
We next examined whether CPC‐derived JAM‐A are involved in the prevention of neutrophil accumulation by RNA interference by using siRNA. We confirmed that siRNA selectively reduced the amount of JAM‐A mRNA and secreted soluble JAM‐A protein in CPCs (Figure 1A and 1B). When the expression levels of Ly6G gene was examined at 1 day after injection of PMs, CPCs+PMs, or CPCs transfected with JAM‐A siRNA (JAM‐AsiCPC)+PMs to MI heart, the expression levels of Ly6G were significantly reduced in CPC+PM–treated group in comparison with PM‐treated group. The suppressive effect on Ly6G gene expression in the CPC+PM–treated group was abrogated when CPCs transfected with JAM‐AsiCPC+PM was injected into MI hearts (PM: 198±54.1; CPC+PM: 49.6±9.27; JAM‐AsiCPC+PM: 164±30.2, PM versus CPC+PM; P<0.05, CPC+PM versus JAM‐AsiCPC+PM; P<0.05) (Figure 4A). Immunohistochemical images of the injured myocardium 1 day after MI showed that accumulation of Ly6G‐positive neutrophils in PM‐treated mice were prevented in CPC+PM–treated mice but not in JAM‐AsiCPC+PM–treated mice (Figure 4B, upper row). Fewer MPO‐positive neutrophils were observed in CPC+PM–treated mice in comparison with PM‐treated mice (Figure 4B, lower row). The reductive effect of CPC injection on the accumulation of MPO‐positive neutrophils was attenuated in JAM‐AsiCPC+PM–treated mice (Figure 4B, lower row). The number of Ly6G positive neutrophils in the infarct area was 474±31/mm2 in PM‐treated, 284±33/mm2 in CPC+PM–treated, and 446±20/mm2 in JAM‐AsiCPC+PM–treated mice (PM versus CPC+PM; P<0.01, CPC+PM versus JAM‐AsiCPC+PM; P<0.05; Figure 4C). The number of MPO‐positive neutrophils in the infarct area was 231±30/mm2 in PM‐treated, 97.7±8.7/mm2 in CPC+PM–treated, and 197±6.4/mm2 in JAM‐AsiCPC+PM–treated mice (PM versus CPC+PM; P<0.01, CPC+PM versus JAM‐AsiCPC+PM; P<0.01; Figure 4D).
Figure 4.
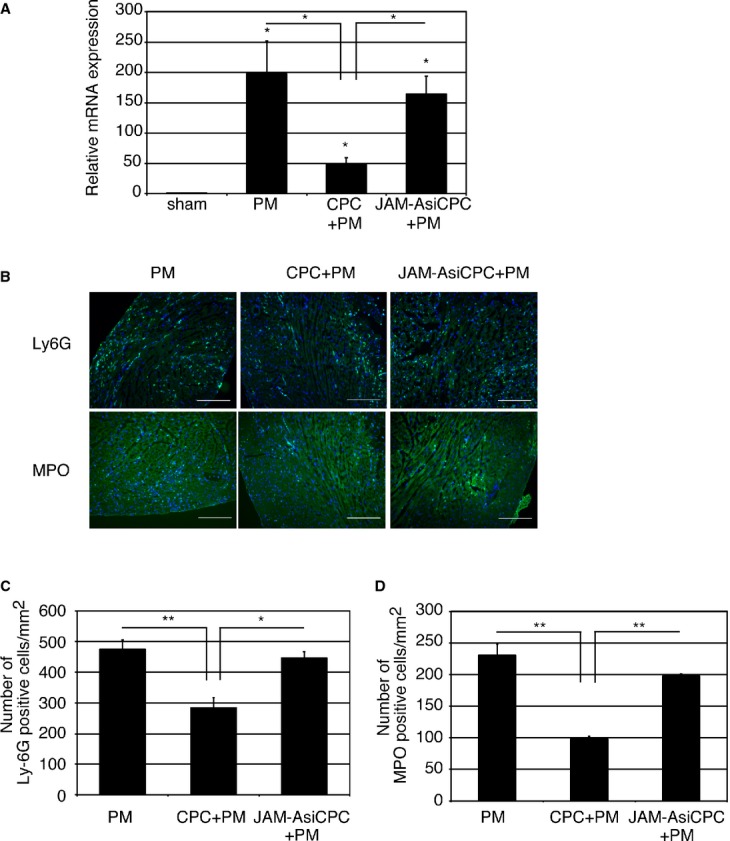
Knockdown of JAM‐A attenuates CPC‐mediated inhibitory effect on neutrophil accumulation. A, Transplantation of CPCs (CPC+PM) but not CPCs transfected with JAM‐A siRNA (JAM‐AsiCPC+PM) reduced relative expression level of Ly6G mRNA. RNA was extracted from left ventricle at 1 day after MI. Expression level was shown as fold changes relative to sham operated mouse using delta delta CT method: sham, n=6; PM, n=7; CPC+PM, n=6; JAM‐AsiCPC+PM, n=5. The Kruskal–Wallis test, followed by the Steel–Dwass test was used for statistical analysis. Asterisks above individual columns indicate significant difference compared with sham. Asterisks above a line spanning 2 columns indicate significant difference between 2 groups (*P<0.05). B, Immunohistochemical images of infarct area stained with anti‐Ly6G and anti‐MPO antibodies at 1 day after MI. Mouse was treated with PM, CPC+PM, or JAM‐AsiCPC+PM. Ly6G and MPO are shown in green and nuclei in blue. Morphology of tissue is shown in nonspecific green fluorescence. Bars were 0.2 mm. C and D, Quantification of neutrophil accumulation in the infarct area of PM‐, CPC+PM–, and JAM‐AsiCPC+PM–treated mice. To obtain the number of Ly6G (C) and that of MPO (D) ‐positive neutrophils/mm2 in infarct area, 2 heart sections at papillary muscle level were examined per mouse. An average of values obtained from 3 mice for each group was presented. One‐way ANOVA–Tukey–Kramer post hoc test was used for statistical analysis. Asterisks indicate statistical significant differences (**P<0.01 and *P<0.05). CPC indicates cardiac progenitor cell; JAM‐A, junctional adhesion molecule‐A; MI, myocardial infarction; MPO, myeloproxidase; siRNA, small interfering RNA.
We then explored whether the injection of soluble JAM‐A protein can inhibit accumulation of neutrophils in MI hearts. When a mixture of recombinant mouse JAM‐A Fc protein and PM was injected into MI hearts, the expression levels of Ly6G were significantly reduced in JAM‐A+PM–treated mice in comparison with PM‐treated mice at 1 day after injection (PM: 108±15; JAM‐A+PM: 43.0±12, P<0.05; Figure 5A). Immunohistochemical images of the injured myocardium 1 day after MI revealed that fewer Ly6G‐positive neutrophils were observed in JAM‐A+PM–treated mice in comparison with PM‐treated mice (Figure 5B, upper row) and that fewer MPO‐positive neutrophils were observed in JAM‐A+PM–treated mice in comparison with PM‐treated mice (Figure 5B, lower row). The number of Ly6G‐positive neutrophils in the infarct area was 430±15/mm2 in PM‐treated and 293±9.2/mm2 in JAM‐A+PM–treated mice (PM versus JAM‐A+PM; P<0.01; Figure 5C). The number of MPO‐positive neutrophils in the infarct area was 197±18/mm2 in PM‐treated and 96.8±20/mm2 in JAM‐A+PM–treated mice (PM versus JAM‐A+PM; P<0.01; Figure 5D). Flow cytometric analysis revealed that percentages of CD45+Ly6G+ neutrophils of total live cells decreased in JAM‐A+PM–treated mice (13.0%) in comparison with PM‐treated mice (23.6%) at 1 day after MI (Figure 6). These results suggest that JAM‐A plays an important role in the prevention of neutrophil accumulation after MI as one of the CPC‐derived paracrine factors.
Figure 5.
Injection of JAM‐A Fc protein prevents neutrophil accumulation in ischemic myocardium. A, Transplantation of JAM‐A Fc protein (JAM‐A+PM) reduced relative expression level of Ly6G mRNA. RNA was extracted from left ventricle of day 1 MI heart. Expression level was shown as fold changes relative to sham operated mouse using delta delta CT method: sham, n=4; PM, n=5; JAM‐A+PM, n=5. The Kruskal–Wallis test, followed by the Steel–Dwass test, was used for statistical analysis. Asterisks above individual columns indicate significant difference compared with sham. Asterisks above a line spanning 2 columns indicate significant difference between 2 groups (*P<0.05). B, Immunohistochemical images of infarct area stained with anti‐Ly6G and anti‐MPO antibodies at 1 day after MI. Mouse was treated with PM or JAM‐A+PM. Ly6G and MPO are shown in green and nuclei in blue. Morphology of tissue is shown in nonspecific green fluorescence. Bars were 0.2 mm. C and D, To obtain the number of Ly6G (C) and that of MPO (D) ‐positive neutrophils/mm2 in infarct area, 2 heart sections at papillary muscle level were examined per mouse. An average of values obtained from 3 mice for each group was presented. Student t test was used for statistical analysis. Asterisks indicate statistical significant differences (**P<0.01 and *P<0.05). JAM‐A, junctional adhesion molecule‐A; MI, myocardial infarction; MPO, myeloproxidase.
Figure 6.
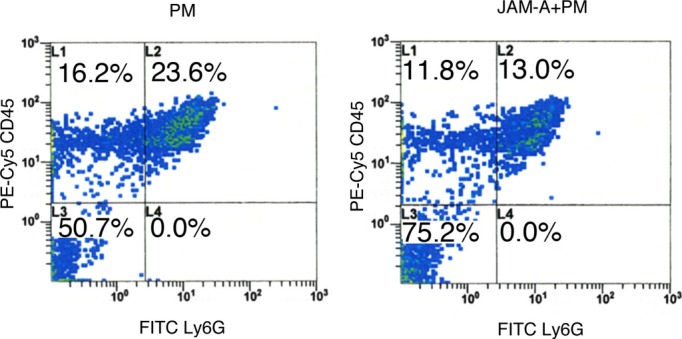
Injection of JAM‐A+PM attenuates myocardial neutrophil infiltration after MI. Representative dot plots from PM‐ or JAM‐A+PM–treated MI hearts. Cell suspensions from PM‐ or JAM‐A+PM–treated MI hearts were stained with anti–Ly‐6G and anti‐CD45 antibodies. Dot plots from a typical experiment of 2 performed are shown. FITC indicates fluorescein isothiocyanate; JAM‐A, junctional adhesion molecule‐A; MI, myocardial infarction.
Soluble JAM‐A Mediates the Reduction of Infarct Size and Prevention of Left Ventricular Remodeling and Enhances Capillary Density
Next, we examined whether the beneficial effect of CPC+PM on the infarct size was mediated by JAM‐A. When a mixture of JAM‐A Fc protein and PM was injected into the infarct area, Masson's trichrome–stained myocardial images at 2 weeks after injection revealed that the infarct area of the heart treated with JAM‐A+PM was smaller than that of PM‐treated heart (Figure 7A, PM and JAM‐A+PM). Average infarct area was 40.4±3.0% in PM‐treated (n=15) and 25.0±3.3% in JAM‐A‐Fc–treated (n=17) hearts (PM versus JAM‐A+PM; P<0.01; Figure 7B). When JAM‐AsiCPC+PM was injected into the infarct area, Masson's trichrome–stained myocardial images at 2 weeks after injection revealed that the infarct area of JAM‐AsiCPC+PM–treated heart was larger than that of CPC+PM–treated heart (Figure 7A, CPC+PM and JAM‐AsiCPC+PM). Average infarct area was 26.1±2.1% in CPC+PM–treated (n=16) and 37.7±3.3% in JAM‐AsiCPC+PM–treated (n=15) hearts (CPC+PM versus JAM‐AsiCPC+PM; P<0.05; Figure 7B). Echocardiographic measurement of hearts at 2 weeks after transplantation revealed that both LVIDD and LVISD of JAM‐A+PM– and CPC+PM–treated hearts were significantly smaller than those of the PM‐treated group. LVISD of CPC+PM–treated hearts was significantly smaller than that of JAMAsiCPC+PM–treated group. LVIDD of CPC+PM–treated hearts tends to be smaller than that of JAMAsiCPC+PM–treated group (Table 4). These reductions were associated with significantly greater value of percent FS of JAM‐A+PM– and CPC+PM–treated hearts in comparison with that of the other 2 groups (Table 4). These findings suggest that JAM‐A is one of the paracrine factors released from CPCs that plays an important role in attenuating cardiac remodeling and dysfunction at 2 weeks after MI through the prevention of neutrophils accumulation.
Figure 7.
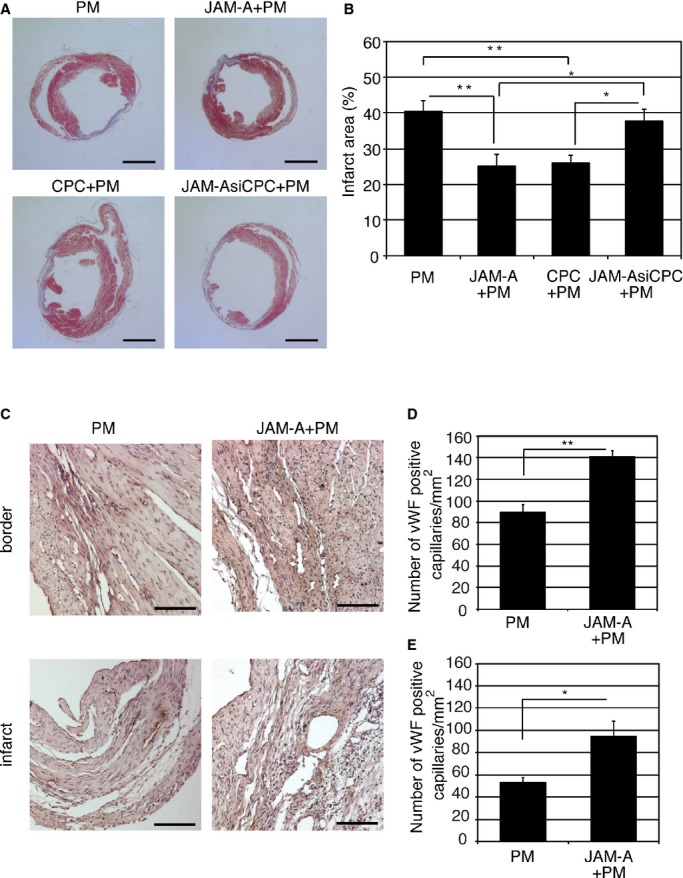
Soluble JAM‐A mediates the reduction of infarct size and prevention of left ventricular remodeling, and enhances capillary density. A, Representative Masson's trichrome–stained myocardial sections from PM‐treated, JAM‐A+PM–treated, CPC+PM–treated, and JAM‐AsiCPC+PM–treated hearts. Bars are 2.5 mm. B, Quantification of infarct size 2 weeks after transplantation. For statistical analysis, 1‐way ANOVA–Tukey–Kramer post hoc test was performed. Significant differences among groups are shown by asterisks (**P<0.01 and *P<0.05). C, Representative images from the sections of PM‐treated and JAM‐A+PM–treated hearts stained for capillaries with anti‐vWF antibodies. Bars are 100 μm. D and E, Quantification of the number of vWF‐positive capillaries in border (D) and infarct (E) area 2 weeks after transplantation. To obtain the number of vWF‐positive capillaries/mm2 in infarct area and border area, 2 heart sections at papillary muscle level were examined per mouse. An average of values obtained from 3 mice for each group was presented. Student t test was used for statistical analysis. Asterisks indicate statistical significant differences (**P<0.01 and *P<0.05). JAM‐A indicates junctional adhesion molecule‐A; vWF, von Willebrand factor.
Table 4.
Echocardiographic Measurement of Hearts 2 Weeks After Transplantation
| Parameter | n | HR, beats/min | SWT, mm | PWT, mm | LVIDD, mm | LVISD, mm | %FS |
|---|---|---|---|---|---|---|---|
| PM | 9 | 672±13 | 0.27±0.05 | 0.30±0.04 | 6.2±0.20 | 5.8±0.26 | 6.9±1.3 |
| JAM‐A+PM | 11 | 711±11 | 0.43±0.05‡‡ | 0.52±0.05**,‡‡ | 4.9±0.32* | 4.2±0.37* | 15±2.0*,‡ |
| CPC+PM | 9 | 683±20 | 0.43±0.08‡ | 0.57±0.04**,‡‡ | 4.9±0.24** | 4.1±0.27**,‡ | 15±2.4*,‡ |
| JAMAsiCPC+PM | 7 | 708±11 | 0.15±0.02 | 0.22±0.05 | 6.2±0.34 | 5.8±0.40 | 6.9±1.4 |
Statistical analysis was performed by 1‐way ANOVA–Tukey–Kramer post hoc test (HR, PWT, and %FS) or Kruskal–Wallis test, followed by Steel–Dwass test (SWT, LVIDD, and LVISD). CPC indicates cardiac progenitor cell; FS, fractional shortening; HR, heart rate; JAMA, junctional adhesion molecule‐A; LVIDD, left ventricular internal diastolic diameter; LVISD, left ventricular internal systolic diameter; PWT, posterior wall thickness; siCPC, CPC transfected with JAM‐A silent interfering RNA; SWT, septal wall thickness.
P<0.05 vs PM, **P<0.01 vs PM, †P<0.05 vs JAM‐AsiCPC+PM, ††P<0.01 vs JAM‐AsiCPC+PM.
To examine the effect of JAM‐A on neovascularization in post‐MI hearts, we examined the number of vWF‐positive capillaries in the ischemic area of PM‐ and JAM‐A+PM–treated hearts. At 2 weeks after transplantation, immunohistochemical staining for vWF indicated that more vWF‐positive capillaries exist in the border and infarct areas of the JAM‐A+PM–treated heart in comparison with the PM‐treated heart (Figure 7C). The number of vWF‐positive capillaries was 90.0±12/mm2 in PM‐treated heart (n=3) and 141±9.7/mm2 in JAM‐A+PM–treated (n=3) hearts in the border area (P<0.01, Figure 7D). In the infarct area, the number of vWF‐positive capillaries was 53.1±4.2/mm2 in PM‐treated heart (n=3) and 94.7±24/mm2 in JAM‐A+PM–treated (n=3) hearts in the border area (P<0.05; Figure 7E).
It has been reported that changes in fibroblastic properties during healing impact scar formation and that accumulation of the different collagens is connected with the impaired contractile function.15 We measured qualitative changes in collagen fibers by staining myocardial sections with picrosirius red at 2 weeks after MI. Sirius red polarization microscopy revealed that the treatment with JAM‐A+PM decreased the ratio of green to yellow‐orange fibers; however, the difference did not reach the statistical significant level against the other 3 groups (Figure 8).
Figure 8.
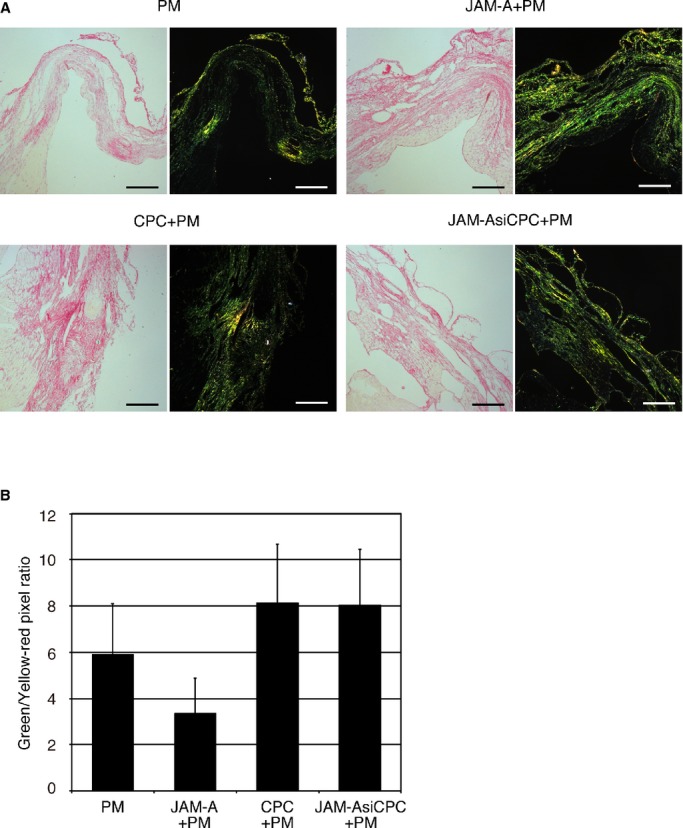
JAM‐A does not affect scar maturation in infarcted myocardium. A, Representative Picro–Sirius red staining and polarization microscopy in PM‐treated, JAM‐A+PM–treated, CPC+PM–treated, and JAM‐AsiCPC+PM–treated hearts. Bars are 200 μm. B, Quantitative analysis of color component in infarct area. Vertical axis indicates the ratio of pixel number of green to that of yellow‐red. A heart section at the level of the largest scar size was examined per mouse. An average of values obtained from 3 mice for each group was presented. For statistical analysis, 1‐way ANOVA–Tukey–Kramer post hoc test was performed. CPC indicates cardiac progenitor cell; JAM‐A, junctional adhesion molecule‐A.
Injection of CPC Prevents Reactive Oxygen Species Production and Early Inflammatory Response
Neutrophils have been implicated as a primary mechanisms underlying ischemic injury.16 Reactive oxygen species (ROS) production is one of the major processes that is involved in inducing tissue injury by neutrophils.17–18 To examine whether prevention of neutrophil accumulation by CPC+PM injection reduces ROS production, measurement of superoxide in myocardium submitted to MI was performed by using DHE. DHE fluorescence staining demonstrated that in the MI area, the DHE fluorescence intensity of the CPC+PM–treated group was lower than that of the PM‐treated group (Figure 9A, PM and CPC+PM). The result of quantitative analysis was shown in Figure 9B (PM: 2.68±0.46; CPC+PM: 2.14±0.20, P<0.05).
Figure 9.
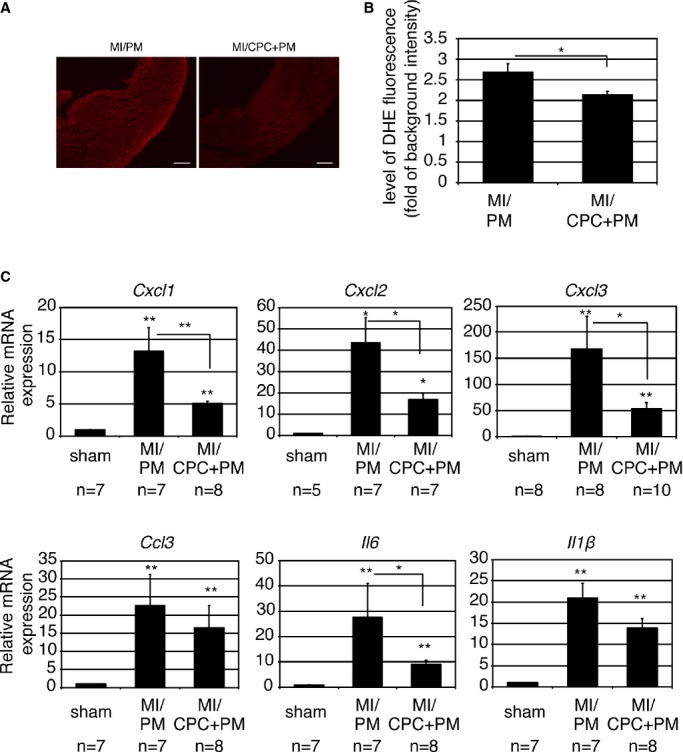
CPC transplantation attenuates ROS production and expression of various proinflammatory cytokines. A, Representative images from PM‐treated and CPC+PM–treated hearts. At 1 day after MI, the heart sections were stained with dihydroethidium. Bars are 2 mm. B, Fluorescence intensity at infarct area. A heart section at the level of the largest infarct size was examined per mouse. An average of values obtained from 5 mice for each group was presented. For statistical analysis, Mann–Whitney U test was performed. Statistically significant differences between the 2 groups are shown by asterisks (*P<0.05). C, Gene expression of various proinflammatory cytokines at 1 day after MI. Expression level was shown as fold changes relative to sham operated mouse using delta delta CT method. The number of mice examined was indicated below each graph. The Kruskal–Wallis test, followed by the Steel–Dwass test, was used for statistical analysis. Asterisks above individual columns indicate significant difference compared with sham. Asterisks above a line spanning 2 columns indicate significant difference between 2 groups (**P<0.01 and *P<0.05). CPC indicates cardiac progenitor cell; DHE, dihydroethidium; MI, myocardial infarction; ROS, reactive oxygen species.
A variety of stimuli, including ROS generation, potentially stimulate induction of chemokines and proinflammatory cytokines. Several inducible chemokines have been reported to stimulate neutrophil chemotaxis and activation in the ischemic heart.19 Induction and release of the proinflammatory cytokines, such as IL‐6, IL‐1β, and TNFα, have been demonstrated in the infarcted myocardium.20 The results of quantitative real‐time PCR revealed that expression of neutrophil chemoattractants and proinflammatory cytokines was upregulated at 24 hours after MI with the injection of PM; however, expression levels of Cxcl1, Cxcl2, Cxcl3, and Il6 were significantly reduced in CPC+PM–treated MI hearts (Figure 9C). Expression levels of Ccl3 and Il1β also tended to be reduced in CPC+PM–treated MI hearts (Figure 9C). There was no significant difference in expression level of Tnf‐α between PM‐ and CPC+PM–treated MI hearts (data not shown). The expression levels of the examined genes were decreased at 72 hours after MI, and no significant difference was observed between PM‐treated and CPC+PM–treated hearts (data not shown).
CPC‐Derived Soluble JAM‐A Reduces Neutrophil Motility
After adhesion and diapedesis through endothelial cells, emigrated neutrophils infiltrate the myocardial tissues toward neutrophil chemotactic factors. Therefore, we next examined whether CPC‐derived soluble JAM‐A reduces the motility of activated neutrophils. In response to the chemotactic factors, neutrophils attach to the substratum and polarize, leading to the establishment of a distinct leading edge (pseudopod) and tail (uropod).21 In order to maintain motility, migrating neutrophils must break adhesive contacts at the uropod and establish new contacts at the pseudopod. When cell migration is impaired, the uropod remained tightly anchored to the substratum and often become flat or elongated tails.22 When neutrophils were preincubated with CPC CM, JAM‐AsiCPC CM, CPC CM+anti–JAM‐A antibody, or JAM‐A Fc and then cultured on the fibronectin‐coated dishes under the stimulation with WKYMV, actin immunofluorescence staining of neutrophils demonstrated that treatment with CPC CM or JAM‐A Fc induced the formation of elongated (white arrowheads) or flattened (white arrow) uropods, whereas treatment with JAM‐AsiCPC CM or CPC CM+anti–JAM‐A antibody preserved short uropods (black arrowheads) as well as nontreated control neutrophils (Figure 10A). Treatment with CPC CM or JAM‐A Fc decreased the percentage of neutrophils with uropod shorter than 5 μm and increased the percentage of neutrophils with uropod longer than 10 μm (Figure 10B). On the contrary, treatment with JAM‐AsiCPC CM or CPC CM+anti–JAM‐A antibody abolished the effects of CPC CM and JAM‐A FC on uropod length, resulting in the distribution of uropod length similar to the control (Figure 10B). These findings suggest that soluble JAM‐A released from injected CPC reduces the motility of neutrophils, preventing their infiltration into myocardial tissue, and ameliorates tissue damage to the infarct heart through the prevention of excessive inflammation.
Figure 10.
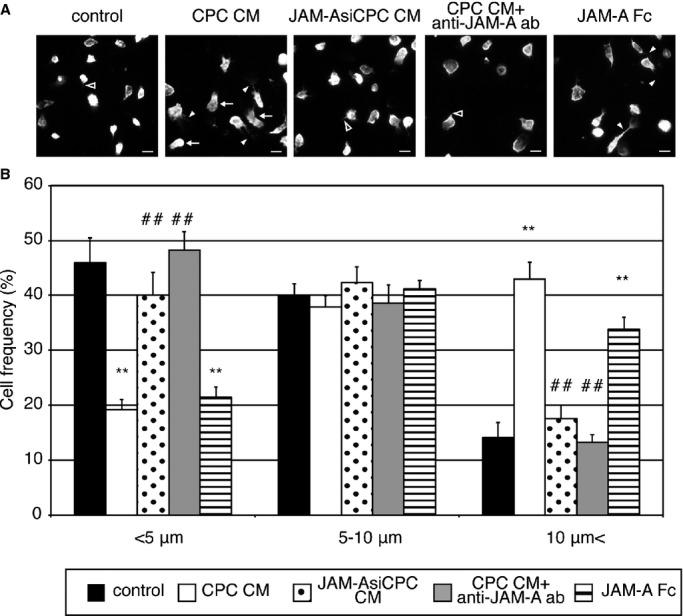
Soluble JAM‐A derived from CPC alters the uropod length of neutrophils. A, Actin immunofluorescence staining of neutrophils pretreated with control medium, CPC CM, JAM‐AsiCPC CM, CPC CM+anti‐JAM‐A antibody, or JAM‐A Fc. Bars are 10 μm. B, The frequency of cells with different uropod length in 1 control and 4 treated groups. The graph shows the mean±SEM of 5 experiments. One‐way ANOVA–Tukey–Kramer post hoc test was used for statistical analysis for different uropod length. Asterisks (**P<0.01) indicate significant differences between the treatment and control groups for different uropod length. Number signs (##P<0.01) indicate significant differences between the treatment and CPC CM groups for different uropod length. CM indicates conditioned medium; CPC, cardiac progenitor cell; JAM‐A, junctional adhesion molecule‐A.
Discussion
We previously reported that CPCs are most effective for cardiac repair in comparison with SMs, BMs, or AMCs.4 The dominance of CPC in part stems from not only their cardiomyogenic capacity but also unique paracrine factors. It has been reported that paracrine factors from transplanted cells are involved in various kinds of beneficial effects such as cytoprotection, angiogenesis, inhibition of fibrosis, and anti‐inflammation.3 Here, we first reported that transplantation of CPC inhibited neutrophil infiltration after MI through the paracrine effect. We identified that JAM‐A is exclusively secreted from CPCs and inhibited the emigration of neutrophils into myocardium, leading to reduction of oxidative stress and inflammatory response after infarction. In addition, we have shown that local delivery of JAM‐A with self‐assembling nanopeptides reduces cardiac remodeling after MI.
JAM‐A is an immunoglobulin‐like, PDZ binding domain containing transmembrane protein that is expressed in tight junction of endothelial and epithelial cells as well as leukocytes, including neutrophils, monocytes, and B‐ and T‐lymphocytes.23 JAM‐A on endothelial cells associates through their extracellular domains with leukocyte function–associated antigen‐1 and supports the adhesion and transendothelial migration of T cells and neutrophils. Although the mechanism by which JAM‐A regulates leukocyte transendothelial migration is still unclear, Woodfin et al reported that JAM‐A mediates neutrophil transmigration independent of other adhesion molecules such as intercellular adhesion molecule‐2 and platelet endothelial cell adhesion molecule‐1 through TNFα‐mediated activation of leukocytes and endothelial cells in vivo.24 Beside the membrane‐tethered JAM‐A, recently a soluble form of JAM‐A and its anti‐inflammatory effect have been identified. Koenen et al reported that soluble JAM‐A is released from endothelial cells via proteolytic shedding by a disintegrin and metalloproteinases 10 and 17. This shedding process is enhanced by endothelial cell activation with TNFα and interferon‐γ, resulting in inhibition of neutrophil transmigration by released soluble JAM‐A.6 Thus, JAM‐A and soluble JAM‐A play important roles in regulating local inflammatory responses mediated by neutrophils.
The interface between neutrophils and endothelial cells appears to be a critical site of actions for soluble JAM‐A, but that is not the case when CPCs or soluble JAM‐A is directly delivered into myocardial tissue. Although a fraction of transplants may be excreted to the blood flow, it has been reported that PM effectively retains the cells or proteins in the injected site.4,25 Therefore, the mechanism through which soluble JAM‐A reduces the number of extravasated neutrophils is not simply due to the inhibition of transendothelial migration. Recently, Cera et al reported that on activated neutrophils, JAM‐A is internalized in intracellular vesicles at the leading edge and uropod where JAM‐A codistributes with β1‐integrin. Neutrophils derived from JAM‐A–null mice were unable to correctly internalize and recycle β1‐integrin during cell migration on chemotactic stimuli, and this caused impaired directional migration.26 We have shown that treatment with soluble JAM‐A reduces the motility of activated neutrophils (Figure 10), suggesting that soluble JAM‐A secreted from CPCs may bind to its homophilic or heterophilic counterparts of JAM‐A or β1‐integrin on neutrophils, leading to failure of the colocalization of JAM‐A and β1‐integrin and correct turnover of β1‐integrin on neutrophils when the migrate through the interstitial tissue of ischemic myocardium.
Immunohistochemical images of normal hearts revealed that JAM‐A and Sca‐1 are colocalized at perivascular cells as well as endothelial cells (Figure 11). The role of JAM‐A–expressing perivascular cells has not been defined yet, but it is conceivable that the Sca‐1–positive perivascular cells, in which Sca‐1–positive CPCs are included, may control the fate of infiltrated leukocytes by secreting soluble JAM‐A and contribute to the regulation of inflammatory response in the heart. It is interesting to know the amount and localization of endogenous JAM‐A following myocardial ischemia. As shown in Figure 12, most of JAM‐A was colocalized with CD31‐positive endothelial cells of capillaries or those of smooth muscle actin–positive arterioles from day 1 to day 14 after MI (Figure 12A, border area; Figure 12B, infarct area). Semiquantitative analysis of fluorescent intensity revealed that the time courses of the expression level of JAM‐A and CD31 were parallel and peaked at 3 day in the border area (Figure 12C). In the infarct area, the time course of the expression level of JAM‐A transiently parted from that of CD31 at day 3 and subsequently both peaked at day 14 (Figure 12D). The reason for the time course difference between JAM‐A and CD31 in infarct area is unclear; however, taking into account the good colocalization of JAM‐A with CD31‐positive endothelial cells, the total amount of JAM‐A in the ischemic area is mostly dependent on the level of vascularization after MI. CD31‐negative nonendothelial JAM‐A–positive cells were observed at day 3 in the border area and at day 14 in the infarct area (white arrowheads in Figure 12A and 12B). Although the identity and the role of these interstitial JAM‐A–positive cells are uncertain, these cells may contribute to an endogenous anti‐inflammatory system in the acute and late phases of ischemia.
Figure 11.
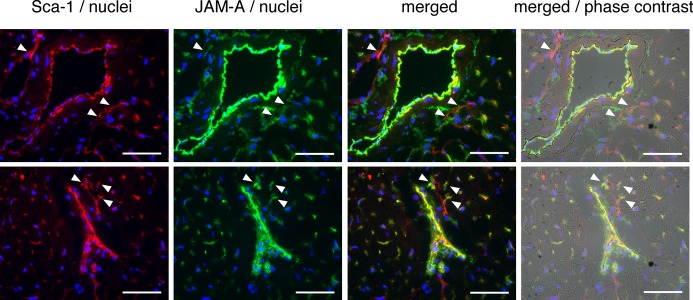
Immunohistochemical images of Sca‐1 and JAM‐A in normal mouse heart. Frozen sections were triple‐stained with Sca‐1 in red, JAM‐A in green, and nuclei in blue. The images overlaid with phase contrast images are shown. White arrowheads indicate perivascular Sca‐1–positive cells, which co‐express JAM‐A. Bars are 50 μm. JAM‐A indicates junctional adhesion molecule‐A.
Figure 12.

Time course of distribution and expression level of JAM‐A following myocardial ischemia. A and B, Frozen sections obtained from border (A) and infarct area (B) 1, 3, 7, and 14 days following MI were stained with CD31 or JAM‐A in red. The samples were costained with smooth muscle actin in green and nuclei in blue. As both CD31 and JAM‐A antibody originated from rat IgG, a pair of serial sections was stained with CD31 and JAM‐A, respectively. White arrowheads indicate CD31 negative nonendothelial JAM‐A–positive cells. Bars are 20 μm. C and D, Semiquantitative immunofluorescence intensity of JAM‐A and CD31 in border (C) and infarct area (D). One mouse was killed at each time point. Two adjacent sections were stained for CD31 and for JAMA separately. The values of immunofluorescence intensity obtained from at least 5 fields were examined. Two main effects and interaction effect were analyzed by using 2‐factor factorial ANOVA (border area [C]; main effect of expressed protein: P=0.27, main effect of time: P=6.5×10−7, interaction effect: P=0.75, infarct area [D]; main effect of expressed protein: P=0.0073, main effect of time: P=1.6×10−9, interaction effect: P=0.038). The difference in the level of expressed proteins between time points was analyzed by 1‐way ANOVA–Tukey–Kramer post hoc test. Asterisks and number signs above individual columns indicate significant difference compared with day 3 in C and with day 14 in (D) (**P<0.01, *P<0.05, ##P<0.01, and #P<0.05). We performed the experiment twice and similar results were obtained. JAM‐A indicates junctional adhesion molecule‐A; MI, myocardial infarction; SMA, smooth muscle actin.
Neutrophils have been implicated as a primary mechanism underlying ischemic injury. The processes involved in inducing tissue injury by neutrophils include oxygen free radical generation, degranulation and release of proteases, and release of proinflammatory mediators.27 After the hypoxic or mechanical stimulus is applied, cytokines such as TNFα and IL‐6 are rapidly released in ischemic and border zones.28–29 The inflammatory cytokines induce the expression of endothelial adhesion molecules, such as selectin families, VCAM‐1, and intercellular adhesion molecule‐1, and promote leukocyte adhesion to the vascular endothelium and subsequent transendothelial migration.30 Neutrophils stimulated by inflammatory cytokines produce superoxide anions and hydroxyl radicals in a respiratory burst.16 These oxygen free radicals promote the release of various kinds of proinflammatory chemokines from endothelial cells, cardiac fibroblasts, and other sources, leading to further enhancement of neutrophil adhesion and infiltration.27,31–32 Among these chemokines, CXC chemokines, including CXCL1, and CXCL2, play a critical role in basal and inflammatory neutrophil locomotion, trafficking, and activation.33–34 Recent studies revealed that neutrophils secrete CXCL1, CXCL2, CCL4, and CCL3 in response to a variety of stimulants, including lipopolysaccharides, zymosan, and substance P, thereby recruiting more neutrophils and activating themselves using the autocrine mechanisms of CXCL1 and CXCL2 secretion, and then attracting monocytes by using CCL4 and CCL3.35 Activated monocytes have been reported to inhibit neutrophil apoptosis through granulocyte‐macrophage colony‐stimulating factor secretion.36 McGettrick et al reported that neutrophils transmigrating through TNFα‐ or IL‐1β–treated HUVECs were strongly protected against apoptosis and that binding of β2‐integrins after transmigration was necessary for the survival signal for neutrophils, suggesting that an inflammatory condition during endothelial transmigration and integrin‐mediated adhesion after transmigration is essential for protection against neutrophil apoptosis.37 In addition, it has been reported that the firm adhesion between cardiomyocytes and neutrophils through β2‐ or α4‐integrins is required for both the release of toxic mediators including ROS and the subsequent injury and dysfunction.17–18 Therefore, reduction of ROS generation and downregulation of inflammatory chemokines expression may inhibit further neutrophil accumulation and survival, resulting in a reduction in neutrophil‐derived toxic mediators and a decrease in the contact between emigrated neutrophils, which may lessen the tissue injury.
It has been reported that JAM‐A is required for basic fibroblast growth factor–induced angiogenesis and is important in αVβ3‐integrin–specific endothelial cell migration on vitronectin.38–39 Recently, Peddibhotla et al proposed a model of angiogenic signaling regulated by JAM‐A and CD9, in which stimulation with basic fibroblast growth factor releases monomeric JAM‐A from the JAM‐A–CD9–αV3β complex, subsequently inducing monomeric JAM‐A to form homodimers that mediate mitogen‐activated protein kinase activation.40 Accordingly, antibodies against JAM‐A, as well as genetic deletion of JAM‐A, inhibited migration of endothelial cells. Considering the inhibitory effect of soluble JAM‐A on neutrophil migration, it is possible that transplanted JAM‐A may inhibit migration of endothelial cells and disturb angiogensis. However, our data revealed that myocardial administration of soluble JAM‐A enhances capillary density (Figure 7C through 7E). We suppose that reduction of neutrophil‐derived ROS may prevent endothelial cells from massive death and maintain their function in the acute phase of MI or that endothelial the JAM‐A–αVβ3 complex may interact with soluble JAMA in a different way from neutrophil JAM‐A/leukocyte function–associated antigen‐1 complex.
We have explored the other mechanisms involved in the reduction of infarct size. It is generally accepted that thin collagen, such as collagen type III, is immature and deposited predominantly during early remodeling, while thick collagen (collagen type I) predominates during late remodeling stages and contributes to scar maturation.41 A sustained high ratio of green to yellow‐red fibers means an excess of immature collagen matrix and correlates with deterioration of cardiac remodeling. As shown in Figure 8, injection of JAM‐A does not have a large effect on the composition of collagen fibers at least 2 weeks after MI.
The adult mammalian heart posses a limited capacity for regeneration, but recently it was reported that several factors can simulate cardiac regeneration after injury.42 We examined whether injection of JAM‐A enhances cardiac regeneration through cardiac differentiation of transplanted CPCs, proliferation of endogenous CPCs, or cardiomyocyte division. When RFP‐expressing CPCs were transplanted with PM or JAM‐A+PM into MI heart, RFP and sarcomeric α‐actinin double‐positive cells were localized to epicardial lesion, where a mixture of CPCs and PM was overlaid (upper panels of Figure 13A). A few double‐positive cells were localized to the border area (lower panels of Figure 13A). These double‐positive cells were small and showed no or immature sarcomere structure. There was no significant difference in the number of double‐positive cells per section between PM‐ and JAM‐A+PM–treated groups (PM: 11.5±5.7 [n=4]; JAM‐A+PM: 24.3±14 [n=3]; Figure 13B). We examined the frequency of BrdU‐positive endogenous CPCs, which were identified as Nkx2.5 positive and sarcomeric α‐actinin–negative cells (Figure 13C). There was no significant difference in the percentage of BrdU‐positive CPCs in total CPCs between PM‐ and JAM‐A+PM–treated mice (PM: 94.4±5.6% [n=3]; JAM‐A+PM: 90.9±5.5% [n=4]; Figure 13D). The percentage of BrdU‐positive cardiomocytes in total cardiomocytes did not show a significant difference between PM‐ and JAM‐A+PM–treated mice (PM: 0.034±0.02% [n=3]; JAM‐A+PM: 0.039±0.02% [n=3]; Figure 13E). These findings suggest that although a fraction of transplanted CPCs were engrafted and acquire cardiac phenotype, a large part of the beneficial effects of CPCs do not stem from cardiac regeneration in our model. In addition, JAM‐A enhances neither the transdifferentiation of CPCs nor the intrinsic cardiac regeneration capacity after injury.
Figure 13.
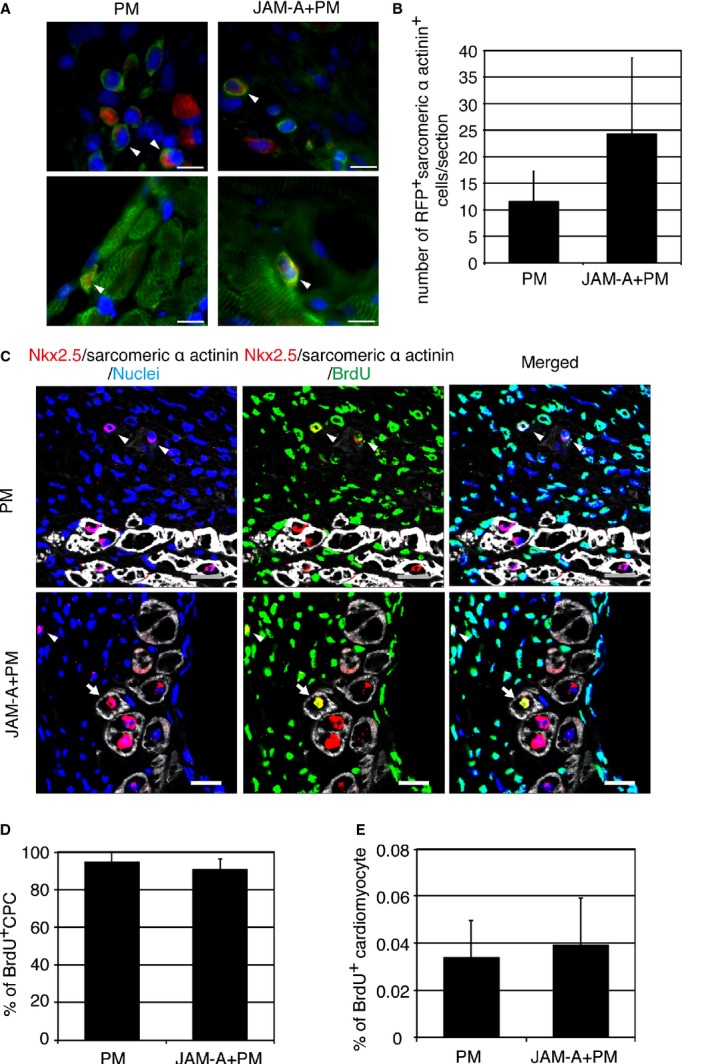
Cardiomyocyte‐like differentiation of transplanted CPCs and proliferation of endogenous CPCs and cardiomyocytes in PM‐ and JAM‐A+PM–treated hearts. A, Representative images of RFP (red) and sarcomeric α‐actinin (green) double‐positive cells (white arrowheads) in epicardial region (upper panels) and border area (lower panels). Nuclei were stained in blue. Bars are 10 μm. B, Number of RFP and sarcomeric α‐actinin double‐positive cells per section of PM‐ and JAM‐A+PM–treated hearts. An average of values obtained from 3 sections per mouse was analyzed: n=3 mice for PM‐ and n=4 mice for JAM‐A+PM–treated group. Student t test was used for statistical analysis. C, Confocal images of BrdU‐positive CPC (white arrowheads) and BrdU‐positive cardiomyocytes (white arrows). Bars are 20 μm. D, Frequency of BrdU‐positive CPCs in total CPCs in PM‐ and JAM‐A+PM–treated hearts. The whole area of LV in a section through the long axis of the heart was examined per mouse: n=3 mice for PM‐ and n=4 mice for JAM‐A+PM–treated group. Student t test was used for statistical analysis. E, Frequency of BrdU‐positive cardiomyocytes in PM‐ and JAM‐A+PM–treated hearts. The whole area of LV in a section through the long axis of the heart was examined per mouse” n=3 mice per group. Student t test was used for statistical analysis. CPC indicates cardiac progenitor cell; JAM‐A, junctional adhesion molecule‐A; LV, left ventricle; RFP, red fluorescent protein.
We have shown that transplantation of CPCs or soluble JAM‐A protein, but not JAM‐A knockdown CPCs, ameliorates cardiac remodeling and expansion of infarct size. This suggests that blocking neutrophil infiltration through the inactivation of JAM‐A could be therapeutically valuable for the ischemic heart disease. Studies in models of myocardial ischemia and hepatic ischemia–reperfusion of JAM‐A–deficient mouse have demonstrated that JAM‐A deficiency in polymorphonuclear cells inhibits transendothelial migration of leukocytes but not their rolling and adhesion to endothelial cells.43–44 Although neutrophil infiltration was inhibited, increasing vascular adherent leukocytes results in the disturbance of blood flow and release of inflammatory mediators, consequently causing more tissue injury in comparison with wild‐type.43–44 Recently, Lakshmi et al reported in a murine model of acute lung injury that JAM‐A knockout and treatment with anti–JAM‐A blocking antibody fail to reduce oxidative stress or cytokine and chemokine levels in whole lung and have no effect on capillary leakage and lung edema, presumably reflecting the histologically observed retention of neutrophils in lung tissue.45 The plausible explanation of discordant results between previous reports and ours are as follows: systemic deletion of JAM‐A or intravenous injection of anti–JAM‐A blocking antibody may subject the circulating neutrophils to lose tuned balance between adhesion and transendothelial migration, whereas myocardial injection of CPCs or soluble JAM‐A protein may specifically affect the extravasated neutrophils without retention of neutrophils in microvasculature in the heart.
A limitation of our study is a limited survival of transplanted CPCs. As shown in our previous report, the frequency of transplanted CPCs was only 0.21% at 7 days after transplantation.4 When cardiac function of PM‐ and CPC+PM–treated groups was examined at 4 weeks after myocardial infarction, both LVIDD and LVISD of CPC+PM–treated group were smaller and percent FS of the CPC+PM–treated group was higher than that of the PM‐treated group, although the measured values did not reach statistical significance (Table 5). The infarct size was 42.2±2.6% in the CPC+PM–treated group (n=9) and 49.1±4.8% in the PM‐treated group (n=6). Although the infarct size of the CPC+PM–treated group was smaller than that of the PM‐treated group, this finding did not reach statistical significance (Student t test). The reason for a lack of beneficial effects in a 4‐week observation may stem from the poor survival of transplanted CPCs for a long period. However, we believe that good correlation between the survival time of transplanted CPCs and the phase of acute neutrophil accumulation indicates that JAM‐A released from CPCs prevents deleterious effects derived from neutrophils during the acute phase of MI.
Table 5.
Echocardiographic Measurement of Hearts 4 Weeks After Transplantation
| Parameter | n | HR, beats/min | SWT, mm | PWT, mm | LVIDD, mm | LVISD, mm | %FS |
|---|---|---|---|---|---|---|---|
| PM | 4 | 689±19 | 0.17±0.01 | 0.49±0.09 | 6.1±0.48 | 5.7±0.59 | 7.7±3.0 |
| CPC+PM | 9 | 690±21 | 0.26±0.05 | 0.64±0.07 | 5.5±0.25 | 4.9±0.27 | 10.7±1.2 |
Statistical analysis was performed by using Student t test (PWT, LVIDD, and LVISD) or Mann–Whitney U test (HR, SWT and %FS). No statistical difference was observed between PM and CPC+PM in each parameter. FS indicates fractional shortening; HR, heart rate; LVIDD, left ventricular internal diastolic diameter; LVISD, left ventricular internal systolic diameter; PWT, posterior wall thickness; SWT, septal wall thickness.
Although many researchers have reported the detrimental effects of infiltrating neutrophils and inflammatory mediators on cardiomyocytes in the infarcted heart, therapy directed to mitigate inflammatory processes has been, in general, unsuccessful in clinical practice.46 The results of methylprednisolone trial have been controversial, in that some have demonstrated efficacy of the drug to limit extension of evolving MI, while other results have been deleterious.47–48 The results of a clinical trial demonstrated that an antibody to CD11/CD18 leukocyte integrin receptor did not reduce infarct size in patients who underwent primary angioplasty after MI.49 Several methods for JAM‐A inhibition, such as genetic inactivation, blocking antibody, and soluble recombinant proteins, prevent inflammatory reactions in meningitis, peritonitis, skin, and ischemic injury of heart and liver in animal models.6,50–51 However, the consequences of JAM‐A targeting inhibition vary in different cell types and tissues, depending on the site (endothelium or leukocytes) and the mechanism (adhesion, diapedesis, or migration) of action. Systemic deletion of JAM‐A gene or intravenous injection of JAM‐A antibody caused retention of neutrophils on the vascular surface and, in some cases, aggregation in the capillaries. The permanence of neutrophils on the endothelial surface leads to the release of oxygen species and lytic enzymes, which aggravated tissue damage.43–45 Here, we have shown that local administration of soluble JAM‐A into ischemic myocardium inhibits neutrophil emigration, probably without disturbing microcirculation, which suggests its usefulness for the clinical application. Although direct injection of JAM‐A into myocardium during the acute ischemic phase is difficult to conduct, recent advances in nanoparticle‐mediated drug deliver system may enable the transport of soluble JAM‐A across capillary endothelial cells by nanoparticle transcytosis following coronary injection.52 In addition, it has been reported that a small inhibitory peptide targeting the JAM‐A–αLβ2 integrin interaction reduced leukocyte recruitment during postischemic inflammation in brain ischemia–reperfusion injury and reduced the lesion size.53 Nanoparticles in which such a peptide inhibitor is encapsulated may be another choice for clinical anti‐inflammatory strategy targeting JAM‐A. The anti‐inflammatory peptides secreted from cardiac dormant cells may become a new candidate for the treatment of cardiovascular disease through the prevention of excess inflammation.
Sources of Funding
This work was supported by a Grant‐in‐Aid for Scientific Research, Developmental Scientific Research, and Scientific Research on Priority Areas from the Ministry of Education, Science, Sports, and Culture and the Takeda Science Foundation.
Disclosures
None.
Acknowledgments
The authors thank A. Furuyama and Y. Otsuki for their excellent technical assistance. We thank 3‐D Matrix, Ltd for providing Puramatrix.
References
- 1.Rosenzweig A. Cardiac cell therapy‐mixed results from mixed cells. N Engl J Med. 2006; 355:1274-1277. [DOI] [PubMed] [Google Scholar]
- 2.Wollert KC, Drexler H. Cell therapy for the treatment of coronary heart disease: a critical appraisal. Nat Rev Cardiol. 2010; 7:204-215. [DOI] [PubMed] [Google Scholar]
- 3.Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms in adult stem cell signaling and therapy. Circ Res. 2008; 103:1204-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tokunaga M, Liu ML, Nagai T, Iwanaga K, Matsuura K, Takahashi T, Kanda M, Kondo N, Wang P, Naito AT, Komuro I. Implantation of cardiac progenitor cells using self‐assembling peptide improves cardiac function after myocardial infarction. J Mol Cell Cardiol. 2010; 49:972-983. [DOI] [PubMed] [Google Scholar]
- 5.Matsuura K, Honda A, Nagai T, Fukushima N, Iwanaga K, Tokunaga M, Shimizu T, Okano T, Kasanuki H, Hagiwara N, Komuro I. Transplantation of cardiac progenitor cells ameliorates cardiac dysfunction after myocardial infarction in mice. J Clin Invest. 2009; 119:2204-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koenen RR, Pruessmeyer J, Soehnlein O, Fraemohs L, Zernecke A, Schwarz N, Reiss K, Sarabi A, Lindbom L, Hackeng TM, Weber C, Ludwig A. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood. 2009; 113:4799-4809. [DOI] [PubMed] [Google Scholar]
- 7.Matsuura K, Nagai T, Nishigaki N, Oyama T, Nishi J, Wada H, Sano M, Toko H, Akazawa H, Sato T, Nakaya H, Kasanuki H, Komuro I. Adult cardiac Sca‐1‐positive cells differentiate into beating cardiomyocytes. J Biol Chem. 2004; 279:11384-11391. [DOI] [PubMed] [Google Scholar]
- 8.Planat‐Bénard V, Menard C, André M, Puceat M, Perez A, Garcia‐Verdugo JM, Penicaud L, Casteilla L. Spontaneous cardiomyocyte differentiation from adipose tissue stroma cells. Circ Res. 2004; 94:223-229. [DOI] [PubMed] [Google Scholar]
- 9.Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell‐mediated gene therapy. J Cell Biol. 1994; 125:1275-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuura K, Wada H, Nagai T, Iijima Y, Minamino T, Sano M, Akazawa H, Molkentin JD, Kasanuki H, Komuro I. Cardiomyocytes fuse with surrounding noncardiomyocytes and reenter the cell cycle. J Cell Biol. 2004; 167:351-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allport JR, Ding HT, Ager A, Steeber DA, Tedder TF, Lucinskas FW. L‐selectin shedding does not regulate human neutrophilattachment, rolling, or transmigration across human vascular endothelium invitro. J Immunol. 1997; 158:4365-4372. [PubMed] [Google Scholar]
- 12.Junqueira LC, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J. 1979; 11:447-455. [DOI] [PubMed] [Google Scholar]
- 13.Rich L, Whittaker PB. Collagen and picrosirius red staining: a polarized light assessment of fibrillar hue and spatial distribution. J Morphol Sci. 2005; 22:97-104. [Google Scholar]
- 14.Seino Y, Ikeda U, Minezaki KK, Funayama H, Kasahara T, Konishi K, Shimada K. Expression of cytokine‐induced neutrophil chemoattractant in rat cardiac myocytes. J Mol Cell Cardiol. 1995; 27:2043-2051. [DOI] [PubMed] [Google Scholar]
- 15.Wei S, Chow LT, Shum IO, Qin L, Sanderson JE. Left and right ventricular collagen type I/III ratios and remodeling post‐myocardial infarction. J Card Fail. 1999; 5:117-126. [DOI] [PubMed] [Google Scholar]
- 16.Jordan JE, Zhao ZQ, Vinten‐Johansen J. The role of neutrophils in myocardial ischemia‐reperfusion injury. Cardiovasc Res. 1999; 43:860-878. [DOI] [PubMed] [Google Scholar]
- 17.Entman ML, Youker K, Shoji T, Kukielka G, Shappell SB, Taylor AA, Smith CW. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18‐ICAM‐1 adherence. J Clin Invest. 1992; 90:1335-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poon BY, Ward CA, Cooper CB, Giles WR, Burns AR, Kubes P. alpha(4)‐integrin mediates neutrophil‐induced free radical injury to cardiac myocytes. J Cell Biol. 2001; 152:857-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frangogiannis NG. The role of the chemokines in myocardial ischemia and reperfusion. Curr Vasc Pharmacol. 2004; 2:163-174. [DOI] [PubMed] [Google Scholar]
- 20.Frangogiannis NG, Youker KA, Rossen RD, Gwechenberger M, Lindsey MH, Mendoza LH, Michael LH, Ballantyne CM, Smith CW, Entman ML. Cytokines and the microcirculation in ischemia and reperfusion. J Mol Cell Cardiol. 1998; 30:2567-2576. [DOI] [PubMed] [Google Scholar]
- 21.Fais S, Malorni W. Leukocyte uropod formation and membrane/cytoskeleton linkage in immune interactions. J Leukoc Biol. 2003; 73:556-563. [DOI] [PubMed] [Google Scholar]
- 22.Eddy RJ, Pierini LM, Matsumura F, Maxfield FR. Ca2+‐dependent myosin II activation is required for uropod retraction during neutrophil migration. J Cell Sci. 2000; 113:1287-1298. [DOI] [PubMed] [Google Scholar]
- 23.Severson EA, Parkos CA. Mechanisms of outside‐in signaling at the tight junction by junctional adhesion molecule A. Ann N Y Acad Sci. 2009; 1165:10-18. [DOI] [PubMed] [Google Scholar]
- 24.Woodfin A, Voisin MB, Imhof BA, Dejana E, Engelhardt B, Nourshargh S. Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM‐2, JAM‐A, and PECAM‐1. Blood. 2009; 113:6246-6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelain F, Unsworth LD, Zhang S. Slow and sustained release of active cytokines from self‐assembling peptide scaffolds. J Control Release. 2010; 145:231-239. [DOI] [PubMed] [Google Scholar]
- 26.Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M, Reichel CA, Krombach F, Pardi R, Dejana E. JAM‐A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci. 2009; 122:268-277. [DOI] [PubMed] [Google Scholar]
- 27.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002; 53:31-47. [DOI] [PubMed] [Google Scholar]
- 28.Irwin MW, Mak S, Mann DL, Qu R, Penninger JM, Yan A, Dawood F, Wen WH, Shou Z, Liu P. Tissue expression and immunolocalization of tumour necrosisfactor‐alpha in post infarction‐dysfunctional myocardium. Circulation. 1999; 99:1492-1498. [DOI] [PubMed] [Google Scholar]
- 29.Ono K, Matsumori A, Shioi T, Furukawa Y, Sasayama S. Cytokine gene expression after myocardial infarction in rat hearts: possible implication in left ventricular remodeling. Circulation. 1998; 98:149-156. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Alcaide P, Liu L, Sun J, He A, Luscinskas FW, Shi GP. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS One. 2011; 6:e14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis MS, Whatley RE, Cain P, McIntyre TM, Prescott SM, Zimmerman GA. Hydrogen peroxide stimulates the synthesis of platelet‐activating factor by endothelium and induces endothelial cell‐dependent neutrophil adhesion. J Clin Invest. 1988; 82:2045-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long CS. The role of interleukin‐1 in the failing heart. Heart Fail Rev. 2001; 6:81-94. [DOI] [PubMed] [Google Scholar]
- 33.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001; 2:108-115. [DOI] [PubMed] [Google Scholar]
- 34.Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol. 2001; 2:123-128. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci. 2008; 13:2400-2407. [DOI] [PubMed] [Google Scholar]
- 36.Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, Buridi A, McLeish KR. Granulocyte‐macrophage colonystimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3‐kinase and extracellular signal‐regulated kinase pathways. J Immunol. 2000; 164:4286-4291. [DOI] [PubMed] [Google Scholar]
- 37.McGettrick HM, Lord JM, Wang KQ, Rainger GE, Buckley CD, Nash GB. Chemokine‐ and adhesion‐dependent survival of neutrophils after transmigration through cytokine‐stimulated endothelium. J Leukoc Biol. 2006; 79:779-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naik MU, Vuppalanchi D, Naik UP. Essential role of junctional adhesion molecule‐1 in basic fibroblast growth factor‐induced endothelial cell migration. Arterioscler Thromb Vasc Biol. 2003; 23:2165-2171. [DOI] [PubMed] [Google Scholar]
- 39.Naik MU, Naik UP. Junctional adhesion molecule‐A‐induced endothelial cell migration on vitronectin is integrin αVβ3 specific. J Cell Sci. 2006; 119:490-499. [DOI] [PubMed] [Google Scholar]
- 40.Peddibhotla SS, Brinkmann BF, Kummer D, Tuncay H, Nakayama M, Adams RH, Gerke V, Ebnet K. Tetraspanin CD9 links junctional adhesion molecule‐A to αvβ3 integrin to mediate basic fibroblast growth factor‐specific angiogenic signaling. Mol Biol Cell. 2013; 24:933-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Res Cardiol. 1994; 89:397-410. [DOI] [PubMed] [Google Scholar]
- 42.Kikuchi K, Poss KD. Cardiac regenerative capacity and mechanisms. Annu Rev Cell Dev Biol. 2012; 28:719-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khandoga A, Kessler JS, Meissner H, Hanschen M, Corada M, Motoike T, Enders G, Dejana E, Krombach F. Junctional adhesion molecule‐A deficiency increases hepatic ischemia‐reperfusion injury despite reduction of neutrophil transendothelial migration. Blood. 2005; 106:725-733. [DOI] [PubMed] [Google Scholar]
- 44.Corada M, Chimenti S, Cera MR, Vinci M, Salio M, Fiordaliso F, De Angelis N, Villa A, Bossi M, Staszewsky LI, Vecchi A, Parazzoli D, Motoike T, Latini R, Dejana E. Junctional adhesion molecule‐A‐deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia‐reperfusion injury. Proc Natl Acad Sci USA. 2005; 102:10634-10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lakshmi SP, Reddy AT, Naik MU, Naik UP, Reddy RC. Effects of JAM‐A deficiency or blocking antibodies on neutrophil migration and lung injury in a murine model of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2012; 303:L758-L766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seropian IM, Toldo S, Van Tassell BW, Abbate A. Anti‐inflammatory strategies for ventricular remodeling following ST‐segment elevation acute myocardial infarction. J Am Coll Cardiol. 2014; 63:1593-1603. [DOI] [PubMed] [Google Scholar]
- 47.LeGal YM, Morrissey LL. Methylprednisolone interventions in myocardial infarction: a controversial subject. Can J Cardiol. 1990; 6:405-410. [PubMed] [Google Scholar]
- 48.Roberts R, DeMello V, Sobel BE. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation. 1976; 53:I204-I206. [PubMed] [Google Scholar]
- 49.Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALTMI study. J Am Coll Cardiol. 2002; 40:1199-1204. [DOI] [PubMed] [Google Scholar]
- 50.Nourshargh S, Krombach F, Dejana E. The role of JAM‐A and PECAM‐1 in modulating leukocyte infiltration in inflamed and ischemic tissues. J Leukoc Biol. 2006; 80:714-718. [DOI] [PubMed] [Google Scholar]
- 51.Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007; 7:467-477. [DOI] [PubMed] [Google Scholar]
- 52.Kreuter J. Drug delivery to the central nervous system by polymeric nanoparticles: what do we know? Adv Drug Deliv Rev. 2014; 71:2-14. [DOI] [PubMed] [Google Scholar]
- 53.Sladojevic N, Stamatovic SM, Keep RF, Grailer JJ, Sarma JV, Ward PA, Andjelkovic AV. Inhibition of junctional adhesion molecule‐A/LFA interaction attenuates leukocyte trafficking and inflammation in brain ischemia/reperfusion injury. Neurobiol Dis. 2014; 67:57-70. [DOI] [PMC free article] [PubMed] [Google Scholar]