

Abstract
Rett syndrome is a dominant X-linked male-lethal disorder largely caused by mutations in the gene encoding methyl-CpG binding protein 2 (MECP2). Clinical manifestations include neurodevelopmental disorder characterized by early-onset intractable seizures, severe developmental delay, intellectual disability, and abnormal electroencephalograms. Afflicted females show normal development until the age of 6 to 18 months, followed by gradual loss of speech abilities, microcephaly, social impairment, ataxia, and stereotypic hand movements. We report a 7-year-old girl who was born of a nonconsanguineous marriage presenting with mental retardation and delayed development. Physical examination revealed loss of speech, repetitive hand-wringing movement, short stature (120 cm), strabismus, microcephaly, and autistic behavior. The diagnosis was confirmed by sequencing MECP2 gene with heterozygous mutation C385A in exon 2. The current study aimed to report the first case of Rett syndrome in the Azeri Turkish population.
Keywords: Rett syndrome, MECP2 mutation, Azeri Turkish population
1. Introduction
Rett syndrome (RTT) is a childhood neurodevelopmental disorder, with an approximate prevalence of 1 in 10,000–15,000 female live births. The syndrome is characterized by normal early development, followed by loss of purposeful use of hands, distinctive hand movements, slowed growth of brain and head, gait abnormalities, seizures, and mental retardation [1], [2], [3]. Infants with RTT show normal growth and development until the age of 6–18 months, followed by backsliding of development and regression of skills and abilities; they often exhibit autistic behaviors in the early stages. Other likely symptoms include toe walking; sleep problems; wide-based gait; teeth grinding and difficulty chewing; slowed growth; seizures; cognitive disabilities; and breathing problems when awake, such as hyperventilation, apnea, and swallowing air [1], [2]. Rett syndrome is a dominant X-linked disorder and a common genetic cause of mental retardation in girls. In males, however, the mutation usually results in severe clinical manifestations and leads to lethality in the male fetus [4].
The discovery of the gene responsible for RTT (GenBank Accession No.: AF030876.2, OMIM: 300005) has permitted studies on the distribution of various mutations in different geographic and ethnic groups [5]. Rett syndrome is caused by mutations in the MECP2 gene, which has been mapped to the locus Xq28. The MECP2 gene encodes the protein methyl-CpG binding protein 2 (MECP2), which functions as one of several biochemical switches; patients with RTT show improper functioning of this gene and insufficient amounts or structurally abnormal forms of the protein [1], [2]. Methyl-CpG binding protein 2 functions as a transcriptional repressor, an activator, and an RNA-binding protein. Systematic studies of the MECP2 gene revealed a range of mutations which were associated with different phenotypes [6]. In vivo studies using mice with MECP2 mutations revealed neuropathological and behavioral deficits similar to those reported for RTT [7], [8], [9].
Mutations in the MECP2 gene were observed in 70%–80% of the girls diagnosed with RTT using current diagnostic techniques. The remaining 20%–30% of the cases were attributed to partial gene deletion and unknown mutations [1], [2].
Mutations in CDKL5 (cyclin-dependent kinase-like 5) gene are associated with clinical manifestations in a few patients with RTT [10]. CDKL5 has been mapped to the locus Xp22.3; different mutations in the gene have been detected in girls (heterozygous mutations) and a few boys, with all subjects presenting early-onset intractable seizures [11], [12]. Recently, a novel mutation has been identified (p.z D263VfsX190) in FOXG1 gene in a patient with a congenital variant of RTT, which results in an altered reading frame of the entire coding sequence downstream of the mutation [13].
2. Case report
2.1. History and examination
A 7-year-old girl (Fig. 1), an only child who was born of a nonconsanguineous marriage and through vaginal delivery, was brought to our Medical Genetic Centre in March 2014 because of mental retardation. At birth, her father and mother were 25 and 23 years of age, respectively. Normal development was observed until the age of 6 months, following which development regressed. Loss of eye contact was observed at the age of 7 months, followed by a gradual change into an introvert. The patient had been previously examined by several doctors, and a diagnosis of autism had been made based on behavior. Finally, because of lack of improvement in symptoms, the patient was referred to our Medical Centre.
Fig. 1.
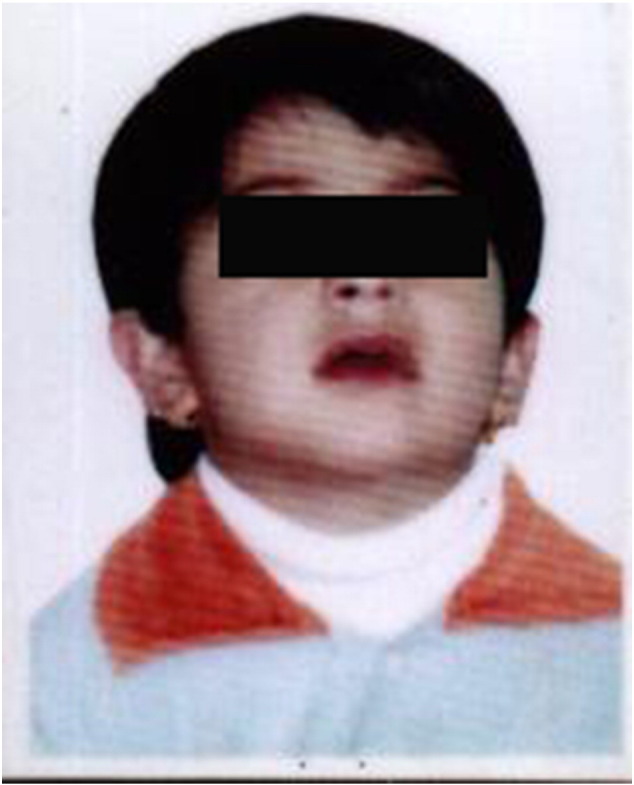
Affected child at 18 months of age.
Clinical examination carried out at the time of initial presentation revealed microcephaly (HC = 49 cm), short stature (120 cm), low-set ears, strabismus, repetitive hand wrestling movement, irritability, sleep disturbances, loss of speech, and autistic behavior. Normal HC was observed at birth, followed by slowing of the rate of head growth from the age of 6 months. This symptom profile led to initial suspicions of inborn errors of metabolism; therefore, chromatography of serum amino acids was carried out, which revealed nonspecific changes.
2.2. Paraclinical findings
Brain CT scan was normal. The clinical and paraclinical findings prompted suspicions of RTT, and a molecular genetic test was carried out. The blood sample was sent to the Laboratory of Houshmand in Tehran, Iran. Polymerase chain reaction (PCR) and bidirectional sequencing of the MECP2 gene revealed a heterozygous mutation C385A (Ala to Asp) in exon 2, indicating positivity for RTT.
After several years, her parents decided to get pregnant again. They were referred to the gynecologist with records of their daughter. During pregnancy, amniocentesis and DNA testing were reported to be normal, and finally, a healthy male child was born.
Coincidentally, during the time we evaluated this family, we were studying the genetic aspects of other genetic diseases [14], [15], [16].
2.3. Follow-up and representation
At her last visit, the patient was 15 years old (Fig. 2, Fig. 3, Fig. 4), and clinical examination revealed microcephaly (HC = 52 cm), short stature (145 cm), sleep disturbances, repetitive hand wrestling movement, rigidity of muscle hands, curved legs, irritability, loss of speech and writing and reading, sleep disturbances, strange noises, and lack of control over urinary and fecal excretion. Their son was a healthy child (Fig. 2, Fig. 3, Fig. 4).
Fig. 2.
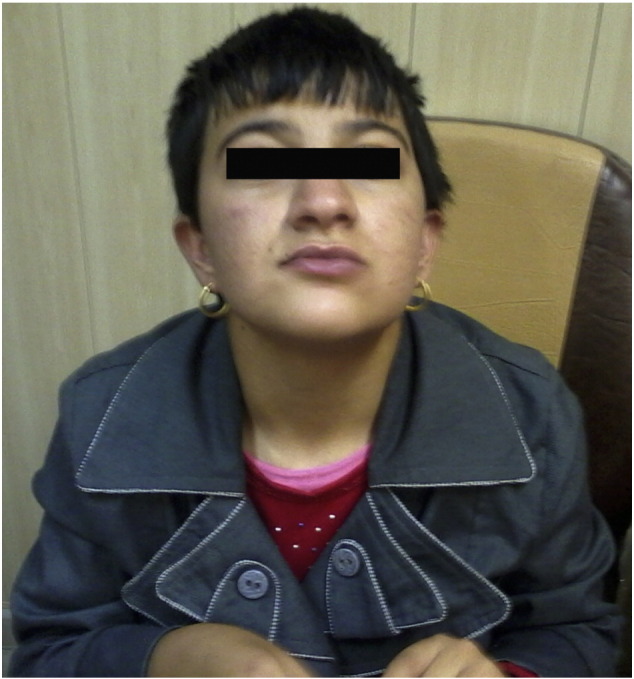
Affected child at 15 years of age.
Fig. 3.

Hand wrestling.
Fig. 4.

The patient in standing position.
3. Discussion
3.1. Clinical prognosis
Rett syndrome was originally described by Dr. Andres Rett of Austria in 1966 and was globally recognized two decades later following the publication of a report that described 35 affected girls from Sweden, Portugal, and France [17], [18]. Rett syndrome is considered the second common cause of mental retardation in females after Down syndrome [19]. Four stages are typically associated with RTT. Stage I, called early onset, generally begins between the ages of 6 and 18 months. This stage is frequently overlooked because the subtle symptoms of slowing development are likely to be somewhat vague and quite possibly missed by parents and specialists. There is reduced eye contact and lesser interest in toys and, possibly, delays in gross motor skills. This stage usually lasts for a few months but can persist for more than a year. Stage II, or the rapid destructive stage, usually begins between the ages of 1 and 4 years. This stage could have either a rapid or a gradual onset as purposeful hand skills and spoken language are lost. Hands are sometimes clasped behind the back or held at the sides. These movements persist while the child is awake but disappear during sleep. Autistic behaviors such as loss of social interaction and communication could also be encountered. Breathing irregularities, general irritability, slowing of head growth, and sleep irregularities are likely to be observed, along with unsteady gait patterns and difficulty in initiating motor movements. Stage III, also called the plateau or the pseudostationary stage, usually begins between the ages of 2 and 10 years. Apraxia, motor problems, and seizures are prominent during this stage. However, improvement in behavior (lesser irritability and crying), alertness, attention span, and communication skills is likely. Several affected females remain in this stage for the greater part of their lives. The last stage, stage IV or the late motor deterioration stage, is characterized by reduced mobility, muscle weakness, rigidity (stiffness), spasticity, and increased dystonia. In general, decline in cognition, communication, or hand skills is not observed in stage IV. Feeding disorders and poor weight gain are common. The clinical criteria required for a diagnosis of RTT are of three kinds: essential, supportive, and exclusion. Essential diagnostic criteria or symptoms include (1) normal HC at birth followed by slowing of the rate of increase in HC between 6 and 18 months of age and apparently normal development until 6–18 months of age, followed by (2) normal HC at birth or slowing of the rate of head growth between 3 months and 4 years of age, with either followed by impaired expressive language; repetitive hand movements; shaking of the torso; and toe-walking or unsteady, wide-based, and stiff-legged gait. Supportive criteria are not required for a diagnosis of RTT; a child with supportive criteria but none of the essential criteria is not diagnosed with RTT. Supportive criteria include breathing difficulties; electroencephalogram (EEG) abnormalities; seizures; muscle rigidity, spasticity, and/or joint contracture; scoliosis; teeth-grinding; small feet in relation to height; growth retardation; decreased body fat and muscle mass; abnormal sleep pattern; irritability or agitation; chewing and/or swallowing difficulty; poor circulation in the lower extremities with cold and bluish-red feet and legs; decreased mobility with age; and constipation. Children with any of the following exclusion criteria do not receive a diagnosis of RTT: enlargement of body organs or other signs of storage disease, vision loss due to retinal disorder or optic atrophy, microcephaly at birth, an identifiable metabolic disorder or other inherited degenerative disorders, an acquired neurological disorder, and evidence of in utero growth retardation or of brain damage acquired after birth [1], [2], [6]. Rett syndrome is most often misdiagnosed as autism, cerebral palsy, or nonspecific developmental delay. Differential diagnosis varies by clinical stage [20], and the list is presented in Table 1 as per the clinical stage.
Table 1.
The list of differential diagnosis with Rett syndrome.
| Stage I Developmental arrest (typically in children 6–18 months of age) |
Stage II Rapid deterioration or regression (typically in children 1–4 years of age) |
Stage III Pseudostationary (typically in children 2–10 years of age) |
Stage IV Late motor deterioration (typically in patients > 10 years) |
|---|---|---|---|
| Benign congenital hypotonia | Autism | Spastic ataxia | Other degenerative disorders |
| Cerebral palsy | Angelman syndrome | Cerebral palsy | |
| Prader–Willi syndrome | Encephalitis | Spinocerebellar | |
| Metabolic disorders (e.g., fetal alcohol syndrome, trisomy 13) | Hearing and/or visual disturbance | degeneration | |
| Landau–Kleffner syndrome | Leukodystrophies | ||
| Psychoses | Neuroaxonal dystrophy | ||
| Slow virus | Lennox–Gastaut syndrome | ||
| Panencephalopathy | Angelman syndrome (likely not Kabuki because patients would have macrocephaly) | ||
| Tuberous sclerosis | |||
| Metabolic disorders (e.g., phenylketonuria, ornithine transcarbamylase deficiency) | |||
| Infantile neuronal ceroid lipofuscinosis |
3.2. Candidate genes
Rett syndrome is caused by mutations (structural alterations or defects) in the MECP2 gene located on the X chromosome and has been detected in 70%–80% of girls diagnosed with RTT. Methyl-CpG binding protein 2 has been mapped to the chromosome Xq28 between the loci IRAK (interleukin-1 receptor-associated kinase) and RCP (red posing gene) [21]. The MECP2 gene spans approximately 76 kb and encodes MECP2 of 487 amino acids. Given that the disorder occurs spontaneously in most individuals with MECP2 mutation, the incidence of asymptomatic carriers of the disorder is an extremely rare possibility.
Certain cases with atypical RTT did not carry any of the reported mutations in MECP2. Mutation in the CDKL5 gene was found to result in an atypical form of RTT in females, called ‘early-onset seizure’ [10]. Of the two X chromosomes in females, only one is active in any given cell. Therefore, in a child with RTT, nearly 50% of the cells in the nervous system show expression of the defective gene, while the rest show expression of the wild type gene and normal amounts of the protein. In males with MECP2 mutation, however, the situation is different: males have a single copy of the X chromosome and, therefore, lack a wild type copy of the gene that could potentially compensate for the defective one, resulting in lethality. Different mutations in the MECP2 gene have been detected in affected males with mental retardation [1], [2], [19], [22], and recent reports have revealed that mild phenotypes are caused by deletions in the locus Xq28 [23].
Thiery Bienvenu et al. reported a total of 30 mutations in MECP2 among 46 girls with typical RTT; these include 12 novel mutations, 5 of which were nonsense mutations [R168X (n = 3), R198X (n = 1), R255X (n = 2), R270X (n = 5), and R294X (n = 3)], 3 were missense mutations [T158M (n = 3), P302R (n = 1), and R306C (n = 1)], 1 insertion [677insA (n = 1)], 4 deletions [1156del17 (n = 1), 1158del10 (n = 1), 1163del26 (n = 1), and 1164del26 + 1165A → T (n = 1)], and 1 silent polymorphism (S194S). The nonsense mutations were most frequently (4 of 5 cases) due to C → T transitions occurring in CpG dinucleotides [3].
The study by Wan et al. reported multiple recurrences of nonsense (R168X, R255X) and missense (R106W, R306C) mutations in MECP2. R168X mutation was observed in 6 unrelated sporadic cases, including 2 affected sisters and their healthy mother. The missense mutations preserved the domain structure of MECP2. All the mutations involved C → T transitions at CpG hotspots. Moreover, a single nucleotide deletion was recognized at codon 137, resulting in L138X mutation within the methyl-binding domain. A deletion (806delG) that resulted in the mutation V288X in the transcription–repression domain was identified in a female patient with X inactivation as well as in her sister and daughter, who were afflicted with classic RTT, and her homozygous son, who died from congenital encephalopathy [24]. Huppke et al. in Germany identified the mutations T158M, R168X, R255X, and R270X in 24 females [25]. Meloni et al. in Italy identified Xq27.2-qter in 2 male patients presenting with severe mental retardation and progressive spasticity, and 2 obligate carrier females were found to simulate an X-linked recessive trait [26].
4. Management
There are currently no known specific treatments for RTT. Clinical care, therefore, comprises genetic counseling (with DNA tests to rule out familial transmission), support and advice for the families, anticonvulsant medication upon the development of epilepsy, and physiotherapeutic measures for alleviating scoliosis development to the extent possible.
During regression, certain features of RTT are similar to those of autism; misdiagnosis of RTT as autism is, therefore, likely. The important role played by genetic factors in these conditions should be considered by individuals concerned with autism spectrum disorders, including psychiatrists, psychologists, or pediatricians. Of note, the current study features the first report of a mutation in RTT among the Azeri Turkish population of Iran.
Ethical standards
Ethical aspects were considered while obtaining permission from the parents of the child for the study, and the name of the child was kept anonymous.
Acknowledgments
We would like to thank the State Welfare Organization of Tabriz and the Liver & Gastrointestinal Disease Research Center.
Conflict of interest
None of the authors have any conflicts of interest.
References
- 1.Chahrour M., Huda Y.Z. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2000;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Rett B.O.A. Autism and Rett syndrome: behavioural investigations and differential diagnosis. Dev Med Child Neurol. 1987;29:429–441. doi: 10.1111/j.1469-8749.1987.tb02503.x. [DOI] [PubMed] [Google Scholar]
- 3.Bienvenu T., Carrie A., de Roux N., Vinet M.C., Jonveaux P., Couvert P. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum Mol Genet. 2000;9:1377–1384. doi: 10.1093/hmg/9.9.1377. [DOI] [PubMed] [Google Scholar]
- 4.Chae J.H., Hwang Y.S., Kim K.J. Mutation analysis of MECP2 and clinical characterization in Korean patients with Rett syndrome. JCN. 2002;17:33–36. doi: 10.1177/088307380201700108. [DOI] [PubMed] [Google Scholar]
- 5.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 6.Kaufmann W.E., Johnston M.V., Blue M.E. MeCP2 expression and function during brain development: implications for Rett syndrome's pathogenesis and clinical evolution. Brain Dev. 2005;27:S77–S87. doi: 10.1016/j.braindev.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Shahbazian M., Young J., Yuva-Paylor L., Spencer C., Antalffy B., Noebels J. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- 8.Chao H.T., Chen H., Samaco R.C., Xue M., Chahrour M., Yoo J. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Filippis B., Ricceri L., Laviola G. Early postnatal behavioral changes in the Mecp2-308 truncation mouse model of Rett syndrome. Genes Brain Behav. 2010;9:213–223. doi: 10.1111/j.1601-183X.2009.00551.x. [DOI] [PubMed] [Google Scholar]
- 10.Stachon A., Assumpcao F.B., Jr., Raskin S. Rett syndrome: clinical and molecular characterization of two Brazilian patients. Arq Neuropsiquiatr. 2007;65(1):36–40. doi: 10.1590/s0004-282x2007000100009. [DOI] [PubMed] [Google Scholar]
- 11.Bahi-Buisson N., Kaminska A., Boddaert N., Rio M., Afenjar, Gérard M.A. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia. 2008;49:1027–1037. doi: 10.1111/j.1528-1167.2007.01520.x. [DOI] [PubMed] [Google Scholar]
- 12.Bahi-Buisson N., Nectoux J., Rosas-Vargas H., Milh M., Boddaert N., Girard B. Key clinical features to identify girls with CDKL5 mutations. Brain. 2008;131:2647–2661. doi: 10.1093/brain/awn197. [DOI] [PubMed] [Google Scholar]
- 13.Das D.K., Jadhav V., Ghattargi V.C., Udani V. Novel mutation in forkhead box G1 (FOXG1) gene in an Indian patient with Rett syndrome. Gene. 2014;538:109–112. doi: 10.1016/j.gene.2013.12.063. [DOI] [PubMed] [Google Scholar]
- 14.Gharesouran J., Rezazadeh M., Mohaddes Ardebili S.M. Investigation of five polymorphic DNA markers associated with late onset Alzheimer disease. Genetika. 2013;45(2):503–514. [Google Scholar]
- 15.Gharesouran J., Rezazadeh M., Khorrami A., Ghojazadeh M., Talebi M. Genetic evidence for the involvement of variants at APOE, BIN1, CR1, and PICALM loci in risk of late-onset Alzheimer's disease and evaluation for interactions with APOE genotypes. J Mol Neurosci. 2014 doi: 10.1007/s12031-014-0377-5. [DOI] [PubMed] [Google Scholar]
- 16.Rezazadeh M., Gharesouran J., Mirabzadeh A., Khorram Khorshid H.R., Biglarian A., Ohadi M. A primate-specific functional GTTT-repeat in the core promoter of CYTH4 is linked to bipolar disorder in human. Prog Neuropsychopharmacol Biol Psychiatry. Sep 18 2014;56C:161–167. doi: 10.1016/j.pnpbp.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Hagberg B. Rett syndrome: Swedish approach to analysis of prevalence and cause. Brain Dev. 1985;7(3):277–280. [PubMed] [Google Scholar]
- 18.Hagberg B., Goutières F., Hanefeld F., Rett A., Wilson J. Rett syndrome: criteria for inclusion and exclusion. Brain Dev. 1985;7:372–373. doi: 10.1016/s0387-7604(85)80048-6. [DOI] [PubMed] [Google Scholar]
- 19.Kliegman R.M., Stanton B.M.D., Schor N.F., St. Geme J., Schor N., Behrman R.E. 19th ed. Saunders; Philadelphia: 2011. Nelson textbook of pediatrics; p. 2075. [Google Scholar]
- 20.Chakrabarti S., Fombonne E. Pervasive developmental disorders in preschool children. JAMA. 2001;285:3093–3099. doi: 10.1001/jama.285.24.3093. [DOI] [PubMed] [Google Scholar]
- 21.Reichwald K., Thiesen J., Wiehe T., Weitzel J., Poustka W.A., Rosenthal A. Comparative sequence analysis of the MECP2-locus in human and mouse reveals new transcribed regions. Mamm Genome. 2000;11:182–190. doi: 10.1007/s003350010035. [DOI] [PubMed] [Google Scholar]
- 22.Hagberg B., Witt-Engerström I. Rett syndrome: a suggested staging system for describing impairment profile with increasing age towards adolescence. AJMG. 1986;25:47–59. doi: 10.1002/ajmg.1320250506. [DOI] [PubMed] [Google Scholar]
- 23.Iourov I.Y., Vorsanova S.G., Voinova V.Y., Kurinnaia O.S., Zelenova M.A., Demidova I.A. Xq28 (MECP2) microdeletions are common in mutation-negative females with Rett syndrome and cause mild subtypes of the disease. Mol Cytogenet. 2013;6:53. doi: 10.1186/1755-8166-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan M., Lee S.S., Zhang X., Houwink-Manville I., Song H.R., Amir R.E. Rett syndrome and beyond: recurrent spontaneous and MECP2 mutations at CPG hotspots. Am J Hum Genet. 1999;65(6):1520–1529. doi: 10.1086/302690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huppke P., Laccone F., Krämer N., Engel W., Hanefeld F. Rett syndrome: analysis of MECP2 and clinical characterization of 31 patients. Hum Mol Genet. 2000;9(9):1369–1375. doi: 10.1093/hmg/9.9.1369. [DOI] [PubMed] [Google Scholar]
- 26.Meloni I., Bruttini M., Longo I., Mari F., Rizzolio F., D'Adamo P. A mutations in the Rett syndrome gene, MECP2, causes X-linked mental retardation and progressive spasticity in males. Am J Hum Genet. 2000;67(4):982–985. doi: 10.1086/303078. [DOI] [PMC free article] [PubMed] [Google Scholar]