

Abstract
Objective
Reducing levels of the microtubule-associated protein tau has shown promise as a potential treatment strategy for diseases with secondary epileptic features such as Alzheimer disease. We wanted to determine whether tau reduction may also be of benefit in intractable genetic epilepsies.
Methods
We studied a mouse model of Dravet syndrome, a severe childhood epilepsy caused by mutations in the human SCN1A gene encoding the voltage-gated sodium channel subunit Nav1.1. We genetically deleted 1 or 2 Tau alleles in mice carrying an Nav1.1 truncation mutation (R1407X) that causes Dravet syndrome in humans, and examined their survival, epileptic activity, related hippocampal alterations, and behavioral abnormalities using observation, electroencephalographic recordings, acute slice electrophysiology, immunohistochemistry, and behavioral assays.
Results
Tau ablation prevented the high mortality of Dravet mice and reduced the frequency of spontaneous and febrile seizures. It reduced interictal epileptic spikes in vivo and drug-induced epileptic activity in brain slices ex vivo. Tau ablation also prevented biochemical changes in the hippocampus indicative of epileptic activity and ameliorated abnormalities in learning and memory, nest building, and open field behaviors in Dravet mice. Deletion of only 1 Tau allele was sufficient to suppress epileptic activity and improve survival and nesting performance.
Interpretation
Tau reduction may be of therapeutic benefit in Dravet syndrome and other intractable genetic epilepsies. Ann Neurol 2014;76:443–456
Despite the development of various antiepileptic drugs over the past 20 years, the efficacy of drug treatments for epilepsy has not substantially improved, and 25 to 40% of patients suffer from drug-resistant seizures.1 New antiepileptic strategies are urgently needed to improve the quality of lives and prevent premature deaths of patients with epilepsy.
Several lines of evidence led us to hypothesize that reduction of the microtubule-associated protein tau2 might be of therapeutic benefit for intractable epilepsy. We previously showed that genetic ablation of tau reduces epileptic activity in human amyloid precursor protein (hAPP) transgenic mice,3,4 which simulate key aspects of Alzheimer disease.5–8 We further found that genetic reduction of tau makes mice with or without hAPP expression more resistant to chemically induced seizures.3 Others have confirmed the antiepileptic effects of tau reduction in other animal models of hyperexcitability.9–11 However, the effectiveness of tau reduction has not yet been investigated in a model of severe human epilepsy. In addition, it is unknown whether various comorbidities of epilepsy such as cognitive and behavioral impairments and sudden death12–14 could also be ameliorated by tau reduction.
We decided to investigate Dravet syndrome, one of the most intractable and severe childhood epilepsies; it is associated with multiple comorbidities and sudden death.15 Dravet syndrome is caused by mutations in the SCN1A gene, which encodes the voltage-gated sodium channel subunit Nav1.1.16 SCN1A mutations are the most common genetic cause of epilepsy, and Dravet syndrome is the most severe form among this spectrum of syndromes.17,18 It typically manifests in the first year of life with febrile seizures that progress to severe partial or generalized tonic–clonic seizures, myoclonic seizures, and episodes of status epilepticus.19 Dravet patients also have poor development of language and motor skills, cognitive stagnation, hyperactivity, and autistic traits. In addition, 14 to 20% of Dravet patients succumb to “sudden unexpected death in epilepsy (SUDEP).”13 The cause of SUDEP is unknown; it may be related to cardiac or respiratory abnormalities.20,21 Dravet syndrome is among the most drug-resistant forms of epilepsy.22,23 Drugs currently used as first-line therapy for Dravet syndrome, valproate and benzodiazepines, have limited efficacy, and other antiepileptic drugs, including lamotrigine, vigabatrin, and carbamazepine, even exacerbate symptoms in Dravet patients.23,24 Alternative new therapies are urgently needed for this highly refractory epilepsy syndrome.
To determine the potential therapeutic benefit of tau reduction in Dravet syndrome, we genetically deleted tau in a mouse model of Dravet syndrome (“Dravet mice”). Dravet mice have a knockin truncation mutation in the Scn1a gene (R1407X) that is identical to a mutation in human Dravet patients, resulting in a null allele.25 Homozygous Scn1aRX/RX mice die shortly after birth; heterozygous Scn1aRX/+ mice develop seizures in the early postnatal period and many of them die by 3 months of age.25 Scn1aRX/+ mice also have behavioral abnormalities reminiscent of the human condition, including impaired learning and memory, hyperactivity, and social dysfunction.26,27 Here we show that complete or partial tau reduction prevents or significantly ameliorates most disease-related phenotypes in this model.
Materials and Methods
Animals
Scn1aRX/+ mice25 on a mixed C3HeB/FeJ × C57BL/6J background (N2 onto C3HeB/FeJ) originally generated by Dr K. Yamakawa (Laboratory for Neurogenetics, RIKEN Brain Science Institute) were obtained from Dr M. H. Meisler (Department of Human Genetics, University of Michigan). This line was crossed onto a Tau−/− C57BL/6J background.28 Mice used in this study were approximately 88 to 98% C57BL/6J (N3–N6 onto C57BL/6J). Mice had free access to food (Picolab Rodent Diet 20; LabDiet, St Louis, MO) and water and were maintained on a regular light/dark (12 hours on/12 hours off) cycle. All experiments were performed on sex-balanced groups. Most cohorts of mice underwent multiple tests, and within each cohort the sequence of tests was the same for all genotypes, as specified in Supplementary Table S1. The Institutional Animal Care and Use Committee of the University of California, San Francisco approved all mouse experiments.
Electroencephalographic Recordings
Freely behaving mice were examined by electroencephalography (EEG) using continuous 24-hour recordings as previously reported29 with minor modifications. For quantification of epileptic spikes, one hour of daytime recording (between noon and 2 PM) during rest or sleep was selected to avoid movement artifacts. Gotman spike detectors from Harmonie were used to automatically identify any epileptic discharges that were 5-fold greater than the average baseline amplitude measured during the 5 seconds preceding the deflection. EEG traces were then inspected manually (with the low and high frequency filters set at 5Hz and 70Hz, respectively) to identify single epileptic spikes, defined as brief (<80 milliseconds), high-voltage deflections on the EEG. To determine the proportion of mice with spontaneous epileptic seizures, the entire 24-hour recording was inspected visually for each mouse. Additional mice underwent long-term video-EEG recording on a PowerLab data acquisition system 16/35 (AD Instruments, Colorado Springs, CO) linked to 6 differential amplifiers (DP-304; Harvard Apparatus, Holliston, MA). Each mouse was recorded for at least 100 hours at a 1kHz sampling rate on this system. EEG traces were analyzed offline on LabChart 7 Pro (AD Instruments) to identify and score spontaneous epileptic seizures. Spontaneous seizures were identified and characterized based on EEG traces and scored behaviorally based on inspection of video recordings as follows: 0 = normal exploratory behavior; 1 = immobility; 2 = brief generalized spasm, tremble, or twitch; 3 = forelimb clonus; 4 = tail extension; 5 = generalized clonus; 6 = aberrant bouncing or running; 7 = full tonic extension; 8 = death (modified from Palop et al30).
Heat-Induced Seizures
Seizures were induced in P30–45 mice using a heat lamp as described,31 except that a 2-minute acclimation period was used.
Hippocampal Slice Preparation and Electrophysiology
Coronal brain slices were prepared from P45–60 mice as described.4 For recording, slices were transferred to a submerged chamber and continuously perfused with warmed (32°C) oxygenated artificial cerebrospinal fluid (ACSF; 126mM NaCl, 3mM KCl, 1.25mM NaH2PO4, 26mM NaHCO3, 2mM CaCl2, 2mM MgCl2, 20mM glucose). Field potential recordings were obtained from the CA1 pyramidal layer using glass electrodes filled with ACSF. To induce seizure activity, slices were perfused with ACSF containing 200μM 4-aminopyridine and 50μM picrotoxin. Burst and spike frequencies were quantified during a 200-second period after 30 minutes of drug perfusion. Data were acquired with WinLTP software (University of Bristol) and analyzed offline with pClamp software (Molecular Devices, Sunnyvale, CA).
Immunohistochemistry
Brain tissues were processed and immunostained as described.32,33 Neuropeptide Y (NPY) and calbindin immunoreactivities were quantitated as described33 except that layer VI of the cortex was used as an internal control because there was significant calbindin depletion in the CA1 region of the hippocampus in Scn1aRX/+ mice.
Protein Extraction
Mice were anesthetized with Avertin (tribromoethanol, 250mg/kg, intraperitoneally) and perfused transcardially with 0.9% saline. Brains were removed, microdissected in ice-cold phosphate-buffered saline (PBS), and homogenized in ice-cold buffer containing 1× PBS (pH 7.4), 1mM dithiothreitol, 0.5mM ethylenediaminetetraacetic acid, 0.5% Triton X-100, 0.1M phenylmethyl sulfonyl fluoride (PMSF), a protease inhibitor cocktail (Roche, Basel, Switzerland), and phosphatase inhibitor cocktails 2 and 3 (Sigma, St Louis, MO). Homogenates were sonicated twice for 5 minutes at an amplitude of 40 using an EpiSonic sonicator (Epigentek, Farmingdale, NY) and centrifuged at 10,000rpm for 10 minutes at 4°C. The supernatants were collected for measurement of protein concentrations by Bradford assay (Bio-Rad, Hercules, CA) and Western blotting.
Western Blotting
Proteins (15µg/well) were separated electrophoretically on a 4 to 12% NuPAGE Bis-Tris gel (Life Technologies, Grand Island, NY) and transferred onto nitrocellulose membranes using a Criterion Blotter (Bio-Rad) at 0.4A for 2.5 hours at 4°C. After blocking for 1 hour in 5% bovine serum albumin diluted in Tris-buffered saline (BSA-TBS), membranes were incubated overnight at 4°C in anti-Nav1.1 (1:1,000; Alomone Labs, Jerusalem, Israel), anti–pan-sodium channel (Pan Nav, 1:1,000; Sigma), anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:10,000; Millipore, Billerica, MA), anti-tau clone Tau-5 (1:3,000; Life Technologies), anti-tau clone EP2456Y (1:1,000; Millipore), anti–phospho-tau Ser 396/404 clone PHF-1 (1:3,000, a gift from Dr P. Davies), anti–phospho-tau Thr231 clone CP9 (1:25, a gift from Dr P. Davies), or anti–phospho-PHF-tau pSer202+Thr205 clone AT8 (1:80; Thermo Scientific, Waltham, MA). Antibodies were diluted in BSA-TBS containing 0.1% Tween 20. Except for CP9 and AT8, membranes were then incubated for 1 hour at room temperature in secondary infrared dye (IRD)-tagged antibodies (IRDye 800CW or 680LT donkey antirabbit or antimouse immunoglobulin [Ig] G, 1:10,000; LI-COR, Lincoln, NE) diluted in Odyssey Blocking Buffer (LI-COR) containing 0.2% Tween 20. Signals were detected by Odyssey CLx (LI-COR) and quantified using Image Studio v2.1.10 (LI-COR). AT8 and CP9 were detected with donkey antimouse IgG horseradish peroxidase (HRP; 1:10,000; Calbiochem, San Diego, CA) and goat antimouse IgM HRP (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA), respectively. Chemiluminescent bands were visualized with an enhanced chemiluminescence system (Pierce, Rockford, IL) and quantified densitometrically using ImageJ.
Behavioral Experiments
General
Seven cohorts of mice were tested behaviorally (see Supplementary Tables S1 and S2 for details on mice used and experiments performed in each cohort). Before all behavioral tests, except fear conditioning, mice were transferred to the testing room and acclimated for 1 hour. All the behavioral equipment was cleaned with 70% ethanol (by volume) before and after testing of each mouse.
Nest Building Behavior
Mice were tested once for nest building ability between 1 and 7 months of age. Group-housed mice were transferred individually into new cages with nest-building material consisting of a 5 × 5cm square of white compressed cotton pads (Nestlets; Ancare, Bellmore, NY) placed in the center of the cage. Nest quality was scored on a scale of 0 to 5, modified from Deacon34 by adding a score of zero if the cotton pad remained intact. Scores were assigned at 2, 6, and 24 hours, and then daily for up to 8 days.
Open Field
Mice were tested for total movements and rearing as described.29 To measure circling, mice were placed into plastic chambers (40 × 40 × 24cm) with 1 clear and 3 white sides, and allowed to explore freely for 30 minutes. Mouse movements were video recorded and analyzed offline using the Topscan tracking system (Clever Sys, Reston, VA).
Barnes Maze
A brightly lit circular platform (91.4cm diameter) with 20 holes around the periphery (5.1cm diameter) was used as described26,27,35 to assess the ability of mice to use extramaze cues to locate an escape tunnel. An escape box was attached to the bottom of 1 of the holes and shallow boxes were attached to the bottom of the other holes. The lights were kept bright (650 lux) to motivate mice to find and enter the dark tunnel. Visual extramaze cues were present on 3 walls adjacent to the maze at a distance of 1.5 to 1.8m from the maze. For all trials, mice were placed individually in a cylindrical black start chamber in the center of the maze for 10 seconds, which was then lifted to start the test. During an adaptation period, mice were guided to the escape tunnel and allowed to stay there for 2 minutes. During a spatial acquisition period, a total of 10 spatial acquisition trials (2 trials per day with an intertrial interval of 15 minutes) were performed; mice were allowed to explore the maze freely for 3 minutes. Each trial ended when the mouse entered the escape tunnel or after 3 minutes had elapsed. Mice that did not find the tunnel were guided to it. All mice were allowed to remain in the tunnel for 1 minute. During a probe trial, conducted 5 days after the last training trial, the escape tunnel was replaced by a shallow box and mice were allowed to explore the maze for 90 seconds. Mice were video recorded and the time (“latency”), path length (“distance”), and path traces to the target location during the 5-day probe trial were analyzed offline using the Topscan tracking system. For mice that did not reach the target location, total testing time (90 seconds) and total distance moved were used for analysis in lieu of latency and distance to target.
Context Fear Conditioning
Associative learning and contextual fear memory were assessed as described.36,37 Training and testing were performed in a set of 4 identical fear conditioning chambers (30 × 25 × 25cm; Med Associates, St Albans, VT) equipped with a Med Associates VideoFreeze system. The floor of each chamber was made up of 16 stainless steel rods that were wired to a shock generator and scrambler (Med-Associates) to deliver foot shocks. Each chamber was placed in a sound-attenuating cubicle. A metal pan containing a thin film of 1% acetic acid was placed underneath the grid floors to provide an olfactory component to the context. On 4 consecutive days, mice were placed individually into the same context chamber for 3 minutes. In the chamber, they received a single 2-second foot shock (0.45mA) on days 1, 2 and 3, but not on day 4, and remained in the chamber for an additional 1 minute following each shock. Their freezing behavior in the chamber was monitored before and after the shock on all days. Freezing behavior, which was defined as the absence of any visible movement (including of the vibrissae, but excluding respiration), was monitored with automated near-infrared video tracking software (VideoFreeze). Video was recorded at 30 frames per second; the software calculated the noise (standard deviation) for each pixel in a frame by comparing its gray scale value to previous and subsequent frames. This produced an “activity unit” score for each frame. Based on previous validation by an experimenter, freezing was defined as subthreshold activity (set at 19 activity units) for >1 second. Percentage freezing was then calculated from the number of seconds the animal was scored as freezing divided by the total time it was monitored. The percentage time mice spent freezing immediately following each shock was taken as a measure of associative learning, and the percentage time mice spent freezing upon re-exposure to the context 24 hours later (before they received another shock) was used as the primary measure of contextual fear memory.38,39
Study Design and Statistical Analyses
Experimenters who obtained the primary data were blinded to the genotype of mice, except for the electrophysiological analysis of acute hippocampal slices and febrile seizure inductions. Sample sizes were determined based on previous studies.4,25–27,29,30,32 For the Barnes maze analysis and fear conditioning, data collection was stopped when learning curves leveled off. Some mice were excluded from the Barnes maze analysis because of an eye injury (1 mouse of cohort 7) or poor exploration (3 mice of cohort 3 and 1 mouse of cohort 7 contacted only 1 of 20 holes). Behavioral findings were replicated in 1 or more independent cohorts using similar conditions (see Supplementary Tables S1 and S2).
Statistical analyses were performed using R.40 Differences among multiple means were assessed by 2-way analysis of variance with post hoc testing of all pairwise comparisons and corrected with the Tukey–Kramer method. Multiple comparisons were corrected using the method of Holm41 except where explicitly indicated otherwise. Data for EEG spikes and brain slice spikes and bursts were log transformed before analysis. Outliers were excluded by the Grubb test based on a priori defined criteria using the GraphPad (San Diego, CA) QuickCalcs Web site: http://www.graphpad.com/quickcalcs/ConfInterval1.cfm. In the linear regression analysis of nest building scores versus epileptic spike frequencies, 1 data point was removed from the Scn1aRX/+/Tau+/+ data set (Cook's distance = 1.5). Null hypotheses were rejected at the 0.05 level. No significant differences were found on any comparisons between Scn1a+/+/Tau+/+ mice or tissues and Scn1a+/+/Tau−/−, Scn1aRX/+/Tau−/−, or Scn1aRX/+/Tau+/− mice or tissues, except as described in results.
Survival and febrile seizure data were analyzed by Cox proportional hazards regression using the R survival package42 and corrected for multiple comparisons with the method of Holm.41
For analysis of drug-induced epileptic activity in brain slices, a linear mixed effects model43 was fitted using the R package lme4.44 Random intercepts were included for each mouse and each genotype such that multiple comparison corrections were not needed due to “partial pooling.”45 Five thousand draws were obtained of parameter estimates, and 95% confidence intervals (CIs) were estimated as the 2.5th and 97.5th quantiles of these draws. Probability values were calculated by inverting the simulated CIs around the differences.46 Analyses of log(spike frequency + 0.1) and log(burst frequency + 0.1) were conducted separately.
For learning curves in the Barnes maze, a linear mixed effects model for censored responses47 was fitted using the R package lmec.48 The model included the following fixed effects: Trial, Scn1a, Tau, Scn1a*Tau, Trial*Scn1a, Trial*Tau, and Trial*Scn1a*Tau. Because 2 trials were conducted per day, a fixed effect for observations from trials 2, 4, 6, 8, and 10 was also included to allow for improvements from the first to the second trial per day. Random mouse-level intercepts and slopes accounted for the correlation among repeated observations.
For nest building analysis, a linear mixed effects model was fitted using the R package lme4.44 Nest building scores were adjusted to increase monotonically over time. To ensure that predictions would fall between 0 and 5, a logit transformation of the score variable was used (logit[(score + 0.1)/5.2] − logit[0.1/5.2]). To allow for a flexible, nonlinear time trend, a 3df natural cubic spline was used. Random intercepts and time splines were included for each mouse to account for the correlation among repeated observations. To insure that trend estimates would pass through the origin (ie, produce an estimated score of 0 at time 0), both fixed and random intercept terms were omitted. Estimates of the area under the curve and probability values were obtained using the fitted model (as described above for drug-induced epileptic activity in brain slices), and corrected for multiple comparisons using the method of Holm.41
Results
Tau Reduction Improves Survival of Scn1aRX/+ Mice
As a first step toward assessing the potential therapeutic benefit of tau reduction in Dravet syndrome, we compared the survival of Scn1a+/+ and Scn1aRX/+ mice with 2, 1, or no functional Tau alleles ( Fig 1). On the Tau wild-type background, Scn1aRX/+ mice showed a high incidence of sudden death, most commonly observed between postnatal days 21 and 73, and a 31-fold higher risk of death than wild-type littermates. By 73 days, only 18% of Scn1aRX/+ mice remained alive. Tau reduction ameliorated the abnormal mortality of Scn1aRX/+ mice in a gene dose-dependent manner. When 1 Tau allele was deleted, survival of Scn1aRX/+ mice markedly improved and was no longer significantly different from that of wild-type littermates. When both Tau alleles were deleted, survival of Scn1aRX/+ mice was indistinguishable from that of wild-type littermates.
FIGURE 1.
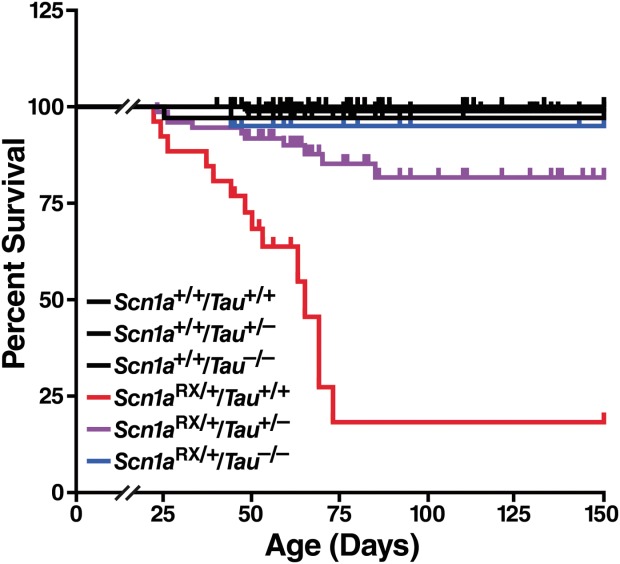
Tau reduction improves survival of Scn1aRX/+ mice in a gene dose-dependent manner. Survival plots of 292 Scn1aRX/+ mice and littermate controls with 2, 1, or no Tau alleles (n = 20–98 mice per genotype) indicate the percentage of live mice between 22 and 150 days postnatally. Scn1aRX/+/Tau+/+ mice differed from Scn1a+/+/Tau+/+ (p = 0.0048, hazard ratio [HR] = 31.0), Scn1aRX/+/Tau+/− (p = 0.00011, HR = 6.1), and Scn1aRX/+/Tau−/− mice (p = 0.026, HR 17.1). Scn1a+/+/Tau+/+ mice did not differ from Scn1aRX/+/Tau+/− mice (p = 0.37, HR = 5.1) or Scn1aRX/+/Tau−/− mice (p = 1.0, HR = 1.8). Gene–dose effect: p = 0.00005, HR = 0.018 for each Tau deletion (Cox proportional hazards regression).
Tau Reduction Lowers Epileptic Activity and Network Hyperexcitability in Scn1aRX/+ Mice
We used video-EEG recordings to examine seizures and interictal epileptic activity in Scn1aRX/+ mice and the effect of tau ablation on these measures. At 2 to 3 months of age, we detected spontaneous seizures by EEG in 43% of Scn1aRX/+ mice (Table 1, Fig 2A). Seizures with motor manifestations typically started with forelimb clonus and progressed to tail extension, generalized clonic activity, and bouncing and running. They lasted on average 34 seconds and reached an average severity of 4.5 on a modified Loscher/Racine scale30,49,50 (see Table 1, Fig 2A). Tau ablation reduced the percentage of Scn1aRX/+ mice with seizures 2.7-fold to 16% without affecting seizure severity or duration in those mice that did develop seizures. No seizures were observed in Scn1a+/+ littermates with or without tau ablation.
TABLE 1.
Incidence, Severity, and Duration of Spontaneous Seizures in 2- to 3-Month-Old Scn1aRX/+ Mice and Littermates with 2 or No Tau Alleles
| Genotype | ||||||
|---|---|---|---|---|---|---|
| Scn1a | Tau | Mice, No. | Hours of EEG Recording | Mice with Seizures, %a | Seizure Severityb | Seizure Duration, sa |
| +/+ | +/+ | 9 | 216 | 0 | N/A | N/A |
| +/+ | −/− | 7 | 168 | 0 | N/A | N/A |
| RX/+ | +/+ | 21 | 1,169 | 43 | 4.5 ± 0.9 | 34.1 ± 10.0 |
| RX/+ | −/− | 25 | 1,233 | 16 | 4.3 ± 1.0 | 33.6 ± 9.2 |
Determined by EEG and observation of mouse behavior.
Seizure severity was scored as described in Materials and Methods.
p < 0.05 versus Scn1aRX/+/Tau+/+ (Fisher exact test).
EEG = electroencephalogram; N/A = not applicable.
FIGURE 2.
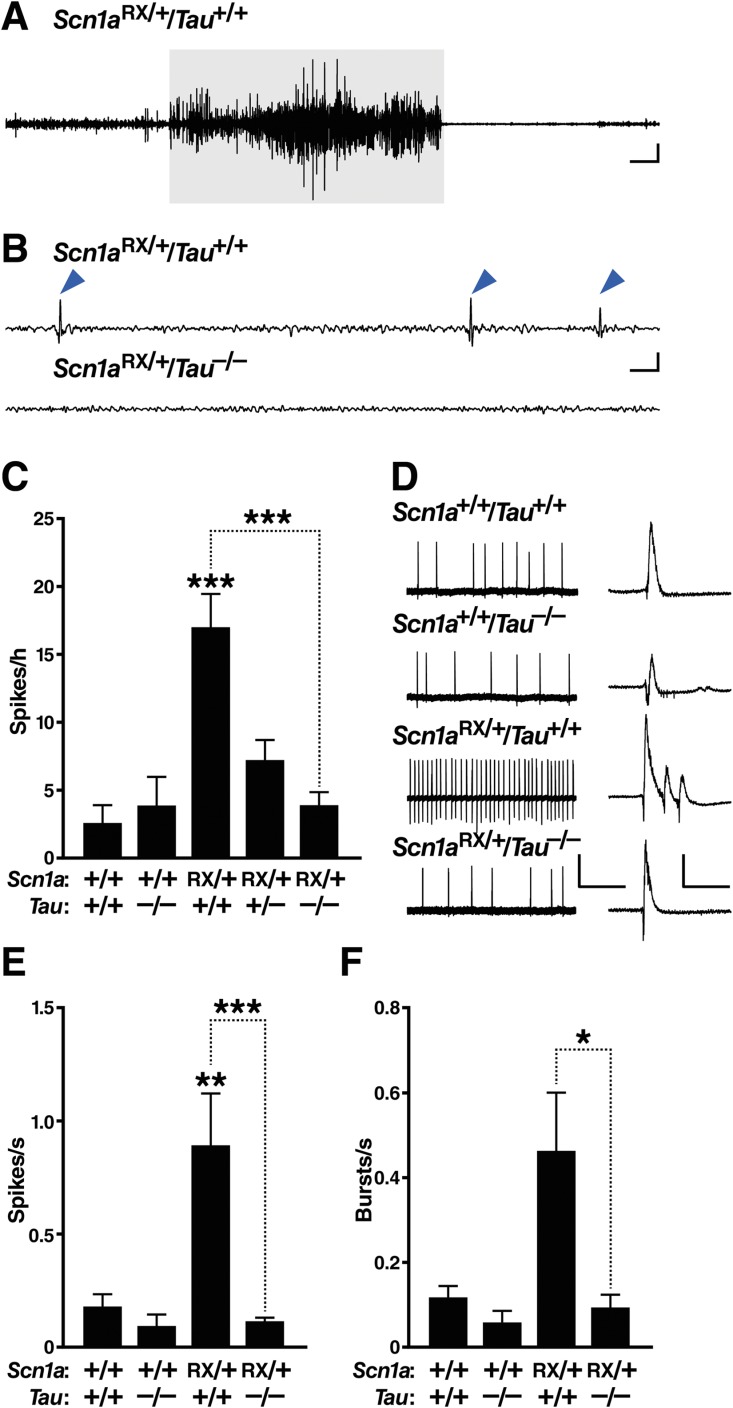
Tau ablation reduces epileptic activity in Scn1aRX/+ mice. (A–C) Subdural electroencephalographic (EEG) recordings were obtained in freely behaving mice of the indicated genotypes at 2 to 3 months of age. For the traces in (A) and (B), the low and high frequency filters were set at 5Hz and 40Hz, respectively. (A) Representative EEG trace depicting a seizure in an Scn1aRX/+ mouse. This seizure (highlighted in gray) lasted 30 seconds and had a severity score of 4 (see Materials and Methods). Scale bars = 3 sec (horizontal), 0.25V (vertical). (B) Representative traces of interictal EEG activity. Note the spikes (arrowheads) in Scn1aRX/+/Tau+/+ mice. Scale bars = 0.3 sec (horizontal), 0.5V (vertical). (C) Quantitation of epileptic spikes during 1 hour of a 24-hour recording session (n = 6–24 mice per genotype). Linear regression: p = 0.0030, F1,58 = 11.08 for an interaction between Scn1a and Tau genotypes. Gene–dose effect of Tau deletion in Scn1aRX/+ mice: p = 0.0000054 (Wald test). Exploratory post hoc 1-tailed t tests without multiple comparison correction indicated that Scn1aRX/+/Tau+/− mice differed from both Scn1aRX/+/Tau+/+ (p = 0.0085) and Scn1a+/+/Tau+/+ mice (p = 0.0087). ***p < 0.00001 versus Scn1a+/+/Tau+/+ mice or as indicated by bracket (Tukey–Kramer test). (D–F) Epileptiform activity in acute hippocampal slices from mice of the indicated genotypes was elicited by superfusion with picrotoxin (50μM) and 4-aminopyridine (200μM) for 30 minutes. (D) Representative field recordings of epileptic activity. Higher resolution traces are shown on the right, depicting individual spikes and a burst of multiple spikes recorded in the Scn1aRX/+ slice. Scale bars in main traces: 20 seconds (horizontal), 0.5mV (vertical); insets: 250 milliseconds (horizontal), 1.0mV (vertical). (E) Quantification of spike frequency. (F) Quantification of burst frequency (n = 3–6 mice per genotype and 17–27 slices per mouse). *p < 0.05, **p < 0.01, ***p < 0.001 versus Scn1a+/+/Tau+/+ slices or as indicated by bracket (linear mixed effects model). Values represent mean ± standard error of the mean. [Color figure can be viewed in the online issue, which is available at http://www.annalsofneurology.org.]
We also found that Scn1aRX/+ mice have an increased susceptibility to heat-induced seizures, as demonstrated previously for Scn1a knockout mice.31 This phenotype is likely related to the febrile seizures characteristically seen at the onset of disease in Dravet patients.19 On the Tau wild-type background, heat-induced seizures were observed in 86% (6 of 7) of Scn1aRX/+ mice at an average internal body temperature of 41.1 ± 0.1°C (mean ± standard error of the mean). Deletion of 1 Tau allele markedly reduced seizure susceptibility in Scn1aRX/+ mice, with only 33% (3 of 9) of Scn1aRX/+/Tau+/− mice showing seizures at 41.3 ± 0.3°C (p = 0.03; hazard ratio = 0.21 vs Scn1aRX/+/Tau+/+ mice by Cox regression). Ablation of both Tau alleles prevented heat-induced seizures in all (n = 3) Scn1aRX/+/Tau−/− mice.
In the interictal period, Scn1aRX/+ mice had more epileptic spikes on EEG recordings than wild-type controls, with an average frequency of ∼17 spikes/h (see Fig 2B, C). Tau ablation reduced spiking activity in Scn1aRX/+ mice in a gene dose-dependent manner; deletion of 1 Tau allele reduced spiking by 60%, and ablation of both Tau alleles reduced spiking by nearly 80%.
To further explore the effects of tau ablation on excitability, we examined the response of acute hippocampal slices from the 4 genotypes of mice to superfusion with picrotoxin and 4-aminopyridine ex vivo. These drugs can be used to elicit epileptic activity in brain slices.51–53 Compared with slices of all other genotypes, Scn1aRX/+/Tau+/+ slices showed a greater frequency of spikes (see Fig 2D, E). Tau ablation reduced both spike and burst frequencies in Scn1aRX/+ slices to control levels (see Fig 2D–F).
Tau Ablation Ameliorates Abnormalities in Seizure-Modulated Proteins in the Hippocampus of Scn1aRX/+ Mice
Seizure activity can lead to prominent remodeling of neuronal circuits and to changes in the expression of diverse proteins.30,32,54 Many of these changes are compensatory and may prevent spreading of epileptic activity. The hippocampus appears to be particularly important for seizure generation in Scn1a knockout mice.55 To look for molecular signatures of seizure activity in Scn1aRX/+ mice and assess the effect of tau ablation on these measures, we examined the hippocampal expression of NPY and calbindin. Scn1aRX/+ mice showed a marked increase of NPY in mossy fibers and in the molecular layer of the dentate gyrus (see Fig 3A–C). They also showed depletion of calbindin in the CA1 region of the hippocampus (stratum radiatum) and the molecular layer of the dentate gyrus (see Fig 3D–F). To our knowledge, these changes have not been previously reported for either Scn1aRX/+ mice or Scn1a knockout mice, although they are characteristic of animal models and patients with epilepsy.56 Tau ablation markedly reduced or fully reversed abnormalities in NPY and calbindin expression in all hippocampal subfields examined.
FIGURE 3.

Tau ablation improves hippocampal abnormalities in neuropeptide Y (NPY) and calbindin expression in Scn1aRX/+ mice. Coronal brain sections of 6- to 10-month-old mice (n = 11–14 per genotype) were immunostained for NPY (A–C) or calbindin (D–F). (A) Photomicrographs illustrating NPY alterations in the hippocampus of Scn1aRX/+ mice and improvement of this measure in mice with tau ablation. (B, C) Densitometric quantitation of NPY in the mossy fiber pathway (B) and the molecular layer of the dentate gyrus (C). (D) Photomicrographs illustrating calbindin alterations in the hippocampus of Scn1aRX/+ mice and improvement of this measure in mice with tau ablation. (E, F) Densitometric quantitation of calbindin in the stratum radiatum of hippocampal region CA1 (E) and the molecular layer of the dentate gyrus (F). Interaction between the Scn1a and Tau genotypes by 2-way analysis of variance: (B) p = 4.66E−07, F1,46 = 34.1; (C) p = 0.00034, F1,46 = 14.9; (E) p = 0.00039, F1,46 = 16.3; and (F) p = 0.0014, F1,46 = 11.6. *p < 0.05, **p < 0.01, ***p < 0.001 versus Scn1a+/+/Tau+/+ mice or as indicated by bracket (Tukey–Kramer test). DG, dentate gyrus. Values are mean ± standard error of the mean. [Color figure can be viewed in the online issue, which is available at http://www.annalsofneurology.org.]
We did not find any changes in the levels or phosphorylation of tau in Scn1aRX/+ mice compared to wild-type controls ( Fig 4). Nav 1. 1 levels were reduced by approximately half in Scn1aRX/+ mice and were not improved by tau ablation ( Fig 5).
FIGURE 4.
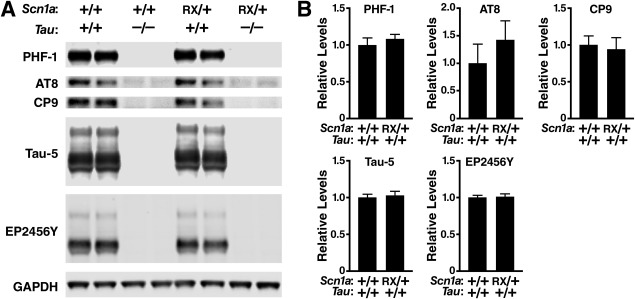
Cortical levels of total and phosphorylated tau are not altered in Scn1aRX/+ mice. Levels of phospho-tau (PHF-1, Ser396/Ser404; AT8, Ser202/Thr205; CP9, Thr231) and total tau (Tau-5, EP2456Y) in the parietal cortex of 8-month-old mice of the indicated genotypes were determined by Western blot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (A) Representative Western blot. (B) Quantification of Western blot signals (n = 5–7 mice per genotype) revealed no statistically significant differences between Scn1aRX/+/Tau+/+ and Scn1a+/+/Tau+/+ mice (Student t test). Average phospho-tau to EP2456Y ratios (PHF-1, AT8, CP9) or average total tau levels (Tau-5, EP2456Y) in Scn1a+/+/Tau+/+ mice were arbitrarily defined as 1.0. Values represent mean ± standard error of the mean.
FIGURE 5.
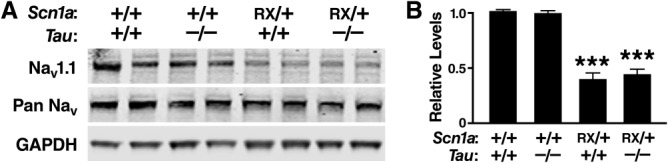
Nav1.1 levels are reduced in Scn1aRX/+ mice, and this reduction is not prevented by tau ablation. Levels of Nav1.1 and total sodium channels (pan Nav) in the parietal cortex of 8-month-old mice were determined by Western blot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels were used as a loading control. (A) Representative Western blot. (B) Quantification of Western blot signals (n = 5–7 mice per genotype). The average Nav1.1 to pan Nav ratio in Scn1a+/+/Tau+/+ mice was arbitrarily defined as 1.0. ***p < 0.001 versus Scn1a+/+/Tau+/+ mice (Tukey–Kramer test). Values represent mean ± standard error of the mean.
Tau Reduction Improves Nest Building and Open Field Behaviors in Scn1aRX/+ Mice
To determine whether tau ablation also modulates behavioral abnormalities in Scn1aRX/+ mice, we evaluated their nest building performance and open field activity. Nest building is a natural behavior of mice that may relate to activities of daily living in human patients,57 and deficits in this behavior are found in several mouse models of autism.58,59 Scn1aRX/+ mice had a profound deficit in nest building (see Fig 6A). Wild-type mice were able to form full nests within 24 hours (and some within 2 hours), whereas Scn1aRX/+ mice showed major delays in accomplishing this task. Most Scn1aRX/+ mice failed to form a complete nest, and several did not attempt nest building at all. Tau reduction effectively reduced these deficits in a gene dose-dependent manner, improving nest building performance in Scn1aRX/+ mice lacking 1 Tau allele and bringing nest building performance of completely tau-deficient Scn1aRX/+ mice to control levels. Interestingly, there was a negative correlation between nest building performance and epileptic activity in both Scn1aRX/+/Tau+/+ (p = 0.02, R2 = 0.33, n = 15) and Scn1aRX/+/Tau−/− mice (p = 0.04, R2 = 0.29, n = 15), indicating that epileptic activity may contribute to nesting deficits.
FIGURE 6.
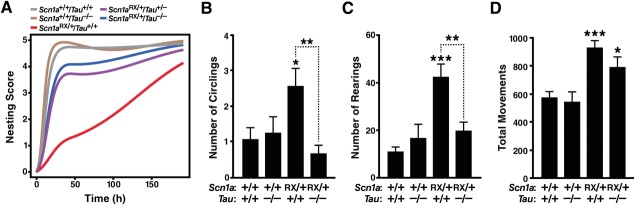
Tau ablation improves alterations in nest building, open field behaviors and social function in Scn1aRX/+ mice. Mice of the indicated genotypes were tested for nest building at 1 to 7 months (n = 11–22 mice per genotype) or open field activity and social approach at 2 to 3 months (n = 8–13 mice per genotype). See Supplementary Table S2 for age balance among groups. (A) Nest building behavior was monitored for up to 8 days and scored as described in Materials and Methods. A linear mixed effects model was used to fit the data and to obtain estimates of the area under the curve as a measure of nest building performance. Scn1aRX/+/Tau+/+ mice differed from Scn1a+/+/Tau+/+ (p = 0.0000004) and Scn1aRX/+/Tau−/− (p = 0.00063) mice, whereas Scn1aRX/+/Tau−/− mice did not differ from Scn1a+/+/Tau+/+ mice (p = 0.21). A gene–dose effect of Tau deletion was present (p = 0.00063). Exploratory post hoc analyses without multiple comparison correction indicated that Scn1aRX/+/Tau+/− mice differed more from Scn1aRX/+/Tau+/+ (p = 0.014) than Scn1a+/+/Tau+/+ (p = 0.069) mice. (B–D) Open field behavior. (B) Circling was recorded for 30 minutes and (C) rearing and (D) total movements during the first 5 minutes. Interaction between Scn1a and Tau genotypes by 2-way analysis of variance: (B) p = 0.015, F1,37 = 6.5; (C) p = 0.0022, F1,38 = 10.8; (D) p = 0.34, F1,38 = 0.93. *p < 0.05, **p < 0.01, ***p < 0.001 versus Scn1a+/+/Tau+/+ mice or as indicated by bracket (Tukey–Kramer test). Data are mean ± standard error of the mean.
Scn1aRX/+ mice also showed more circling and rearing in the open field than wild-type controls (see Fig 6B, C). Tau ablation prevented these behavioral abnormalities. Consistent with previous studies,27 Scn1aRX/+ mice were hyperactive in the open field (see Fig 6D). Tau ablation tended to ameliorate the hyperactivity of Scn1aRX/+ mice, but this trend did not reach statistical significance.
Tau Ablation Improves Learning and Memory Deficits in Scn1aRX/+ Mice
To determine whether tau ablation also improves cognitive deficits in Scn1aRX/+ mice, we assessed mice in 2 paradigms: the Barnes maze, which tests spatial learning and memory, and context-dependent fear conditioning, which tests associative learning and memory. Both Scn1a mutant and Scn1a knockout mice have deficits in the Barnes maze,26,27 whereas deficits in fear conditioning appear to have been reported only for Scn1a knockout mice.26
In the Barnes maze, mice are placed on a flat platform with 20 holes near the perimeter and are motivated by the presence of bright lights to learn the location of a single target hole with a dark escape tunnel. Mice of all 4 genotypes tested were able to learn this task ( Fig 7A). However, in a probe trial carried out 5 days after training, Scn1aRX/+ mice required increased time and path lengths to find the target location (see Fig 7B, C), suggesting a long-term memory deficit. During this probe trial, the majority of Scn1aRX/+ mice showed longer and less directed paths than the other groups, indicating random rather than serial or target-oriented search strategies (see Fig 7D, E). Tau ablation prevented these deficits, bringing latency, distance, and strategy measures in Scn1aRX/+ mice to control levels. We confirmed the beneficial effects of tau ablation on Barnes maze performance of Scn1aRX/+ mice in 2 additional cohorts (cohorts 3 and 7; n = 17–23 mice per genotype; 5 months of age; see Supplementary Tables S1 and S2, and data not shown).
FIGURE 7.

Tau ablation ameliorates deficits of Scn1aRX/+ mice in the Barnes maze and in a fear conditioning task. (A–E) Mice (n = 6–7 mice per genotype) were tested in the Barnes maze at 4 months of age. (A) Learning curves in the Barnes maze did not differ significantly among genotypes (linear mixed effects model analysis). (B, C) Latency (B) and distance traveled (C) to reach the target location during a probe trial 5 days after training in the Barnes maze. Interaction between Scn1a and Tau genotypes by 2-way analysis of variance: (B) p = 0.020, F1,22 = 6.3 and (C) p = 0.0073, F1,22 = 8.7. *p < 0.05, **p < 0.01, ***p = 0.001 versus Scn1a+/+/Tau+/+ mice or as indicated by bracket (Tukey–Kramer test). (D, E) Search strategies (D) and composite of paths (E) during a probe trial 5 days after training in the Barnes maze. Dots in (E) indicate the target location. (F–H) Mice were tested in context-dependent fear conditioning at 6 months of age (n = 8–16 mice per genotype). On each of 3 consecutive days, mice received a single foot shock 3 minutes after being placed individually into the same context chamber. Their freezing behavior before and after the shock was monitored on each of the 3 days, and also on a fourth, no-shock day. (F) Maximum motion index calculated based on movements immediately following the first shock mice received. (G, H) Percentage of time mice spent freezing 24 hours after receiving a shock when placed back into the same context, but prior to receiving the next shock (G) or during the first 1 minute after receiving a shock (H). Interaction between Scn1a and Tau genotypes by 2-way analysis of variance: p = 0.0046, F1,38 = 9.1 for day 2 in G. *p < 0.05, ***p = 0.001 versus Scn1a+/+/Tau+/+ mice or as indicated by bracket (Tukey–Kramer test). Data are mean ± standard error of the mean.
In the contextual fear conditioning test, mice learn to associate a specific, initially neutral and nonaversive context with receiving a foot shock. We used a gradual learning paradigm by administering 1 relatively mild foot shock on each of 3 consecutive days in the same context chamber (see Fig 7). Learning and memory were assessed by measuring freezing behavior after reintroducing mice into the same context 24 hours after each shock. Mice of all genotypes showed a similar immediate reaction (jumping and running) to the first shock, demonstrating that all groups were able to perceive the shock. However, Scn1aRX/+ mice showed a significant impairment when their associative memory was tested on day 2, 24 hours after receiving the first foot shock. This impairment was ameliorated by tau ablation. Compared to the other groups, Scn1aRX/+ mice also had an impaired conditioned fear response, showing less freezing immediately after they received the first foot shock on day 1. This deficit, which may represent a deficit in associative learning, was not seen in Scn1aRX/+ mice lacking tau. We confirmed the beneficial effects of tau ablation on fear conditioning performance of Scn1aRX/+ mice in an independent cohort (cohort 4; n = 6–7 mice per genotype; 5 months of age; see Supplementary Table S1 and S2, and data not shown). Thus, tau ablation prevents or ameliorates deficits in spatial memory and associative learning and memory in Scn1aRX/+ mice.
Discussion
In this study, we demonstrated several beneficial effects of tau reduction in a mouse model of Dravet syndrome, including markedly reduced occurrence of sudden death and epileptic manifestations and significant improvements in cognitive and behavioral performance. To our knowledge, this is the first demonstration that endogenous wild-type tau may fulfill an enabling role in the pathogenesis of a severe human epilepsy.
One of the most robust findings was that reducing tau conferred a dose-dependent survival advantage to Scn1aRX/+ mice. This result most likely reflects the antiepileptogenic effect of tau reduction. Sudden death in Dravet patients and related mouse models is thought to be caused directly or indirectly by seizure activity.20,21,60 In an elegant study of SUDEP by Kalume et al, sudden death in Scn1a knockout mice was shown to occur immediately following generalized tonic–clonic seizures in all monitored mice that died.21 In the same study, death could be predicted by a high frequency of seizures 24 hours before death, but not by seizure duration or severity. Extrapolating from these findings, our Scn1aRX/+ mice most likely also died primarily of seizures, and tau ablation prevented sudden death by decreasing their seizure frequency. Similarly to Kalume et al,21 we found no differences in seizure duration or severity in mice that died (most Scn1aRX/+ mice) or lived (Scn1aRX/+ mice with tau ablation), suggesting that tau participates in an early epileptogenic process but does not substantially modulate seizure activity once it has been triggered.
Notably, tau reduction can block diverse epileptogenic processes, including those initiated by pathological elevation of hAPP/Aβ,3,4,11 pharmacological blockade of γ-aminobutyric acid (GABA)A channels,3,10 genetic ablation of the voltage-gated potassium channel subunit Kv1.1,9 depletion of ethanolamine kinase or of the K+–Cl− cotransporter,9 and depletion of the voltage-gated sodium channel subunit Nav1.1 (this study). The precise mechanisms by which endogenous wild-type tau enables or promotes epileptogenesis triggered by such diverse factors is under intense investigation. Published evidence suggests that these mechanisms could involve alterations in GABAergic neurotransmission and the excitation/inhibition balance,4 in synaptic activity-related signaling pathways,2,11 and in the axonal transport of proteins that affect neuronal excitability.61 Regardless, the mechanisms underlying the antiepileptic effects of tau ablation may well be distinct from those of currently available therapies,2,62,63 and thus might offer new avenues for treating drug-resistant epilepsy.
Interestingly, tau ablation prevented or ameliorated not only epileptic activity and premature mortality in Scn1aRX/+ mice, but also their deficits in learning/memory, nest building, and other behaviors. The most parsimonious explanation of these findings is that the behavioral alterations are directly or indirectly caused by tau-dependent epileptic activity. Dravet syndrome is classified as an epileptic encephalopathy, based on the hypothesis that epileptic activity is the main cause of cognitive and behavioral alterations in Dravet patients.64–66 Both subclinical and clinical epileptic activity can cause cognitive and behavioral impairments in a variety of settings.67–71 Conversely, interventions that reduce epileptic activity can have beneficial effects on cognition and behavior in both humans and animal models.29,72,73 However, cognitive and behavioral abnormalities in patients with Dravet syndrome often do not improve upon treatment with antiepileptic medications,12–14,64 which could simply be due to currently available antiepileptic drugs not being very effective at suppressing epileptic activity in this syndrome.23,24 In addition, the frequency of convulsive seizures does not appear to correlate with cognitive outcomes in patients with Dravet syndrome,74–76 which makes it interesting to consider additional mechanisms that could underlie the beneficial effects of tau reduction in Scn1aRX/+ mice. Such mechanisms may include alterations in excitation/inhibition balance4,77 and brain rhythms76,78–80 and deserve to be explored in future studies.
The robust protective effects of tau reduction in Scn1aRX/+ mice revealed by our study have potentially important therapeutic implications for the treatment of Dravet syndrome and possibly other epilepsy syndromes that are refractory to currently available treatments. Uncontrolled seizures adversely impact the quality of life of patients, increase the burden on caregivers, and greatly increase the chance for multiple comorbidities, including cognitive impairment, injury, and death.81 In regard to tau-lowering approaches, it is encouraging that even partial reduction of tau made mice more resistant to epileptic activity when the reduction was either constitutive3,4 or initiated during adulthood.10 Similarly, in the current study, Scn1aRX/+ mice with deletion of only 1 Tau allele showed a substantial improvement in survival, epileptic activity, and nesting performance compared to Scn1aRX/+ mice on the Tau wild-type background.
It is also important to note in this context that genetic ablation of tau is well tolerated,3,4,11,82–84 as is antisense oligonucleotide-mediated knockdown and methylene blue–induced reduction of tau in adult mice.10,85 Similarly, in our study, the behavioral performance of Scn1a+/+ mice with or without tau ablation was indistinguishable.
Nonetheless, some studies have cautioned against using tau reduction as a therapeutic approach. For example, acute tau knockdown during embryonic development delayed neuronal migration and cell-autonomously reduced neuronal complexity and connectivity,86 problems that appear to be restricted to early developmental stages. In contrast to the beneficial effects of tau reduction we documented in 3 lines of hAPP transgenic mice3,4 and Ittner and colleagues independently demonstrated in a fourth hAPP transgenic line on a different tau knockout background,11 tau reduction has been reported to worsen behavioral deficits in the Tg2576 line of hAPP transgenic mice.87 Another report suggested that genetic tau ablation causes iron accumulation resulting in loss of dopaminergic neurons and severe motor deficits in aged mice.88 However, we were unable to replicate these findings.82 Thus, although the safety of tau-lowering treatments should clearly be further tested, most of the available experimental evidence suggests a rather attractive risk/benefit ratio and makes the identification of tau-lowering drugs an important objective.
Methylene blue and antisense oligonucleotides against tau represent 2 potential tau-lowering approaches currently under study. Methylene blue can reduce tau aggregation and lower soluble tau levels in mice and in cell culture assays.85,89 Treatment with this compound ameliorated learning and memory deficits in tau transgenic mice.85 However, methylene blue has diverse activities,90 complicating the interpretation of these findings. Antisense oligonucleotides to knock down tau expression represent an alternative approach to lowering tau levels in the brain. Intracerebroventricular infusion of such compounds in adult wild-type mice was well tolerated, lowered tau, and made mice more resistant to drug-induced seizures; tau levels correlated positively with epileptic severity.10 The further development of such compounds and related small-molecule drugs should make it possible to evaluate the beneficial effects of tau reduction in the clinical setting before long. In light of the encouraging findings obtained here and in the studies discussed above, the therapeutic potential of tau reduction deserves to be further explored in regard to intractable epilepsy as well as other conditions involving neuronal hyperexcitability and network dysrhythmias.
Acknowledgments
The study was supported by NIH (NINDS) grants NS066930 (A.L.G.), NS041787 (L.M.), and NS065780 (L.M.), by NIH (NCRR) grant RR18928, and by a gift from the S. D. Bechtel, Jr Foundation.
We thank Drs K. Yamakawa and M. H. Meisler for the Scn1aRX/+ mice; Drs M. Morris, J. Palop, and L. Verret for helpful advice on experimental design; Drs K. Vossel, S. Maeda, and M. Morris for comments on the manuscript; K. Bummer, J. Kang, X. Wang, and G.-Q. Yu for excellent technical assistance; O. Zhang and D. Nathaniel for analysis of electrophysiological recordings; I. Lo and A. Davis for behavioral testing; J. Carroll, T. Roberts, and C. Goodfellow for figure preparation; and M. Dela Cruz and A. Cheung for administrative assistance.
Authorship
All authors were involved in study design and data analysis. In addition, A.L.G., B.D., R.J.C., K.H., and W.G. performed experiments; P.E.S. contributed analytic tools; L.M. supervised the study; and A.L.G. and L.M. wrote the article.
Potential Conflicts of Interest
L.M.: grants, Bristol-Myers Squibb, Takeda Pharmaceuticals; SAB member, iPierian, Neuropore Therapies; consultancy, Catenion, Johnson & Johnson; speaking fees, Isis Pharmaceuticals; patent, PCT Pub WO/2008/124066 (licensee, Bristol-Myers Squibb).
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- 1.Wilcox KS, Dixon-Salazar T, Sills GJ, et al. Issues related to development of new antiseizure treatments. Epilepsia. 2013;54(suppl 4):24–34. doi: 10.1111/epi.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 4.Roberson ED, Halabisky B, Yoo JW, et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chin J, Scharfman HE. Shared cognitive and behavioral impairments in epilepsy and Alzheimer's disease and potential underlying mechanisms. Epilepsy Behav. 2013;26:343–351. doi: 10.1016/j.yebeh.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vossel KA, Beagle AJ, Rabinovici GD, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holth JK, Bomben VC, Reed JG, et al. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci. 2013;33:1651–1659. doi: 10.1523/JNEUROSCI.3191-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeVos SL, Goncharoff DK, Chen G, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33:12887–12897. doi: 10.1523/JNEUROSCI.2107-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ittner LM, Ke YD, Delerue F, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 12.Li BM, Liu XR, Yi YH, et al. Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav. 2011;21:291–295. doi: 10.1016/j.yebeh.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 13.Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. 2011;52(suppl 2):44–49. doi: 10.1111/j.1528-1167.2011.03001.x. [DOI] [PubMed] [Google Scholar]
- 14.Brunklaus A, Ellis R, Reavey E, et al. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain. 2012;135(pt 8):2329–2336. doi: 10.1093/brain/aws151. [DOI] [PubMed] [Google Scholar]
- 15.Dravet C, Oguni H. Dravet syndrome (severe myoclonic epilepsy in infancy) Handb Clin Neurol. 2013;111:627–633. doi: 10.1016/B978-0-444-52891-9.00065-8. [DOI] [PubMed] [Google Scholar]
- 16.Helbig I, Lowenstein DH. Genetics of the epilepsies: where are we and where are we going? Curr Opin Neurol. 2013;26:179–185. doi: 10.1097/WCO.0b013e32835ee6ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol. 2010;588(pt 11):1849–1859. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meisler MH, O'Brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol. 2010;588(pt 11):1841–1848. doi: 10.1113/jphysiol.2010.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52(suppl 2):3–9. doi: 10.1111/j.1528-1167.2011.02994.x. [DOI] [PubMed] [Google Scholar]
- 20.Surges R, Sander JW. Sudden unexpected death in epilepsy: mechanisms, prevalence, and prevention. Curr Opin Neurol. 2012;25:201–207. doi: 10.1097/WCO.0b013e3283506714. [DOI] [PubMed] [Google Scholar]
- 21.Kalume F, Westenbroek RE, Cheah CS, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123:1798–1808. doi: 10.1172/JCI66220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dravet C. Dravet syndrome history. Dev Med Child Neurol. 2011;53(suppl 2):1–6. doi: 10.1111/j.1469-8749.2011.03964.x. [DOI] [PubMed] [Google Scholar]
- 23.Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol. 2011;53(suppl 2):16–18. doi: 10.1111/j.1469-8749.2011.03967.x. [DOI] [PubMed] [Google Scholar]
- 24.Plosker GL. Stiripentol: in severe myoclonic epilepsy of infancy (Dravet syndrome) CNS Drugs. 2012;26:993–1001. doi: 10.1007/s40263-012-0004-3. [DOI] [PubMed] [Google Scholar]
- 25.Ogiwara I, Miyamoto H, Morita N, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han S, Tai C, Westenbroek RE, et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489:385–390. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito S, Ogiwara I, Yamada K, et al. Mouse with Na(v)1.1 haploinsufficiency, a model for Dravet syndrome, exhibits lowered sociability and learning impairment. Neurobiol Dis. 2012;49C:29–40. doi: 10.1016/j.nbd.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Dawson HN, Ferreira A, Eyster MV, et al. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114(pt 6):1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez PE, Zhu L, Verret L, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–E2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palop JJ, Chin J, Roberson ED, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oakley JC, Kalume F, Yu FH, et al. Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc Natl Acad Sci U S A. 2009;106:3994–3999. doi: 10.1073/pnas.0813330106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palop JJ, Jones B, Kekonius L, et al. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palop JJ, Mucke L, Roberson ED. Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer's disease: depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. Methods Mol Biol. 2011;670:245–262. doi: 10.1007/978-1-60761-744-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deacon RM. Assessing nest building in mice. Nat Protoc. 2006;1:1117–1119. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- 35.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- 36.Young SL, Fanselow MS. Associative regulation of Pavlovian fear conditioning: unconditional stimulus intensity, incentive shifts, and latent inhibition. J Exp Psychol Anim Behav Process. 1992;18:400–413. doi: 10.1037//0097-7403.18.4.400. [DOI] [PubMed] [Google Scholar]
- 37.Sanders MJ, Kieffer BL, Fanselow MS. Deletion of the mu opioid receptor results in impaired acquisition of Pavlovian context fear. Neurobiol Learn Mem. 2005;84:33–41. doi: 10.1016/j.nlm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Fanselow MS. Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci. 1980;15:177–182. doi: 10.1007/BF03001163. [DOI] [PubMed] [Google Scholar]
- 39.Fanselow MS. Associative vs topographical accounts of the immediate shock-freezing deficit in rats: implications for the response selection rules governing species-specific defensive reactions. Learn Motiv. 1986;17:16–39. [Google Scholar]
- 40.R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]
- 41.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- 42.Therneau TM. Survival analysis. R package version 2.37–4 ed2013.
- 43.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- 44.Bates D, Maechler M, Bolker B. lme4: linear mixed-effects models using S4 classes. R package version 0.999999-0 ed2012.
- 45.Gelman A, Hill J, Yajima M. Why we (usually) don't have to worry about multiple comparisons. J Res Educ Eff. 2012;5:189–211. [Google Scholar]
- 46.Altman DG, Bland JM. How to obtain the P value from a confidence interval. BMJ. 2011;343:d2304. doi: 10.1136/bmj.d2304. [DOI] [PubMed] [Google Scholar]
- 47.Vaida F, Fitzgerald AP, Degruttola V. Efficient hybrid EM for linear and nonlinear mixed effects models with censored response. Comput Stat Data Anal. 2007;51:5718–5730. doi: 10.1016/j.csda.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaida F, Liu L. lmec: linear mixed-effects models with censored responses. R package version 1.0 ed2009.
- 49.Loscher W, Honack D, Fassbender CP, Nolting B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. III. Pentylenetetrazole seizure models. Epilepsy Res. 1991;8:171–189. doi: 10.1016/0920-1211(91)90062-k. [DOI] [PubMed] [Google Scholar]
- 50.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 51.Zahn RK, Tolner EA, Derst C, et al. Reduced ictogenic potential of 4-aminopyridine in the perirhinal and entorhinal cortex of kainate-treated chronic epileptic rats. Neurobiol Dis. 2008;29:186–200. doi: 10.1016/j.nbd.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 52.Ziburkus J, Cressman JR, Schiff SJ. Seizures as imbalanced up states: excitatory and inhibitory conductances during seizure-like events. J Neurophysiol. 2013;109:1296–1306. doi: 10.1152/jn.00232.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cymerblit-Sabba A, Schiller Y. Network dynamics during development of pharmacologically induced epileptic seizures in rats in vivo. J Neurosci. 2010;30:1619–1630. doi: 10.1523/JNEUROSCI.5078-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vezzani A, Sperk G. Overexpression of NPY and Y2 receptors in epileptic brain tissue: an endogenous neuroprotective mechanism in temporal lobe epilepsy? Neuropeptides. 2004;38:245–252. doi: 10.1016/j.npep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 55.Liautard C, Scalmani P, Carriero G, et al. Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia. 2013;54:1251–1261. doi: 10.1111/epi.12213. [DOI] [PubMed] [Google Scholar]
- 56.Scharfman HE. Alzheimer's disease and epilepsy: insight from animal models. Future Neurol. 2012;7:177–192. doi: 10.2217/fnl.12.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deacon R. Assessing burrowing, nest construction, and hoarding in mice. J Vis Exp. 2012;(59):e2607. doi: 10.3791/2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moretti P, Bouwknecht JA, Teague R, et al. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum Mol Genet. 2005;14:205–220. doi: 10.1093/hmg/ddi016. [DOI] [PubMed] [Google Scholar]
- 59.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ogiwara I, Iwasato T, Miyamoto H, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet. 2013;22:4784–4804. doi: 10.1093/hmg/ddt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vossel KA, Zhang K, Brodbeck J, et al. Tau reduction prevents Aβ-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bialer M. Chemical properties of antiepileptic drugs (AEDs) Adv Drug Deliv Rev. 2012;64:887–895. doi: 10.1016/j.addr.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 63.Gotz J, Ittner A, Ittner LM. Tau-targeted treatment strategies in Alzheimer's disease. Br J Pharmacol. 2012;165:1246–1259. doi: 10.1111/j.1476-5381.2011.01713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Catarino CB, Liu JY, Liagkouras I, et al. Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain. 2011;134(pt 10):2982–3010. doi: 10.1093/brain/awr129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Engel J., Jr A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42:796–803. doi: 10.1046/j.1528-1157.2001.10401.x. [DOI] [PubMed] [Google Scholar]
- 66.Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 67.Kleen JK, Scott RC, Lenck-Santini PP, Holmes GL. Cognitive and behavioral co-morbidities of epilepsy. In: Noebels JL, Avoli M, Rogawski MA, et al., editors. Jasper's Basic Mechanisms of the Epilepsies. Bethesda, MD: National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
- 68.Kleen JK, Scott RC, Holmes GL, Lenck-Santini PP. Hippocampal interictal spikes disrupt cognition in rats. Ann Neurol. 2010;67:250–257. doi: 10.1002/ana.21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kleen JK, Scott RC, Holmes GL, et al. Hippocampal interictal epileptiform activity disrupts cognition in humans. Neurology. 2013;81:18–24. doi: 10.1212/WNL.0b013e318297ee50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Veran O, Kahane P, Thomas P, et al. De novo epileptic confusion in the elderly: a 1-year prospective study. Epilepsia. 2010;51:1030–1035. doi: 10.1111/j.1528-1167.2009.02410.x. [DOI] [PubMed] [Google Scholar]
- 71.Nicolai J, Ebus S, Biemans DP, et al. The cognitive effects of interictal epileptiform EEG discharges and short nonconvulsive epileptic seizures. Epilepsia. 2012;53:1051–1059. doi: 10.1111/j.1528-1167.2012.03491.x. [DOI] [PubMed] [Google Scholar]
- 72.Pressler RM, Robinson RO, Wilson GA, Binnie CD. Treatment of interictal epileptiform discharges can improve behavior in children with behavioral problems and epilepsy. J Pediatr. 2005;146:112–117. doi: 10.1016/j.jpeds.2004.08.084. [DOI] [PubMed] [Google Scholar]
- 73.Mintz M, Legoff D, Scornaienchi J, et al. The underrecognized epilepsy spectrum: the effects of levetiracetam on neuropsychological functioning in relation to subclinical spike production. J Child Neurol. 2009;24:807–815. doi: 10.1177/0883073808330762. [DOI] [PubMed] [Google Scholar]
- 74.Ragona F, Granata T, Dalla Bernardina B, et al. Cognitive development in Dravet syndrome: a retrospective, multicenter study of 26 patients. Epilepsia. 2011;52:386–392. doi: 10.1111/j.1528-1167.2010.02925.x. [DOI] [PubMed] [Google Scholar]
- 75.Akiyama M, Kobayashi K, Yoshinaga H, Ohtsuka Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia. 2010;51:1043–1052. doi: 10.1111/j.1528-1167.2009.02466.x. [DOI] [PubMed] [Google Scholar]
- 76.Holmes GL, Bender AC, Wu EX, et al. Maturation of EEG oscillations in children with sodium channel mutations. Brain Dev. 2012;34:469–477. doi: 10.1016/j.braindev.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yizhar O, Fenno LE, Prigge M, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Verret L, Mann EO, Hang GB, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bender AC, Morse RP, Scott RC, et al. SCN1A mutations in Dravet syndrome: impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav. 2012;23:177–186. doi: 10.1016/j.yebeh.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bender AC, Natola H, Ndong C, et al. Focal Scn1a knockdown induces cognitive impairment without seizures. Neurobiol Dis. 2013;54:297–307. doi: 10.1016/j.nbd.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.England MJ, Liverman CT, Schultz AM, Strawbridge LM. Epilepsy across the spectrum: promoting health and understanding. A summary of the Institute of Medicine report. Epilepsy Behav. 2012;25:266–276. doi: 10.1016/j.yebeh.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morris M, Hamto P, Adame A, et al. Age-appropriate cognition and subtle dopamine-independent motor deficits in aged Tau knockout mice. Neurobiol Aging. 2013;34:1523–1529. doi: 10.1016/j.neurobiolaging.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morris M, Koyama A, Masliah E, Mucke L. Tau reduction does not prevent motor deficits in two mouse models of Parkinson's disease. PLoS One. 2011;6:e29257. doi: 10.1371/journal.pone.0029257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Andrews-Zwilling Y, Bien-Ly N, Xu Q, et al. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30:13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O'Leary JC, III, Li Q, Marinec P, et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sapir T, Frotscher M, Levy T, et al. Tau's role in the developing brain: implications for intellectual disability. Hum Mol Genet. 2012;21:1681–1692. doi: 10.1093/hmg/ddr603. [DOI] [PubMed] [Google Scholar]
- 87.Dawson HN, Cantillana V, Jansen M, et al. Loss of tau elicits axonal degeneration in a mouse model of Alzheimer's disease. Neuroscience. 2010;169:516–531. doi: 10.1016/j.neuroscience.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lei P, Ayton S, Finkelstein DI, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- 89.Wischik CM, Edwards PC, Lai RY, et al. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schirmer RH, Adler H, Pickhardt M, Mandelkow E. “Lest we forget you—methylene blue…”. Neurobiol Aging. 2011;32:2325. doi: 10.1016/j.neurobiolaging.2010.12.012. e2327–e2316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information