

Abstract
Background
Identifying individuals at risk for sudden cardiac death (SCD) is of critical importance. Electrocardiographic (ECG) deep terminal negativity of P wave in V1 (DTNPV1), a marker of left atrial abnormality, has been associated with increased risk of all‐cause and cardiovascular mortality. We hypothesized that DTNPV1 is associated with increased risk of sudden cardiac death (SCD).
Methods and Results
This analysis included 15 375 participants (54.1±5.8 years, 45% men, 73% whites) from the Atherosclerosis Risk in Communities (ARIC) study. DTNPV1 was defined from the resting 12‐lead ECG as presence of biphasic P wave (positive/negative) in V1 with the amplitude of the terminal negative phase >100 μV, or one small box on ECG scale. After a median of 14 years of follow‐up, 311 cases of SCD occurred. In unadjusted Cox regression, DTNPV1 was associated with an 8‐fold increased risk of SCD (HR 8.21; [95%CI 5.27 to 12.79]). Stratified by race and study center, and adjusted for age, sex, coronary heart disease (CHD), and ECG risk factors, as well as atrial fibrillation (AF), stroke, CHD, and heart failure (HF) as time‐updated variables, the risk of SCD associated with DTNPV1 remained significant (2.49, [1.51–4.10]). DTNPV1 improved reclassification: additional 3.4% of individuals were appropriately reclassified into a higher SCD risk group, as compared with traditional CHD risk factors alone. In fully adjusted models DTNPV1 was associated with increased risk of non‐fatal events: AF (5.02[3.23–7.80]), CHD (2.24[1.43–3.53]), HF (1.90[1.19–3.04]), and trended towards increased risk of stroke (1.88[0.99–3.57]).
Conclusion
DTNPV1 is predictive of SCD suggesting its potential utility in risk stratification in the general population.
Keywords: electrocardiogram, risk stratification, sudden cardiac death
Introduction
In spite of declining overall cardiovascular mortality,1 the incidence of sudden cardiac arrest (SCA) remains high.2 Only a minority of the out‐of‐hospital SCA victims achieved meaningful survival.3–4 In more than half of the cases, SCA is the first manifestation of the cardiovascular disease (CVD).5 Therefore, identifying individuals at risk of sudden death, and subsequently implementing prevention strategies are of critical importance.6 Ventricular fibrillation remains the main cause of SCA.3,7–8 While the electrophysiological substrate of SCA has been extensively studied during the last 3 decades, knowledge of cardiac arrhythmia mechanisms has not been adequately translated into risk stratification for SCA.
Electrocardiographic abnormal P terminal force in V1 (PTFV1) is a measure of compromised inter‐atrial conduction9 due to LA abnormalities.10–11 Previous studies have shown that the presence of abnormal PTFv1 is predictive of AF12 and stroke.13 In addition to associations with the diseases of the upper chambers of the heart and their consequences, left ventricular hypertrophy (LVH)14 is known to be associated with abnormal PTFV1. Recently we showed that diffuse interstitial LV fibrosis (likely via associated LA fibrosis15) can affect and impair interatrial conduction, leading to characteristically abnormal PTFV1.16 PTFV1 is associated with heart failure (HF) hospitalizations and death.17 Development of fibrosis in both upper and lower chambers of the heart could be a unifying mechanism, characterizing the structural heart disease continuum. We recently found that a simplified ECG metric of abnormal PTFV1, namely deep terminal negativity of P wave in V1 (DTNPV1), was independently associated with increased risk of all cause and CVD mortality, as well as death due to ischemic heart disease.18 We hypothesized that DTNPV1 would also predict risk of SCD.
Methods
Study Population
The Atherosclerosis Risk in Communities (ARIC) study is a prospective cohort study designed to identify risk factors, progression, and outcomes of atherosclerosis in the community. From 1987 to 1989, 15 792 male and female participants aged 45 to 64 were recruited by probability sampling from 4 US communities (Forsyth County, NC; suburban Minneapolis, MN; Washington County, MD; and Jackson, MS). Details of the enrollment process and study procedures are fully described elsewhere.19 We excluded participants with reported race other than white or black, blacks in the Minnesota and Washington County cohorts (for appropriate stratification by race and study center) (n=47), prevalent AF or flutter (n=37), atrial pacing, unreadable ECGs, and missing clinical covariates relevant to the current analysis (n=333). A final study population consisted of 15 375 participants. The study was approved by the institutional review boards of all participating institutions, and all participants gave informed consent.
Definition of Prevalent Cardiovascular Disease and Diabetes Mellitus
Prevalent CHD at baseline was defined as a history of intermittent claudication, angina, or myocardial infarction (MI), diagnosed by Rose questionnaire,20 a physician diagnosis of myocardial infarction or stroke, history of coronary revascularization, ECG evidence of MI as defined by Minnesota code.21 Prevalent HF was defined as a self‐reported current intake of HF medication or evidence of manifest HF as defined by the Gothenburg criteria22 stage 3, which require the presence of specific cardiac and pulmonary symptoms, as well as medical treatment of HF. Prevalent MI was defined as self‐reported MI, or ECG evidence of MI as defined by Minnesota code.21 Prevalent stroke was diagnosed by the ARIC stroke and transient ischemic attack (TIA) diagnostic algorithm.23 Prevalent diabetes mellitus was defined as a non‐fasting glucose level of at least 140 mg/dL, a history of diabetes, or the current use of diabetes medications.
ECG Recording and Measurement of Deep Terminal Negativity of P Wave in V1
Standard 12‐lead ECGs were digitally acquired using MAC Personal Computer electrocardiograph (Marquette Electronics, Milwaukee, WI) at Epidemiological Cardiology Research Center (EPICARE), Wake Forest School of Medicine, Winston Salem, NC. All ECGs were initially inspected visually for technical errors and inadequate quality, then automatically processed with the GE Marquette 12‐SL program 2001 version (GE Marquette, Milwaukee, WI). DTNPV1 was defined as the presence of biphasic P wave (positive/negative) in V1 with the amplitude of the terminal negative phase >100 μV, or one small box on ECG scale (Figure 1A). Third‐degree interatrial conduction block (IACB III) was diagnosed11 if (1) P duration was ≥120 ms and (2) biphasic (+/−) P wave morphology in any 2 out of 3 inferior leads (II, III, aVF) with negative P prime deflection of any amplitude (<0 μV) was present. The sex‐adjusted Cornell product (QRS duration times the Cornell voltage) was calculated (Cornell voltage=RaVL+SV3, with 6 mm [0.6 mV] added in women) to estimate ECG left ventricular hypertrophy (LVH).24–25
Figure 1.

A. Measurement of P prime amplitude in V1. B, Scatterplot of P terminal force in V1 (Y) against P prime amplitude in V1 (X). C, Scatterplot of P prime amplitude in V1 (Y) against P prime duration in V1 (X). D, Scatterplot of P duration (Y) against P prime amplitude in V1 (X).
Incident CVD Events During Follow‐Up
Participants were followed up with annual telephone calls, surveillance of hospitals in the community, National Death Index, and 3 triennial field visits through 2002; details have been previously reported.26 Incident events (AF, stroke, CHD, HF) occurring through December 31, 2002 were included in the analysis as time‐dependent covariates. Incident AF was diagnosed from 3 sources27: (1) 12‐lead ECG performed during follow‐up exams; (2) hospital discharge records (ICD‐9 code 427.3 or ICD‐10 code I‐48); (3) death certificates. AF occurring during the cardiac surgery hospitalization event was not considered an incident AF event and follow‐up was continued beyond cardiac surgery‐related AF event. Incident stroke included definite or probable cases, defined as sudden or rapid onset of neurological symptoms that lasted for 24 hours in the absence of another cause.28 Incident HF event was diagnosed as first HF hospitalization with ICD‐9 code 428 or ICD‐10 code I50 in any position of the hospital discharge list,29 and included fatal HF cases. Incident CHD was defined as a definite or probable MI, silent ECG evidence of MI as defined by Minnesota code,21 or coronary revascularization procedure (bypass surgery or coronary angioplasty), including fatal CHD cases. All potential clinical CHD events were validated by the ARIC Morbidity and Mortality Classification Committee.26
Outcomes Definition
SCD served as the primary outcome in this study. SCD was adjudicated in a 2‐step process involving 2 independent death adjudication committees.30 First all deaths were reviewed and adjudicated by the ARIC Morbidity and Mortality Classification Committee using established criteria to determine whether or not the death was attributed to CHD.26 Then, both definite and possible CHD deaths were reviewed by an independent SCD Adjudication Committee to determine if the death was a sudden, presumably arrhythmic SCD, as previously described.31 Secondary outcomes included non‐sudden fatal CHD, non‐CHD death, all‐cause mortality, and non‐fatal events (incident AF, incident HF, incident stroke, incident non‐fatal CHD) as defined above.
Statistical Analysis
Data were analyzed using STATA 13 (StataCorp LP) statistical software and a 2‐tailed P<0.05 was considered statistically significant. We used t test and chi‐square test, as appropriate, to compare clinical and demographic characteristics of study participants with and without DTNPV1. Pairwise correlations between P wave parameters were measured by Pearson's correlation coefficient r. Linear regression models were built to study associations between amplitude of P prime deflection in V1, and (1) P prime duration in V1, (2) P terminal force in V1, (3) amplitude of P prime in II, III, aVF. Association between amplitude of terminal negative P wave deflection in V1 and SCD was evaluated through the use of fully adjusted Cox regression model incorporating quadratic splines with 4 knots (at P primeV1 amplitude of −0.073, −0.039, −0.024, and 0 mV). Cox proportional‐hazards models were used to quantify the association between the DTNPV1 and SCD. We constructed 4 models to adjust for covariates. Model 1 was stratified by race and study center, and adjusted by age and sex. Model 2 was further adjusted for prevalent CVD (CHD, MI, HF, stroke), and traditional cardiovascular risk factors (systolic blood pressure, antihypertensive medication, diabetes mellitus, smoking status (current vs. never/former), total cholesterol, high‐density lipoprotein cholesterol (HDL), triglycerides, body mass index, leisure activity index), as well as use of QT‐prolonging drugs and beta‐blockers. Model 3 incorporated all variables in Model 2 plus ECG characteristics: heart rate, QRS duration, P duration, P axis, QTc, spatial QRS‐T angle, sex‐specific Cornell product, and third‐degree inter‐atrial conduction block. Model 4 was further adjusted for AF, stroke, CHD, and HF entered as time‐updated covariates. Schoenfeld residuals were evaluated to test the assumption of the hazards proportionality. In addition, the multivariate (model 3) Fine and Gray's subhazards analyses32 were performed for 3 competing outcomes: SCD, non‐sudden fatal CHD, non‐CHD death.
To evaluate whether the association was consistent across the subgroups, we examined the association of DTNPV1 with SCD in subgroups defined by baseline age (cut‐point 55 years), sex, race, CVD status (defined as CHD, MI, HF, stroke, QRS duration ≥120 ms), ECG‐LVH by Cornell product (defined as sex‐specific Cornell product >2440 mm×ms), hypertension, diabetes, ECG‐left atrial enlargement per ROMICAT33 criteria (defined as P duration >110 ms), third‐degree intratrial conduction block. We evaluated interactions of DTNPV1 with these subgroups in model similar to the fully adjusted Cox model 4 used in the main analysis.
In CVD‐free participants, we further investigated associations of DTNPV1 with non‐fatal events, known mediators of SCD: CHD, HF, AF, and stroke. Model 1 was adjusted for sex and age, stratified by race and study center. Model 2 in addition was adjusted by traditional risk factors: total cholesterol, triglycerides, high density lipoprotein (HDL), current smoking, diabetes mellitus, body mass index, leisure activity index, systolic blood pressure, use of blood‐lowering medications, QT‐prolonging drugs, beta‐blockers. Model 3 in addition was adjusted by ECG characteristics: heart rate, QRS duration, P duration, P axis, QTc, QRS‐T angle, sex‐specific Cornell product, third‐degree inter‐atrial conduction block.
In CVD‐free participants, we calculated sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) for SCD and combined endpoint of either death or non‐fatal CVD event (MI, revascularization (PCI or CABG), stroke, HF hospitalization, AF). The predictive value of DTNPV1 was evaluated by comparing the model with DTNPV1 to the baseline SCD risk model. The baseline SCD risk model was created with the components of the CHD Framingham risk score: age, gender, systolic blood pressure, diabetes, HDL and total cholesterol, smoking, and blood pressure‐lowering therapy. Calculated Framingham risk scores were not directly used due to possible issues of the applicability to different ethnic groups34 and were adjusted for race. Net reclassification improvement (NRI) and integrated discrimination improvement (IDI) were calculated as described by Pencina et al.35 NRI was calculated from the Framingham predicted risk cut points of 5% and 15% at 10 years. NRI and IDI were calculated by an STATA add‐on developed by the Uppsala clinical research center.36
Nonparametric unadjusted receiver operating characteristic (ROC) analysis was performed for SCD in 4 subgroups: white men, white women, black men, and black women. Total area under the ROC curve (AUC) was estimated and AUCs of the following ECG parameters were compared: PTFV1, P‐prime amplitude in V1, P duration, heart rate, QTc, and ECG LVH.
Results
Biphasic P wave with negative P prime was present in 67.4 % of the cohort (n=10 358). The median amplitude of terminal negative P wave deflection in V1 was −39 μV (interquartile range [IQR] −53 to −29 μV). DTNPV1 was observed in 167 individuals (1.09%). P prime amplitude in V1 strongly correlated with P terminal force (Figure 1B) and P prime duration in V1 (r=−0.840; P<0.0001; Figure 1C). In all cases of the terminal negative portion of PV1 amplitude ≥1.00 mm (100 μV) in depth, duration of that terminal negative PV1 portion exceed 0.04 s (Figure 1C). Third‐degree interatrial block was observed more rarely (in 85 subjects; 0.55%) than DTNPV1, and was more frequent in participants with DTNPV1 (Table 1). There was no meaningful correlation between P prime amplitudes in leads II and V1 (r=0.010), III and V1 (r=0.078), aVF and V1 (r=0.060). Correlation between P prime amplitude in V1 and average P duration was minor (Figure 1D); it was smaller as compared with the correlation between P terminal force and average P duration (r=−0.272; P<0.0001).
Table 1.
Comparison of Clinical, Demographic, and ECG Characteristics of ARIC Participants With and Without Deep Terminal Negativity of P Wave in V1 (Negative Amplitude >0.1 mV)
| No DTNPV1 (n=15 209) | Yes DTNPV1 (n=167) | P Value | |
|---|---|---|---|
| Men, n (%) | 6779 (44.6) | 86 (51.5) | 0.73 |
| Whites, n (%) | 11 167 (73.4) | 81 (48.5) | <0.0001 |
| Age (SD), y | 54.1 (5.8) | 56.3 (5.6) | <0.0001 |
| Systolic blood pressure (SD). mmHg | 121.1 (18.7) | 132.0 (25.3) | <0.0001 |
| Total cholesterol (SD), mmol/L | 5.58 (1.08) | 5.42 (1.18) | 0.159 |
| High density lipoprotein (SD), mmol/L | 1.34 (0.44) | 1.29 (0.46) | 0.195 |
| Triglycerides (SD), mmol/L | 1.48 (1.02) | 1.64 (1.57) | 0.208 |
| Body mass index (SD), kg/m2 | 27.7 (5.4) | 27.2 (5.5) | 0.257 |
| Diabetes mellitus, n (%) | 1457 (9.7) | 37 (22.7) | <0.0001 |
| Prevalent coronary heart disease, n (%) | 669 (4.5) | 43 (26.4) | <0.0001 |
| Prevalent heart failure, n (%) | 673 (4.5) | 32 (19.5) | <0.0001 |
| Prevalent myocardial infarction, n (%) | 567 (3.8) | 41 (25.0) | <0.0001 |
| Prevalent stroke, n (%) | 250 (1.7) | 12 (7.3) | <0.0001 |
| Hypertension, n (%) | 3781 (25.0) | 80 (47.9) | <0.0001 |
| Use of QT‐prolonging drugs, n (%) | 1407 (9.3) | 32 (19.2) | <0.0001 |
| Use of beta‐blockers, n (%) | 1316 (8.7) | 28 (16.8) | <0.0001 |
| Leisure activity index, mean (SD) | 2.36 (0.57) | 2.19 (0.59) | 0.195 |
| Current smoker, n (%) | 3941 (25.9) | 60 (36.1) | 0.003 |
| Mean heart rate (SD), bpm | 66.2 (10.2) | 72.6 (12.3) | <0.0001 |
| P duration (SD), ms | 106.4 (13.2) | 115.0 (12.5) | <0.0001 |
| PR interval (SD), ms | 163.2 (25.5) | 169.1 (29.6) | 0.011 |
| P axis (SD), deg | 48.0 (21.6) | 53.9 (19.5) | 0.0001 |
| Interatrial block III, n (%) | 78 (0.51) | 7 (4.19) | <0.0001 |
| QRS‐axis (SD), deg | 29.8 (33.3) | 23.8 (43.2) | 0.076 |
| T‐axis (SD), deg | 39.0 (31.2) | 59.7 (52.9) | <0.0001 |
| QRS‐T angle (SD), deg | 56.5 (33.2) | 80.0 (43.9) | <0.0001 |
| QRS duration (SD), ms | 91.7 (12.5) | 94.1 (15.7) | 0.055 |
| Cornell productsex, (SD), mV×ms | 1458 (619) | 1885 (1011) | <0.0001 |
| QTc (SD), ms | 421.1 (20.5) | 435.2 (28.4) | <0.0001 |
DTNPV1 indicates deep terminal negativity of P wave in V1.
Participants with DTNPV1 were more likely to be older, black, to have CVD (CHD, HF, MI, stroke), to be on anti‐hypertensive, QT‐prolonging drugs and beta‐blockers, with risk factors (hypertension, diabetes, smoking), and ECG abnormalities (faster heart rate, wider P wave, longer QTc, larger Cornell product, and QRS‐T angle) than those without DTNPV1 (Table 1).
During a median follow‐up period of 14 years, there were 1998 cases of CHD, 687 cases of stroke, 1166 cases of incident HF, 958 cases of incident AF, and 311 SCD outcomes. Out of 167 participants with DTNPV1 there were 21 cases of SCD (12.6%), whereas 290 SCD cases occurred in 15 209 participants without DTNPV1 (1.9%). Cumulative unadjusted SCD was higher in participants with DTNPV1 (Figure 2A). In adjusted analysis, there was a dose‐response relationship between P prime amplitude in V1 and SCD (Figure 2B).
Figure 2.
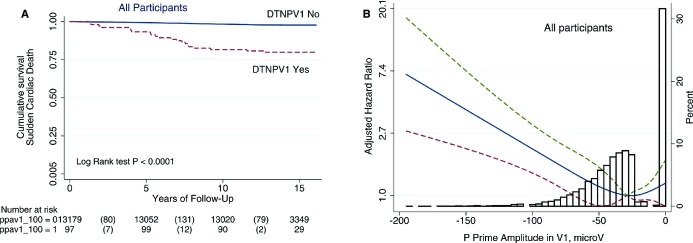
A. Unadjusted Kaplan–Meier curves for the probability of sudden cardiac death in patients with and without deep terminal negativity of P wave in V1. B, Multivariate‐adjusted Hazard Ratio with 95% confidence interval for sudden cardiac death, associated with P prime amplitude in V1, modeled as a continuous variable using quadratic splines. DTNPV1 indicates deep terminal negativity of P wave in V1.
In unadjusted Cox regression, compared with participants without DTNPV1, individuals with DTNPV1 had 8‐times higher risk of SCD (Table 2). Adjusted for age, sex, and stratified by race and study center, individuals with DTNPV1 demonstrated a 5‐fold increased risk of SCD. Further adjustment for potential confounders (prevalent CVD and clinical risk factors) substantially attenuated the association of DTNPV1 with SCD, to a hazard ratio of 2.75. This association did not change appreciably after further adjustment for other ECG abnormalities or AF, stroke, CHD an HF as time‐updated variables, (Table 2). Association of DTNPV1 with all‐cause mortality was similar to that of SCD, but weaker. In competing analyses (Table 2) DTNPV1 was associated with SCD, but not with non‐sudden fatal CHD, or non‐CHD death (Figure 3A through 3C). Adjusted competing analysis showed consistent dose‐response relationship between P prime amplitude in V1 and SCD, but not other outcomes (Figure 3D through 3F).
Table 2.
Deep Terminal Negativity of P Wave in V1 and Risk of Sudden Cardiac Death, Fatal Coronary Heart Disease (CHD), Non‐CHD Death, and All‐Cause Mortality
| SCD (n=311) | Fatal CHD (n=370) | Non‐CHD Death (=1753) | All‐Cause Death (n=2197) | |||||
|---|---|---|---|---|---|---|---|---|
| HR (95%CI) | P Value | HR (95%CI) | P Value | HR (95%CI) | P Value | HR (95%CI) | P Value | |
| Unadjusted | 8.21 (5.27 to 12.79) | <0.0001 | 7.23 (4.70 to 11.13) | <0.0001 | 3.34 (2.25 to 4.44) | <0.0001 | 4.15 (3.30 to 5.22) | <0.0001 |
| Cox model 1 | 5.12 (3.27 to 8.02) | <0.0001 | 4.62 (2.98 to 7.14) | <0.0001 | 2.35 (1.76 to 3.13) | <0.0001 | 2.85 (2.26 to 3.59) | <0.0001 |
| Cox model 2 | 2.75 (1.72 to 4.38) | <0.0001 | 2.18 (1.39 to 3.42) | 0.001 | 1.60 (1.17 to 2.18) | 0.003 | 1.81 (1.41 to 2.31) | <0.0001 |
| Cox model 3 | 2.37 (1.44 to 3.91) | 0.001 | 1.74 (1.07 to 2.84) | 0.026 | 1.31 (0.95 to 1.81) | 0.100 | 1.44 (1.11 to 1.87) | 0.006 |
| Cox model 4 | 2.49 (1.51 to 4.10) | <0.0001 | 1.66 (1.09 to 2.55) | 0.019 | 1.25 (0.93 to 1.69) | 0.135 | 1.41 (1.11 to 1.79) | 0.004 |
| Competing risk model 3 | 2.45 (1.42 to 4.23) | 0.002 | 1.69 (0.97 to 2.95) | 0.066 | 1.15 (0.80 to 1.65) | 0.453 | — | — |
Model 1 adjusted for sex and age, stratified by race and study center. Model 2 in addition adjusted by clinical characteristics: prevalent coronary heart disease, myocardial infarction, heart failure, stroke, total cholesterol, triglycerides, high density lipoprotein, current smoking, diabetes mellitus, body mass index, leisure activity index, systolic blood pressure, use of blood‐lowering medications, QT‐prolonging drugs, beta‐blockers. Model 3 in addition adjusted by ECG characteristics: heart rate, QRS duration, P duration, P axis, QTc, QRS‐T angle, sex‐specific Cornell product, third degree inter‐atrial conduction block. Model 4 in addition adjusted by time‐dependent incidence of atrial fibrillation, stroke, heart failure, and CHD. Competing risk model included all variables as in model 3. CHD indicates coronary heart disease; SCD, sudden cardiac death.
Figure 3.
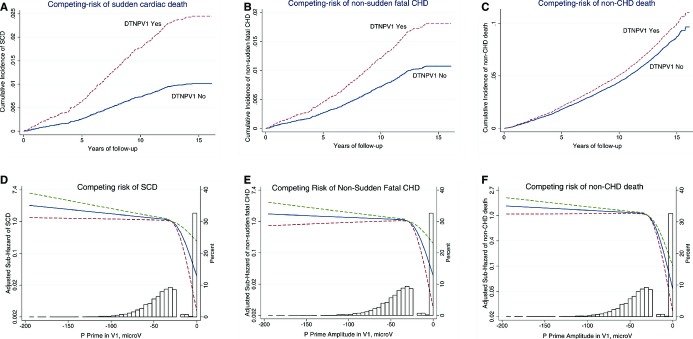
Competing risks. Multivariate‐adjusted cumulative incidence functions for SCD (A), non‐sudden fatal CHD (B), and non‐CHD death (C), associated with deep terminal negativity of P wave in V1 (DTNPV1). Multivariate‐adjusted sub‐hazard ratios (SHRs) for SCD (D), non‐sudden fatal CHD (E), and non‐CHD death (F), associated with P prime amplitude in V1, modeled as a continuous variable using 4 quadratic splines. CHD indicates coronary heart disease; DTNPV1, deep terminal negativity of P wave in V1; SCD, sudden cardiac death.
Finally, we examined this association in various subgroups. Table 3 shows fully adjusted models for all study participants, as well as for patients with and without prevalent CVD at baseline. Table 4 shows that the results were largely consistent across different subgroups of ARIC participants (all P values for interaction >0.05). Importantly, DTNPV1 was independently associated with SCD in participants without diagnosed CVD at baseline.
Table 3.
Risk of SCD, Adjusted by Demographic, Clinical and ECG Risk Factors (Model 3)
| All Participants (n=15 376) | CVD‐Free Participants (n=13 278) | Prevalent CVD (n=2098) | ||||
|---|---|---|---|---|---|---|
| HR (95%CI) | P Value | HR (95%CI) | P Value | HR (95%CI) | P (95%CI) | |
| DTNPV1 | 2.37 (1.44 to 3.91) | 0.001 | 3.51 (1.72 to 7.13) | 0.001 | 2.05 (1.11 to 3.70) | 0.022 |
| Female | 0.56 (0.41 to 0.77) | <0.0001 | 0.45 (0.32 to 0.68) | <0.0001 | 0.68 (0.41 to 1.11) | 0.122 |
| Age, y | 1.006 (1.003 to 1.008) | <0.0001 | 1.07 (1.04 to 1.10) | <0.0001 | 1.04 (1.00 to 1.08) | 0.057 |
| CHD | 0.30 (0.04 to 2.19) | 0.234 | — | 0.82 (0.19 to 3.54) | 0.417 | |
| MI | 15.81 (2.17 to 114.96 | 0.006 | — | 6.22 (1.52 to 25.49) | 0.011 | |
| HF | 0.80 (0.53 to 1.23) | 0.318 | — | 1.01 (0.63 to 1.60) | 0.983 | |
| Stroke | 1.80 (1.15 to 2.83) | 0.010 | — | 2.22 (1.40 to 3.52) | 0.001 | |
| BMI, kg/m2 | 1.00 (0.97 to 1.02) | 0.848 | 0.99 (0.96 to 1.02) | 0.647 | 1.01 (0.97 to 1.05) | 0.568 |
| Total cholesterol, mmol/L | 1.21 (1.09 to 1.34) | <0.0001 | 1.18 (1.04 to 1.35) | 0.009 | 1.27 (1.08 to 1.50) | 0.003 |
| HDL, mmol/L | 0.57 (0.39 to 0.84 | 0.005 | 0.56 (0.36 to 0.89) | 0.014 | 0.68 (0.35 to 1.31) | 0.258 |
| Triglycerides, mmol/L | 0.98 (0.88 to 1.08) | 0.679 | 0.97 (0.85 to 1.11) | 0.663 | 0.96 (0.80 to 1.14) | 0.637 |
| Leisure activity index | 0.87 (0.69 to 1.08) | 0.209 | 0.75 (0.57 to 0.99) | 0.046 | — | |
| Diabetes mellitus | 2.24 (1.68 to 3.00) | <0.0001 | 2.82 (1.98 to 4.00) | <0.0001 | 1.56 (1.00 to 2.43) | 0.048 |
| Current smoking | 2.00 (1.54 to 2.58) | <0.0001 | 2.13 (1.55 to 2.93) | <0.0001 | 1.68 (1.12 to 2.50) | 0.011 |
| Systolic blood pressure, mmHg | 1.01 (1.01 to 1.02) | <0.0001 | 1.02 (1.01 to 1.02) | <0.0001 | 1.01 (1.01 to 1.02) | 0.002 |
| BP lowering drugs | 1.35 (1.03 to 1.77) | 0.029 | 1.74 (1.24 to 2.44) | 0.001 | 0.95 (0.62 to 1.44) | 0.794 |
| QT‐prolonging drugs | 1.05 (0.72 to 1.53) | 0.787 | 1.01 (0.61 to 1.69) | 0.958 | 1.03 (0.62 to 1.73) | 0.903 |
| Beta‐blockers | 0.96 (0.67 to 1.53) | 0.824 | 0.55 (0.30 to 1.01) | 0.054 | 1.23 (0.78 to 1.94) | 0.367 |
| Cornell productsex, mm×ms | 1.00 (1.00 to 1.00) | 0.519 | 1.00 (1.00 to 1.00) | 0.361 | — | |
| QRS duration, ms | 1.00 (0.99 to 1.01) | 0.411 | 0.98 (0.96 to 1.00) | 0.098 | 1.02 (1.00 to 1.03) | 0.004 |
| Heart rate, bpm | 1.01 (0.99 to 1.02) | 0.367 | 1.00 (0.99 to 1.02) | 0.621 | 1.02 (1.00 to 1.04) | 0.027 |
| QTc, ms | 1.008 (1.003 to 1.014) | 0.004 | 1.009 (1.001 to 1.017) | 0.022 | 1.00 (0.99 to 1.01) | 0.531 |
| P duration, ms | 1.00 (0.99 to 1.01) | 0.462 | 0.99 (0.98 to 1.00) | 0.131 | 1.01 (1.00 to 1.03) | 0.046 |
| P axis, deg | 1.00 (0.99 to 1.00) | 0.331 | 0.99 (0.99 to 1.00) | 0.129 | — | |
| QRS‐T angle, deg | 1.005 (1.002 to 1.009) | 0.001 | 1.00 (1.00 to 1.01) | 0.062 | — | |
| Itraatrial block third degree | 1.20 (0.43 to 3.37) | 0.727 | 1.38 (0.33 to 5.78) | 0.660 | — | |
CHD indicates coronary heart disease; DTNPV1, deep terminal negativity of P wave in V1; HF, heart failure; MI, myocardial infarction.
Table 4.
Terminal Negativity of P Wave in V1 and Fully Adjusted Risk of SCD Across Subgroups
| HR (95%CI) | P Value | |
|---|---|---|
| Male (199 SCDs/6865) | 2.36 (1.27 to 4.38) | 0.007 |
| Female (112 SCDs/8511) | 3.37 (1.44 to 7.86) | 0.005 |
| Interaction | 0.375 | |
| Age ≥55 y (207 SCDs/7244) | 2.42 (1.28 to 4.57) | 0.006 |
| Age<55 y (104 SCDs/8132) | 2.84 (1.21 to 6.67) | 0.017 |
| Interaction | 0.929 | |
| White (177 SCDs/11248) | 1.82 (0.87 to 3.80) | 0.111 |
| Black (134 SCDs/4128) | 3.33 (1.67 to 6.65) | 0.001 |
| Interaction | 0.716 | |
| CVD Yes (129 SCDs/2098) | 2.06 (1.07 to 3.96) | 0.030 |
| CVD‐Healthy (182 SCDs/13278) | 3.59 (1.62 to 7.97) | 0.002 |
| Interaction | 0.801 | |
| Diabetes Yes (102 SCDs/1494) | 3.93 (1.57 to 9.82) | 0.003 |
| Diabetes No (208 SCDs/13766) | 1.85 (0.99 to 3.46) | 0.056 |
| Interaction | 0.659 | |
| Hypertension Yes (150 SCDs/3861) | 2.71 (1.35 to 5.44) | 0.005 |
| Hypertension No (159 SCDs/11436) | 2.67 (1.28 to 5.65) | 0.010 |
| Interaction | 0.326 | |
| ECG LVH Yes (52 SCDs/863) | 4.32 (1.56 to 11.92) | 0.005 |
| ECG LVH No (259 SCDs/14513) | 2.10 (1.13 to 3.93) | 0.019 |
| Interaction | 0.192 | |
| P duration >110 ms (155 SCDs/5955) | 2.02 (1.06 to 3.84) | 0.034 |
| P duration ≤110 ms (156 SCDs/9421) | 4.65 (2.10 to 10.28) | <0.0001 |
| Interaction | 0.064 | |
| IACB third degree Yes# (4 SCDs/85) | 6.62 (0.62 to 70.42) | 0.117 |
| IACB third degree No (307 SCDs/15291) | 2.95 (1.79 to 4.85) | <0.0001 |
| Interaction | 0.821 |
CVD is defined as a presence of either coronary heart disease, or heart failure, or stroke, or QRS duration ≥120 ms. Left ventricular hypertrophy defined as sex‐specific Cornell product >2440 mm×ms. IACB, interatrial conduction block. # model was adjusted by sex, race, age, and CVD only. CHD indicates coronary heart disease; CVD, cardiovascular disease; LVH, left ventricular hypertrophy SCD, sudden cardiac death.
In CVD‐free participants, in fully adjusted models DTNPV1 was associated with a 5‐fold increased risk of AF, and about 2‐fold increased risk of non‐fatal CHD and HF (Table 5). Trend towards increasing risk of non‐fatal stroke was observed.
Table 5.
Risk of Non‐Fatal CHD, HF, Stroke, AF Events in CVD‐Healthy Participants With and Without DTNPV1
| Model | Non‐Fatal CHD (N=1422) | Heart Failure (N=910) | Stroke (N=505) | Atrial Fibrillation (N=699) | ||||
|---|---|---|---|---|---|---|---|---|
| HR (95%CI) | P Value | HR (95%CI) | P Value | HR (95%CI) | P Value | HR (95%CI) | P Value | |
| U | 2.61 (1.68 to 4.05) | <0.0001 | 4.57 (2.99 to 6.98) | <0.0001 | 3.97 (2.18 to 7.21) | <0.0001 | 6.35 (4.15 to 9.71) | <0.0001 |
| 1 | 2.27 (1.45 to 3.54) | <0.0001 | 2.88 (1.88 to 4.40) | <0.0001 | 2.43 (1.33 to 4.44) | 0.004 | 5.60 (3.64 to 8.61) | <0.0001 |
| 2 | 2.21 (1.41 to 3.46) | 0.001 | 2.29 (1.46 to 3.62) | <0.0001 | 2.26 (1.23 to 4.14) | 0.009 | 5.51 (3.58 to 8.50) | <0.0001 |
| 3 | 2.24 (1.43 to 3.53) | <0.0001 | 1.90 (1.19 to 3.04) | 0.007 | 1.88 (0.99 to 3.57) | 0.054 | 5.02 (3.23 to 7.80) | <0.0001 |
U, unadjusted; 1, Model 1 is adjusted for sex and age, stratified by race and study center. 2, Model 2 in addition adjusted by traditional risk factors: total cholesterol, triglycerides, high density lipoprotein, current smoking, diabetes mellitus, body mass index, leisure activity index, systolic blood pressure, use of blood‐lowering medications, QT‐prolonging drugs, beta‐blockers. 3, Model 3 in addition adjusted by ECG characteristics: heart rate, QRS duration, P duration, P axis, QTc, QRS‐T angle, sex‐specific Cornell product, third degree inter‐atrial conduction block. CHD indicates coronary heart disease; CVD, cardiovascular disease.
In CVD‐free participants (Table 6), DTNPV1 demonstrated 3.8% sensitivity, 99.3% specificity, 7.5% PPV, and 98.7% NPV for prediction of SCD. For the prediction of the combined endpoint (death, or non‐fatal CHD, HF, stroke, or AF event), DTNPV1 showed 1.6% sensitivity, 99.6% specificity, 55.9% PPV, and 74.5% NPV.
Table 6.
Two‐by‐Two Tables for SCD and Combined Endpoint (Death or Non‐Fatal CHD, HF, Stroke, AF Events) in CVD‐Healthy at Baseline Participants
| Sudden Cardiac Death | Death or Non‐Fatal Event (CHD, HF, Stroke, AF) | ||||||
|---|---|---|---|---|---|---|---|
| Yes | No | Total | Yes | No | Total | ||
| DTNPV1 (+) | 7 | 86 | 93 | DTNPV1 (+) | 56 | 37 | 93 |
| DTNPV1 (−) | 175 | 13 010 | 13 185 | DTNPV1 (−) | 3368 | 9817 | 13 185 |
| Total | 182 | 13 096 | 13 278 | Total | 3424 | 9854 | 13 278 |
AF indicates atrial fibrillation CHD, coronary heart disease; CVD, cardiovascular disease; SCD, sudden cardiac death.
DTNPV1 has shown borderline significant discrimination ability between 2 models (Table 7), with NRI estimate =0.028 (P=0.06). Notably, DTNPV1 appropriately reclassified participants with SCD outcome into the higher risk categories: 3.4% of individuals were appropriately reclassified into a higher risk group, whereas only 0.3% was reclassified into a higher risk group inappropriately. In addition, in the subgroup of CVD‐free at baseline participants we compared unadjusted AUCs of several ECG markers of SCD: P prime amplitude in V1, PTFV1, P duration, ECG LVH, heart rate, and QTc. AUCs of P prime amplitude in V1, PTFV1, ECG LVH, heart rate, and QTc were similar and significantly larger, as compared with AUC of P duration. There were no significant differences in AUCs ROCs in 4 race‐sex subgroups.
Table 7.
Comparison of Proportions of Subjects in Low‐ (<5%), Medium‐ (5 to 20%), and High‐(>20%) Risk Categories, Presented Separately for SCD Events and Nonevents (SCD‐Free)
| Framingham CHD risk factors+ DTNPV1 | ||||
|---|---|---|---|---|
| <5% | 5 to 20% | ≥20% | Total | |
| Framingham CHD risk factors | ||||
| SCD events | ||||
| <5% | 135 | 3 (2.2%) | 138 | |
| 5% to 20% | 1 (2.6%) | 34 | 3 (7.8%) | 38 |
| ≥20% | 3 | 3 | ||
| Total | 136 | 37 | 6 | 179 |
| SCD non‐events | ||||
| <5% | 12 298 | 27 (0.22%) | 12 325 | |
| 5% to 20% | 35 (6.64%) | 485 | 7 (1.33%) | 527 |
| ≥20% | 1 (5.56%) | 17 | 18 | |
| Total | 12 333 | 513 | 24 | 12 870 |
CHD indicates coronary heart disease SDC, sudden cardiac death.
Discussion
In this large bi‐racial prospective cohort, ECG sign of DTNPV1 was independently associated with SCD in individuals with and without prevalent at baseline CVD, even after adjustment for CHD risk factors, known ECG predictors of ventricular and atrial arrhythmias, and time‐updated CHD, HF, AF, and stroke events. Associated with DTNPV1 risk of SCD exceeded risk of non‐fatal CHD, HF, and stroke; only risk of AF exceeded risk of SCD. This finding suggests that DTNPV1 is an intermediate marker on the pathway linking cardiac fibrosis16 (both in LA and LV) with atrial and ventricular arrhythmias and SCD. Our finding of an independent association between DTNPV1 and SCD supports the unifying hypothesis of the key role of fibrosis16 in arrhythmogenesis of both atrial and ventricular arrhythmias.37–38 DTNPV1 represents an epiphenomenon of advanced fibrotic disease process and damaged heart.
DTNPV1 is an easily recognizable ECG sign. DTNPV1 added incremental value to the traditional CHD risk factors for the prediction of SCD events. In CVD‐free individuals, DTNPV1 improved risk prediction over what was achieved by a model with Framingham CHD risk factors alone. Importantly, validation of our findings in another study is needed before DTNPV1 could be considered as a component of a SCD risk score.
Deep Terminal Negativity of P Wave in V1
Morris et al14 in 1964 developed P terminal force metric as a product of amplitude and duration of a P prime deflection in V1. They defined an abnormal P terminal force as “a terminal portion of the P wave at V1 1 box in depth (−1.0 mm.) and 1 box in duration (0.04 sec.),” yielding a P terminal force of −0.04. Our study showed that in all cases of the terminal negative portion of PV1 amplitude ≥1.00 mm in depth, duration of that terminal negative PV1 portion always exceeded 1 box in duration, or 0.04 seconds (Figure 1). Therefore, one can ignore duration and measure only amplitude in order to derive DTNPV1 metric. We confirmed strong correlation between P terminal force and amplitude of P prime in V1. Moreover, virtually identical AUCs for P prime amplitude in V1 and PTFV1 confirmed that the risk assessment by amplitude only is adequate.
Historically investigators considered several underlying mechanisms resulting in the appearance of DTNPV1. Romhilt et al39 regarded DTNPV1 as a “sign of left atrium involvement” in patients with LVH. In the past abnormal PTFV1 was considered a sign of LA enlargement, until Josephson et al established that abnormal PTFV1 represents an inter‐atrial conduction defect that can be produced by a variety of factors.9 Posteriorly directed LA depolarization vector is primarily responsible for the appearance of P wave terminal negative deflection in V1. We speculate that intraatrial fat infiltration40–41 and gradual development of atrial fibrosis could lead to the interruption of interatrial conduction over tiny (and therefore more susceptible) posterior interatrial connections, resulting in predominant conduction via the Bachmann's bundle, associated with the anterior‐to‐posterior direction of left atrial activation. Pulmonary vein isolation could modify P wave morphology in V1.42 Moreover, extensive ablation of septal areas could result in electrical isolation of LA.43
LVH and LV fibrosis characterize pathophysiological substrate for SCD due to VF in patients early in the continuum of structural heart disease. Development of fibrosis both in LA and LV is likely a main mechanism of DTNPV1 manifestation on ECG. Emerging experimental data showed44 that fibrosis disrupts the normal electrical connectivity of cardiac tissue, alters source–sink relationships, facilitates the emergence of after depolarization‐induced premature ventricular complexes and, therefore, increases the vulnerability to arrhythmias. In clinical studies, the presence and amount of LV fibrosis were associated with documented ventricular arrhythmias and SCD.45 Gradually increasing risk of SCD, associated with the increasing amplitude of terminal negative P wave deflection in V1 illustrates the continuum of structural heart disease development, consistent with gradual progression of LA and LV fibrosis.16 In this study, we chose a threshold of P prime amplitude in V1 (1 mm, or 0.1 mV) not only due to historical reasons, but also because of convenience and simplicity of its use. This threshold identified individuals at high risk of SCD. At the same time, we demonstrated the continuity of the risk (Figure 2B) and, therefore, future studies could explore other thresholds of P prime amplitude in V1 to target desired sensitivity and specificity.
Resting 12‐Lead ECG for Prediction of SCD
In this study DTNPV1 predicted SCD, and demonstrated significant 60% precision for prediction of the combined endpoint (death, or non‐fatal CHD, HF, stroke, or AF event). Importantly, DTNPV1 improved discrimination of SCD above traditional CHD risk factors (Framingham CHD risk score). While there is no well‐accepted risk score of SCD in the general population, SCD is included in the CHD classification as one of the manifestations of CHD. It is important to emphasize that while DTNPV1 is suggestive of added value, it has to be validated prospectively before considering implementation of DTNPV1 into clinical practice.
Strength and Weakness of the Study
Strengths of this study include standardized ECG recording procedures, automated central processing of ECG data, long‐term follow‐up data, and well‐ascertained outcomes in a large population‐based cohort. Nonetheless, there are several limitations that merit attention. As with any observational study, we cannot rule out the possibility of residual confounding despite rigorous adjustment for potential confounders. Identification of incident HF relied on International Classification of Diseases codes abstracted from hospital records. Reliance on hospital discharge codes could result in underestimation of HF incidence. Similarly, incident AF diagnosis was based on hospital records and 10‐s, 12‐lead ECGs, recorded during follow‐up visits. Absence of long‐term ECG monitoring likely resulted in an underestimation of AF incidence.46 While adjudication of SCD was performed according to the rigorous protocol, ECG of the exact moment of SCD was not available, and, therefore, causative arrhythmia underlying the SCD event is unknown. This is the common limitation of the SCD investigation field. We reported ROC results for the race and sex subgroups, even if we did not find meaningful interaction with race and sex in this study.
Acknowledgment
The authors thank the staff and participants of the ARIC study for their important contributions.
Sources of Funding
The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). This work was supported by the 1R01HL118277 (Tereshchenko).
Disclosures
None.
References
- 1.Roger VL, Go AS, Lloyd‐Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012; 125:e2-e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kong MH, Fonarow GC, Peterson ED, Curtis AB, Hernandez AF, Sanders GD, Thomas KL, Hayes DL, Al‐Khatib SM. Systematic review of the incidence of sudden cardiac death in the United States. J Am Coll Cardiol. 2011; 57:794-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herlitz J, Andersson E, Bang A, Engdahl J, Holmberg M, Lindqvist J, Karlson BW, Waagstein L. Experiences from treatment of out‐of‐hospital cardiac arrest during 17 years in Goteborg. Eur Heart J. 2000; 21:1251-1258. [DOI] [PubMed] [Google Scholar]
- 4.Rea TD, Eisenberg MS, Becker LJ, Murray JA, Hearne T. Temporal trends in sudden cardiac arrest: a 25‐year emergency medical services perspective. Circulation. 2003; 107:2780-2785. [DOI] [PubMed] [Google Scholar]
- 5.Myerburg RJ, Junttila MJ. Sudden cardiac death caused by coronary heart disease. Circulation. 2012; 125:1043-1052. [DOI] [PubMed] [Google Scholar]
- 6.Goldberger JJ, Basu A, Boineau R, Buxton AE, Cain ME, Canty JM, Jr, Chen PS, Chugh SS, Costantini O, Exner DV, Kadish AH, Lee B, Lloyd‐Jones D, Moss AJ, Myerburg RJ, Olgin JE, Passman R, Stevenson WG, Tomaselli GF, Zareba W, Zipes DP, Zoloth L. Risk stratification for sudden cardiac death: a plan for the future. Circulation. 2014; 129:516-526. [DOI] [PubMed] [Google Scholar]
- 7.Cobb LA, Fahrenbruch CE, Olsufka M, Copass MK. Changing incidence of out‐of‐hospital ventricular fibrillation, 1980‐2000. JAMA. 2002; 288:3008-3013. [DOI] [PubMed] [Google Scholar]
- 8.Kuisma M, Repo J, Alaspaa A. The incidence of out‐of‐hospital ventricular fibrillation in Helsinki, Finland, from 1994 to 1999. Lancet. 2001; 358:473-474. [DOI] [PubMed] [Google Scholar]
- 9.Josephson ME, Kastor JA, Morganroth J. Electrocardiographic left atrial enlargement. Electrophysiologic, echocardiographic and hemodynamic correlates. Am J Cardiol. 1977; 39:967-971. [DOI] [PubMed] [Google Scholar]
- 10.Tsao CW, Josephson ME, Hauser TH, O'Halloran TD, Agarwal A, Manning WJ, Yeon SB. Accuracy of electrocardiographic criteria for atrial enlargement: validation with cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2008; 10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Bayes LA, Platonov P, Cosio FG, Cygankiewicz I, Pastore C, Baranowski R, Bayes‐Genis A, Guindo J, Vinolas X, Garcia‐Niebla J, Barbosa R, Stern S, Spodick D. Interatrial blocks. A separate entity from left atrial enlargement: a consensus report. J Electrocardiol. 2012; 45:445-451. [DOI] [PubMed] [Google Scholar]
- 12.Soliman EZ, Prineas RJ, Case LD, Zhang ZM, Goff DC., Jr Ethnic distribution of ECG predictors of atrial fibrillation and its impact on understanding the ethnic distribution of ischemic stroke in the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2009; 40:1204-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohsaka S, Sciacca RR, Sugioka K, Sacco RL, Homma S, Di Tullio MR. Electrocardiographic left atrial abnormalities and risk of ischemic stroke. Stroke. 2005; 36:2481-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris JJ, Jr, Estes EH, Jr, Whalen RE, Thompson HK, Jr, Mcintosh HD. P wave analysis in valvular heart disease. Circulation. 1964; 29:242-252. [DOI] [PubMed] [Google Scholar]
- 15.Beinart R, Khurram IM, Liu S, Yarmohammadi H, Halperin HR, Bluemke DA, Gai N, van der Geest RJ, Lima JA, Calkins H, Zimmerman SL, Nazarian S. Cardiac magnetic resonance T1 mapping of left atrial myocardium. Heart Rhythm. 2013; 10:1325-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Win TT, Venkatesh BA, Volpe GJ, Mewton N, Rizzi P, Sharma RK, Strauss DG, Lima JA, Tereshchenko LG. Associations of electrocardiographic P wave characteristics with left atrial structure, function and diffuse left ventricular fibrosis defined by cardiac magnetic resonance: the PRIMERI Study. Heart Rhythm. 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu G, Tamura A, Torigoe K, Kawano Y, Shinozaki K, Kotoku M, Kadota J. Abnormal P wave terminal force in lead V1 is associated with cardiac death or hospitalization for heart failure in prior myocardial infarction. Heart Vessels. 2013; 28:690-695. [DOI] [PubMed] [Google Scholar]
- 18.Tereshchenko LG, Shah AJ, Li Y, Soliman EZ. Electrocardiographic deep terminal negativity of the P Wave in V1 and risk of mortality: the national health and nutrition examination survey III. J Cardiovasc Electrophysiol. 2014; 25:1242-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.TA Investigators. The Atherosclerosis Risk in Community (ARIC) Study: design and objectives. Am J Epidemiol. 1989; 129:687-702. [PubMed] [Google Scholar]
- 20.Rose GA, Blackburn H. Cardiovascular survey methods. Monogr Ser World Health Organ. 1968; 56:1-188. [PubMed] [Google Scholar]
- 21.Blackburn H, Keys A, Simonson E, Rautaharju P, Punsar S. The electrocardiogram in population studies. A classification system. Circulation. 1960; 21:1160-1175. [DOI] [PubMed] [Google Scholar]
- 22.Eriksson H, Caidaul K, Larsson B, Ohlson LO, Welin L, Wilhelmsen L, Svärdsudd K. Cardiac and pulmonary causes of dyspnoea—validation of a scoring test for clinical‐epidemiological use: The Study of Men Born in 1913. Eur Heart J. 1987; 8:1007-1014. [DOI] [PubMed] [Google Scholar]
- 23.Toole JF, Chambless LE, Heiss G, Tyroler HA, Paton CC. Prevalence of stroke and transient ischemic attacks in the atherosclerosis risk in communities (ARIC) study. Ann Epidemiol. 1993; 3:500-503. [DOI] [PubMed] [Google Scholar]
- 24.Okin PM, Roman MJ, Devereux RB, Borer JS, Kligfield P. Electrocardiographic diagnosis of left ventricular hypertrophy by the time‐voltage integral of the QRS complex. J Am Coll Cardiol. 1994; 23:133-140. [DOI] [PubMed] [Google Scholar]
- 25.Okin PM, Roman MJ, Devereux RB, Kligfield P. Gender differences and the electrocardiogram in left ventricular hypertrophy. Hypertension. 1995; 25:242-249. [DOI] [PubMed] [Google Scholar]
- 26.White AD, Folsom AR, Chambless LE, Sharret AR, Yang K, Conwill D, Higgins M, Williams OD, Tyroler HA. Community surveillance of coronary heart disease in the Atherosclerosis Risk in Communities (ARIC) Study: methods and initial two years' experience. J Clin Epidemiol. 1996; 49:223-233. [DOI] [PubMed] [Google Scholar]
- 27.Alonso A, Agarwal SK, Soliman EZ, Ambrose M, Chamberlain AM, Prineas RJ, Folsom AR. Incidence of atrial fibrillation in whites and African‐Americans: the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2009; 158:111-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosamond WD, Folsom AR, Chambless LE, Wang CH, McGovern PG, Howard G, Copper LS, Shahar E. Stroke incidence and survival among middle‐aged adults: 9‐year follow‐up of the Atherosclerosis Risk in Communities (ARIC) cohort. Stroke. 1999; 30:736-743. [DOI] [PubMed] [Google Scholar]
- 29.Matsushita K, Blecker S, Pazin‐Filho A, Bertoni A, Chang PP, Coresh J, Selvin E. The association of hemoglobin a1c with incident heart failure among people without diabetes: the atherosclerosis risk in communities study. Diabetes. 2010; 59:2020-2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soliman EZ, Prineas RJ, Case LD, Russell G, Rosamond W, Rea T, Sotoodehnia N, Post WS, Siscovick D, Psaty BM, Burke GL. Electrocardiographic and clinical predictors separating atherosclerotic sudden cardiac death from incident coronary heart disease. Heart. 2011; 97:1597-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kao WH, Arking DE, Post W, Rea TD, Sotoodehnia N, Prineas RJ, Bishe B, Doan BQ, Boerwinkle E, Psaty BM, Tomaselli GF, Coresh J, Siscovick DS, Marban E, Spooner PM, Burke GL, Chakravarti A. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community‐based populations. Circulation. 2009; 119:940-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999; 94:496-509. [Google Scholar]
- 33.Truong QA, Charipar EM, Ptaszek LM, Taylor C, Fontes JD, Kriegel M, Irlbeck T, Mahabadi AA, Blankstein R, Hoffmann U. Usefulness of electrocardiographic parameters as compared with computed tomography measures of left atrial volume enlargement: from the ROMICAT trial. J Electrocardiol. 2011; 44:257-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D'Agostino RB, Sr, Grundy S, Sullivan LM, Wilson P. Validation of the Framingham coronary heart disease prediction scores: results of a multiple ethnic groups investigation. JAMA. 2001; 286:180-187. [DOI] [PubMed] [Google Scholar]
- 35.Pencina MJ, D'Agostino RB, Sr, D'Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008; 27:157-172. [DOI] [PubMed] [Google Scholar]
- 36.Sundstrom J, Byberg L, Gedeborg R, Michaelsson K, Berglund L. Useful tests of usefulness of new risk factors: tools for assessing reclassification and discrimination. Scand J Public Health. 2011; 39:439-441. [DOI] [PubMed] [Google Scholar]
- 37.de Jong S, van Veen TA, van Rijen HV, de Bakker JM. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011; 57:630-638. [DOI] [PubMed] [Google Scholar]
- 38.Wong TC, Piehler K, Meier CG, Testa SM, Klock AM, Aneizi AA, Shakesprere J, Kellman P, Shroff SG, Schwartzman DS, Mulukutla SR, Simon MA, Schelbert EB. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short‐term mortality. Circulation. 2012; 126:1206-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romhilt DW, Estes EH., Jr A point‐score system for the ECG diagnosis of left ventricular hypertrophy. Am Heart J. 1968; 75:752-758. [DOI] [PubMed] [Google Scholar]
- 40.Murthy S, Rizzi P, Mewton N, Strauss DG, Liu CY, Volpe GJ, Marchlinski FE, Spooner P, Berger RD, Kellman P, Lima JA, Tereshchenko LG. Number of P wave fragmentations on P SAECG correlates with infiltrated atrial fat. Ann Noninvasive Electrocardiol. 2014; 19:114-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tereshchenko LG, Rizzi P, Mewton N, Volpe GJ, Murthy S, Strauss DG, Liu CY, Marchlinski FE, Spooner P, Berger RD, Kellman P, Lima JA. Infiltrated atrial fat characterizes underlying atrial fibrillation substrate in patients at risk as defined by the ARIC atrial fibrillation risk score. Int J Cardiol. 2014; 172:196-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janin S, Wojcik M, Kuniss M, Berkowitsch A, Erkapic D, Zaltsberg S, Ecarnot F, Hamm CW, Pitschner HF, Neumann T. Pulmonary vein antrum isolation and terminal part of the P wave. Pacing Clin Electrophysiol. 2010; 33:784-789. [DOI] [PubMed] [Google Scholar]
- 43.Ning M, Dong JZ, Ma CS. Left atrium electrical isolation as a complication of catheter ablation of persistent atrial fibrillation. Acta Cardiol. 2010; 65:271-273. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen TP, Qu Z, Weiss JN. Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol. 2014; 70C:83-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perazzolo MM, De LM, Zorzi A, Migliore F, Zilio F, Calore C, Vettor G, Tona F, Tarantini G, Cacciavillani L, Corbetti F, Giorgi B, Miotto D, Thiene G, Basso C, Iliceto S, Corrado D. Impact of the presence and amount of myocardial fibrosis by cardiac magnetic resonance on arrhythmic outcome and sudden cardiac death in nonischemic dilated cardiomyopathy. Heart Rhythm. 2014; 11:856-863. [DOI] [PubMed] [Google Scholar]
- 46.O'Neill MD, Wright M, Knecht S, Jais P, Hocini M, Takahashi Y, Jonsson A, Sacher F, Matsuo S, Lim KT, Arantes L, Derval N, Lellouche N, Nault I, Bordachar P, Clementy J, Haissaguerre M. Long‐term follow‐up of persistent atrial fibrillation ablation using termination as a procedural endpoint. Eur Heart J. 2009; 30:1105-1112. [DOI] [PubMed] [Google Scholar]