

Abstract
Background
The p.Gln554X mutation in desmocollin‐2 (DSC2) is prevalent in ≈10% of the Hutterite population. While the homozygous mutation causes severe biventricular arrhythmogenic right ventricular cardiomyopathy, the phenotypic features and prognosis of heterozygotes remain incompletely understood.
Methods and Results
Eleven homozygotes (mean age 32±8 years, 45% female), 28 heterozygotes (mean age 40±15 years, 50% female), and 22 mutation‐negatives (mean age 43±17 years, 41% female) were examined. Diagnostic testing was performed as per the arrhythmogenic right ventricular cardiomyopathy modified Task Force Criteria. Inverted T waves in the right precordial leads on ECG were seen in all homozygotes but not in their counterparts (P<0.001). Homozygotes had higher median daily premature ventricular complex burden than did heterozygotes or mutation‐negatives (1407 [IQR 1080 to 2936] versus 2 [IQR 0 to 6] versus 6 [IQR 0 to 214], P=0.0002). Ventricular tachycardia was observed in 60% of homozygotes but in none of the remaining individuals (P<0.001). On cardiac magnetic resonance imaging, homozygotes had significantly larger indexed end‐diastolic volumes (right ventricular: 122±24 versus 83±17 versus 83±12 mL/m2, P<0.0001; left ventricular: 93±18 versus 76±13 versus 80±11 mL/m2, P=0.0124) and lower ejection fraction values compared with heterozygotes and mutation‐negatives (right ventricular ejection fraction: 41±9% versus 59±9% versus 61±6%, P<0.0001; left ventricular ejection fraction: 53±8% versus 65±5% versus 64±5%, P<0.0001). Most affected individuals lacked right ventricular wall motion abnormalities. Thus, few met cardiac magnetic resonance imaging task force criteria.
Conclusions
The ECG reliably identifies homozygous p.Gln554X carriers and may be useful as an initial step in the screening of high‐risk Hutterites. The cardiac phenotype of heterozygotes appears benign, but further prospective follow‐up of their arrhythmic risk is needed.
Keywords: arrhythmogenic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy/dysplasia, ECG screening, Hutterite population, risk stratification, sudden cardiac death
Introduction
Arrhythmogenic right ventricular (RV) cardiomyopathy (ARVC) is an inherited disease of cardiac muscle associated with ventricular arrhythmias and predominantly RV structural abnormalities.1–3 The prevalence of ARVC in the general population is at least 1:5000,1,4 and it is a leading cause of sudden cardiac death (SCD).2–3 ARVC is caused primarily by mutations in the 5 desmosomal proteins―plakoglobin, desmoplakin, desmoglein‐2, plakophillin‐2, and desmocollin‐2 (DSC2)―and is inherited in an autosomal dominant manner in the majority of cases.5–12 Less frequently, a recessive inheritance pattern is observed and is associated with cardiocutaneous manifestations such as occur in Naxos disease and Carvajal syndrome.13–15 Biventricular and left ventricular (LV) dominant forms of ARVC are being increasingly recognized16–18; hence it is also broadly referred to as arrhythmogenic cardiomyopathy (AC).
The c.1660C>T (p.Gln554X) mutation in DSC2 is associated with AC and is prevalent in ≈10% of Hutterites, a genetically isolated population originating in 16th‐century Europe who are descendants from <100 founders.19–22 There are 3 branches (leuts) of Hutteries: Dariusleut, Lehrerleut, and Schmiedeleut; marriages between leuts are rare.20 It is estimated that >45 000 Hutterites reside in North America. While the homozygous p.Gln554X mutation is associated with a severe biventricular arrhythmogenic cardiomyopathy in the absence of cutaneous manifestations, the phenotypic features and prognosis of heterozygote carriers remain incompletely understood.22 Guidelines for screening of carriers follow the ARVC Modified Task Force Criteria (TFC)23 and consist of extensive and costly testing. We sought to characterize the phenotypic features of the p.Gln554X mutation carriers to optimize screening strategies and to determine their arrhythmic risk.
Methods
Patients
All members of 3 large Hutterite families (representing all 3 leuts) aged >14 years followed at the University of Calgary Inherited Arrhythmia Clinic who had been genotyped for the p.Gln554X mutation and had clinical investigations were included in the study. Clinical evaluation included 12‐lead ECG, 24‐hour ambulatory monitoring, exercise testing using the Bruce protocol, signal‐averaged ECG (SAECG), and cardiac magnetic resonance imaging (CMR). The diagnosis of ARVC was as per 2010 TFC.23 The presence of ventricular tachycardia (VT) was determined from all ECG data (rest recordings, ambulatory monitoring, and exercise testing) and was defined as ≥3 consecutive QRS complexes of ventricular origin >100 bpm. Epsilon waves were defined as discrete low‐amplitude complexes occurring after the end of the QRS in leads V1 through V3.24 All study participants provided informed consent, and the study was approved by the University of Calgary Ethics Review Board (ID‐23441).
Genotyping
Genotyping of study participants was performed as described previously.22 Study participants underwent targeted screening for the c.1660C>T (p.Gln554X) mutation in DSC2 via PCR amplification of exon 11 followed by direct Sanger sequencing.
CMR Examination
CMR was performed by using a 1.5‐ or 3.0‐T scanner (Avanto/Skyra; Siemens) equipped with a 32‐channel cardiac coil. LV systolic function determination was obtained from 6 radial long‐axis and 3 short‐axis steady state free precession cine images (gated, 15 to 26 seconds breath‐hold, slice thickness 10 mm). RV systolic function was determined from short‐axis, cine images acquired along the axis of the RV (slice thickness 6 mm). Analysis of RV structure was also performed on sagittal RV steady state free precession cine imaging. Late gadolinium enhancement imaging was performed using a standard phase sensitive inversion recovery sequence 10 to 15 minutes after 0.2 mmol/kg gadolinium chelate administration. Because detailed assessment of LV morphology is not part of the standard CMR ARVC protocol at our institution, all studies were reread by 2 expert readers (S.G.W. and L.K.), blinded to the participant genotypes.
Data Analysis
Continuous variables are expressed as mean±SD or median and IQR, if the data were not normally distributed. Categorical variables are expressed as frequencies. Three group comparisons of continuous data were performed by using ANOVA or the Kruskal–Wallis test if the data were not normally distributed. Two group comparisons of continuous data were performed by using the Student t test or the Wilcoxon rank‐sum test if the data were not normally distributed. Fisher's exact test was used for comparison of categorical variables. Two‐tailed probability values <0.05 were considered significant. The Bonferroni correction was used for subgroup comparisons when 3 group comparisons were statistically significant. Statistical analysis was performed using Stata version 13.0 (StataCorp LP).
Results
Patients
One hundred eleven individuals from 3 large Hutterite families were initially screened, with a total of 64 patients meeting inclusion criteria. Thirteen were homozygous for the p.Gln554X mutation (homozygotes), 29 were heterozygous (heterozygotes), and 22 did not carry the mutation (mutation‐negatives). A total of 4 patients had an SCD event: 3 homozygotes and 1 presumed heterozygous (obligate) carrier. Among the 4 deceased individuals, 1 homozygote had clinical investigations performed and was included in the study. Our study population thus consisted of 11 homozygotes (mean age 32±8 years, 45% female), 28 heterozygotes (mean age 40±15 years, 50% female), and 22 mutation‐negatives (mean age 43±17, 41% female). All homozygotes with an SCD event underwent autopsy (ages 14, 15, and 28), confirming the diagnosis of ARVC. An autopsy was not performed on the heterozygous obligate carrier (a 54‐year‐old man) due to family refusal, and cause of death could not be determined.
Repolarization and Depolarization Abnormalities
Repolarization abnormalities are summarized in Table 1. Inverted T waves in the right precordial leads (V1 to V3) were seen in all homozygotes but in none of the heterozygotes or mutation‐negatives (P<0.001). Further, all heterozygotes and mutation‐negatives lacked repolarization abnormalities, with the one exception being a heterozygous individual who had inverted T waves in V1 to V2. All homozygotes met major TFC for repolarization abnormalities compared with none of the heterozygotes or mutation‐negatives (sensitivity and specificity both 100%). Only 1 heterozygote and 0 of the mutation‐negatives met minor TFC. Table 2 summarizes TFC ECG depolarization abnormalities. The presence of an epsilon wave, the only major TFC in this category, was observed in 27% of homozygotes but in none of the remaining individuals (P=0.006). Delayed terminal QRS activation ≥55 ms was seen in 64% of homozygotes but in only 12% of heterozygotes and in none of the mutation‐negatives (P<0.001). Although homozygotes had greater evidence of late potentials on SAECG, 5 heterozygotes and 1 noncarrier had at least 1 abnormality on SAECG. The majority (82%) of homozygotes met minor TFC for depolarization, compared with only 5 heterozygotes and 1 mutation‐negative (P<0.001).
Table 1.
Repolarization Abnormalities in the Study Population as per 2010 ARVC Modified Task Force Criteria
| Category | Homozygotes (n=11) | Heterozygotes (n=28) | Mutation‐Negatives (n=22) | P Value |
|---|---|---|---|---|
| Inverted T waves V1 to V3, n (%) | 11/11 (100) | 0/25 (0) | 0/21 (0) | <0.001 |
| Inverted T waves V1 to V2, n (%) | 11/11 (100) | 1/25 (4) | 0/21 (0) | <0.001 |
| Inverted T waves V1 to V3+,* n (%) | 8/11 (73) | 0/25 (0) | 0/21 (0) | <0.001 |
| Inverted T waves V4 to V6, n (%) | 5/11 (45) | 0/25 (0) | 0/21 (0) | <0.001 |
| RBBB+inverted T waves V1 to V4, n (%) | 5/11 (45) | 0/25 (0) | 0/21 (0) | <0.001 |
| Inverted T waves V2 to V3, n (%) | 11/11 (100) | 0/25 (0) | 0/21 (0) | <0.001 |
| TFC repolarization (major), n (%) | 11/11 (100) | 0/25 (0) | 0/21 (0) | <0.001 |
| TFC repolarization (minor), n (%) | 0/11 (0) | 1/25 (4) | 0/21 (0) | 1.0 |
ARVC indicates arrhythmogenic right ventricular cardiomyopathy; RBBB, right bundle branch block; TFC, Task Force Criteria.
T‐wave inversion in V1 to V3 and beyond.
Table 2.
Depolarization Abnormalities in the Study Population as per 2010 ARVC Modified Task Force Criteria
| Category | Homozygotes (n=11) | Heterozygotes (n=28) | Mutation‐Negatives (n=22) | P Value |
|---|---|---|---|---|
| ECG | ||||
| Epsilon wave, n (%) | 3/11 (27) | 0/25 (0) | 0/21 (0) | 0.006 |
| Terminal QRS ≥55 ms, n (%) | 7/11 (64) | 3/25 (12) | 0/21 (0) | <0.001 |
| SAECG | ||||
| Filtered QRS duration, ms | 123±16 | 102±10 | 97±13 | 0.0013 |
| Terminal QRS <40 μV, ms | 51±16 | 30±12 | 26±6 | 0.0025 |
| RMS voltage terminal 40 ms, μV | 12±5 | 42±23 | 56±41 | 0.016 |
| Task Force Criteria | ||||
| Depolarization (major), n (%) | 3/11 (27) | 0/25 (0) | 0/21 (0) | 0.006 |
| Depolarization (minor), n (%) | 9/11 (82) | 5/25 (20) | 1/21 (5) | <0.001 |
Continuous variables presented as mean±SD. ARVC indicates arrhythmogenic right ventricular cardiomyopathy; SAECG, signal‐averaged ECG; RMS, root‐mean‐square.
Ventricular Arrhythmias
The presence of ventricular arrhythmias in the study population is summarized in Table 3. Homozygotes had a significantly higher median premature ventricular complex (PVC) burden compared with heterozygotes or mutation‐negatives (1407 [IQR 1080 to 2936] versus 2 [IQR 0 to 6] versus 6 [IQR 0 to 214] PVCs/24 h, respectively; P=0.0002). In addition, nearly all homozygotes (89%) had >500 PVCs/24 h, a minor TFC, while only 1 heterozygote and 1 mutation‐negative had such a finding (P<0.001). Overall, 60% of homozygotes had documented VT, compared with none of the heterozygotes or mutation‐negatives (P<0.001). If those with SCD are included in the analysis, the prevalence of VT among homozygotes increased to 69%. Among those individuals with VT, only 1 had left bundle branch block with superior axis morphology VT, which is the only major TFC in the arrhythmia category. Overall, 90% of homozygotes met minor TFC compared with only 1 heterozygote and 1 mutation‐negative (P<0.001).
Table 3.
Presence of Ventricular Arrhythmias in the Study Population as per 2010 ARVC Modified Task Force Criteria
| Category | Homozygotes (n=11) | Heterozygotes (n=28) | Mutation‐Negatives (n=22) | P Value |
|---|---|---|---|---|
| Holter | ||||
| PVCs/24 h, median (IQR) | 1407 (1080, 2936) | 2 (0, 6) | 6 (0, 214) | 0.0002 |
| >500 PVCs/24 h, n (%) | 8/9 (89) | 1/24 (4) | 1/7 (14) | <0.001 |
| Ventricular tachycardia | ||||
| Any morphology, n (%) | 6/10 (60) | 0/26 (0) | 0/7 (0) | <0.001 |
| LBBB, superior axis, n (%) | 1/10 (10) | 0/26 (0) | 0/7 (0) | 0.395 |
| LBBB, inferior axis, n (%) | 3/10 (30) | 0/26 (0) | 0/7 (0) | 0.013 |
| RBBB, n (%) | 2/10 (20) | 0/26 (0) | 0/7 (0) | 0.073 |
| Task Force Criteria | ||||
| Major, n (%) | 1/10 (10) | 0/26 (0) | 0/7 (0) | 0.395 |
| Minor, n (%) | 9/10 (90) | 1/26 (4) | 1/7 (14) | <0.001 |
ARVC indicates arrhythmogenic right ventricular cardiomyopathy; PVC, premature ventricular complex; LBBB, left bundle branch block; RBBB, right bundle branch block.
Global/Regional Dysfunction and Structural Alterations
CMR findings are summarized in Table 4. Indexed RV end‐diastolic volume was significantly greater in homozygotes, with 82% having abnormal values as per TFC compared with 25% of heterozygotes and 33% of mutation‐negatives (P=0.006). Similarly, homozygotes had larger indexed LV end‐diastolic volume values than their counterparts. RV and LV ejection fractions were significantly decreased in homozygotes compared with heterozygotes or mutation‐negatives (RV ejection fraction: 41±9% versus 59±9% versus 61±6%, P<0.0001 and LV ejection fraction: 53±8% versus 65±5% versus 64±5%, P<0.0001). Furthermore, 73% of homozygotes had an abnormal RV ejection fraction as per TFC compared with none of the heterozygotes or mutation‐negatives (P<0.001). Despite these significant differences, regional RV akinesis, dyskinesis, or dyssynchrony was observed in the minority of patients (4 homozygotes, 4 heterozygotes, and 0 of the mutation‐negatives, P=0.2). As a result, only 3 homozygotes and 2 heterozygotes met major CMR TFC, while 0 met minor criteria. Interestingly, 73% of homozygotes had significant regional LV findings such as focal areas of myocardial thinning and the presence of dyskinetic/aneurysmal segments particularly involving the lateral wall. LV structural findings currently do not form part of the CMR TFC. Overall, heterozygotes lacked the profound RV and LV structural abnormalities observed in homozygotes. LV structural abnormalities were observed in 25% of heterozygotes with the typical CMR findings including apical thinning with microaneurysms (Figures 1 and 2). Similarly, 33% of heterozygotes had other abnormal RV findings, with the presence of microaneurysms and trabecular hypertrophy being the most common. Biventricular abnormalities were seen in 8%. A small, hypokinetic LV apical diastolic bulge was the sole structural abnormality found among mutation‐negatives. Among heterozygotes, there was no apparent temporal association between the appearance of electrical abnormalities as seen on ECG, Holter, or SAECG, and the structural abnormalities observed on CMR. In many cases, abnormal CMR findings appeared to precede electrical abnormalities (Table 5). Older age or male sex was not associated with an increased frequency of structural or electrical abnormalities in heterozygotes (Table 6), although sample size likely limited our statistical power to detect a difference.
Table 4.
CMR Findings in the Study Population
| Category | Homozygotes (n=11) | Heterozygotes (n=28) | Mutation‐Negatives (n=22) | P Value |
|---|---|---|---|---|
| RV EDV, mL | 224±59 | 163±37 | 147±21 | 0.0005 |
| RV ESV, mL | 139±52 | 68±23 | 59±15 | <0.0001 |
| BSA, m2 | 1.8±0.2 | 2.0±0.2 | 1.8±0.3 | 0.198 |
| RV EDV/BSA, mL/m2 | 122±24 | 83±17 | 83±12 | <0.0001 |
| RV ESV/BSA, mL/m2 | 74±23 | 34±10 | 33±7 | <0.0001 |
| RVEF, % | 41±9 | 59±9 | 61±6 | <0.0001 |
| TFC major volume,* n (%) | 8/11 (73) | 4/24 (17) | 0/6 (0) | 0.002 |
| TFC minor volume,* n (%) | 1/11 (9) | 2/24 (8) | 2/6 (33) | 0.311 |
| TFC major RVEF,* n (%) | 5/11 (45) | 0/24 (0) | 0/6 (0) | 0.001 |
| TFC minor RVEF,* n (%) | 3/11 (27) | 0/24 (0) | 0/6 (0) | 0.033 |
| Any RV WMA, n (%) | 4/11 (36) | 4/24 (17) | 0/6 (0) | 0.2 |
| RV akinesia, n (%) | 3/11 (27) | 1/24 (4) | 0/6 (0) | 0.106 |
| RV dyskinesia, n (%) | 2/11 (18) | 1/24 (4) | 0/6 (0) | 0.221 |
| RV dyssynchrony, n (%) | 0/11 (0) | 2/24 (8) | 0/6 (0) | 1.0 |
| Any RV non‐WMA, n (%) | 6/8 (75) | 8/24 (33) | 2/6 (33) | 0.155 |
| Microaneurysm, n (%) | 2/8 (25) | 3/21 (13) | 0/6 (0) | 0.51 |
| Segmental dilation, n (%) | 1/6 (17) | 1/24 (4) | 1/6 (17) | 0.253 |
| Accordion sign, n (%) | 2/5 (40) | 2/24 (8) | 0/6 (0) | 0.135 |
| Fibrofatty replacement, n (%) | 2/6 (33) | 0/24 (0) | 0/6 (0) | 0.045 |
| Trabecular hypertrophy, n (%) | 6/8 (75) | 4/24 (17) | 2/6 (33) | 0.008 |
| LV EDV, mL | 169±36 | 147±27 | 142±17 | 0.0972 |
| LV ESV, mL | 82±28 | 52±12 | 51±10 | 0.0002 |
| LV EDV/BSA, mL/m2 | 93±18 | 76±13 | 80±11 | 0.0124 |
| LVEF, % | 53±8 | 65±5 | 64±5 | <0.0001 |
| Any LV abnormality, n (%) | 8/11 (73) | 6/24 (25) | 1/6 (17) | 0.014 |
| LV wall thinning | 7/11 (64) | 3/24 (13) | 0/6 (0) | 0.004 |
| LV apical thinning | 3/11 (27) | 4/24 (17) | 0/6 (0) | 0.526 |
| LV hypokinesia/akinesia | 4/11 (36) | 0/24 (0) | 1/6 (17) | 0.005 |
| LV aneurysm | 7/11 (64) | 1/24 (4) | 0/6 (0) | <0.001 |
| CMR TFC major, n (%) | 3/11 (27) | 2/24 (8) | 0/6 (0) | 0.311 |
| CMR TFC minor, n (%) | 0/11 (0) | 0/24 (0) | 0/6 (0) | 1.0 |
Continuous variables expressed as mean±SD. RV indicates right ventricular; EDV, end‐diastolic volume; ESV, end‐systolic volume; BSA indicates body surface area; EF, ejection fraction; LV, left ventricular; TFC, Task Force Criteria; WMA, wall motion abnormality.
Exceeds major cardiac magnetic resonance imaging (CMR) criteria cut‐off for volume.
Exceeds minor CMR criteria cut‐off for volume.
Exceeds major CMR criteria cut‐off for RVEF.
Exceeds minor CMR criteria cut‐off for RVEF.
Figure 1.
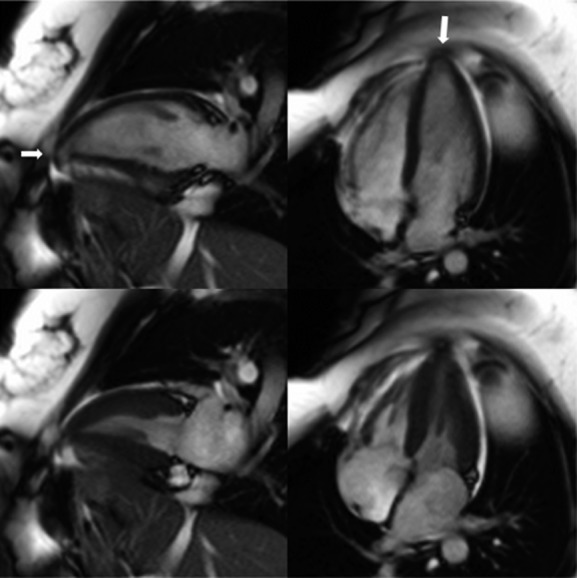
Cardiac magnetic resonance imaging 2‐chamber and 4‐chamber views at end‐diastole (top panels) and end‐systole (bottom panels) of a desmocollin‐2 (DSC2) p.Gln554X heterozygote showing left ventricular thinning and a small aneurysm at the apex (arrows). There were no right ventricular abnormalities.
Figure 2.
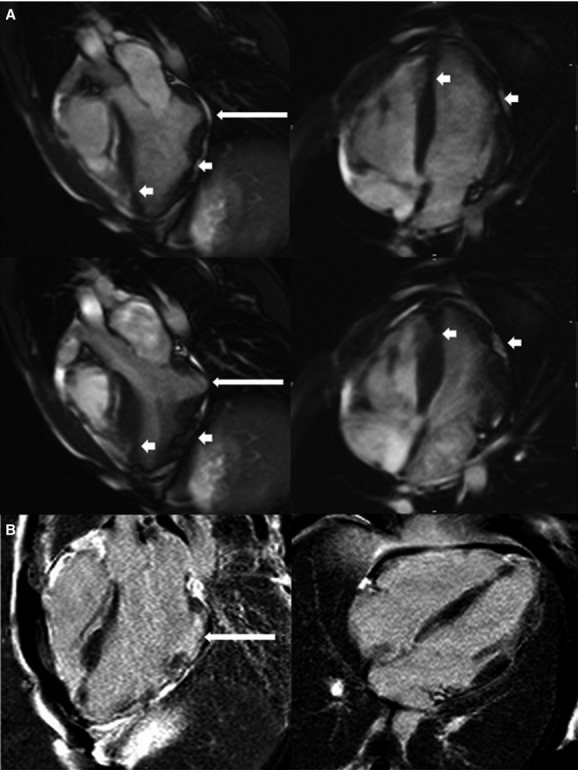
A, Cardiac magnetic resonance imaging 3‐chamber and 4‐chamber views at end‐diastole (top panels) and end‐systole (bottom panels) of a homozygote showing a basal inferolateral aneurysm (long arrow) with additional focal areas of thinning and akinesis in the apex, apical septum, apical anterior, and mid anterolateral walls (short arrows). There were no right ventricular abnormalities. B, Corresponding views showing small regions of late gadolinium enhancement at the edge of the basal inferolateral aneurysm (long arrow).
Table 5.
CMR Findings in Heterozygous Carriers Stratified by the Presence of Electrical Abnormalities
| Category | No Electrical Abnormality* (n=17) | Electrical Abnormality* (n=7) | P Value |
|---|---|---|---|
| RV EDV, mL | 165±42 | 156±22 | 0.591 |
| RV ESV, mL | 72±23 | 56±22 | 0.123 |
| RV EDV/BSA, mL/m2 | 82±18 | 86±15 | 0.654 |
| RV ESV/BSA, mL/m2 | 36±10 | 31±12 | 0.307 |
| RVEF, % | 57±7 | 64±11 | 0.067 |
| TFC major volume,* n (%) | 3/17 (18) | 1/7 (14) | 1.0 |
| TFC minor volume,* n (%) | 1/17 (6) | 1/7 (14) | 0.507 |
| TFC major RVEF,* n (%) | 0/17 (0) | 0/7 (0) | 1.0 |
| TFC minor RVEF,* n (%) | 0/17 (0) | 0/7 (0) | 1.0 |
| Any RV WMA, n (%) | 4/17 (24) | 0/7 (0) | 0.283 |
| RV akinesia, n (%) | 1/17 (6) | 0/7 (0) | 1.0 |
| RV dyskinesia, n (%) | 1/17 (6) | 0/7 (0) | 1.0 |
| RV dyssynchrony, n (%) | 2/17 (12) | 0/7 (0) | 1.0 |
| Any RV non‐WMA, n (%) | 7/17 (41) | 1/7 (14) | 0.352 |
| Microaneurysm, n (%) | 2/17 (12) | 1/7 (14) | 1.0 |
| Segmental dilation, n (%) | 1/17 (6) | 0/7 (0) | 1.0 |
| Accordion sign, n (%) | 2/17 (12) | 0/7 (0) | 1.0 |
| Fibrofatty replacement, n (%) | 0/17 (0) | 0/7 (0) | 1.0 |
| Trabecular hypertrophy, n (%) | 3/17 (18) | 1/7 (14) | 1.0 |
| LV EDV, mL | 148±29 | 146±24 | 0.844 |
| LV ESV, mL | 52±12 | 53±12 | 0.904 |
| LV EDV/BSA, mL/m2 | 74±13 | 80±16 | 0.32 |
| LVEF, % | 65±5 | 64±4 | 0.677 |
| Any LV finding, n (%) | 4/17 (24) | 2/7 (29) | 1.0 |
| LV wall thinning, n (%) | 3/17 (19) | 0/7 (0) | 0.526 |
| LV apical thinning, n (%) | 2/17 (12) | 2/7 (29) | 0.557 |
| LV hypokinesia/akinesia, n (%) | 0/17 (0) | 0/7 (0) | 1.0 |
| LV aneurysm, n (%) | 0/17 (0) | 1/7 (14) | 0.304 |
| CMR TFC major, n (%) | 2/17 (12) | 0/7 (0) | 1.0 |
| CMR TFC minor, n (%) | 0/17 (0) | 0/7 (0) | 1.0 |
Continuous variables expressed as mean±SD. RV indicates right ventricular; EDV, end‐diastolic volume; BSA, body surface area; ESV, end‐systolic volume; EF, ejection fraction; TFC, Task Force Criteria; WMA, wall motion abnormality; LV, left ventricular.
Electrical abnormality was defined as presence of any major or minor Task Force Criteria in depolarization, repolarization, or arrhythmia category.
Exceeds major cardiac magnetic resonance imaging (CMR) criteria cut‐off for volume.
Exceeds minor CMR criteria cut‐off for volume.
Exceeds major CMRI criteria cut‐off for RVEF.
Exceeds minor CMRI criteria cut‐off for RVEF.
Table 6.
Sex and Age Associations With RV Structural and Electrical Abnormalities in Heterozygotes
| Category | Male (n=14) | Female (n=14) | P Value | Age <50 y (n=18) | Age >50 y (n=10) | P Value |
|---|---|---|---|---|---|---|
| Electrical abnormalities,* n (%) | 3/14 (21) | 4/13 (31) | 0.678 | 3/17 (18) | 4/10 (40) | 0.365 |
| Any structural abnormality, n (%) | 6/12 (50) | 3/12 (25) | 0.4 | 4/15 (27) | 5/9 (56) | 0.212 |
| Regional WMA,* n (%) | 3/12 (25) | 1/12 (8) | 0.59 | 3/15 (20) | 1/9 (11) | 1.0 |
| Non‐WMA,* n (%) | 5/12 (42) | 3/12 (25) | 0.667 | 3/15 (20) | 5/9 (56) | 0.099 |
| RVEF, % | 57±11 | 61±6 | 0.219 | 56±5 | 63±13 | 0.103 |
RVEF indicates right ventricular ejection fraction.
Electrical abnormalities defined as presence of any major or minor Task Force Criteria in depolarization, repolarization, or arrhythmia categories.
WMA, wall motion abnormality (any of akinesis, dyskinesis, or dyssynchrony).
Presence of any of RV microaneurysms, segmental RV dilation, accordion sign, fibrofatty replacement, or trabecular hypertrophy.
Overall TFC Score
There were no statistically significant differences between heterozygotes and mutation‐negatives in all aforementioned categories (not shown). All homozygotes and heterozygotes received 1 major criterion for being mutation carriers. Seven mutation‐negatives received a major criterion for being first‐degree relatives of an individual with ARVC, while 15 received a minor criterion for having an affected second‐degree relative. Thus, all homozygotes met TFC for definite ARVC compared with only 2 heterozygotes and 0 of the mutation‐negatives (Table 7). Seven heterozygotes and 2 mutation‐negatives had borderline scores for ARVC. The median TFC major score in homozygotes was 3 (IQR 2 to 3) compared with 1 (IQR 1 to 1) in heterozygotes and 0 (IQR 0 to 1) in mutation‐negatives (P<0.001). In addition, the median TFC minor score in homozygotes was 2 (IQR 1 to 2) versus 0 (IQR 0 to 0.5) in heterozygotes and 1 (IQR 1 to 1) in mutation‐negatives (P<0.001).
Table 7.
Summary of 2010 ARVC Task Force Criteria for the Study Population
| ARVC Task Force Criteria | Homozygotes (n=11) | Heterozygotes (n=28) | Mutation‐Negatives (n=22) | P Value |
|---|---|---|---|---|
| Repolarization major, n (%) | 11/11 (100) | 0/25 (0) | 0/21 (0) | <0.001 |
| Repolarization minor, n (%) | 0/11 (0) | 1/25 (4) | 0/21 (0) | 1.0 |
| Depolarization major, n (%) | 3/11 (27) | 0/25 (0) | 0/21 (0) | 0.006 |
| Depolarization minor, n (%) | 9/11 (82) | 5/25 (20) | 1/21 (5) | <0.001 |
| Arrhythmia major, n (%) | 1/10 (10) | 0/26 (0) | 0/7 (0) | 0.395 |
| Arrhythmia minor, n (%) | 9/10 (90) | 1/26 (4) | 1/7 (14) | <0.001 |
| CMR major, n (%) | 3/11 (27) | 2/24 (8) | 0/6 (0) | 0.311 |
| CMR minor, n (%) | 0/10 (0) | 0/24 (0) | 0/6 (0) | 1.0 |
| Family history major, n (%) | 11/11 (100) | 28/28 (100) | 7/22 (32) | <0.001 |
| Family history minor, n (%) | 0/11 (0) | 0/28 (0) | 15/22 (68) | <0.001 |
| Total major score, median [IQR] | 3 [2 to 3] | 1 [1 to 1] | 0 [0 to 1] | <0.001 |
| Total minor score, median [IQR] | 2 [1 to 2] | 0 [0 to 0.5] | 1 [1 to 1] | <0.001 |
| Definite TFC, n (%) | 11/11 (100) | 2/28 (7) | 0/22 (0) | <0.001 |
| Borderline TFC, n (%) | 0/11 (0) | 7/28 (25) | 2/22 (9) | 0.113 |
ARVC indicates arrhythmogenic right ventricular cardiomyopathy; CMR, cardiac magnetic resonance imaging; TFC, Task Force Criteria.
Discussion
The key findings in this study of the clinical characterization of DSC2 p.Gln554X mutation carriers in Hutterites are (1) repolarization abnormalities were seen in all homozygotes but rarely in heterozygotes and mutation‐negatives; the presence of T‐wave inversions in V1 to V3 was 100% specific and sensitive for the homozygous mutation; (2) depolarization abnormalities were not frequently seen in heterozygotes or mutation‐negatives but were observed in a large proportion of homozygotes; (3) heterozygotes and mutation‐negatives had no documented VT, while it was common among homozygotes; (4) although cardiac structural abnormalities were relatively frequent, only a minority met CMR TFC; (5) CMR abnormalities in heterozygotes did not correlate with and appeared to precede electrical abnormalities; and (6) all homozygotes met definite TFC criteria for ARVC compared with only a few heterozygotes and no mutation‐negatives.
AC is caused primarily by mutations in the 5 desmosomal proteins: plakoglobin, desmoplakin, desmoglein‐2, plakophillin‐2), and desmocollin‐2.6–11,25 To date, disease‐causing AC mutations on DSC2 have been linked, for the most part, to an autosomal dominant pattern of inheritance.5–6 This is not surprising because such mutations are likely to cause significant protein structural alterations such that a single copy of the defective gene is expected to have deleterious effects on desmosomal function. Interestingly, the DSC2 p.Gln554X mutation under study causes a severe biventricular arrhythmogenic cardiomyopathy in the homozygous state, much like in Naxos disease and Carvajal syndrome.13–15,22 In contrast, the phenotype of heterozygote carriers remains uncertain. To the best of our knowledge, there has been only 1 study in patients with Naxos disease or Carvajal syndrome focusing on the phenotypic findings of heterozygous carriers. Antoniades et al reported on the phenotypic characterization of 46 heterozygous carriers of the 2–base pair deletion in plakoglobin (2157del2) associated with Naxos disease.26 In this study, repolarization abnormalities were observed in 19% of heterozygotes, while regional wall motion abnormalities as identified by echocardiography and frequent PVCs were seen in 2 and 1 individual, respectively. Depolarization abnormalities, enlarged RV volumes, and VT were not observed. None of the heterozygotes fulfilled ARVC TFC using original criteria. Similarly, in our study, abnormal findings were infrequent in heterozygotes. Depolarization abnormalities were seen in 20%, while the presence of repolarization abnormalities and frequent PVCs was rare. Structural abnormalities appeared to be more prevalent in our heterozygous population with an abnormally enlarged RV volume seen in 25%, RV regional wall motion abnormalities (akinesia, dyskinesia, or dyssynchrony) observed in 17%, and other RV and LV structural abnormalities seen in 33% and 25%, respectively. One caveat is that we used CMR to identify structural alterations in our cohort, which is a more sensitive imaging technique than that used by Antoniades. Notwithstanding, structural abnormalities appeared out of proportion with electrical abnormalities, suggesting that CMR findings may precede electrical ones. Overall, 2 heterozygotes met definite ARVC TFC, while 7 met borderline ARVC criteria. Despite this, 0 of the heterozygotes had documented VT.
Although long‐term follow‐up data are lacking, the heterozygous phenotype of the p.Gln554X mutation appears to be benign. To date, we are only aware of 1 SCD event in a heterozygote: a 54‐year‐old smoker and hypertensive man in whom coronary artery disease is also a likely culprit. Unfortunately, the family did not permit an autopsy and the cause of death was never determined. We have previously estimated the prevalence of the DSC2 p.Gln554X mutation to be 9.4% of the Hutterite population.22 Given that there are ≈45 000 Hutterites in North America, the potential number of gene carriers would consist of nearly 4500 individuals who would require serial clinical testing as per ARVC TFC. Our data showing a relatively benign phenotype in heterozygotes are helpful in that they allow us to counsel affected family members about their presumably low risk for SCD provided that high‐risk homozygous carriers can be identified efficiently and cost‐effectively. While the availability of genetic testing helps to identify homozygous carriers, Hutterites' propensity for living in rural areas and their variable cultural acceptance for genetic testing limit its utility as the initial step in the screening of high‐risk individuals. For these reasons, noninvasive and easily accessible tests that are highly sensitive and specific for the presence of the homozygous mutation would be extremely useful. In our study, we found that the presence of T‐wave inversions in V1 to V3 was both 100% sensitive and specific for the homozygous mutation. Further, the presence of >500 PVCs/24 h had a specificity of 89% and sensitivity of 94%. We propose that these inexpensive, noninvasive tests could be used as a first step to screen the Hutterite population to identify high‐risk homozygotes. Those with abnormal testing results would then proceed to full clinical evaluation as per TFC, while those with normal testing results would undergo repeat yearly ECGs. If successful, this strategy would not only be extremely cost‐effective but also increase Hutterites' access to health evaluation, which can be limited due to their rural livelihoods. Prospective validation of such a screening approach is needed and is currently under way.
Our study also illustrates some of the limitations with the current ARVC TFC. Only a minority (27%) of homozygotes met CMR TFC under structural alterations, despite having significant biventricular findings. Further, while 60% of homozygotes had documented VT, only one individual met major TFC under the arrhythmia category by way of left‐bundle branch block, superior‐axis VT. These TFC shortcomings are likely secondary to the biventricular nature of the arrhythmogenic cardiomyopathy seen in p.Gln554X homozygotes and reflect the current lack of recognition of LV‐dominant and biventricular forms of ARVC in the current guidelines. As the latter forms of ARVC are being increasingly recognized, it is imperative that future TFC iterations be inclusive of them.
This study has several limitations. First, our findings are based on a small number of patients and at times lacked the statistical power needed to show certain associations. Second, the retrospective nature of the study led to some missing data, which could have introduced an element of bias in the interpretation of the results. Third, since our investigation protocol for evaluating mutation‐negatives typically consists of only a 12‐lead ECG, complete clinical evaluation was only available for a small proportion in this subgroup. Finally, long‐term follow‐up data of heterozygotes are not currently available and we cannot rule out the manifestation of a latent phenotype of significance among heterozygotes. Despite these limitations, our study provides valuable insights into the phenotype and arrhythmogenic risk of DSC2 p.Gln554X heterozygotes, which will benefit from continued long‐term follow‐up.
In conclusion, heterozygote carriers of the DSC2 p.Gln554X mutation have few abnormalities as per TFC compared with homozygotes and rarely do individuals meet definite diagnostic criteria for ARVC. Noninvasive testing by way of rest and ambulatory ECG may be useful in the screening of Hutterite colonies. Despite the noted TFC abnormalities, the prognosis of heterozygotes appears benign, but prospective, long‐term data are needed.
Sources of Funding
This study was supported by funds from the Libin Cardiovascular Institute of Alberta, Alberta Innovates Health Solutions, and the Canadian Institutes for Health Research.
Disclosures
None.
Acknowledgments
The authors would like to thank Linda Ellis for performing the SAECGs and Stephanie Desmarais, Priyana Sharma, Julia Tagoe, and Raechel Ferrier for their helpful assistance with genetic counseling and family recruitment.
References
- 1.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom‐Lundqvist C, Wlodarska EK, Fontaine G, Camerini F. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997; 30:1512-1520. [DOI] [PubMed] [Google Scholar]
- 2.Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982; 65:384-398. [DOI] [PubMed] [Google Scholar]
- 3.Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988; 318:129-133. [DOI] [PubMed] [Google Scholar]
- 4.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009; 373:1289-1300. [DOI] [PubMed] [Google Scholar]
- 5.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010; 107:700-714. [DOI] [PubMed] [Google Scholar]
- 6.den Haan AD, Tan BY, Zikusoka MN, Llado LI, Jain R, Daly A, Tichnell C, James C, Amat‐Alarcon N, Abraham T, Russell SD, Bluemke DA, Calkins H, Dalal D, Judge DP. Comprehensive desmosome mutation analysis in north Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet. 2009; 2:428-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse‐Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze‐Bahr E, Thierfelder L. Mutations in the desmosomal protein plakophilin‐2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004; 36:1162-1164. [DOI] [PubMed] [Google Scholar]
- 8.Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse‐Klaassen S, Thierfelder L, MacRae CA, Gerull B. Mutant desmocollin‐2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006; 79:1081-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green KJ, Simpson CL. Desmosomes: new perspectives on a classic. J Invest Dermatol. 2007; 127:2499-2515. [DOI] [PubMed] [Google Scholar]
- 10.Bhuiyan ZA, Jongbloed JD, van der Smagt J, Lombardi PM, Wiesfeld AC, Nelen M, Schouten M, Jongbloed R, Cox MG, van Wolferen M, Rodriguez LM, van Gelder IC, Bikker H, Suurmeijer AJ, van den Berg MP, Mannens MM, Hauer RN, Wilde AA, van Tintelen JP. Desmoglein‐2 and desmocollin‐2 mutations in Dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: results from a multicenter study. Circ Cardiovasc Genet. 2009; 2:418-427. [DOI] [PubMed] [Google Scholar]
- 11.Cox MG, van der Zwaag PA, van der Werf C, van der Smagt JJ, Noorman M, Bhuiyan ZA, Wiesfeld AC, Volders PG, van Langen IM, Atsma DE, Dooijes D, van den Wijngaard A, Houweling AC, Jongbloed JD, Jordaens L, Cramer MJ, Doevendans PA, de Bakker JM, Wilde AA, van Tintelen JP, Hauer RN. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index‐patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype‐phenotype follow‐up study. Circulation. 2011; 123:2690-2700. [DOI] [PubMed] [Google Scholar]
- 12.Gehmlich K, Syrris P, Peskett E, Evans A, Ehler E, Asimaki A, Anastasakis A, Tsatsopoulou A, Vouliotis AI, Stefanadis C, Saffitz JE, Protonotarios N, McKenna WJ. Mechanistic insights into arrhythmogenic right ventricular cardiomyopathy caused by desmocollin‐2 mutations. Cardiovasc Res. 2011; 90:77-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000; 355:2119-2124. [DOI] [PubMed] [Google Scholar]
- 14.Norgett EE, Hatsell SJ, Carvajal‐Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP. Recessive mutation in desmoplakin disrupts desmoplakin‐intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000; 9:2761-2766. [DOI] [PubMed] [Google Scholar]
- 15.Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 2004; 13:185-194. [DOI] [PubMed] [Google Scholar]
- 16.Norman M, Simpson M, Mogensen J, Shaw A, Hughes S, Syrris P, Sen‐Chowdhry S, Rowland E, Crosby A, McKenna WJ. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation. 2005; 112:636-642. [DOI] [PubMed] [Google Scholar]
- 17.Sen‐Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left‐dominant arrhythmogenic cardiomyopathy: an under‐recognized clinical entity. J Am Coll Cardiol. 2008; 52:2175-2187. [DOI] [PubMed] [Google Scholar]
- 18.van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, Cox MG, van Lochem LT, de Boer RA, Hofstra RM, Christiaans I, van Spaendonck‐Zwarts KY, Lekanne dit Deprez RH, Judge DP, Calkins H, Suurmeijer AJ, Hauer RN, Saffitz JE, Wilde AA, van den Berg MP, van Tintelen JP. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012; 14:1199-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin AO. The founder effect in a human isolate: evolutionary implications. Am J Phys Anthropol. 1970; 32:351-367. [DOI] [PubMed] [Google Scholar]
- 20.Hostetler JA. History and relevance of the Hutterite population for genetic studies. Am J Med Genet. 1985; 22:453-462. [DOI] [PubMed] [Google Scholar]
- 21.Chong JX, Ouwenga R, Anderson RL, Waggoner DJ, Ober C. A population‐based study of autosomal‐recessive disease‐causing mutations in a founder population. Am J Hum Genet. 2012; 91:608-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerull B, Kirchner F, Chong JX, Tagoe J, Chandrasekharan K, Strohm O, Waggoner D, Ober C, Duff HJ. Homozygous founder mutation in desmocollin‐2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ Cardiovasc Genet. 2013; 6:327-336. [DOI] [PubMed] [Google Scholar]
- 23.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Circulation. 2010; 121:1533-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nasir K, Bomma C, Tandri H, Roguin A, Dalal D, Prakasa K, Tichnell C, James C, Spevak PJ, Marcus F, Calkins H. Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity: a need to broaden diagnostic criteria. Circulation. 2004; 110:1527-1534. [DOI] [PubMed] [Google Scholar]
- 25.Syed SE, Trinnaman B, Martin S, Major S, Hutchinson J, Magee AI. Molecular interactions between desmosomal cadherins. Biochem J. 2002; 362:317-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antoniades L, Tsatsopoulou A, Anastasakis A, Syrris P, Asimaki A, Panagiotakos D, Zambartas C, Stefanadis C, McKenna WJ, Protonotarios N. Arrhythmogenic right ventricular cardiomyopathy caused by deletions in plakophilin‐2 and plakoglobin (Naxos disease) in families from Greece and Cyprus: genotype‐phenotype relations, diagnostic features and prognosis. Eur Heart J. 2006; 27:2208-2216. [DOI] [PubMed] [Google Scholar]