

Introduction
Tuberous sclerosis complex (TSC) is a genetic syndrome with a highly variable phenotype that may affect several organ systems. The central nervous system findings were the first to be described, and the classic triad of cognitive impairment, facial angiofibromas, and seizures was delineated shortly thereafter.1–2 As the variability and extent of organ involvement were appreciated, diagnostic criteria evolved to include major and minor criteria that taken together would lead to a definite, probable, or possible clinical diagnosis.3–4 Since the most recent refinement of the diagnostic criteria, dramatic advances have been made in understanding the genetic basis and pathogenesis of TSC, and new treatment strategies have been established, significantly affecting all aspects of coordinated care for TSC patients.
The Tuberous Sclerosis Alliance (www.tsalliance.org) convened a Consensus Conference composed of 8 working groups that generated Revised Diagnostic Criteria5 and new Surveillance and Management Guidelines6 with the intention of creating “living documents” to accommodate rapid advances and the need for coordination of care. The conference was informed in part by a recent constituency survey of key opinion leaders, which summarized interim progress, areas in need of further research, unmet medical needs, and barriers to progress.7 The goals of this report are to highlight the new diagnostic criteria and management guidelines as they pertain to cardiology and to expand consideration of the issues relevant to optimal cardiac care of patients with TSC.
TSC is characterized by widespread hamartomas, or abnormal growth of normal tissues. Cardiac rhabdomyomas are hamartomatous growths or benign tumors composed of cardiac myocytes, and they represent the classic neonatal manifestation of cardiac disease in TSC. Additional cardiac diseases such as arrhythmia occur later in life, underscoring the importance of ongoing cardiology care. Here, we review what is known about the natural history of cardiac manifestations in TSC with an emphasis on diagnostic testing, surveillance, and treatment.
The Revised Diagnostic Criteria Include Clinical Genetic Testing
Significant changes have been implemented in the revised diagnostic criteria.5 For example, clinical genetic testing has been added as an independent criterion, sufficient to make the diagnosis of TSC. Since TSC1 and TSC2, the genes that encode hamartin and tuberlin, were identified as the cause of TSC,8–9 substantial strides have been made in defining the pathogenesis of TSC. Mutations in the genes TSC1 and TSC2 cause 75% to 90% of cases (Figure A). Given the increasing appreciation for disease variability and an assortment of mild disease phenotypes that may be on the TSC spectrum, the inclusion of a molecular test represents an important change in the approach to diagnosis. While approximately one‐third of cases have a positive family history, this has not been included as diagnostic criteria but remains informative given the various challenges with performing genetic testing. Importantly, the designation of a definite, probable, or possible clinical diagnosis has been simplified to either “definite” or “possible.” Additional changes were made in several of the clinical criteria (Table 1), and changes regarding cardiovascular features are considered next in detail.
Figure 1.
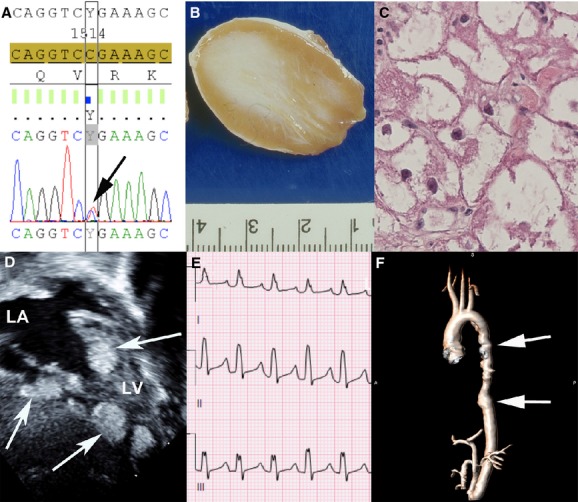
Genetic basis, pathology, and early and late cardiovascular manifestations of TSC. Sequencing of TSC2 demonstrates a missense mutation 1513C>T known to cause TSC (A). Gross pathology shows a discrete well‐demarcated nonencapsulated cardiac rhabdomyomas with heterogeneous tissue (B). Histopathologic examination of the rhabdomyomas demonstrates the classic finding of spider cells representing abnormal myocardial cells (C). Echocardiography shows multiple cardiac rhabdomyomas in the ventricular myocardium (D). ECG shows supraventricular tachycardia with aberrant conduction that can result from cardiac rhabdomyomas or in isolation (E). MRI shows thoracoabdominal aortic aneurysm (arrows) with tortuosity of the descending thoracic aorta (F). LA indicates left atrium; LV, left ventricle; MRI, magnetic resonance imaging; TSC, tuberous sclerosis complex.
Table 1.
Revised Diagnostic Criteria for TSC
| Genetic diagnostic criteria |
| The identification of either a TSC1 or TSC2 pathogenic mutation in DNA from normal tissue is sufficient to make a definite diagnosis of TSC. A pathogenic mutation is defined as a mutation that clearly inactivates the function of the TSC1 or TSC2 proteins (eg, out of frame indel or nonsense mutation), prevents protein synthesis (eg, large genomic deletion), or is a missense mutation whose effect on protein function has been established by functional assessment (www.lovd.nl/TSC1, www.lovd.nl/TSC2,10–11). Other TSC1 or TSC2 variants whose effect on function is less certain do not meet these criteria and are not sufficient to make a definite diagnosis of TSC. Note that 10% to 25% of TSC patients have no mutation identified by conventional genetic testing, and a normal result does not exclude TSC, or have any effect on the use of Clinical Diagnostic Criteria to diagnose TSC. |
| Clinical diagnostic criteria |
| Major features |
| 1. Hypomelanotic macules (≥3, at least 5‐mm diameter) |
| 2. Angiofibromas (≥3) or fibrous cephalic plaque |
| 3. Ungual fibromas (≥2) |
| 4. Shagreen patch |
| 5. Multiple retinal hamartomas |
| 6. Cortical dysplasias* |
| 7. Subependymal nodules |
| 8. Subependymal giant cell astrocytoma |
| 9. Cardiac rhabdomyoma |
| 10. Lymphangioleiomyomatosis (LAM)* |
| 11. Angiomyolipomas (≥2)* |
| Minor features |
| 1. “Confetti” skin lesions |
| 2. Dental enamel pits (>3) |
| 3. Intraoral fibromas (≥2) |
| 4. Retinal achromic patch |
| 5. Multiple renal cysts |
| 6. Nonrenal hamartomas |
Definite diagnosis: 2 major features or 1 major feature with ≥2 minor features. Possible diagnosis: either 1 major feature or ≥2 minor features. TSC indicates tuberous sclerosis complex.
Includes tubers and cerebral white matter radial migration lines.
A combination of the 2 major clinical features LAM and angiomyolipomas without other features does not meet criteria for a definite diagnosis.
Reproduced with permission from Northrup et al.5
Overall Recommendations Have Shifted to Careful Surveillance and Early Intervention
Guidelines for the management and surveillance of TSC patients were comprehensively addressed in a companion article to the revised diagnostic criteria.6 Given the successful clinical trials establishing mammalian target of rapamycin (mTOR) inhibition as a new pharmacologic treatment strategy, a variety of surveillance issues have been considered (Tables 2 and 3). The addition of genetic testing to the diagnostic criteria has implications for screening that were addressed as well. These recommendations affect cardiologists directly with respect to surveillance and potentially in rare circumstances with respect to medical therapy. There is an increasing appreciation for latent cardiovascular phenotypes, indicating a need for continued surveillance of these patients. As the natural history of disease in the cardiovascular system is better understood, continued care in adulthood needs to be defined, underscoring efforts to transition care from pediatric to adult cardiology and to maintain surveillance vigilance in adulthood.
Table 2.
Surveillance and Management Recommendations for Newly Diagnosed or Suspected TSC Summary Table
| Organ System or Specialty Area | Recommendation |
|---|---|
| Genetics |
|
| Brain |
|
| Kidney |
|
| Lung |
|
| Skin |
|
| Teeth |
|
| Heart |
|
| Eye |
|
MRI indicates magnetic resonance imaging; TSC, tuberous sclerosis complex.
Reproduced with permission from Krueger et al.6
Table 3.
Surveillance and Management Recommendations for Patients Already Diagnosed With Definite or Possible TSC Summary Table
| Organ System or Specialty Area | Recommendation |
|---|---|
| Genetics |
|
| Brain |
|
| Kidney |
|
| Lung |
|
| Skin |
|
| Teeth |
|
| Heart |
|
| Eye |
|
TSC indicates tuberous sclerosis complex; MRI, magnetic resonance imaging; SEGA, subependymal giant cell astrocytoma; mTOR, mammalian target of rapamycin; TAND, TSC‐associated neuropsychiatric disorder; EEG, electroencephalography; ACTH, adrenocorticotropic hormone; LAM, lymphangioleiomyomatosis; HRCT, high‐resolution chest computed tomography; PFT, pulmonary function tests; GFR, glomerular filtration rate.
Reproduced with permission from Krueger et al.6
The Natural History and Diagnosis of TSC
The Natural History of Cardiac Rhabdomyomas
Cardiac tumors are rare, and ascertaining incidence is difficult.12–13 Based on clinical studies and autopsy series, primary cardiac tumors occur in 0.2% of children.14 Cardiac rhabdomyomas are by far the most common primary cardiac tumor in childhood.15–16 After the advent of echocardiography, but before clinical genetic testing was available, studies estimated that up to 70% to 90% of children with rhabdomyomas have TSC,17–19 and at least 50% of children with TSC have rhabdomyomas,18 representing a significant increase in the proportion of cardiac rhabdomyomas attributed to TSC compared with historic clinical data.20–22 In 1 study, Allan et al analyzed 52 cardiac tumor cases, among which 44 (86%) were rhabdomyomas.23 Tumors are diagnosed more frequently in fetal series than in postnatal series, resulting in an increased sensitivity when examining fetal echocardiograms.18–19 Cardiac rhabdomyomas tend to appear at 20 to 30 weeks' gestation, with the earliest diagnosis having been made at 15 weeks at the current technical limits of ultrasonography,24 suggesting rhabdomyomas may be present earlier in development. The frequency of fetal detection is increasing dramatically; therefore, it is reasonable to anticipate that the rate of fetal diagnosis will increase significantly.
Fetal cardiac tumors may present in utero as a mass on ultrasonography, irregular heart rhythm, hydrops fetalis, or pericardial effusion. Cardiac rhabdomyomas can increase in size during the second half of gestation, and this has been attributed to maternal hormonal changes associated with pregnancy. When larger tumors result in hemodynamic compromise in utero, intrauterine fetal demise may occur. Fetal loss has been reported to be ≈11% in 1 small series of 44 cases.23 Cardiac rhabdomyomas do not cause symptoms or hemodynamic compromise in the vast majority of patients but may become symptomatic shortly after birth or in the first year of life. Tumors may obstruct inflow or outflow, which can cause ventricular dysfunction and heart failure, as well as redirection of flow across the foramen ovale.19,25–26 Nearly 100% of fetuses with multiple rhabdomyomas have TSC, underscoring the practical importance of identifying additional tumors at the time of fetal assessment for diagnosis and prognosis.17,27 In light of emerging human genetic and molecular knowledge, it is a possibility that the underlying pathogenesis of all rhabdomyomas is a result of a spectrum of TSC disease.
Cardiac rhabdomyomas are typically well circumscribed and nonencapsulated (FigureB). Micropathologic examination demonstrates abnormal myocyte architecture, including vacuolization and pathognomonic “spider cells” (FigureC). The individual cardiac rhabdomyomas range in size from a few millimeters to several centimeters and are multiple in number in 90% of cases. There is an equal predilection for left, right. and septal ventricular myocardium.26,28 Tumors are typically located in the ventricles, where they can compromise ventricular function and on occasion interfere with valve function or result in outflow obstruction. Tumors may be located in the atria, where they can compress the coronary arteries, leading to myocardial ischemia.29
Diagnosis of Cardiac Rhabdomyoma
Echocardiography is the imaging modality of choice for assessing cardiac involvement in TSC. Cardiac rhabdomyomas can be detected prenatally or postnatally. In prenatal life, ultrasound detection of multiple cardiac tumors is often the first sign of TSC.30 Typically, cardiac rhabdomyomas are visible as multiple, echogenic, nodular masses embedded in the ventricular myocardium, sometimes protruding into the involved chamber (FigureD). They are homogeneous and hyperechoic compared with normal myocardium. Diagnosis of cardiac rhabdomyomas is made easily when these typical features are present, but differentiation from other cardiac tumors may be difficult when there is a large solitary tumor or when tumors are located in an atypical location, such as the atria. Doppler echocardiography is also useful in assessing the presence of obstruction to ventricular inflow or outflow. Echocardiography is also used to assess ventricular function, which may be impaired by multiple confluent tumors.
Cardiac rhabdomyomas are seen readily in fetal life after 20 weeks of gestation and are seen in a majority of infants with TSC. Rhabdomyomas can enlarge significantly in size during gestation and may be seen later in gestation when they are not visible prior to 20 weeks. Cardiac rhabdomyomas regress spontaneously in a large majority of patients during the first year of life and as a result are seen with decreasing frequency in patients with TSC after 2 years of age.31 There is some suggestion that the incidence of identifiable cardiac rhabdomyomas in TSC increases during adolescence,31 but this observation has not been validated in additional studies.
Sensitivity and Specificity of Echocardiography to Identify Cardiac Rhabdomyomas
This varies with patient age, related to the previous discussion. In fetal life, of patients diagnosed with cardiac rhabdomyomas by echocardiography, 75% to 80% are found to satisfy criteria for TSC postnatally.32–33,24 The presence of multiple ventricular tumors seems to be the finding best associated with TSC.24 The presence of a family history of TSC also increases the likelihood of TSC.34 In fetuses with a single ventricular tumor, only 30% satisfy criteria for TSC postnatally.24 Because a diagnosis of TSC during fetal life is often prompted by the presence of cardiac rhabdomyomas, the negative predictive value of fetal echocardiography is not established.
In early infancy, the predictive value of echocardiography is similar to that in fetal life, with ≈80% of infants with cardiac rhabdomyomas eventually satisfying a diagnosis of TSC. Conversely, 80% to 85% of children with confirmed TSC have identifiable rhabdomyomas when younger than 2 years.31 Beyond 2 years of age, the incidence of identifiable rhabdomyomas is significantly lower (≈20% to 25%), although if they are readily seen on echocardiography, the likelihood of TSC likely remains high. In late childhood and adolescence (9 to 14 years of age), the incidence of cardiac rhabdomyomas in patients with confirmed TSC seems to increase again (≈40%) in small series.31 It has been speculated that this may be related to hormonal changes.
Alternative Imaging Modalities for Cardiac Rhabdomyomas
Cardiac magnetic resonance imaging (MRI) can also be used to detect cardiac rhabdomyomas; however, its strength lies in more specific tissue characterization.35 It can be a useful adjunct to echocardiography in situations where it is unclear whether a cardiac tumor represents a rhabdomyoma (eg, in patients with a large solitary tumor). In addition, MRI is more accurate than echocardiography in delineating the proximity of cardiac tumors to normal myocardium and the great vessels36–37 and therefore may be a useful adjunct to surgical planning once a decision to operate has been made. It can also provide a more reliable and reproducible estimate of ventricular systolic function. Cardiac MRI in infants and young children (<8 years of age) requires general anesthesia or sedation and, hence, its use should be limited by necessity.
Cardiac Arrhythmia Is a Significant Problem in TSC
While arrhythmia is relatively common in individuals with TSC, the range of arrhythmic substrates is wide and not sufficiently specific to form a specific diagnostic criterion. Reported cases of arrhythmia associated with TSC from slow to irregular to fast heart rhythms. Bradycardia mechanisms have been associated with both sinus and atrioventricular (AV) nodal dysfunction. Tachycardia mechanisms have been related to atrial, accessory AV connection reentrant, and ventricular tachycardia.38–42 Ventricular preexcitation associated with abnormal AV connections has also been commonly reported and has been noted to participate in rapid potentially life‐threatening anomalous AV conduction during atrial fibrillation.43–45 The mechanisms of arrhythmia have often been directly linked to the location of specific cardiac rhabdomyomas.46 Indeed, abnormal AV connections associated with TSC have been shown histologically to be directly related to rhabdomyomas tumor tissue connecting the atrium to the ventricle, rather than a “typical” accessory pathway.
In addition to the wide range of mechanisms of arrhythmia in these individuals, the effects of the arrhythmias can be extremely varied. Isolated atrial or ventricular ectopy may remain without symptoms for a lifetime. Bradycardia, depending on its severity, may also remain without symptoms but may result in fatigue or syncope. Differentiation of fatigue related to bradycardia from other organ system dysfunction associated with tuberous sclerosis may be challenging.47 Syncope may also have similar presentation to “drop attacks” and other neurologic events seen with TSC. Sustained tachyarrhythmia may result in palpitations or, in some instances, in syncope or cardiac arrest and sudden death.48 Developmentally delayed individuals may not report symptomatic palpitations associated with hemodynamically stable sustained tachyarrhythmia and may present with signs and symptoms of heart failure due to tachycardia‐mediated cardiomyopathy. Recurrent syncope may be mistaken for seizures or “drop attacks,” and thus the warning signs of impending cardiac arrest may not be attended.
The presence of a diagnosis of TSC does not alter treatment recommendations for any arrhythmia. Observation and treatment of episodes of tachycardia as they occur, antiarrhythmic medications, catheter and surgical ablation, and implanted pacemakers and defibrillators remain options for treatment as they do in all individuals. Catheter ablation appears to have less frequent success than in those without TSC, probably due to the size of the tumor and possible participation of the entire tumor in the arrhythmia mechanism.
Cardiology Changes to the Diagnostic Criteria
The presence of cardiac rhabdomyomas remains a major criterion (Table 4). There is no longer a need to specify 1 versus >1 rhabdomyoma. Importantly, because cardiac rhabdomyomas are often the presenting manifestation of TSC, it is important to emphasize the need for pediatric cardiologists to initiate and facilitate the TSC evaluation. Specifically, the pediatric cardiologist making a new diagnosis of rhabdomyomas should obtain clinical genetic testing and make the appropriate subspecialty referrals; typically human genetics and neurology, depending on available local resources. Genetic testing practices may vary by center, prompting a need to be familiar with local processes and the closest tertiary center with specialized care for patients with TSC. Clinical genetic testing should be obtained in all multiple cardiac rhabdomyomas and most isolated or possible rhabdomyomas. Because there is benefit to early diagnosis and potential added morbidity to late diagnosis, a proactive approach is warranted.
Table 4.
New Cardiology‐Specific Recommendations for Tuberous Sclerosis Complex
| Cardiac rhabdomyomas remain a major diagnostic criterion |
| Echocardiogram at the time of diagnosis If fetal diagnosis, then serial observation and at least 1 postnatal echocardiogram Surveillance studies until regression demonstrated |
| Electrocardiogram at the time of diagnosis Surveillance studies every 3 to 5 years Holter monitor as indicated for appropriate signs and symptoms |
| Cardiology consultation at time of diagnosis Ongoing cardiology surveillance as indicated Medical and surgical intervention as indicated Referral to genetics and neurology when cardiology makes initial diagnosis Pediatric to adult transition plan with ongoing cardiology surveillance |
The Management of Cardiac Manifestations of TSC
Medical Treatment for Heart Failure
Cardiac rhabdomyomas can lead to hemodynamic compromise and congestive heart failure, and while this occurrence is rare, it remains one of the most frequent causes of death among TSC children <10 years old.49 Heart failure occurs in 2% to 5% of infants and children with TSC‐associated rhabdomyomas.50–51 Pharmacology‐based therapy for congestive heart failure due to TSC‐associated rhabdomyomas is typically not needed; however, on occasion, medical management for CHF, including digitalis, angiotensin‐converting enzyme inhibition, and diuresis, may be indicated. When the cause of heart failure is arrhythmia, then the appropriate antiarrhythmic treatment is indicated and effective.52 However, when the cause of heart failure is inflow or outflow obstruction, typically “watchful waiting” is used with the anticipation that most cardiac rhabdomyomas will spontaneously regress over a period of months. If the heart failure is refractory, then surgery is indicated. When there is hemodynamic compromise in the neonate, prostaglandin E may be initiated to stabilize the critically ill newborn. A critically ill neonate with hemodynamic compromise due to cardiac rhabdomyomas at the time of diagnosis should be transferred to a tertiary center with cardiac intensive care infrastructure and the ability to perform surgery if needed.
New Treatment Modalities May Have a Role in Cardiology Management
mTOR inhibitors have been successfully used for TSC‐associated tumors in different organ systems,53–55 and limited observations to date suggest they may also be efficacious in reducing the size of cardiac rhabdomyomas.56–57 Because mTOR inhibitors are not benign drugs and rhabdomyomas tend to regress, therapy should be considered only in situations of hemodynamic compromise where there is the potential to avoid surgery with their use. Given the low frequency of surgical resection for cardiac rhabdomyomas, it will be challenging to study in a controlled manner. However, there may be opportunities to use mTOR inhibitors, such as sirolimus (rapamycin) or everolimus, to induce tumor regression. Based on limited observations, there do not appear to be significant cardiovascular side effects associated with mTOR inhibitors. In general, side effects are considered manageable in adults, but because mTOR inhibitors affect the immune system and cells' ability to grow and proliferate, there may be an increased risk for infections and malignant tumors over the long term. Case reports using sirolimus56 in the context of an infant with cardiac rhabdomyomas and refractory heart failure and everolimus57 in the context of hemodynamic instability demonstrate benefit and avoided surgery, suggesting that application in cases of malignant arrhythmia may also be therapeutic and avoid the need for invasive intervention. More studies are needed to define the role of mTOR inhibitors in this situation, as well as for arrhythmias, aneurysms, and latent left ventricular dysfunction.
Surgical Intervention for Complete or Partial Cardiac Rhabdomyoma Resection is Indicated in Rare Circumstances
Because most cardiac rhabdomyomas are asymptomatic and the natural history is spontaneous regression, surgical resection is not required for the vast majority of infants with TSC. Among the 2% to 5% of cases that do present with heart failure and/or hemodynamic instability, only a small proportion require surgery.58 Surgical series have reported a rate as high as 20%, but this is likely a reflection of referral bias.26,59 Because the infant with heart failure requiring surgery may be critically ill, these patients are relatively high‐risk surgical candidates. However, partial resection is typically adequate if complete excision would sacrifice vital structures or myocardial mass. Orthotopic heart transplantation can be considered in extreme cases, such as in the rare event that tumor replaces myocardium; however, the necessary medical regimen is associated with significant medical risks. Specifically, the seizure threshold is lowered, and the risk of infection and malignant cancer is increased. Excellent short‐ and long‐term results have been reported in multiple series, but cases of early death have been reported.19,25–26 To date, late fetal surgical resection has not been reported, but ex utero intrapartum treatment technology suggests this may be feasible in select situations.
Recommedations Expand Surveillance Efforts
Cardiology Changes to Surveillance Recommendations
Due to the rise of diagnosis on fetal echocardiography, serial imaging during gestation is now indicated to monitor disease severity and postnatal imaging is indicated to confirm anatomy and determine the status of disease after birth. Surveillance is now recommended until regression is demonstrated (Table 4). Because cardiac rhabdomyomas are often the presenting manifestation of TSC, it is important to emphasize the need for pediatric cardiologists to make the appropriate subspecialty referrals, typically human genetics and neurology, depending on available local resources. Given the increasing appreciation for cardiology issues later in life, including arrhythmias, ECGs are now recommended every 3 to 5 years. A lower index of suspicion is required during adolescent ages. Importantly, increasing efforts are required to facilitate transition from pediatric to adult care.60
Recommendations for Imaging Surveillance
In fetal life, echocardiography is recommended if there is a positive family history of TSC in a first‐degree relative or if there is suspicion for TSC based on other criteria. If cardiac rhabdomyomas are identified, evaluation should include assessment for inflow or outflow obstruction that may lead to hemodynamic compromise postnatally, evaluation for arrhythmias and ventricular dysfunction, and evidence of hydrops fetalis. The presence of a complicating factor requires close follow‐up during the pregnancy along with careful coordination and planning of prenatal and postnatal care with involvement of specialists from maternal‐fetal medicine, cardiology, and cardiac surgery. Even in the absence of complicating factors, if cardiac rhabdomyomas are diagnosed or suspected on fetal echocardiography, consultation with maternal‐fetal medicine and genetics is recommended to counsel the family regarding the likelihood of TSC and long‐term prognosis. Because rhabdomyomas can enlarge during gestation, follow‐up imaging later during gestation (30 to 35 weeks) is recommended.
After birth and in the first 2 years of life, echocardiography is recommended for any child with a suspected diagnosis of TSC, because of the high correlation between the presence of cardiac rhabdomyomas and TSC in this age group. In addition, hemodynamic compromise due to outflow or inflow obstruction is most likely in this age group and can be easily assessed on echocardiography. If echocardiography is conclusive of the diagnosis of rhabdomyomas, no further imaging is recommended. In patients who are suspected but not confirmed to have TSC and have a cardiac tumor on echocardiography but the diagnosis of rhabdomyoma is uncertain, referral to a tertiary pediatric cardiac center for cardiac MRI under sedation or general anesthesia should be considered for tissue characterization. However, this decision should be made jointly by experts in cardiology, neurology, and/or genetics to consider the risk.
In typical cases, and in the absence of inflow/outflow obstruction and ventricular dysfunction, follow‐up echocardiography is not recommended in the first year of life but may be considered once between 1 and 3 years of age to document regression of the tumors. Once regression of tumor size has been documented, follow‐up echocardiography is not recommended, unless new cardiac concerns such as arrhythmia or syncope arise, and, in these cases, should be performed in consultation with a pediatric cardiologist. Closer follow‐up should be considered in atypical cases. In patients with inflow/outflow obstruction, ventricular dysfunction, or large solitary tumors, more frequent repeat echocardiography may be necessary and should be coordinated in consultation with a pediatric cardiologist. In patients suspected with TS beyond 2 years of age, echocardiography should be considered although the yield is significantly lower. Echocardiography is recommended if the physical examination is consistent with outflow tract obstruction (rare in this age group) or if there is concern for arrhythmia or syncope.
Recommendations for Electrophysiologic Surveillance
All individuals with tuberous sclerosis, regardless of age, should have a 12‐ to 15‐lead ECG performed at the time of diagnosis. Subsequently, in an individual with tuberous sclerosis and no cardiac symptomatology, a repeat study every 3 to 5 years may be prudent. Evaluation of symptomatic palpitations should include cardiac event monitoring as appropriate. Episodes of “drop attacks” or “seizures” that cannot definitively exclude cardiac syncope should be evaluated with monitors with “looping” memory, either external or implanted. Particularly concerning cases of syncope or episodes in individuals with other concerning cardiac manifestations should be evaluated with invasive cardiac electrophysiology study.
Sudden deaths in individuals with TSC are reported at all ages and have potentially diverse etiologies, including not only arrhythmia but also epilepsy, intratumor hemorrhage, obstructive hydrocephalus, and aneurysmal rupture. It remains unclear whether surveillance with ambulatory ECGs for occult arrhythmia will be able to predict and prevent the arrhythmic deaths. Periodic ambulatory ECGs seem prudent until the question can be answered definitively.
Future Research Directions and Unresolved Clinical Issues
Animal Models of TSC Provide Potential Insight Into Mechanisms of Tumor Regression
Both TSC1‐ and TSC2‐deficient mice are embryonic lethal with ventricular dysfunction potentially contributing to death.61–62 Heterozygous and conditional mice appear to recapitulate some of TSC phenotypes, with the Tsc2+/− mouse demonstrating more severe overall disease. Importantly, these mice are responsive to sirolimus.63 Meikle et al examined ventricular myocytes of mice with Tsc1 insufficiency (haploinsufficiency) conditionally restricted to the myocardium and demonstrated cardiomyopathy with cell findings reminiscent of human cardiac rhabdomyomas.64 However, most preclinical studies have not focused on cardiac findings, so evaluating the cardiac phenotype in these mice may provide special insight into early disease processes. For example, general mechanisms of hypertrophy may be elucidated.65 In addition, because rhabdomyomas tend to regress spontaneously, mechanistic insights into regression may be elucidated, potentially identifying new therapeutic targets. The mice would also provide a mechanism to preclinically test the benefit of mTOR inhibitors controlling for regression, which may underscore limited observations in human studies.
Unresolved Issues Warrant Consideration for Future Investigation
Several unresolved issues have been identified and require careful examination (Table 5). These research questions require substantial organization. Strategies that may enhance these efforts include future clinical studies examining mTOR inhibitor effects on the cardiovascular system. By adding cardiac end points to longitudinal clinical studies, we will gain insight into various natural history questions. Some of these issues may be addressed by using the TSC Alliance Clinical Registry, which has collected comprehensive clinical data on >1000 TSC patients from 17 centers (Table 6). The TSC Alliance is organized to function as a network for this purpose but requires subspecialty commitments from investigators at large centers where TSC patients are cared for (not necessarily participating already within the Alliance), highlighting the importance of coordinated multidisciplinary care and standardized approaches to care. Transitioning the patient from pediatric to adult cardiology care remains a challenging and important goal, with a need for careful monitoring and a low index of suspicion for latent manifestations of cardiovascular disease, including arrhythmias.
Table 5.
Cardiology‐Specific Future Research Directions
| Why do cardiac rhabdomyomas regress and other hamartomas do not? |
| Do cardiac rhabdomyomas completely resolve? |
| What is the incidence of sudden death? Malignant arrhythmia? |
| Do TSC1 and TSC2 genotypes predict cardiac phenotype or outcome? |
| Does treatment with mTOR inhibitors decrease the long‐term risk of arrhythmia? |
| What is the incidence of latent left ventricular hypertrophy and/or dysfunction? |
| What is the incidence and natural history of lipidemia in TSC? |
mTOR indicates mammalian target of rapamycin; TSC, tuberous sclerosis complex.
Table 6.
Cardiology Variables Maintained in the TSC Alliance Clinical Registry
| Medical history, physical examination, family history |
| Current medications |
| ECG, CXR, echocardiogram, MRI, CT |
| Pathology if available |
| Other cardiac conditions (malformation, hypertension, lipidemia, aneurysm) |
| Number, size, and location of cardiac rhabdomyomas |
TSC, tuberous sclerosis complex; CXR, chest radiography; MRI, magnetic resonance imaging; CT, computed tomography.
TSC is a multisystem genetic disorder characterized by variable abnormalities, such that patients carrying mutations may not fulfill clinical criteria for diagnosis, raising questions regarding familial screening. For example, does the presence of fetal cardiac rhabdomyomas warrant a recommendation for family screening, which is not presently indicated? In mutation‐positive children, parents and siblings can undergo specific mutation testing as screening, but in parents and siblings of phenotypically diagnosed children, it may be prudent to perform ECG in parents and both ECG and echocardiography in children <3 years old. Some studies have demonstrated that cardiac rhabdomyomas are more frequent in those with TSC2 (54%) versus TSC1 (20%) mutations.49 Currently, there is insufficient evidence of absolute risks to recommend surveillance by TSC1‐ and TSC2‐associated cardiac disease. Variability in pathology or natural history based on presentation with TSC1 and TSC2 mutations is unclear but potentially clinically significant. Genetic testing will facilitate early identification and provide opportunities for disease stratification and early intervention.
Author Contributions
The authors participated in the Cardiology Working Group for the TSC Alliance Consensus Conference (Drs Hinton, Prakash, Romp, and Knilans), from which the concept and design for this report were conceived (Drs Hinton, Prakash, Romp, and Knilans). The manuscript was drafted by Drs Hinton, Prakash, and Knilans and revised by Drs Hinton, Prakash, Romp, Knilans, and Krueger. All authors approved the final manuscript.
Sources of Funding
This manuscript was supported in part by the Tuberous Sclerosis Alliance.
Disclosures
None.
Acknowledgments
The authors would like to thank the TS Alliance Consensus Conference organizers and Jo Anne Nakagawa, director of clinical projects for the TS Alliance, for sharing information and insights about the TSC Natural History Database.
References
- 1.Bourneville DM. Scerose tuberereuse des circonvultions cerebrales: idiotie et epilepsie hemiplegique. Arch Neurol (Paris). 1880; 1:81-91. [Google Scholar]
- 2.Vogt H. Zue pathologie und pathologishcen anatomie der verschiedenen idiotieform. Monatsschr Psychiatr Neurol. 1908; 24:106-150. [Google Scholar]
- 3.Gomez MR. In: Gomez MR. (ed.). Criteria for diagnosis. Tuberous Sclerosis. 19882nd edNew York, NY: Raven Press; 75-87. [Google Scholar]
- 4.Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998; 13:624-628. [DOI] [PubMed] [Google Scholar]
- 5.Northrup H, Krueger DAInternational Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Consensus Conference. Pediatr Neurol. 2013; 49:243-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krueger DA, Northrup HInternational Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013; 49:255-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993; 75:1305-1315. [DOI] [PubMed] [Google Scholar]
- 8.van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A, Halley D, Young J, Burley M, Jeremiah S, Woodward K, Nahmias J, Fox M, Ekong R, Osborne J, Wolfe J, Povey S, Snell RG, Cheadle JP, Jones AC, Tachataki M, Ravine D, Sampson JR, Reeve MP, Richardson P, Wilmer F, Munro C, Hawkins TL, Sepp T, Ali JB, Ward S, Green AJ, Yates JR, Kwiatkowska J, Henske EP, Short MP, Haines JH, Jozwiak S, Kwiatkowski DJ. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997; 277:805-808. [DOI] [PubMed] [Google Scholar]
- 9.Whittemore VH. Unlocking a Cure for Tuberous Sclerosis Complex: An Assessment of Scientific Progress and Research Needs. A White Paper. Tuberous Sclerosis Alliance. 2011Silver Spring, MD: Silver Springs [Google Scholar]
- 10.Hoogeveen‐Westerveld M, Ekong R, Povey S, Karbassi I, Batish SD, den Dunnen JT, van Eeghen A, Thiele E, Mayer K, Dies K, Wen L, Thompson C, Sparagana SP, Davies P, Aalfs C, van den Ouweland A, Halley D, Nellist M. Functional assessment of TSC1 missense variants identified in individuals with tuberous sclerosis complex. Hum Mutat. 2012; 33:476-479. [DOI] [PubMed] [Google Scholar]
- 11.Hoogeveen‐Westerveld M, Ekong R, Povey S, Mayer K, Lannoy N, Elmslie F, Bebin M, Dies K, Thompson C, Sparagana SP, Davies P, van Eeghen AM, Thiele EA, van den Ouweland A, Halley D, Nellist M. Functional assessment of TSC2 variants identified in individuals with tuberous sclerosis complex. Hum Mutat. 2013; 34:167-175. [DOI] [PubMed] [Google Scholar]
- 12.Marx GR, Moran AM. In: Allen HD, Driscoll DJ, Shaddy RE, Feltes TF. (eds.). Cardiac tumors. Moss and Adams' Heart Disease in Infants, Children and Adolescents. 20087th edNew York: Lippincott Williams and Wilkins; 1479-1495. [Google Scholar]
- 13.Marx GR, Flyer DC. In: Keane JF, Lock JE, Fyler DC. (eds.). Cardiac tumors. Nadas' Pediatric Cardiology. 20062nd edNew York: Elsevier; 825-832. [Google Scholar]
- 14.Freedom RM, Lee KJ, MacDonald C, Taylor G. Selected aspects of cardiac tumors in infancy and childhood. Pediatr Cardiol. 2000; 21:299-316. [DOI] [PubMed] [Google Scholar]
- 15.Jozwiak S, Kawalec W, Diuzewska J, Dazkowska J, Mirkowicz‐Malek M, Michalowicz R. Cardiac tumors in tuberous sclerosis: their incidence and course. Eur J Pediatr. 1994; 153:155-157. [DOI] [PubMed] [Google Scholar]
- 16.Smythe JF, Dyck JD, Smallhorn JF, Freedom RM. Natural history of cardiac rhabdomyomas in infancy and childhood. Am J Cardiol. 1990; 66:1247-1249. [DOI] [PubMed] [Google Scholar]
- 17.Harding CO, Pagon RA. Incidence of tuberous sclerosis in patients with cardiac rhabdomyoma. Am J Med Genet. 1990; 37:443-446. [DOI] [PubMed] [Google Scholar]
- 18.Holley DG, Martin GR, Brenner JI, Fyfe DA, Huhta JC, Kleinman CS, Ritter SB, Silverman NH. Diagnosis and management of fetal cardiac tumors: a multicenter experience and review of published reports. J Am Coll Cardiol. 1995; 26:516-520. [DOI] [PubMed] [Google Scholar]
- 19.Beghetti M, Gow RM, Haney I, Mawson J, Williams WG, Freedom RM. Pediatric primary benign cardiac tumors: a 15‐year review. Am Heart J. 1997; 134:1107-1114. [DOI] [PubMed] [Google Scholar]
- 20.Smith HC, Watson GH, Patel RG, Super M. Cardiac rhabdomyomata in tuberous sclerosis: their course and diagnostic value. Arch Dis Child. 1989; 64:196-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nadas AS, Ellison RC. Cardiac tumors in infancy. Am J Cardiol. 1968; 21:363-366. [DOI] [PubMed] [Google Scholar]
- 22.Silverman NA. Primary cardiac tumors. Ann Surg. 1980; 191:127-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allan LD, Sharland GK, Milburn A, Lockhart SM, Groves AM, Anderson RH, Cook AC, Fagg NL. Prospective diagnosis of 1006 consecutive cases of congenital heart disease in the fetus. J Am Coll Cardiol. 1994; 23:1452-1458. [DOI] [PubMed] [Google Scholar]
- 24.Tworetzky W, McElhinney DB, Margossian R, Moon‐Grady AJ, Sallee D, Goldmuntz E, van der Velde ME, Silverman NH, Allan LD. Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol. 2003; 92:487-489. [DOI] [PubMed] [Google Scholar]
- 25.Takach TJ, Reul GJ, Ott DA, Cooley DA. Primary cardiac tumors in infants and children: immediate and long‐term operative results. Ann Thorac Surg. 1996; 62:559-564. [PubMed] [Google Scholar]
- 26.Black MD, Kadletz M, Smallhorn JF, Freedom RM. Cardiac rhabdomyomas and obstructive left heart disease: histologically but not functionally benign. Ann Thorac Surg. 1998; 65:1388-1390. [DOI] [PubMed] [Google Scholar]
- 27.Groves AM, Fagg NL, Cook AC, Allan LD. Cardiac tumours in intrauterine life. Arch Dis Child. 1992; 67:1189-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McAllister JA, Fenoflio JJ. In: Hartman WH, Cowan WR. (eds.). Tumors of the cardiovascular system. Atlas of Tumor Pathology. 1978Washington DC: Armed Forces Institute of Pathology; 956-978. [Google Scholar]
- 29.Geva T, Santini F, Pear W, Driscoll SG, Van Praagh R. Cardiac rhabdomyoma. Rare cause of fetal death. Chest. 1991; 99:139-142. [DOI] [PubMed] [Google Scholar]
- 30.Datta AN, Hahn CD, Sahin M. Clinical presentation and diagnosis of tuberous sclerosis complex in infancy. J Child Neurol. 2008; 23:268-273. [DOI] [PubMed] [Google Scholar]
- 31.Jozwiak S, Schwartz RA, Janniger CK, Bielicka‐Cymerman J. Usefulness of diagnostic criteria of tuberous sclerosis complex in pediatric patients. J Child Neurol. 2000; 15:652-659. [DOI] [PubMed] [Google Scholar]
- 32.Bader RS, Chitayat D, Kelly E, Ryan G, Smallhorn JF, Toi A, Hornberger LK. Fetal rhabdomyoma: Prenatal diagnosis, clinical outcome, and incidence of associated tuberous sclerosis complex. J Pediatr. 2003; 143:620-624. [DOI] [PubMed] [Google Scholar]
- 33.Degueldre SC, Chockalingam P, Mivelaz Y, Di Bernardo S, Pfammatter JP, Barrea C, Sekarski N, Jeannet PY, Fouron JC, Vial Y, Meijboom EJ. Considerations for prenatal counselling of patients with cardiac rhabdomyomas based on their cardiac and neurologic outcomes. Cardiol Young. 2010; 20:18-24. [DOI] [PubMed] [Google Scholar]
- 34.Chao AS, Chao A, Wang TH, Chang YC, Chang YL, Hsieh CC, Lien R, Su WJ. Outcome of antenatally diagnosed cardiac rhabdomyoma: case series and a meta‐analysis. Ultrasound Obstet Gynecol. 2008; 31:289-295. [DOI] [PubMed] [Google Scholar]
- 35.Beroukhim RS, Prakash A, Buechel ER, Cava JR, Dorfman AL, Festa P, Hlavacek AM, Johnson TR, Keller MS, Krishnamurthy R, Misra N, Moniotte S, Parks WJ, Powell AJ, Soriano BD, Srichai MB, Yoo SJ, Zhou J, Geva T. Characterization of cardiac tumors in children by cardiovascular magnetic resonance imaging: a multicenter experience. J Am Coll Cardiol. 2011; 58:1044-1054. [DOI] [PubMed] [Google Scholar]
- 36.Freedberg RS, Kronzon I, Rumancik WM, Liebeskind D. The contribution of magnetic resonance imaging to the evaluation of intracardiac tumors diagnosed by echocardiography. Circulation. 1988; 77:96-103. [DOI] [PubMed] [Google Scholar]
- 37.Berkenblit R, Spindola‐Franco H, Frater RW, Fish BB, Glickstein JS. MRI in the evaluation and management of a newborn infant with cardiac rhabdomyoma. Ann Thorac Surg. 1997; 63:1475-1477. [DOI] [PubMed] [Google Scholar]
- 38.Hirakubo Y, Ichihashi K, Shiraishi H, Momoi MY. Ventricular tachycardia in a neonate with prenatally diagnosed cardiac tumors: a case with tuberous sclerosis. Pediatr Cardiol. 2005; 26:655-657. [DOI] [PubMed] [Google Scholar]
- 39.Scurry J, Watkins A, Acton C, Drew J. Tachyarrhythmia, cardiac rhabdomyomata and fetal hydrops in a premature infant with tuberous sclerosis. J Paediatr Child Health. 1992; 28:260-262. [DOI] [PubMed] [Google Scholar]
- 40.Kathare PA, Muthuswamy KS, Sadasivan J, Calumbar N, Koneti NR. Incessant ventricular tachycardia due to multiple cardiac rhabdomyomas in an infant with tuberous sclerosis. Indian Heart J. 2013; 65:111-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu CT, Chen MR, Hou SH. Neonatal tuberous sclerosis with cardiac rhabdomyomas presenting as fetal supraventricular tachycardia. Jpn Heart J. 1997; 38:133-137. [DOI] [PubMed] [Google Scholar]
- 42.Enbergs A, Borggrefe M, Kurlemann G, Fahrenkamp A, Scheld HH, Jehle J, Breithardt G. Ventricular tachycardia caused by cardiac rhabdomyoma in a young adult with tuberous sclerosis. Am Heart J. 1996; 132:1263-1265. [DOI] [PubMed] [Google Scholar]
- 43.Mas C, Penny DJ, Menahem S. Pre‐excitation syndrome secondary to cardiac rhabdomyomas in tuberous sclerosis. J Paediatr Child Health. 2000; 36:84-86. [DOI] [PubMed] [Google Scholar]
- 44.O'Callaghan FJ, Clarke AC, Joffe H, Keeton B, Martin R, Salmon A, Thomas RD, Osborne JP. Tuberous sclerosis complex and Wolff‐Parkinson‐White syndrome. Arch Dis Child. 1998; 78:159-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mehta AV. Rhabdomyoma and ventricular preexcitation syndrome. A report of two cases and review of literature. Am J Dis Child. 1993; 147:669-671. [DOI] [PubMed] [Google Scholar]
- 46.Venugopalan P, Babu JS, Al‐Bulushi A. Right atrial rhabdomyoma acting as the substrate for Wolff‐Parkinson‐White syndrome in a 3‐month‐old infant. Acta Cardiol. 2005; 60:543-545. [DOI] [PubMed] [Google Scholar]
- 47.Cowley CG, Tani LY, Judd VE, Shaddy RE. Sinus node dysfunction in tuberous sclerosis. Pediatr Cardiol. 1996; 17:51-52. [DOI] [PubMed] [Google Scholar]
- 48.Byard RW, Blumbergs PC, James RA. Mechanisms of unexpected death in tuberous sclerosis. J Forensic Sci. 2003; 48:172-176. [PubMed] [Google Scholar]
- 49.Shepherd CW, Gomez MR, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc. 1991; 66:792-796. [DOI] [PubMed] [Google Scholar]
- 50.Nir A, Tajik AJ, Freeman WK, Seward JB, Offord KP, Edwards WD, Mair DD, Gomez MR. Tuberous sclerosis and cardiac rhabdomyoma. Am J Cardiol. 1995; 76:419-421. [DOI] [PubMed] [Google Scholar]
- 51.Jóźwiak S, Kotulska K, Kasprzyk‐Obara J, Domańska‐Pakieła D, Tomyn‐Drabik M, Roberts P, Kwiatkowski D. Clinical and genotype studies of cardiac tumors in 154 patients with tuberous sclerosis complex. Pediatrics. 2006; 118:e1146-e1151. [DOI] [PubMed] [Google Scholar]
- 52.De Rosa G, De Carolis MP, Pardeo M, Bersani I, Tempera A, De Nisco A, Caforio L, Romagnoli C, Piastra M. Neonatal emergencies associated with cardiac rhabdomyomas: an 8‐year experience. Fetal Diagn Ther. 2011; 29:169-177. [DOI] [PubMed] [Google Scholar]
- 53.Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, Witt O, Kohrman MH, Flamini JR, Wu JY, Curatolo P, de Vries PJ, Whittemore VH, Thiele EA, Ford JP, Shah G, Cauwel H, Lebwohl D, Sahmoud T, Jozwiak S. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST‐1): a multicentre, randomised, placebo‐controlled phase 3 trial. Lancet. 2013; 381:125-132. [DOI] [PubMed] [Google Scholar]
- 54.Krueger DA, Care MM, Agricola K, Tudor C, Mays M, Franz DN. Everolimus long‐term safety and efficacy in subependymal giant cell astrocytoma. Neurology. 2013; 80:574-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisbury S, Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008; 358:140-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tiberio D, Franz DN, Phillips JR. Regression of a cardiac rhabdomyoma in a patient receiving everolimus. Pediatrics. 2011; 127:e1335-e1337. [DOI] [PubMed] [Google Scholar]
- 57.Mlczoch E, Hanslik A, Luckner D, Kitzmüller E, Prayer D, Michel‐Behnke I. Prenatal diagnosis of a gigantic cardiac rhabdomyoma in tuberous sclerosis complex‐ a new therapeutic option with everolimus. Ultrasound Obstet Gynecol. 2014 [DOI] [PubMed] [Google Scholar]
- 58.Foster ED, Spooner EW, Farina MA, Shaher RM, Alley RD. Cardiac rhabdomyoma in the neonate: surgical management. Ann Thorac Surg. 1984; 37:249-253. [DOI] [PubMed] [Google Scholar]
- 59.Jacobs JP, Konstantakos AK, Holland FW, II, Herskowitz K, Ferrer PL, Perryman RA. Surgical treatment for cardiac rhabdomyomas in children. Ann Thorac Surg. 1994; 58:1552-1555. [DOI] [PubMed] [Google Scholar]
- 60.Warnes CA, Williams RG, Bashore TM, Child JS, Connolly HM, Dearani JA, del Nido P, Fasules JW, Graham TP, Jr, Hijazi ZM, Hunt SA, King ME, Landzberg MJ, Miner PD, Radford MJ, Walsh EP, Webb GD. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. Circulation. 2008; 118:e714-e833. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E, Noda T, Hino O. A germ‐line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc Natl Acad Sci USA. 2001; 98:8762-8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest. 1999; 104:687-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008; 63:444-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meikle L, McMullen JR, Sherwood MC, Lader AS, Walker V, Chan JA, Kwiatkowski DJ. A mouse model of cardiac rhabdomyoma generated by loss of Tsc1 in ventricular myocytes. Hum Mol Genet. 2005; 14:429-435. [DOI] [PubMed] [Google Scholar]
- 65.Malhowski AJ, Hira H, Bashiruddin S, Warburton R, Goto J, Robert B, Kwiatkowski DJ, Finlay GA. Smooth muscle protein‐22‐mediated deletion of Tsc1 results in cardiac hypertrophy that is mTORC1‐mediated and reversed by rapamycin. Hum Mol Genet. 2011; 20:1290-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]