

Abstract
Background
The compensatory actions of the endogenous natriuretic peptide system require adequate processing of natriuretic peptide pro‐hormones into biologically active, carboxyl‐terminal fragments. Natriuretic peptide pro‐peptide processing is accomplished by corin, a transmembrane serine protease expressed by cardiomyocytes. Brain natriuretic peptide (BNP) processing is inadequate in advanced heart failure and is independently associated with adverse outcomes; however, the molecular mechanisms causing impaired BNP processing are not understood. We hypothesized that the development of endoplasmic reticulum stress in cardiomyocytes in advanced heart failure triggers inositol‐requiring protein 1 (IRE1)‐dependent corin mRNA decay, which would favor a molecular substrate favoring impaired natriuretic peptide pro‐peptide processing.
Methods and Results
Two independent samples of hearts obtained from patients with advanced heart failure at transplant demonstrated that corin RNA was reduced as Atrial natriuretic peptide (ANP)/BNP RNA increased. Increases in spliced X‐box protein 1, a marker for IRE1‐endoribonuclease activity, were associated with decreased corin RNA. Moreover, ≈50% of the hearts demonstrated significant reductions in corin RNA and protein as compared to the nonfailing control sample. In vitro experiments demonstrated that induction of endoplasmic reticulum stress in cultured cardiomyocytes with thapsigargin activated IRE1's endoribonuclease activity and time‐dependent reductions in corin mRNA. In HL‐1 cells, overexpression of IRE1 activated IRE1 endoribonuclease activity and caused corin mRNA decay, whereas IRE1‐RNA interference with shRNA attenuated corin mRNA decay after induction of endoplasmic reticulum stress with thapsigargin. Pre‐treatment of cells with Actinomycin D to inhibit transcription did not alter the magnitude or time course of thapsigargin‐induced corin mRNA decline, supporting the hypothesis that this was the result of IRE1‐mediated corin mRNA degradation.
Conclusions
These data support the hypothesis that endoplasmic reticulum stress‐mediated, IRE1‐dependent targeted corin mRNA decay is a mechanism leading to corin mRNA resulting in corresponding corin protein deficiency may contribute to the pathophysiology of impaired natriuretic peptide pro‐hormone processing in humans processing in humans with advanced systolic heart failure.
Keywords: atrial natriuretic factor, genes, heart failure, molecular biology, myocytes, natriuretic peptides
Introduction
The natriuretic peptide system (NPS) is activated in heart failure and provides compensatory systemic and cardiac autocrine/paracrine actions that oppose the development and progression of heart failure including vasodilation, attenuating activation of the renin–angiotensin–aldosterone and sympathetic systems, and augments natriuresis.1–3 Cardiac autocrine paracrine actions of the NPS oppose excessive hypertrophy and reduce excessive interstitial fibrosis.4–6 Corin is a transmembrane serine protease, abundantly expressed by cardiomyocytes, that has an extracellular protease domain that interacts with both proANP and proBNP and cleaves them into biologically active, carboxyl‐terminal peptides.7–8 In the case of human BNP (1‐108), corin processes it into a 32 amino acid, biologically active, carboxyl‐terminal fragment BNP (77‐108), also referred to as BNP32 and the biologically inactive 76 amino acid, amino terminal fragment, fragment BNP (1‐76).9 It is demonstrated that BNP processing is impaired in heart failure, and the degree of impaired processing positively correlates with New York Heart Association class.10 We demonstrated that within a cohort of patients with predominantly New York Heart Association Class III heart failure, there was significant interindividual variation in the severity of impaired BNP processing and that more severe impairment of BNP processing was independently associated with an increased risk for all‐cause mortality or cardiac transplant.11 In vitro studies demonstrate that BNP (1‐108) less effectively activates its cognate receptor, the type A guanylate‐cyclase receptor, as compared to BNP32.12–13 Therefore, inefficient BNP processing in heart failure may contribute to attenuation of the NPS and heart failure progression.14–15 However, the molecular mechanisms leading to impaired BNP (1‐108) processing in heart failure are not well understood and if elucidated might provide a novel therapeutic target to promote compensation and delay progression of heart failure.
We hypothesized that impaired natriuretic peptide processing in advanced heart failure might result from corin deficiency caused by regulated, inositol‐requiring‐1 (IRE1)–dependent messenger RNA decay (RIDD),16–17 which is a component of generalized cellular endoplasmic reticulum (ER) stress response. It is characterized by IRE1‐mediated degradation of messenger RNAs that are targeting proteins to the ER for post‐translational modifications and folding. Our hypothesis developed by extrapolating from work examining the role of ER stress in diabetes in which pancreatic β‐islet cells demonstrate evidence of RIDD insulin mRNA decay.18 In a similar manner, corin is an ER‐targeted protein, and failing cardiomyocytes demonstrate ER stress due to a variety of factors including secretory demand (natriuretic peptides), oxidative stress, and calcium overload in the sarcoplasmic reticulum.19–21 Consistently, another report demonstrated that in human heart failure left ventricular samples, BNP gene expression, a marker for secretory demand, was positively correlated with GRP78, a marker of ER stress.22 Lastly, we previously reported the observation of an unexpected inverse relationship between BNP mRNA and corin mRNA in hearts from patients with end‐stage heart failure obtained at the time of cardiac transplant.23 To test this hypothesis, we embarked upon an examination of gene expression profiles including markers of ER stress, ANP, BNP, and corin in 2 independent libraries consisting of end‐stage heart‐failure hearts obtained at the time of cardiac transplantation and a series of in vitro experiments examining the role of ER stress‐mediated IRE1 activation and corin mRNA stability. Together, these data support the hypothesis that RIDD is operative in advanced human heart failure and may contribute to corin deficiency both at the mRNA and protein level. We reason that this may contribute to the recognized phenomenon of impaired natriuretic peptide pro‐hormone activation processing in advanced human heart failure and attenuation of the compensatory actions of the endogenous NPS.
Materials and Methods
Human Hearts Obtained From End‐Stage Human Cardiomyopathy
UPENN database
Analysis was conducted using human left ventricle tissue samples from the explanted heart from a total of 14 unmatched donors without history of cardiac disease (nonfailing control hearts) and 205 end‐stage heart‐failure patients (failing hearts) undergoing cardiac transplantation. In this sample, hearts from patients with an ischemic and nonischemic etiology were included. Heart samples were frozen in liquid nitrogen and stored at −80°C until use.
UK heart collection
The study was approved by the Royal Brompton and Harefield ethical review committee, and informed consent was obtained from the patients. The investigation conforms to the principles outlined in the Declaration of Helsinki. The UK population comprised 73 ventricular samples as follows: 36 transmural left ventricle samples collected from patients with end‐stage heart failure at the time of cardiac transplant or left‐ventricular assist device implantation (33 cases were dilated cardiomyopathy and 3 ischemic cardiomyopathy); 20 left ventricle samples (transmural and endomyocardial biopsies) collected from left‐ventricular assist device–supported end‐stage heart‐failure Dilated cardiomyopathy (DCM) patients at the time of left‐ventricular assist device explantation; 9 transmural left ventricle samples taken from donor hearts unsuitable for cardiac transplantation due to poor systolic function; and 8 right ventricle endomyocardial biopsies taken from donor hearts used for cardiac transplantation. Tissue was flash‐frozen in liquid nitrogen immediately after collection and stored at −80°C. The rationale for using this assembly of samples was to maximize our sample size of samples from advanced heart failure but also to include some samples from near‐normal hearts.
UPENN heart collection
Total cellular RNA was extracted from frozen left ventricle specimens (35 to 50 mg) using the RNeasy Fibrosis Tissue mini Kit (Qiagen, Valencia, CA) following the manufacturer's protocol. The frozen heart tissue was homogenized by using Qiagen Tissuelyser (Qiagen). The RNA was precipitated and dissolved in RNase‐free water. The concentration of total RNA was measured spectrophotometrically. RNA was utilized to assess gene expression using the Affymetrix GeneChip as described below.
UK heart collection
For quantitative polymerase chain reaction (PCR) analysis, total RNA was extracted, DNase‐treated, and quantified using a protocol optimized for maximal recovery from myocardial biopsies.24 The cDNA was prepared using a maximum of 50 ng of total RNA per 10 μL reaction volume with random hexamers and diluted to the RNA equivalent of 2 ng/μL. Quantitative PCR was performed on Applied Biosystems' 7500 Fast real‐time PCR system using TaqMan chemistry and 3 μL of diluted cDNA. Assays for ANP and BNP were designed in house,25 whereas assays for 18S ribosomal RNA, spliced X box protein 1 (XBP1s), corin, and GRP78 were purchased from Life Technologies (catalogue numbers: 4310893E, Hs03929085_g1, Hs00198141_m1, and Hs00946084_g1, respectively).
Sample Preparation and Chip Hybridization (UPENN Collection)
The sample preparation and processing procedure was performed as described in the Affymetrix GeneChip Expression Analysis manual. For these studies, we used the Affymetrix HG_U133 Human Chipset, which includes 22 242 sequences (including 2484 expressed sequence tags (ESTs) on the A chip and 22 577 sequences (including 13 921 ESTs) on the B chip. In accordance with Minimum Information About a Microarray Experiment (MIAME) guidelines, the annotation for the Affymetrix microarray is available at: http://www.mged.org/Workgroups/MIAME/miame_1.1.html.
Quantitative Real‐Time PCR From In Vitro Cell Experiments
Total RNA was extracted from cellular homogenates using the cell protocol for the RNeasy kit. First‐strand cDNA was reverse transcribed from 1 μg of total RNA with oligo(dT) using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). The resulting cDNA was used for real‐time PCR analysis by TaqMan® Gene Expression Assays or Custom Gene Expression Assays (Applied Biosystems) (Table). Data were normalized to 18S ribosomal RNA and analyzed using the comparative Ct or standard curve method. Results are given as mean±SEM. Significance was tested using nonparametric paired and unpaired t tests and nonparametric correlation tests (*P≤0.05, **P≤0.01).
Table 1.
TaqMan Probes in Human, Mouse, and Rat Gene Expression Assay
| Origin | Target | Probe Sequence |
|---|---|---|
| Human | ANP | FAM‐5′‐GCTGTAACAGCTTCCGGTACCGAAG‐3′‐TAMRA |
| BNP | FAM‐5′‐CTGGGCTGCAAAGTGCTGAGGCGGC‐3′‐TAMRA | |
| XBP1 spliced | FAM‐5′‐CCGCAGCAGGTGCAG‐3′‐TAMRA | |
| Corin | FAM‐5′‐TCCCTCCTTGCAGGGCATTGTGTGA‐3′‐TAMRA | |
| 18S/Rps18 | FAM‐5′‐GGAGGGCAAGTCTGGTGCCAGCAGC‐3′‐TAMRA | |
| Mouse | GRP78 | FAM‐5′‐TGGGAAAGAAGGTTACCCATGCAGT‐3′‐TAMRA |
| GRP94 | FAM‐5′‐GCTGACCTTCGGGTTCGTCAGAGCT‐3′‐TAMRA | |
| ATF4 | FAM‐5′‐CTCAGACACCGGCAAGGAGGATGCC‐3′‐TAMRA | |
| CHOP | FAM‐5′‐CCACACCTGAAAGCAGAACCTGGTC‐3′‐TAMRA | |
| XBP1 spliced | FAM‐5′‐CCGCAGCAGGTGCAG‐3′‐TAMRA | |
| Corin | FAM‐5′‐TGATGGCGATGACAGGCATGGTCTT‐3′‐TAMRA | |
| IRE1 α | FAM‐5′‐CGGCCCCGGGAGTTTTGGAAGAACC‐3′‐TAMRA | |
| 18S/Rps18 | FAM‐5′‐TGCGCCACTTTTGGGGCCTTCGTGT‐3′‐TAMRA | |
| Rat | GRP78 | FAM‐5′‐CAATCAAGGTCTACGAAGGTGAACG‐3′‐TAMRA |
| GRP94 | FAM‐5′‐GCTGACCTTCGGGTTCGTCAGAGCT‐3′‐TAMRA | |
| ATF4 | FAM‐5′‐CTCAGACACCGGCAAGGAGGATGCC‐3′‐TAMRA | |
| CHOP | FAM‐5′‐GGAGAGAGAAACCGGTCCAATTACA‐3′‐TAMRA | |
| XBP1 spliced | FAM‐5′‐CCGCAGCAGGTGCAG‐3′‐TAMRA | |
| Corin | FAM‐5′‐TGCTGGTGCACAGGCGGTCCTGCCC‐3′‐TAMRA | |
| IRE1 α | FAM‐5′‐ACTGGCTTCTCATAGGACACCATGA‐3′‐TAMRA | |
| 18S/Rps18 | FAM‐5′‐TGCGCCACTTTTGGGGCCTTCGTGT‐3′‐TAMRA |
ANP indicates Atrial natriuretic peptide; ATF, activating transcription factor; BNP, Brain natriuretic peptide; CHOP, C/EBP homology protein; IRE1, inositol‐requiring protein 1; XBP1, X‐box protein 1.
HL‐1 (Immortalized Murine Adult Atrial Cardiomyocyte) Cell Culture
HL‐1 cells are a cardiac muscle cell line, derived from the AT‐1 mouse, atrial cardiomyocyte tumor lineage; they demonstrate contractile function and adult atrial cardiomyocyte gene expression profiles, and were maintained as previously described.26 Briefly, HL‐1 cells were plated in gelatin/fibronectin‐coated 30‐mm culture dishes or 6‐well plates and were maintained in Claycomb medium (JRH Biosciences) supplemented with 10% fetal bovine serum, 0.1 mmol/L norepinephrine, 2 mmol/l‐glutamine, and 100 U/mL penicillin/streptomycin. Cells were then incubated at 37°C in 5% CO2.
Lentiviral Production and Transduction Experiment
A total of 5×106 HEK293T cells were seeded in a 10‐cm‐diameter dish 24 hours prior to transfection in the Dulbecco's Modified Eagle's medium (DMEM) supplement with 10% heat‐inactivated fetal bovine serum, penicillin (100 IU/mL), and streptomycin (100 μg/mL) in a 5% CO2 incubator. The culture medium was replaced by the antibiotic‐free medium 2 hours prior to transfection. A total of 12 μg of plasmid DNA (3 μg of expressing plasmid, 9 μg of packaging plasmids) and 36 μL of Lipofectamine2000 were diluted in 1.5 mL of Opti‐MEM1 medium without serum. Plasmids and diluted Lipofectamine2000 were mixed together and incubated for another 20 minutes; after that, the mixture was added to culture dishes dropwise, and incubated overnight. On the second day, fresh complete medium was fed into cells containing sodium pyruvate. The medium was collected after 62‐hour transfection following centrifugation and filtered (0.45‐μm filter; Millex‐HA; Millipore). The supernatants were stored at −80°C. Titers were 5 to 10×106 transduction U/mL, as determined in the 293T cells.
XBP1 Splicing Assay Using Restriction Endonuclease Approach
The abundance of XBP1 versus spliced XBP1 (XBP1s) can also be measured using PCR and the restriction endonuclease Pst1. Unspliced XBP1 mRNA contains an intron that contains an Pst1 restriction site, whereas XBP1s mRNA, defined by the removal of this intron, does not contain this Pst1 site. Therefore, in rats Pst1 digestion cleaves XBP1 into 2 smaller fragments (186 and 290 bp), whereas XBP1s is not cleaved by Pst1 and migrates as a 450‐bp fragment. Total RNA isolated from cultured neonatal rat cardiomyocytes and first‐strand cDNA synthesis were analyzed on a 2% agarose gel containing ethidium bromide before and after exposure to Pst1 to quantify XBP1 splicing.
Corin Protein Measurement in Explanted Human Hearts Using Human Corin ELISA
Human left ventricle total proteins were extracted from 5 nonfailing hearts, 9 high corin mRNA failing hearts (corin mRNA higher than nonfailing control hearts), and 5 low corin mRNA failing hearts (corin mRNA lower than nonfailing control hearts) that were randomly selected from the original sample of 100 after stratification into low/high corin from real‐time PCR analysis. Human corin protein was measured in left ventricular lysates using the Human Corin Quantikine ELISA Kit (R&D Systems, Minneapolis, MN).
Cell Culture Systems
Two independent cell‐culture systems were utilized to determine the effect of ER stress on corin messenger RNA:neonatal rat cardiomyocytes and immortalized adult murine atrial myocytes (HL‐1) cells. For induction of ER stress, the cells were treated with 0.1 μmol/L TG (Sigma Chemical Co, St Louis, MO). Serum‐free DMEM/M‐199 was used as control treatment. After the indicated time of treatment, the cells were harvested for extraction of total RNA and reverse‐transcription into cDNA.
IRE1 Overexpression in HL‐1 Cells
HIV‐based lentiviral overexpression of mouse IRE1 plasmid EX‐Mn12120‐Lv141 and control vector EX‐eGFP‐Lv105 were purchased from GeneCopoeia (Rockville, MD). Lentiviral plasmids were propagated in and purified from Stbl2 competent cells (Invitrogen, Carlsbad, CA) and co‐transfected with the lentiviral packaging plasmids pLP1, pLP2, pLP/VSVG into HEK293T cells for virus production. ViralPower packaging plasmids pLP1, pLP2, pLP/VSVG were kind gifts from Dr Frank S. Lee (University of Pennsylvania). The control vector expressing green fluorescent protein was used as a measure of transduction efficiency. Neonatal mouse cardiomyocytes were plated in 6‐well plates with complete medium 1 day before infection. On the second day, cells were infected with IRE1 overexpressed lentivirus containing 8 μg/mL polybrene (Sigma Chemical Co, St Louis, MO) and incubated overnight. After that, the cells were fed with fresh complete medium. Cells were harvested 72 hours after transduction and the total RNA was extracted as previously described.
IRE1 Gene Knockdown With shRNA
Mouse IRE1 SureSilencing shRNA plasmid and nontargeted negative control (KM36937G) were purchased from Qiagen. HL‐1 cells were seeded onto 6‐well dishes at the recommended density and grown to 60% to 80% confluence before transfection. IRE1 shRNAs and the nontargeted negative control plasmids were transfected into cells by using Lipofectamine2000 as described above.
Use of Actinomycin D to Arrest Transcription Prior to TG
In order to determine whether the decrease in corin mRNA after induction of ER stress was related to alterations in gene transcription or RIDD‐mediated RNA degradation, we performed experiments in which we utilized actinomycin D to halt transcription. Actinomycin D was administered to HL‐1 cells at a concentration of 1 μg/mL for a total of 3 hours prior to the administration of TG. Control cells did not receive actinomycin D. After this period, cells were exposed to TG added to medium at a concentration of 0.1 μmol/L for a total of 6 hours. Cells were harvested and RNA extracted and converted to cDNA for analysis of corin and XBP1s relative abundance. The relative mRNA abundance of genes of interest was compared to cells at baseline that did not receive TG.
Statistical Analysis
Gene expression data in the UK database
Messenger RNA transcripts of the genes were quantified using real‐time PCR with TaqMan chemistry. Gene expression was normalized to 18S ribosomal RNA levels and data are presented as dCt values.
Gene expression data in UPENN explanted heart database
The gene expression data from the UPENN explanted heart database is expressed as normalized RNA. Reported in this study, we used a combination of Affymetrix normalization and GeneExpress MAS 5.0 (Gene Logic, Inc). Affymetrix normalization is a global scaling method, where the overall intensity of the chip affects the scaling factor. The top and bottom 2% of all expression intensity values on the chip are discarded, and the remaining 96% of values are used to compute the “trimmed mean.” The scaling factor is then calculated using this adjusted mean (scaling factor=100/trimmed mean), and this single scale factor is applied to the expression values for every fragment on a given chip to produce normalized expression intensity. The MAS 5.0 normalization was designed by Affymetrix to more accurately reflect the distribution of data from microarray experiments. Expression values are calculated based on the hybridization.
Gene expression correlation analysis
In both the UPENN and UK Heart databases, nonparametric correlation analysis of gene expression was performed using Spearman's rank test and Graph Pad's Prism 6 software.
Target gene expression determination and analysis in cell‐based experiments
Messenger RNA transcripts of the genes of interest were determined using real‐time PCR with TaqMan chemistry. Each experiment was performed in triplicate. Data were normalized to 18S ribosomal RNA and analyzed using the comparative Ct or standard curve method. Results are given as mean± SEM. Data were compared using unpaired parametric t tests assuming unequal variance. In the TG experiments, gene‐specific relative messenger RNA abundance was compared at each time point to the corresponding gene‐specific relative messenger RNA abundance at baseline (prior to TG exposure). Each time point was considered a separate experiment and we employed unpaired parametric t tests to assess the significance of differences in relative mRNA abundance at these time points. A 2‐sided P‐value ≤0.05 was considered statistically significant.
Results
Corin mRNA Is Negatively Associated With ANP, BNP, and XBP1s mRNA
The relationship of corin mRNA versus ANP or BNP mRNA was plotted for the UPENN sample (Figure 1A) and UK sample (Figure 1B). In both the ischemic and nonischemic samples, there was a significant inverse correlation between BNP mRNA and corin mRNA; more specifically, as BNP messenger RNA increased, there was a corresponding associated decrease in corin RNA. A similar finding was identified in the UK sample for both ANP as well as BNP and its inverse correlation with corin mRNA (Figure 1B). Also demonstrated in the UK heart collection, BNP mRNA increases were positively correlated with GRP78 mRNA. XBP contains an intron that is excised by the endoribonuclease activity of IRE1 into XBP1s. Thus, XBP1s RNA abundance is a marker for IRE1 activation. In the UK sample, there was a significant inverse correlation between corin mRNA and XBP1s (Figure 1B). When we limited our analysis in the UK collection to 20 samples taken from left‐ventricular assist device–supported patients at the time of cardiac transplantation, similar trends were observed as in the entire sample, although correlation testing did not achieve formal statistical significance.
Figure 1.
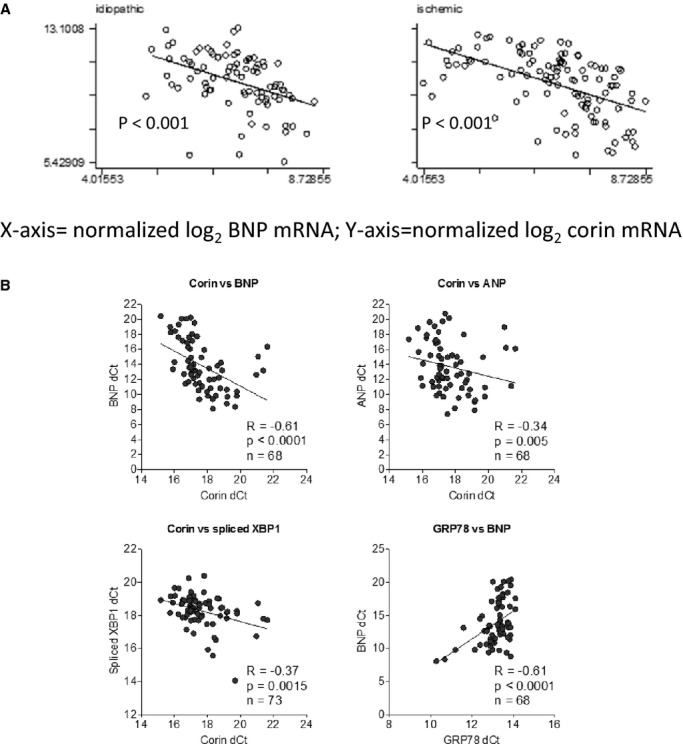
Corin mRNA inversely correlates with Atrial natriuretic peptide (ANP )/Brain natriuretic peptide (BNP ) mRNA in chronic, advanced human systolic heart failure. We examined the relationship of corin mRNA to ANP and BNP RNA (A) in the UPENN database in the nonischemic cardiomyopathy (n=120) and nonischemic (n=80) samples. Data are expressed as log2 normalized RNA abundance (corin on x‐axis; BNP on y‐axis.) Corin mRNA was significantly inversely correlated with either ANP or BNP mRNA. The UK heart collection demonstrated a similar significant inverse relationship between corin mRNA and ANP or BNP mRNA (B). In addition, BNP mRNA was significantly positively correlated with GRP78 mRNA, and corin mRNA was significantly inversely correlated with spliced X‐box protein 1 (XBP1).
Reduced Corin RNA Is Associated With Highest BNP mRNA Levels
Using 100 randomly sampled left ventricle transmural samples hearts from the Penn Explanted Heart Library, we analyzed ANP, BNP, and corin expression using real‐time PCR as compared to 10 nonfailing control hearts. We discovered that in 53 of the samples, corin mRNA was increased (high corin) and in the remainder (N=47) corin mRNA was decreased (low corin group) (Figure 2A). The low‐corin group demonstrated significantly higher expression of ANP (Figure 2B) and BNP (Figure 2C) when compared to the high‐corin group. Corin protein abundance normalized to total protein in samples from left ventricle apex measures using the commercially available ELISA was significantly lower in the low‐corin RNA as compared to high‐corin RNA group (Figure 2D). When comparing the low‐corin to the high‐corin group, there was evidence of increased ER stress in the low‐corin group as demonstrated by significantly higher expression of ATF4 (P<0.05) and CHOP (P<0.05), and corin mRNA was significantly negatively correlated with CHOP mRNA in the total group (R=−0.3267; P=0.0084).
Figure 2.

Corin RNA and protein in heart failure compared to nonfailing controls. Using a sample of 100 randomly selected samples of transmural sample hearts from the UPENN collection consisting of left‐ventricular (LV) tissue, we compared Atrial natriuretic peptide (ANP), Brain natriuretic peptide (BNP), and corin gene expression compared to nonfailing controls (n=10) using real‐time polymerase chain reaction. We discovered that corin mRNA was increased in 53 of the hearts compared to nonfailing controls (high corin mRNA group) and decreased in 47 hearts (low corin mRNA group) (A). The low corin mRNA group, when compared to the high corin mRNA group, demonstrated significantly higher expression of ANP (B) and BNP (C). Corin protein in samples from LV apex was significantly lower in the low corin RNA as compared to the high corin RNA group (D), demonstrating that changes in corin mRNA resulted in expected changes in corin protein. *P<0.05, **P<0.01, ***P<0.001. Error bars are SEM.
TG‐Induced ER Stress Causes Time‐Dependent Corin mRNA Decline
As demonstrated in Figure 3, TG induced ER stress in neonatal rat cardiomyocytes as demonstrated by significant time‐dependent increases in GRP78, GRP94, CHOP, and ATF4, including evidence of IRE1 endoribonuclease activity as evidenced by a significant increase in XBP1 splicing (XBP1s mRNA). In contrast, there was a highly significant, time‐dependent decrease in corin mRNA. We also examined IRE1 activation measuring XBP1s using restriction endonuclease (Pst1) digestion and PCR amplification to distinguish nonspliced XBP1 from spliced (XBP1s) (Figure 3G and 3H), which demonstrated significant increases in XBP1s in response to TG. Using an identical experimental design, TG induced ER stress in immortalized, adult murine atrial cardiomyocytes (HL‐1 cells) (Figure 4) as indicated by post TG increases in GRP78, GRP94, CHOP, ATF4, and XBP1s. There was a significant time‐dependent decrease in corin mRNA. In all 3 cell lines, when we repeated the experiments using tunicamycin rather than TG to induce ER stress, we observed similar results (data not shown).
Figure 3.
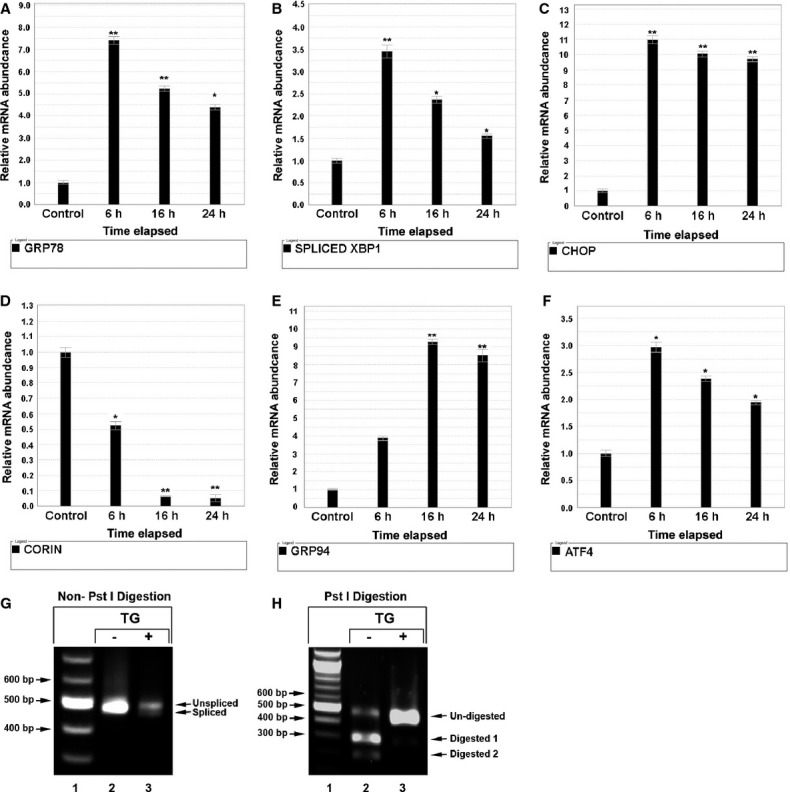
Thapsigargin (TG)‐induced ER stress causes and time‐dependent corin mRNA decay in neonatal rat cardiomyocytes. Neonatal rat cardiomyocytes were plated in 6‐well plates. Twenty‐four hours later, cells were cultured in serum‐free DMEM‐M199 (4:1) with and without 0.1 μmol/L TG for 6, 16, 24, and 30 hours, separately. Total RNA was extracted and reverse‐transcribed into cDNA and gene expression of the following genes was assessed by RT‐PCR (A through F): (A) GRP78; (B) XBP1 splicing; (C) CHOP; (D) corin; (E) GRP94; and (F) ATF4. Compared with non‐TG‐treated controls, cells treated with TG demonstrated increased ER stress as evidenced by significant increases in GRP78, GRP94, ATF4, and CHOP mRNA, with a corresponding significant increase in XBP1 splicing based on the increase in the spliced form (intron removed) XBP1s mRNA; *P<0.05, **P<0.01. Corin mRNA demonstrated a significant, time‐dependent decrease in the cells treated with TG. We also analyzed XBP splicing using PCR, restriction digestion with Pst1, and gel electrophoresis. Fragments of XBP1 were amplified and resolved in 2% agarose gel. PCR products were not digested by Pst I (G). Lane 1: DNA ladder; Lane 2: TG untreated; Lane 3: TG treated. XBP1 fragment: Spliced=450 bp; unspliced=476 bp. PCR products were digested by Pst I (H). Lane 1: DNA ladder; Lane 2: TG untreated; Lane 3: TG treated. Spliced rat XBP1 fragment was 450 bp, while unspliced rat XBP1 appears as 2 fragments of 186 and 290 bp due to the presence of the Pst I restriction site, which is contained in the intronic segment that is removed by IRE1 endonuclease activity. Error bars represent SEM. ATF6 indicates activating transcription factor‐6; ER, endoplasmic reticulum; IRE1, inositol‐requiring protein 1; RT‐PCR, real‐time polymerase chain reaction; CHOP, C/EBP homology protein; DMEM, Dulbecco's Modified Eagle's medium; XBP1, X‐box protein 1.
Figure 4.

Thapsigargin (TG)‐induced ER stress and time‐dependent corin mRNA decay in adult mouse atrial cardiomyocytes (HL‐1) cells. HL‐1 cells were plated in 6‐well plates. Twenty‐four hours later, cells were cultured in serum‐free Claycomb medium with and without thapsigargin (TG) 0.1 μmol/L for 6, 16, 24, 30 hours, separately. Total RNA was extracted and reverse‐transcribed into cDNA and mRNA abundance of the following genes was assessed by RT‐PCR (Figure 4A through 4F): (A) GRP78; (B) XBP1 splicing; (C) CHOP; (D) corin; (E) GRP94; (F) ATF4. Compared with non‐TG‐treated HL‐1 control cells, HL‐1 cells treated with TG demonstrated increased ER stress as evidenced by significant increases in GRP78, GRP94, ATF4 and CHOP mRNA, with a corresponding significant increase in XBP1 splicing based on the increase in the spliced form (intron removed) XBP1s mRNA; *P<0.05, **P<0.01. Corin mRNA demonstrated a significant, time‐dependent decrease in the TG‐treated cells. Error bars represent standard error of the mean. ATF indicates activating transcription factor; ER, endoplasmic reticulum; HL‐1, adult murine atrial myocytes; RT‐PCR, real‐time polymerase chain reaction; XBP1, X‐box protein 1.
IRE1 Overexpression Activates IRE1 Endoribonuclease Activity, Resulting in Decreased Corin mRNA
Overexpression of IRE1 leads to dimerization, transphosphorylation, and activation.27 As demonstrated in Figure 5, cells infected with IRE1 demonstrated significant increases in ER stress (increased GRP78, GRP94, ATF4, and CHOP), and increased IRE1 endoribonuclease activity as evidenced by a significant increase in XBP1s. This was also accompanied by a significant decrease in corin mRNA.
Figure 5.
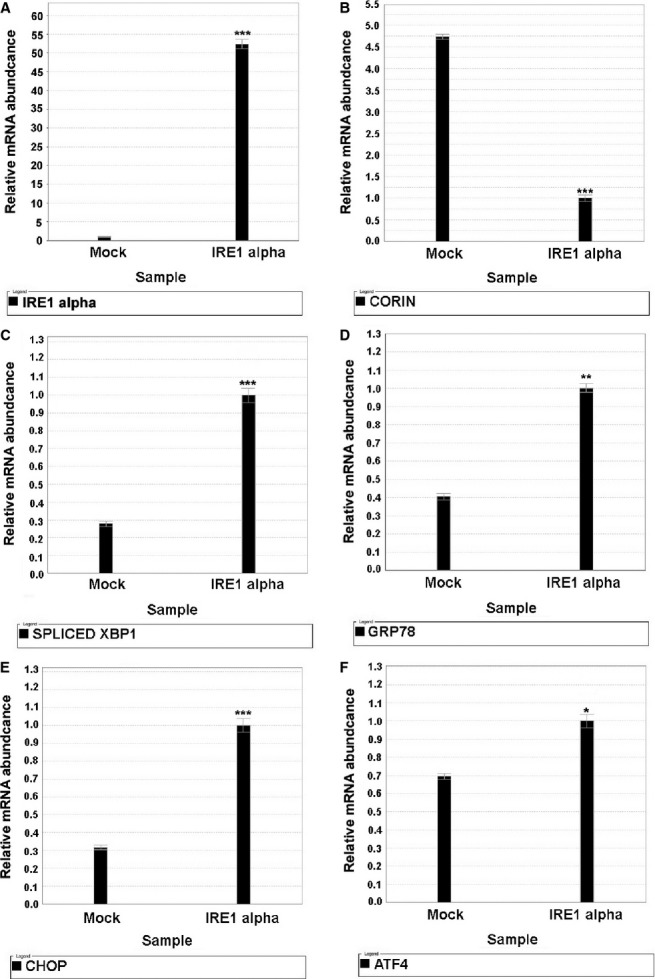
IRE1‐gene overexpression. Neonatal murine cardiomyocytes were plated in 6‐well plates with complete medium 1 day before infection. On the second day, cells were infected with IRE1‐overexpressed lentivirus containing 8 μg/mL polybrene and incubated overnight. After that, the cells were fed with fresh complete medium. Cells were harvested at 72 hours after transduction and the total RNA was extracted. Quantitative polymerase chain reaction was conducted using specific TaqMan mRNA probes: (A) IRE1 alpha; (B) Corin; (C) Spliced XBP1; (D) GRP78; (E) CHOP; (F) ATF4 mRNA were quantified by normalization to 18S ribosomal RNA. IRE1‐overexpression activated IRE1 endoribonuclease activity, resulting in a significant increase in XBP1s and a parallel significant decrease in corin mRNA. Note: Compared with mock group, *P<0.05; **P<0.01; ***P<0.001. Mock: cells transduced with empty lentivirus vehicle; IRE1‐: cells transduced with IRE1‐overexpressed lentivirus. Error bars represent standard error of the mean. ATF indicates activating transcription factor; CHOP, ; IRE1, inositol‐requiring protein 1; XBP1, X‐box protein 1.
IRE1 Knockdown Attenuates the Severity of Corin RNA Decay After Induction of ER Stress With TG
HL‐1 cells were transfected with 2 μg mammalian expression plasmid containing IRE1 shRNA or nontargeted negative control. Cells were then treated for 12 hours with TG. As demonstrated in Figure 6, IRE1 shRNA silencing reduced XBP1s and attenuated corin mRNA decay in response to exposure of cells to TG.
Figure 6.
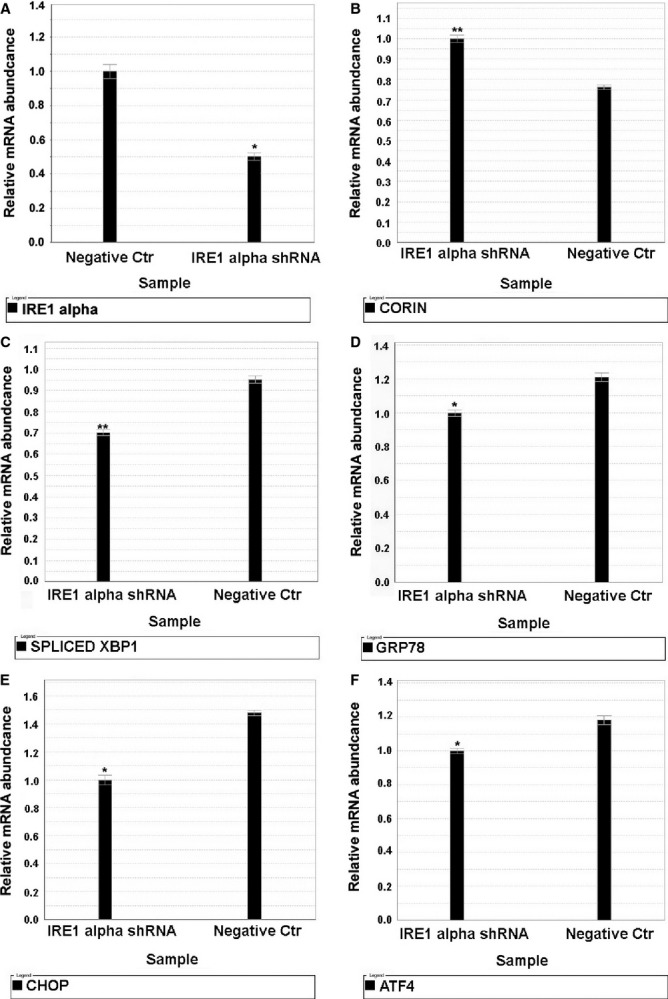
IRE1 shRNA knockdown attenuates corin mRNA decay. HL‐1 cells were transfected with 2 μg mammalian expression plasmid containing IRE1 shRNA or nontargeted negative control (Negative Ctr), separately. Cells were exposed to thapsigargin 0.1 μmol/L for 12 hours. Thereafter, cellular total RNA was isolated and quantitative polymerase chain reaction was conducted using specific TaqMan probes: (A) IRE1 alpha; (B) Corin; (C) Spliced XBP1; (D) GRP78; (E) CHOP; (F) ATF4 mRNA were quantified by normalization to 18S ribosomal RNA. Cells with IRE1 shRNA knockdown prior to exposure to thapsigargin (12 hours) demonstrated less XBP1 splicing and increased corin mRNA values compared to cells transfected with control plasmid and also exposed to 12 hours thapsigargin. *P<0.05 and **P<0.01. Error bars represent SEM. ATF indicates activating transcription factor; CHOP, C/EBP homology protein; HL‐1, adult murine atrial myocytes; IRE1, inositol‐requiring protein 1; XBP1, X‐box protein 1.
Similar Corin mRNA Decline in Cells Pretreated With Actinomycin D
As demonstrated in Figure 7, the decrease in corin mRNA was similar in cells pretreated or not pretreated with actinomycin D for a total of 3 hours prior to the administration of TG for 12 hours. The cells pretreated with actinomycin D demonstrated lower abundance of XBP1s mRNA as compared to cells not pretreated with actinomycin D. At 12 hours, both cells demonstrated an increase in XBP1s. As demonstrated in Figure 7A and 7B, the decrease in corin mRNA was similar in cells pretreated or not pretreated with actinomycin D for a total of 3 hours prior to the administration of TG for 12 hours. The cells pretreated with actinomycin D demonstrated lower abundance of XBP1s mRNA as compared to cells not pretreated with actinomycin D.
Figure 7.
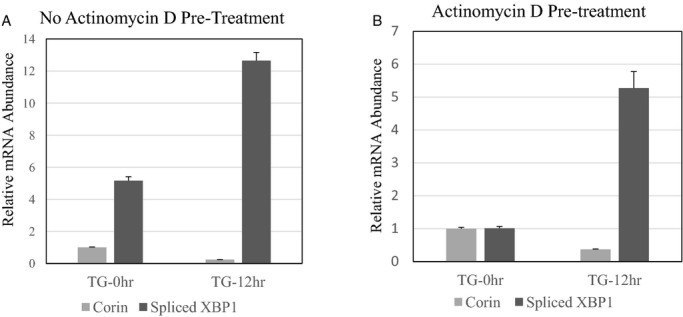
Effect of actinomycin D prior to thapsigargin (TG). HL‐1 cells were incubated with or without actinomycin D at a concentration of 1 μg/μL 3 hours prior to administration of TG (0.1 μmol/L) for 12 hours. Cells were harvested and the relative abundance of corin mRNA and XBP1s (normalized to 18S rRNA) was compared to cells not receiving TG. We compared the changes in corin and XBP1s in cells not treated (A) or treated with actinomycin D (B) prior to the administration of TG to the medium. We compared relative mRNA abundance (normalized to 18S ribosomal RNA) of corin and XBP1s 12 hours after TG as compared to baseline. As demonstrated in (B), the decrease in corin mRNA and corresponding increase in XBP1s (P<0.001), a marker of IRE1 endoribonuclease activity, was similar in cells pretreated with actinomycin D as compared to cells not pretreated with actinomycin D (B) prior to the induction of ER stress with TG. Baseline XBP1s was higher in cells not pretreated with actinomycin D. The error bars represent SEM for the relative mRNA expression. (P<0.001 for baseline compared to 12 hours). Error bars represent SEM. ER indicates endoplasmic reticulum; HL‐1, adult murine atrial myocytes; IRE1, inositol‐requiring protein 1; XBP1s, spliced X‐box protein 1.
Evidence of RIDD‐Substrate mRNA Decay in Advanced Human Systolic Heart Failure
We examined the expression profile of the GEO explanted heart database to determine the pattern of messenger RNAs significantly reduced at least 2‐fold as compared to nonfailing control hearts. We identified a total of 267 mRNAs that were reduced 2‐fold or greater as compared to controls. Utilizing a database analysis, we characterized the cellular location of these mRNAs. As demonstrated in Figure 8, a significant majority (73.8%) of these mRNAs encoded membrane proteins that are targeted to the ER and presumptive targets for RIDD.
Figure 8.
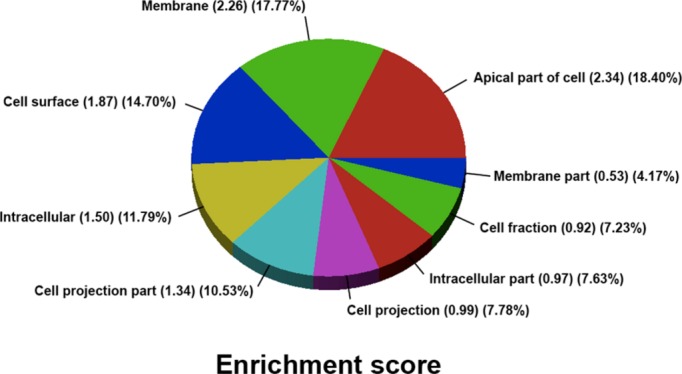
We examined the expression profile of the UPENN Explanted Heart Database consisting of 200 explanted heart samples divided between nonischemic (n=120) and nonischemic (n=80) samples to determine the pattern of messenger RNAs significantly reduced at least 2‐fold as compared to nonfailing control hearts. We identified a total of 267 mRNAs that were reduced 2‐fold or greater as compared to controls. Utilizing a database analysis, we characterized the cellular location of these mRNAs. As demonstrated in this figure, a significant majority (73.8%) of these mRNAs encoded membrane proteins that are targeted to the endoplasmic reticulum and presumptive targets for regulated, inositol‐requiring‐1‐dependent messenger RNA decay.
Discussion
The data from our in vitro experiments support the hypothesis that ER stress can activate the targeted IRE1‐dependent mRNA decay pathway, and this can lead to corin mRNA decay. The data from the human heart libraries demonstrate that BNP transcription is positively correlated with ER stress (GRP78) and that XBP1s, a marker of IRE1 endoribonuclease activity, is inversely correlated with corin mRNA.
The in vitro experiments were conducted in 3 different myocyte cell lines: neonatal mouse cardiomyocytes, neonatal rat cardiomyocytes, and HL‐1 cells. The latter cells represent an adult atrial cardiomyocyte lineage to extend the results from neonatal to adult cardiomyocytes. When we induced ER stress by the addition of TG, we observed marked activation of the ER stress response and a time‐dependent reduction in corin mRNA. To determine whether this represented mRNA decay as opposed to altered transcriptional events, we next pretreated HL‐1 cells with actinomycin D to halt transcription prior to inducing ER stress by TG. We observed a similar magnitude of corin mRNA decay, suggesting that the reduction was related to decay or destruction of corin mRNA already produced. The dependence on IRE1‐α was supported by the ability to replicate this corin mRNA decay by overexpressing IRE1‐α, which results in IRE1‐α endoribonuclease activation by autodimerization and transphosphorylation as evidenced by the significant increase in XBP1s. Likewise, IRE1 knockdown (incomplete) by hnRNA directed at IRE1‐α RNA was attenuated to the degree of corin mRNA decay after inducing ER stress. Together with our observations from the transcriptomes of the explanted human hearts, these data suggest that regulated, IRE1‐dependent decay (RIDD) contributes to corin mRNA and protein deficiency in advanced human heart failure. We propose that this may be a contributing mechanism of impaired processing of proBNP and propose that this deficiency in natriuretic peptide processing is an important element of heart failure progression in humans. Moreover, we observed a significant reduction in a family of mRNAs (not just corin mRNA) in advanced heart failure heavily weighted toward mRNAs that encode membrane proteins and are targeted to the ER prior to membrane insertion. This suggests that RIDD may be globally operative in advanced heart failure, and may influence other signaling cascades other than the natriuretic peptide family.
The unfolded protein response, also referred to as the ER stress response, is an evolutionarily conserved system that allows cells to cope with the demands of unfolded proteins.28 The ER stress response is coordinated by 3 proteins that are anchored in the ER membrane and each capable of “sensing” misfolded protein accumulation in the ER lumen:protein kinase RNA‐like ER kinase.29 Protein kinase RNA‐like endoplasmic reticulum kinase (PERK), IRE1, and activating transcription factor‐6 (ATF6)29: The accumulation of misfolded proteins in the ER lumen activates IRE1, resulting in activation of its endoribonuclease domain that is located on the cytosolic side of the ER membrane, allowing it to remove an intron in XBP mRNA, resulting in XBP1s. XBP1s devoid of the intron segment is translated effectively into a transcription factor that activates ER protein chaperone genes that assist protein folding in the ER lumen, such as GRP78 and GRP94, protein‐modification enzymes, and others assisting degradation of misfolded proteins.30
RIDD is a recently recognized component of the ER stress response.16–17,31 Sustained degrees of moderate ER stress lead to increased scaffolding of IRE1 and complex interactions with adaptor and moderator proteins that increases the promiscuity of IRE1's endoribonuclease activity.27 At this stage, IRE1‐endonucleocytic activity loses specificity for unspliced XBP1 and targets other messenger RNA's attached to ER‐membrane‐bound ribosomes to reduce global demands on protein folding in the ER lumen, and also targets mRNA's attached to ER‐membrane‐bound ribosomes. Our data suggest that corin mRNA is susceptible to this process and that activation of IRE1 and the unfolded protein response is present in advanced human heart failure. We hypothesize, therefore, that RIDD‐mediated corin mRNA degradation leads to corin protein deficiency, creating a molecular milieu favoring impaired natriuretic peptide pro‐hormone processing due to rate limitation of corin‐mediated pro‐natriuretic peptide pro‐hormone processing. We have not directly demonstrated the relationship between cardiac corin mRNA and protein deficiency and the degree of impaired natriuretic pro‐hormone processing in vivo in the present article, but offer it as a molecular hypothesis for a phenomenon (impaired natriuretic peptide pro‐hormone processing) that may contribute to heart failure progression by attenuating the compensatory actions of the endogenous NPS in persons with systolic heart failure.
Clinical Implications
Although some have argued to the contrary,32 others argue that adequate NP pro‐peptide processing is essential to the compensatory systemic and autocrine/paracrine actions of the NPS.9 A recent murine genetic model of heart failure examined the effect of corin overexpression on heart failure mortality and cardiac function.33 That study demonstrated that the corin overexpression mouse had a lower mortality, less adverse ventricular remodeling, improved cardiac function, and histologically less cardiac fibrosis than the wild‐type controls. Interestingly, in their control group, which developed heart failure but did not have corin overexpressed, the mice had lower corin RNA as compared to non‐heart‐failure controls. The authors interpreted this as reflecting reduced corin expression, but perhaps it also was the result of RIDD corin mRNA decay.
It is interesting that in the sample of 100 randomly sampled hearts from the UPENN Explanted Heart Collection, we demonstrated that approximately half of the hearts demonstrated a significant reduction in corin mRNA compared to nonfailing controls, whereas the remainder demonstrated an increase in corin mRNA as compared to the nonfailing controls. We hypothesize that the difference is explained by the presence of a more significant degree of overall ER stress and corresponding activation of RIDD in the hearts demonstrating corin mRNA reduction. A variety of factors contribute to ER stress in advanced heart failure including oxidative stress, excessive calcium in the cardiomyocyte cytoplasm due to reduced reuptake by the sarcoplasmic reticulum, and increased secretory demands, such as secretion of natriuretic peptides. It is interesting that the hearts with reduced corin had significantly higher BNP mRNA levels compared to the hearts with increased corin mRNA, which is consistent with increased secretory demands. This is supported by the finding in the UK Database that BNP relative mRNA abundance was significantly and positively correlated with GRP78 relative mRNA abundance; GRP78 is a marker for ER stress. This raises the intriguing possibility of identifying patients with excessive RIDD and corin mRNA decay using circulating ratios of processed/unprocessed BNP in plasma; perhaps modulating the degree of ER stress in such patients might be beneficial and improve natriuretic processing, thus restoring the compensatory actions of the endogenous NPS. Chemical chaperones have been demonstrated to moderate ER stress. Molecular chaperones, such as 4‐phenylbutyric acid and taurine ursodeoxycholic acid, are capable of assisting in protein folding and reducing ER stress.34–38 It has been demonstrated in animal models that the administration of either 4‐phenylbutyric acid or taurine ursodeoxycholic acid to a murine model of type II diabetes mellitus opposed the development of diabetes mellitus in association with reductions in β‐islet‐cell ER stress and improved insulin secretion in part due to a reduction in regulated IRE1‐dependent insulin mRNA decay in β‐islet cells.39
Conclusions
In summary, we present data supporting the hypothesis that sustained cardiomyocyte ER stress in advanced heart failure leads to corin mRNA decay due to the process of RIDD. Based upon these data, we hypothesize that RIDD‐mediated corin deficiency in advanced heart failure may contribute to impairing BNP (1‐108) processing and attenuated compensatory actions of the endogenous NPS as heart failure progresses.
Sources of Funding
This research was funded by the National Institutes of Health, R01HL091663. The research was supported by the NIHR Biomedical Research Unit in Cardiovascular Disease at Royal Brompton & Harefield NHS Foundation Trust and Imperial College London, and by Heart Research UK.
Disclosures
None.
Acknowledgments
We are grateful to our colleagues Thomas Cappola, MD and Kenneth Margulies, MD from the Perlman School of Medicine, University of Pennsylvania (UPENN) for providing heart tissue from the UPENN Human Heart Tissue Bank.
References
- 1.Burnett JC., Jr Novel therapeutic directions for the natriuretic peptides in cardiovascular diseases: what's on the horizon. J Cardiol. 2006; 48:235-241. [PMC free article] [PubMed] [Google Scholar]
- 2.Kuhn M. Cardiac and intestinal natriuretic peptides: insights from genetically modified mice. Peptides. 2005; 26:1078-1085. [DOI] [PubMed] [Google Scholar]
- 3.Potter LR, Abbey‐Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate‐dependent signaling functions. Endocr Rev. 2006; 27:47-72. [DOI] [PubMed] [Google Scholar]
- 4.Calvieri C, Rubattu S, Volpe M. Molecular mechanisms underlying cardiac antihypertrophic and antifibrotic effects of natriuretic peptides. J Mol Med. 2012; 90:5-13. [DOI] [PubMed] [Google Scholar]
- 5.Holtwick R, van Eickels M, Skryavin Baba HA, Bubikat A, Begrow F, Schneider MD, Garbers DL, Kuhn M. Pressure independent cardiac hypertrophy in mice with cardiomyocyte‐restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase A. J Clin Invest. 2003; 111:1399-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhn M. Molecular physiology of natriuretic peptide signalling. Basic Res Cardiol. 2004; 99:76-82. [DOI] [PubMed] [Google Scholar]
- 7.Knappe S, Wu F, Masikat MR, Morser J, Wu Q. Functional analysis of the transmembrane domain and activation cleavage of human corin: design and characterization of a soluble corin. J Biol Chem. 2003; 278:52363-52370. [DOI] [PubMed] [Google Scholar]
- 8.Wu Q. The serine protease corin in cardiovascular biology and disease. Front Biosci. 2007; 12:4179-4190. [DOI] [PubMed] [Google Scholar]
- 9.Dries DL. Process matters: emerging concepts underlying impaired natriuretic peptide system function in heart failure. Circ Heart Fail. 2011; 4:107-110. [DOI] [PubMed] [Google Scholar]
- 10.Giuliani I, Rieunier F, Larue C, Delagneau JF, Granier C, Pau B, Ferriere M, Saussine M, Cristol JP, Dupuy AM, Merigeon E, Merle D, Villard S. Assay for measurement of intact B‐type natriuretic peptide prohormone in blood. Clin Chem. 2006; 52:1054-1061. [DOI] [PubMed] [Google Scholar]
- 11.Dries DL, Ky B, Wu AH, Rame JE, Putt ME, Cappola TP. Simultaneous assessment of unprocessed ProBNP1‐108 in addition to processed BNP32 improves identification of high‐risk ambulatory patients with heart failure. Circ Heart Fail. 2010; 3:220-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heublein DM. Immunoreactivity and cyclic GMP activating actions of various molecular forms of human B‐type natriuretic peptide. Hypertension. 2007; 49:1114-1119. [DOI] [PubMed] [Google Scholar]
- 13.Liang F, O'Rear J, Schellenberger U, Tai L, Lasecki M, Schreiner GF, Apple FS, Maisel AS, Pollitt NS, Protter AA. Evidence for functional heterogeneity of circulating B‐type natriuretic peptide. J Am Coll Cardiol. 2007; 49:1071-1078. [DOI] [PubMed] [Google Scholar]
- 14.Tsutamoto T, Kanamori T, Wada A, Kinoshita M. Uncoupling of atrial natriuretic peptide extraction and cyclic guanosine monophosphate production in the pulmonary circulation in patients with severe heart failure. J Am Coll Cardiol. 1992; 20:541-546. [DOI] [PubMed] [Google Scholar]
- 15.Tsutamoto T, Wada A, Maeda K, Hisanaga T, Maeda Y, Fukai D, Ohnishi M, Sugimoto Y, Kinoshita M. Attenuation of compensation of endogenous cardiac natriuretic peptide system in chronic heart failure: prognostic role of plasma brain natriuretic peptide concentration in patients with chronic symptomatic left ventricular dysfunction. Circulation. 1997; 96:509-516. [DOI] [PubMed] [Google Scholar]
- 16.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1‐dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009; 186:323-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollien J, Weissman JS. Decay of endoplasmic reticulum‐localized mRNAs during the unfolded protein response. Science. 2006; 313:104-107. [DOI] [PubMed] [Google Scholar]
- 18.Lipson KL, Ghosh R, Urano F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta‐cells. PLoS One. 2008; 3:e1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ackermann U, Deliva RD. Reduced baroreceptor sensitivity during hypotension in ANP‐knockout mice. Can J Physiol Pharmacol. 2001; 79:201-205. [PubMed] [Google Scholar]
- 20.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004; 110:705-712. [DOI] [PubMed] [Google Scholar]
- 21.Toth A, Nickson P, Mandl A, Bannister ML, Toth K, Erhardt P. Endoplasmic reticulum stress as a novel therapeutic target in heart diseases. Cardiovasc Hematol Disord Drug Targets. 2007; 7:205-218. [DOI] [PubMed] [Google Scholar]
- 22.Sawada T, Minamino T, Fu HY, Asai M, Okuda K, Isomura T, Yamazaki S, Asano Y, Okada K, Tsukamoto O, Sanada S, Asanuma H, Asakura M, Takashima S, Kitakaze M, Komuro I. X‐box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE‐like element in cardiomyocytes. J Mol Cell Cardiol. 2010; 48:1280-1289. [DOI] [PubMed] [Google Scholar]
- 23.Dries DL, Margulies KM, Cappola TP. Corin gene expression inversely related to increased BNP expression in advanced cardiomyopathy: setting the stage for inefficient natriuretic peptide processing in heart failure? Circulation. 2005; 112:637 [Google Scholar]
- 24.Felkin LE, Taegtmeyer AB, Barton PJ. Real‐time quantitative polymerase chain reaction in cardiac transplant research. Methods Mol Biol. 2006; 333:305-330. [DOI] [PubMed] [Google Scholar]
- 25.Felkin LE, Lara‐Pezzi EA, Hall JL, Birks EJ, Barton PJ. Reverse remodelling and recovery from heart failure are associated with complex patterns of gene expression. Cardiovasc Transl Res. 2011; 4:321-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr Hl‐1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:2979-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hetz C, Glimcher LH. Fine‐tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell. 2009; 35:551-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007; 8:519-529. [DOI] [PubMed] [Google Scholar]
- 29.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000; 101:451-454. [DOI] [PubMed] [Google Scholar]
- 30.Merksamer PI, Papa FR. The UPR and cell fate at a glance. J Cell Sci. 2010; 123:1003-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009; 457:736-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickey DM, Potter LR. ProBNP(1‐108) is resistant to degradation and activates guanylyl cyclase‐A with reduced potency. Clin Chem. 2011; 57:1272-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gladysheva IP, Wang D, McNamee RA, Houng AK, Mohamad AA, Fan TM, Reed GL. Corin overexpression improves cardiac function, heart failure, and survival in mice with dilated cardiomyopathy. Hypertension. 2013; 61:327-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arakawa T, Ejima D, Kita Y, Tsumoto K. Small molecule pharmacological chaperones: from thermodynamic stabilization to pharmaceutical drugs. Biochim Biophys Acta. 2006; 1764:1677-1687. [DOI] [PubMed] [Google Scholar]
- 35.Asmellash S, Stevens JL, Ichimura T. Modulating the endoplasmic reticulum stress response with trans‐4,5‐dihydroxy‐1,2‐dithiane prevents chemically induced renal injury in vivo. Toxicol Sci. 2005; 88:576-584. [DOI] [PubMed] [Google Scholar]
- 36.Cao SS, Zimmermann EM, Chuang BM, Song B, Nwokoye A, Wilkinson JE, Eaton KA, Kaufman RJ. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology. 2013; 144:989-1000.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Almeida SF, Picarote G, Fleming JV, Carmo‐Fonseca M, Azevedo JE, de Sousa M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J Biol Chem. 2007; 282:27905-27912. [DOI] [PubMed] [Google Scholar]
- 38.Hu WK, Liu R, Pei H, Li B. Endoplasmic reticulum stress‐related factors protect against diabetic retinopathy. Exp Diabetes Res. 2012; 2012:507986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006; 313:1137-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]