

ABSTRACT
Hepatitis C virus (HCV) exploits host membrane cholesterol and its metabolism for progeny virus production. Here, we examined the impact of targeting cellular squalene synthase (SQS), the first committed enzyme for cholesterol biosynthesis, on HCV production. By using the HCV JFH-1 strain and human hepatoma Huh-7.5.1-derived cells, we found that the SQS inhibitors YM-53601 and zaragozic acid A decreased viral RNA, protein, and progeny production in HCV-infected cells without affecting cell viability. Similarly, small interfering RNA (siRNA)-mediated knockdown of SQS led to significantly reduced HCV production, confirming the enzyme as an antiviral target. A metabolic labeling study demonstrated that YM-53601 suppressed the biosynthesis of cholesterol and cholesteryl esters at antiviral concentrations. Unlike YM-53601, the cholesterol esterification inhibitor Sandoz 58-035 did not exhibit an antiviral effect, suggesting that biosynthesis of cholesterol is more important than that of cholesteryl esters for HCV production. YM-53601 inhibited transient replication of a JFH-1 subgenomic replicon and entry of JFH-1 pseudoparticles, suggesting that at least suppression of viral RNA replication and entry contributes to the antiviral effect of the drug. Collectively, our findings highlight the importance of the cholesterol biosynthetic pathway in HCV production and implicate SQS as a potential target for antiviral strategies against HCV.
IMPORTANCE Hepatitis C virus (HCV) is known to be closely associated with host cholesterol and its metabolism throughout the viral life cycle. However, the impact of targeting cholesterol biosynthetic enzymes on HCV production is not fully understood. We found that squalene synthase, the first committed enzyme for cholesterol biosynthesis, is important for HCV production, and we propose this enzyme as a potential anti-HCV target. We provide evidence that synthesis of free cholesterol is more important than that of esterified cholesterol for HCV production, highlighting a marked free cholesterol dependency of HCV production. Our findings also offer a new insight into a role of the intracellular cholesterol pool that is coupled to its biosynthesis in the HCV life cycle.
INTRODUCTION
Hepatitis C virus (HCV) is a causative agent of acute and chronic hepatitis, which can eventually lead to cirrhosis and hepatocellular carcinoma. HCV infection is recognized as a major threat to global public health, with 130 to 150 million people worldwide being infected with the virus (1). Over the last decade, the standard therapy for chronic HCV infection has been a combination of pegylated interferon alpha and ribavirin (2), but that has greatly changed after the emergence of first direct-acting antivirals that selectively target HCV, i.e., telaprevir and boceprevir (3, 4). These drugs, both used in combination with pegylated interferon and ribavirin, have brought significant benefits to patients who did not respond to the conventional therapy. In addition, recent clinical data on the newly approved direct-acting antivirals simeprevir and sofosbuvir have provided novel insights on combination therapies with inhibitors of multiple targets (5). However, direct-acting antivirals are frequently associated with the emergence of drug-resistant HCV variants, likely leading to treatment failure (6). Thus, development of host-targeted agents, which are expected to have a high genetic barrier to resistance, should be encouraged to expand treatment options for chronic hepatitis C.
HCV is an enveloped, positive-sense, single-stranded RNA virus belonging to the Hepacivirus genus of the Flaviviridae family. The HCV genome is 9.6 kb in length and contains a single open reading frame encoding a large polyprotein of approximately 3,000 amino acids. Translation of the polyprotein is directed by an internal ribosome entry site (IRES) located mostly in the highly conserved 5′ untranslated region (7). The polyprotein is co- and posttranslationally processed into three structural proteins (core, E1, and E2), a small ion channel protein (p7), and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) by cellular and viral proteases (8–10). The nonstructural proteins assemble on the endoplasmic reticulum (ER)-derived membranes and recruit the viral genome into an RNA replication complex (11, 12).
Several lines of evidence suggest that HCV is closely associated with cholesterol and its metabolism throughout the viral life cycle in hepatocytes (13). In a previous study using a cholesterol-extracting drug, methyl-β-cyclodextrin, HCV entry was found to be in part dependent on the host membrane cholesterol content (14). Biochemical studies suggest that HCV RNA replication takes place on lipid rafts (15–17), i.e., detergent-resistant membrane microdomains enriched in cholesterol and sphingolipids (18). Lipid rafts also appear to be involved in HCV virion assembly because the viral structural proteins are associated with them (19, 20). Virion assembly occurs at the ER membranes immediately adjacent to the lipid droplet (21, 22), a major storage organelle for cholesteryl esters and triglycerides. Subsequent maturation and release of viral particles are tightly linked to the very-low-density lipoprotein (VLDL) secretion pathway (reference 22 and references therein; 23). Indeed, the lipid composition of secreted viral particles resembles that of VLDLs and low-density lipoproteins (LDLs), with a large amount of cholesteryl esters (24). The viral particles are also enriched in cholesterol and sphingomyelin, both of which are important for particle maturation and infectivity (19).
Cholesterol is synthesized from acetyl coenzyme A (acetyl-CoA) via a series of enzymatic reactions shown in Fig. 1. The rate-limiting enzyme of the cholesterol biosynthetic pathway is 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase, which catalyzes the synthesis of mevalonate (25). Previous studies have shown that HMG-CoA reductase inhibitors or statins (26) block viral RNA replication in HCV genotype 1b replicon cells (27–29). Although statins are widely used as cholesterol-lowering drugs (30), their anti-HCV effect has been attributed not to a decrease in cholesterol content but rather to decreases of the nonsterol isoprenoids geranylgeranyl lipids (27, 28), the biosynthetic pathway of which shares early steps with that of cholesterol (Fig. 1). Although recent studies have shown that downstream enzymes in the cholesterol biosynthetic pathway, such as oxidosqualene cyclase, lanosterol C14-demethylase, 24-dehydrocholesterol reductase, and 7-dehydrocholesterol reductase, are required for HCV production (31–33), the role of the committed steps of cholesterol biosynthesis in the HCV life cycle is not fully understood.
FIG 1.
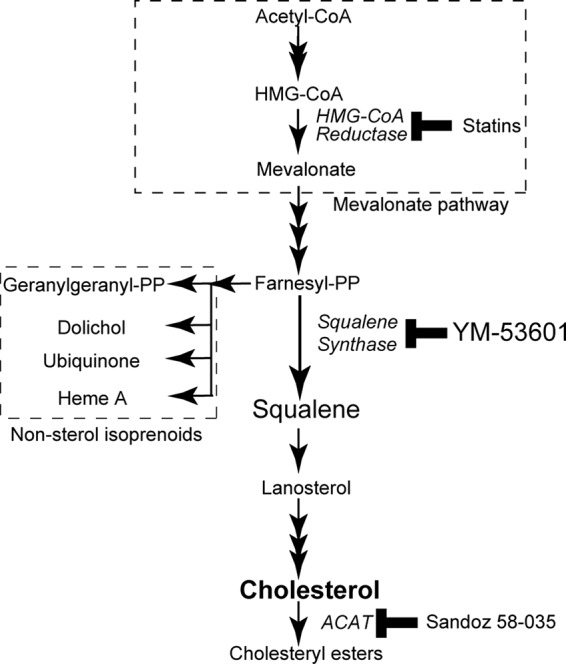
Cholesterol biosynthetic pathway in mammalian cells. Enzymes are shown in italics; inhibitors for the enzymes are shown next to the enzymes. Abbreviations: CoA, coenzyme A; HMG, 3-hydroxy-3-methylglutaryl; PP, pyrophosphate; ACAT, acyl-CoA:cholesterol acyltransferase.
In this study, we focused on squalene synthase (SQS), which is the first committed enzyme in cholesterol biosynthesis (34) (Fig. 1). We examined the impact of SQS inhibition on HCV production by using an HCV cell culture system with human hepatoma Huh-7.5.1-derived cells and an HCV genotype 2a isolate, JFH-1 (35–37). We present data showing that SQS-mediated cholesterol biosynthesis is important for viral production, and we propose that SQS is a potential anti-HCV target.
MATERIALS AND METHODS
Reagents.
YM-53601 (38), zaragozic acid A (39), and Sandoz 58-035 (40) were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA) and dissolved in dimethyl sulfoxide (DMSO). A Stealth RNA interference (RNAi) small interfering RNA (siRNA) for human SQS, HSS103617 (siSQS), and a Stealth RNAi negative-control low-GC duplex (siCONT) were purchased from Life Technologies Corp. (Carlsbad, CA, USA). Human low-density lipoprotein was purchased from Biomedical Technologies, Inc. (Stoughton, MA, USA). [methyl-3H]acetate was purchased from Moravek Biochemicals, Inc. (Brea, CA, USA).
Cell culture.
A highly HCV-permissive subclonal cell line derived from Huh-7.5.1 cells (36), Huh-7.5.1-8 (37), was maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium that contained 10% fetal bovine serum, 0.1 mM nonessential amino acids, 100 units/ml penicillin G, and 100 μg/ml streptomycin sulfate (referred to as “complete medium”). Serum-free culture was performed as described previously (41) with slight modifications: Huh-7.5.1-8 cells were incubated at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium that contained 1% Nutridoma-SP (Roche Applied Science, Penzberg, Upper Bavaria, Germany) and 25 μg/ml gentamicin (referred to as “serum-free medium”).
Virus stock and infection.
HCV JFH-1 was prepared from culture supernatants of Huh-7.5.1-8 cells that had been transfected with in vitro-transcribed JFH-1 RNA as previously described (35). After serial passages of the JFH-1 virus in naive Huh-7.5.1-8 cells, infectious culture supernatants were collected and used as viral stocks in this study. Virus titers were determined by fluorescent-focus assays as previously described (42). For infection, cells were incubated with the virus at a multiplicity of infection of 4 fluorescent-focus-forming units/cell in complete medium for 2 h at 37°C.
Immunoblotting.
Cells were lysed in NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Life Technologies Corp.) that contained 0.05 M dithiothreitol (DTT) and then heated at 95°C for 5 min. The resultant lysates were subjected to SDS-polyacrylamide gel electrophoresis on NuPAGE 4 to 12% Bis-Tris gels (Life Technologies Corp.) and then transferred to Immun-Blot polyvinylidene difluoride membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA) according to the manufacturer's protocols. After being blocked with 5% (wt/vol) skim milk in TBS-T (0.05 M Tris-HCl [pH 7.6], 0.15 M NaCl, 0.1% [vol/vol] Tween 20), the membranes were probed with 1:5,000 dilutions of anti-HCV core monoclonal antibody (B2; Anogen, Yes Biotech Laboratories, Ltd., Mississauga, Ontario, Canada) or anti-HCV NS3 monoclonal antibody (8G-2; Abcam, Plc., Cambridge, United Kingdom) or with a 1:10,000 dilution of anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) monoclonal antibody (6C5; Abcam, Plc.), followed by a 1:5,000 dilution of horseradish peroxidase-conjugated AffiniPure goat anti-mouse IgG(H+L) (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) in TBS-T that contained 2% (wt/vol) skim milk. For detection of SQS, the membranes were probed with a 1:5,000 dilution of anti-SQS polyclonal antibody (B01; Abnova Corp., Taipei City, Taiwan) followed by a 1:5,000 dilution of horseradish peroxidase-conjugated AffiniPure goat anti-rabbit IgG(H+L) (Jackson ImmunoResearch Laboratories, Inc.). Each protein band complexed with the antibody on the membrane was visualized with an enhanced chemiluminescence immunoblotting detection system (GE Healthcare, UK Ltd., Little Chalfont, United Kingdom; Merck Millipore, Billerica, USA) and quantified using Image J 1.440 software (National Institutes of Health, Bethesda, MD, USA).
RT-qPCR analysis.
Total RNA was isolated from cells by using an RNeasy Plus minikit (Qiagen, GmbH, Hilden, Germany) and reverse transcribed by random hexamer primers using a Transcriptor first-strand cDNA synthesis kit (Roche Applied Science) according to the manufacturer's protocols. Quantitative reverse transcription-PCR (RT-qPCR) was carried out on the LightCycler system (Roche Applied Science) using LightCycler FastStart DNA Master SYBR green I (Roche Applied Science) and specific primers for the core sequence (5′-CGCAACGTGGGTAAAGTCATCG-3′ and 5′-CGGGTAGGTTCCCTGTTGCATAA-3′), the NS5B sequence (5′-CAAGGGTCAAACCTGCGGTTACA-3′ and 5′-TGACTACTAGGTCATCGCCGCATAC-3′), or the human GAPDH sequence (Search–LC, GmbH, Heidelberg, Germany). The relative amounts of viral RNA were calculated by dividing the copy number of a viral transcript by that of a GAPDH transcript in the same sample.
Metabolic labeling of lipids with radioactive acetate and TLC.
Cells were plated at 1 × 105 cells per well of a 6-well plate 1 day before labeling and then incubated with [3H]acetate (1.85 MBq/well) in serum-free medium for various periods. The cells were washed and harvested with phosphate-buffered saline (PBS), and a lipid fraction was extracted from the cells according to the method of Bligh and Dyer (43). The lipid fraction was spotted on a silica gel 60 plate (Merck Millipore) and separated by thin-layer chromatography (TLC) using hexane-diethyl ether-acetate (70:30:1, vol/vol/vol). The incorporation of 3H radioactivity into each lipid was quantified using a BAS-1800 Bio-Image Analyzer (Fujifilm Corp.) or a Typhoon FLA 7000 biomolecular imager (GE Healthcare, UK Ltd.) and then normalized with the protein levels.
Determination of cholesterol and cholesteryl ester contents.
Cells were disrupted in PBS by sonication. The lipid fraction was extracted from the cells as described above. The content of cholesterol in the lipid fraction was determined by an enzymatic colorimetric method using the Wako free cholesterol E test (Wako Pure Chemical Industries, Ltd., Osaka, Japan) according to the manufacturer's protocol and then normalized with the protein levels. The content of cholesteryl esters in the lipid fraction was determined by a direct measurement method for the enzymatic determination of cholesteryl esters as described elsewhere (44) and then normalized with the protein levels.
siRNA transfection.
Cells were plated at 3 × 104 cells per well in 24-well plates 2 days before transfection and grown in complete medium. siRNA was complexed with Lipofectamine RNAiMAX transfection reagent (Life Technologies Corp.) according to the manufacturer's protocol and then added to the cells at a final concentration of 5 nM. After 5 h of incubation, the cells were washed and then placed in serum-free medium.
Subgenomic replicon plasmids.
The HCV subgenomic replicon plasmids used in this study contain the T7 promoter followed by a bicistronic replicon sequence; the first is a part of the core region fused to either the luciferase (luc) gene of the firefly Photinus pyralis or the neomycin phosphotransferase (neo) gene translated under the control of the HCV IRES, and the second is the NS3-NS5B-coding region translated under the control of the encephalomyocarditis virus (EMCV) IRES. Subgenomic replicon plasmids of the JFH-1 strain, pSGR-JFH1/Luc and pSGR-JFH1/Luc-GND (45), carry the luc gene; the latter contains a GDD-to-GND mutation in NS5B, which abolishes RNA polymerase activity. Subgenomic replicon plasmids of the Con-1 strain (genotype 1b), pFK-I389Luci/NS3-3′/NK5.1 and pFK-I389neo/NS3-3′/NK5.1/ΔGDD (46), were kindly provided by Ralf Bartenschlager (University of Heidelberg, Germany) and carry the luc gene and the neo gene, respectively; the latter contains a deletion in the GDD active site of NS5B that abolishes RNA polymerase activity. A replication-incompetent mutant of pFK-I389Luci/NS3-3′/NK5.1 was prepared by replacing an AscI-PmeI fragment that codes for the neo gene of pFK-I389neo/NS3-3′/NK5.1/ΔGDD with the corresponding fragment that codes for the luc gene from pFK-I389Luci/NS3-3′/NK5.1 (referred to as pFK-I389/Luci/NS3-3′/NK5.1/ΔGDD).
In vitro transcription of RNA.
Linearization of plasmids, in vitro transcription with T7 RNA polymerase, and RNA purification were performed as previously described (47) except that the AmpliScribe T7 high-yield transcription kit (Epicentre Biotechnologies Corp., Madison, WI, USA) was used.
Transfection with in vitro-transcribed RNA.
Electroporation was performed as described previously (48) with slight modifications. Cells (1 × 107 to 2 × 107) were mixed with 20 to 25 μg of in vitro-transcribed RNA in K-PBS (30 mM NaCl, 120 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, and 5 mM MgCl2, pH 7.9) and then pulsed at 975 μF and 290 V in a cuvette with a gap width of 0.4 cm by using a Gene Pulser Xcell system (Bio-Rad Laboratories, Inc.). For lipofection, cells were plated at 3 × 104 cells per well in a 24-well plate 2 days before transfection. The cells were then transfected with 0.5 μg of in vitro-transcribed RNA for 3 h using the TransMessenger transfection reagent (Qiagen, GmbH) according to the manufacturer's protocol.
Luciferase assay.
Cells were lysed with cell culture lysis reagent (Promega Corp., Madison, WI, USA). Five microliters of the lysate was mixed with 25 μl of luciferase assay reagent (Promega Corp.), and then luciferase activity in the lysate was measured by using a Luminescencer-PSN luminometer (Atto Corp., Tokyo, Japan).
Preparation of HA-tagged NS4B-expressing cells.
An expression plasmid that encodes NS4B protein N-terminally fused to a hemagglutinin (HA) tag sequence followed by a tobacco etch virus (TEV) protease cleavage site, pCXN2/HA-TEV-NS4B, was previously described (49). Huh-7.5.1-8 cells were transfected with pCXN2/HA-TEV-NS4B using FuGENE 6 transfection reagent (Roche Applied Science) and grown in the presence of 500 μg/ml of G418. G418-resistant cells were cloned by limiting dilution, and expression of HA-tagged NS4B protein in each clone was confirmed by immunoblotting with a rat anti-HA monoclonal antibody (clone 3F10; Roche Applied Science). Similarly, Huh-7.5.1-8 cells were transfected with a backbone plasmid, a modified version of pCXN2 (50, 51). The resultant G418-resistant cells were cloned and used as a negative control.
Immunofluorescence analysis.
Cells grown on collagen-coated coverslips (Asahi Glass Co., Ltd., Japan) were fixed with 4% paraformaldehyde phosphate buffer solution (Wako Pure Chemical Industries, Ltd.) for 15 min at room temperature. After being washed with 30 mM glycine in PBS, the cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature and then blocked with 3% (wt/vol) bovine serum albumin (BSA) in PBS. The cells were incubated with the anti-HA rat monoclonal antibody diluted 1:500 with 1% (wt/vol) BSA in PBS followed by an Alexa Fluor 488 goat anti-rat IgG(H+L) antibody (Life Technologies Corp.) diluted 1:300 with the same solution. The cells were mounted with ProLong Diamond antifade mountant with 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies Corp.) and observed using a confocal microscope (LSM 700; Carl Zeiss Microscopy, GmbH, Jena, Germany) equipped with an oil immersion objective lens (Plan-Apochromat 40×/1.4 oil DIC M27; Carl Zeiss Microscopy, GmbH).
Preparation of and infection with HCVpp.
HCV pseudoparticles (HCVpp) were prepared as described previously (52, 53) with slight modifications. Briefly, HEK293T cells were transfected with a Gag-Pol packaging plasmid (Gag-Pol 5349), a reporter (luciferase) plasmid (Luc 126), and a pcDNA3.1(+) (Life Technologies Corp.)-based expression plasmid that encodes HCV envelope proteins (E1 and E2) of the JFH-1 strain (genotype 2a) or the TH strain (54) (genotype 1b) for 24 h using the X-treme Gene HP DNA transfection reagent (Roche Applied Science), and then the medium was replaced with serum-free medium that contained 0.1 mM nonessential amino acids. HCVpp-containing medium was collected after additional 24 to 36 h of culture and used as HCVpp stock. In parallel, HEK293T cells were similarly transfected, except that the envelope protein-expressing plasmid was replaced with pcDNA3.1(+), and their culture medium was used as a negative control. For infection, Huh-7.5.1-8 cells were plated at 6 × 104 cells per well of a 48-well plate and grown in serum-free medium for 2 days. The cells were then infected with HCVpp for 6 h at 37°C. After being washed, the cells were grown in complete medium for an additional 3 days and assayed for luciferase activity.
Other methods.
Protein concentrations were measured using the bicinchoninic acid (BCA) protein assay reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA) with BSA as a standard. The amount of viral core protein in a culture supernatant, which is the hallmark of the secreted virus level, was quantified using the Ortho HCV antigen enzyme-linked immunosorbent assay (ELISA) (Ortho-Clinical Diagnostics, Inc., Raritan, NJ, USA). Cell viability was determined by using the XTT cell proliferation kit II (Roche Applied Science). The 50% inhibitory concentration (IC50) was calculated by using the equation “log (inhibitor) versus normalized response” of the nonlinear regression model included in GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). Statistical analysis was performed by Student's t test using the GraphPad calculator (QuickCalcs); differences with a P value of <0.05 were considered statistically significant.
RESULTS
Anti-HCV effect of YM-53601.
We began to explore the role of the cholesterol biosynthetic pathway in the HCV life cycle by using an SQS inhibitor, YM-53601. We first examined whether HCV JFH-1 can replicate efficiently in Huh-7.5.1-8 cells grown under serum-free conditions where cellular cholesterol requirements are met only through de novo synthesis. The amounts of viral core and NS3 proteins in JFH-1-infected cells clearly increased from the fourth to sixth day postinfection, compared with that of GAPDH protein (Fig. 2A). Similarly, the amount of secreted viral particles increased during the time course (Fig. 2B). These results indicate that the virus replicates efficiently under the serum-free conditions.
FIG 2.
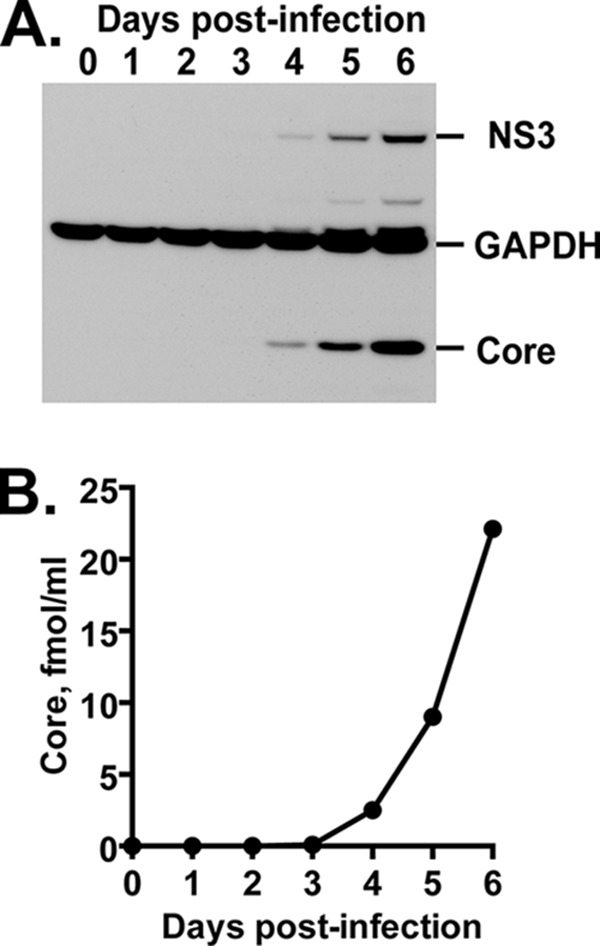
HCV production in Huh-7.5.1-8 cells grown under serum-free conditions. Huh-7.5.1-8 cells were infected with HCV JFH-1 and then cultured in serum-free medium. (A) The cells were harvested at the indicated time points. Each cell lysate (15 μg of protein) was subjected to immunoblotting for core, NS3, and GAPDH proteins. (B) Culture supernatants were harvested at the indicated time points. The amount of secreted viral particles in each supernatant was determined by measuring the amount of core protein by ELISA. The results from one of two independent experiments with similar results are shown.
We next examined the effect of YM-53601 on JFH-1 virus production in Huh-7.5.1-8 cells grown under serum-free conditions. The amounts of core (Fig. 3A) and NS3 (Fig. 3B) proteins relative to that of GAPDH protein in infected cells were decreased by the drug treatment in a dose-dependent manner and nearly reached the background level at ≥1 μM. Similarly, the relative amount of intracellular viral RNA (Fig. 3C) and the amount of secreted viral particles (Fig. 3D) were decreased by the drug treatment. The IC50 for virus secretion calculated from multiple experiments was 0.16 ± 0.10 μM (n = 4). In contrast, cell viability was not affected by the drug at up to 1.5 μM (Fig. 3E). These results indicate that YM-53601 inhibits HCV production in Huh-7.5.1-8 cells without affecting cell viability. The drug also inhibited HCV production from Huh-7.5.1-8 cells grown in serum-containing medium (data not shown) with a slightly higher value of IC50 (0.57 ± 0.66 μM; n = 4).
FIG 3.

YM-53601 inhibits HCV production without affecting cell viability. Huh-7.5.1-8 cells were infected with HCV JFH-1 and then treated with increasing concentrations of YM-53601 (0 to 1.5 μM) in serum-free medium. The cells and culture supernatants were harvested on the fifth day postinfection. (A and B) An equal portion of each cell lysate was subjected to immunoblotting for core, NS3, and GAPDH proteins. The results from one representative experiment performed in triplicate are shown. Similar results were obtained in four independent experiments. For panel A, samples were run on two blots and are partially redundant. (C) Total RNA fractions were prepared from cells, and then viral and GAPDH RNAs were quantified by RT-qPCR analysis using specific primers for the core and GAPDH sequences, respectively. The amounts of viral RNA relative to that of GAPDH mRNA are expressed as a percentage of the control value and plotted as a function of the drug concentration. (D) The amount of secreted viral particles in each culture supernatant was determined by ELISA for the core protein and plotted as in panel C. (E) Cell viability was determined by XTT assay and expressed and plotted as in panel C. The data in each graph are means ± standard deviations (SD) for triplicate samples from one representative experiment. Similar results were obtained in two or more independent experiments.
To evaluate the effect of YM-53601 on the infectivity of progeny virus, we inoculated naive Huh-7.5.1-8 cells with culture supernatants that contained viral particles secreted from drug-treated (1 μM) and untreated cells. When the inoculum dose was adjusted to contain an equal amount of core protein, these supernatants yielded almost the same amount of progeny viral particles (data not shown), indicating that the drug does not alter the infectivity of progeny virus.
YM-53601 inhibits cholesterol biosynthesis.
To test whether YM-53601 inhibits de novo synthesis of cholesterol, we pretreated Huh-7.5.1-8 cells with a 1 μM concentration of the drug in serum-free medium for 24 h and then labeled them with [3H]acetate for up to 18 h in the same medium. The drug treatment led to a 40 to 60% reduction in the incorporation of [3H]acetate into cellular cholesterol (Fig. 4A) and its major metabolites, cholesteryl esters (Fig. 4B), compared with the control treatment. In contrast, the incorporation into triglycerides was not affected by the drug (Fig. 4C), indicating a specific effect of YM-53601 on cholesterol biosynthesis. We also determined the cellular contents of cholesterol and cholesteryl esters in Huh-7.5.1-8 cells treated with 1 μM YM-53601 in serum-free medium. The contents of cholesterol (Fig. 4D) and cholesteryl esters (Fig. 4E) in the drug-treated cells were significantly decreased to 93.5% and 38.0%, respectively, of those in the untreated cells. Collectively, these results confirm that YM-53601 inhibits de novo cholesterol biosynthesis at the antiviral concentrations.
FIG 4.

YM-53601 inhibits de novo synthesis of cholesterol and cholesteryl esters. (A to C) Huh-7.5.1-8 cells were pretreated with 1 μM YM-53601 (white bars) or its vehicle, DMSO (black bars), in serum-free medium for 24 h. The cells were subsequently labeled using [3H]acetate in the same medium as for the pretreatment for the indicated periods of time. The lipid fractions were extracted from the cells and analyzed by TLC. The incorporation of [3H]acetate into cholesterol (A), cholesteryl esters (B), and triglycerides (C) was quantified and expressed as photostimulated luminescence (PSL) values per μg cellular protein. (D and E) Huh-7.5.1-8 cells were treated with 1 μM YM-53601 (white bars) or DMSO (black bars) for 7 days in serum-free medium. The lipid fraction was extracted from the cells, and the contents of cholesterol (D) and cholesteryl esters (E) were determined. The values for cholesteryl esters (E) are expressed as the cholesterol content in the fraction. Data are means ± SD for triplicate samples from one representative experiment. Similar results were obtained in two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
SQS is an anti-HCV target.
To further test whether cellular SQS is important for HCV production, we first investigated the effects of another SQS inhibitor, zaragozic acid A (squalestatin S1), on HCV production. The drug decreased the amounts of core and NS3 proteins and viral RNA (data not shown) and progeny virus production (Fig. 5A) without affecting cell viability (Fig. 5B), indicating that zaragozic acid A inhibits HCV production as well as YM-53601.
FIG 5.
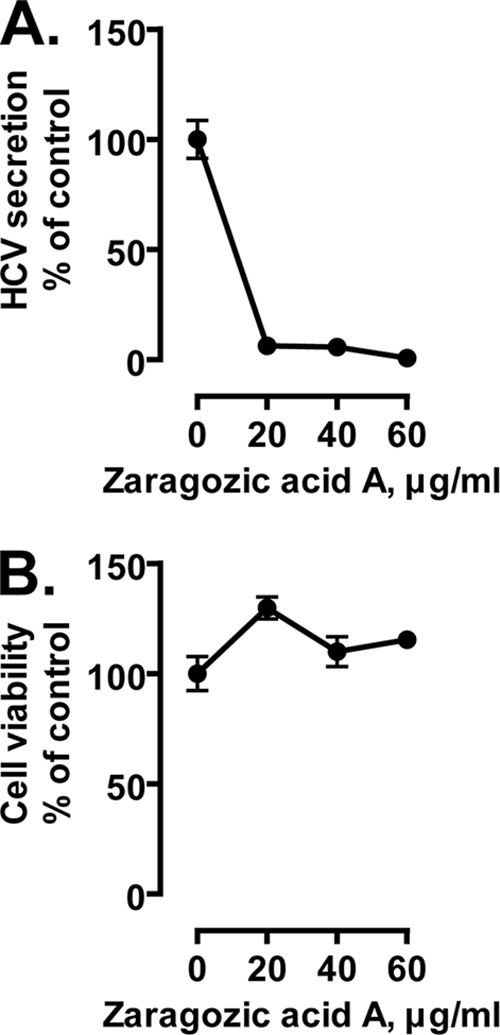
Anti-HCV effect of another SQS inhibitor, zaragozic acid A. Huh-7.5.1-8 cells were infected with HCV JFH-1 and then treated with 0 to 60 μg/ml of zaragozic acid A for 4 days. In this experiment, drug treatment was performed in complete medium because the drug was toxic to cells in serum-free medium. (A) The amount of secreted viral particles in each culture supernatant was determined by ELISA for the core protein. (B) Cell viability was determined by XTT assay. Data in each graph are expressed as a percentage of the control value (without the drug) and are means ± SD for triplicate samples from one representative experiment. Similar results were obtained in two independent experiments. Some error bars are not visible due to their small sizes.
We next investigated the effect of siRNA-mediated knockdown of SQS on HCV production. Huh-7.5.1-8 cells were infected with HCV JFH-1 and then transfected with either an siRNA against SQS (siSQS) or a control siRNA (siCONT). Transfection with siSQS resulted in almost complete depletion of cellular SQS compared with transfection with siCONT (Fig. 6A, bottom row). The amounts of core and NS3 proteins relative to GAPDH protein in the siSQS-transfected cells were markedly lower than those in the control cells (Fig. 6A, compare lanes 4 to 6 with lanes 1 to 3). The relative amounts of intracellular viral RNA (Fig. 6B, left pair of bars) and secreted viral particles (Fig. 6C, left pair of bars) were also decreased in the siSQS-transfected cells. A similar antiviral effect was observed with another siRNA for SQS (a Stealth RNAi siRNA, HSS103616) (data not shown). These results indicate that siRNA-mediated knockdown of SQS leads to reduced HCV production.
FIG 6.
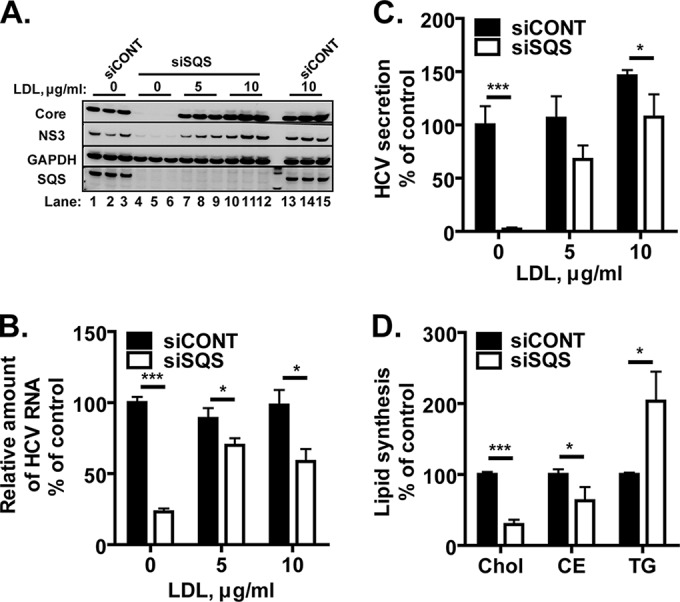
Anti-HCV effect of siRNA-mediated knockdown of SQS and its reversal by the addition of LDL. (A to C) Huh-7.5.1-8 cells were infected with HCV JFH-1 and then incubated in complete medium. On the first day postinfection, the cells were transfected with an siRNA for SQS (siSQS) or a control siRNA (siCONT), and then they were further incubated in serum-free medium that contained 0, 5, and 10 μg/ml of LDL for 4 days. (A) An equal portion of each cell lysate was subjected to immunoblotting for core, NS3, GAPDH, and SQS proteins. The results from one representative experiment performed in triplicate are shown. Similar results were obtained in two independent experiments. (B) Total RNA fractions were isolated from siSQS-transfected (white bars) or siCONT-transfected (black bars) cells, and then HCV RNA was quantified by RT-qPCR analysis using specific primers for the NS5B sequence. The amount of viral RNA relative to that of GAPDH mRNA is expressed as a percentage of the control value (siCONT without LDL) and plotted as a function of the LDL concentration. (C) The amount of secreted viral particles in each culture supernatant was determined by ELISA for the core protein. The concentration of core protein is expressed and plotted as in panel B. (D) Huh-7.5.1-8 cells were transfected with siSQS (white bars) or siCONT (black bars) and subsequently labeled using [3H]acetate in serum-free medium for 18 h. The lipid fractions were extracted from the cells and analyzed by TLC. The incorporation of [3H]acetate into cholesterol (Chol), cholesteryl esters (CE), and triglycerides (TG) was quantified and expressed as a percentage of the control value. Note that the value for cholesteryl esters of siSQS-transfected cells includes in part the incorporation of unidentified metabolites located immediately below the cholesteryl esters on a TLC plate. These metabolites accumulated only in siSQS-transfected cells for unknown reasons. The data in panels B, C, and D are means ± SD for triplicate samples from one representative experiment. Similar results were obtained in at least two independent experiments. *, P < 0.05; ***, P < 0.001.
A metabolic labeling experiment with [3H]acetate confirmed that transfection with siSQS causes a specific decrease in cholesterol biosynthesis (Fig. 6D). To rescue the defective cholesterol biosynthesis, we added LDL (final concentrations, 5 and 10 μg/ml) to the medium of HCV-infected cells after siSQS transfection. The inhibition of virus production by siSQS was reversed by the addition of LDL (Fig. 6A to C), suggesting that the antiviral effect is attributable to a decrease in cellular contents of cholesterol and/or cholesteryl esters. Consistent with this, the inhibition of virus production by YM-53601 was also reversed significantly by the addition of LDL as judged by intracellular viral protein levels (Fig. 7).
FIG 7.

The anti-HCV effect of YM-53601 is significantly reversed by LDL. Huh-7.5.1-8 cells were infected with HCV JFH-1 and then treated with 1 μM YM-53601 or DMSO in serum-free medium that contained 0 to 100 μg/ml of LDL for 5 days. An equal portion of each cell lysate was subjected to immunoblotting for core, NS3, and GAPDH proteins. The results from one representative experiment performed in triplicates are shown. Similar results were obtained in two independent experiments.
Collectively, these results demonstrate that SQS is a potential target for anti-HCV strategies.
An ACAT inhibitor does not show an anti-HCV effect.
The degree of decrease in the cholesteryl ester content after treatment with YM-53601 (Fig. 4E) was substantially higher than that in the cholesterol content (Fig. 4D), raising the possibility that biosynthesis of cholesteryl esters is more important for HCV production than that of cholesterol. To test this possibility, we examined the effect of Sandoz 58-035, an inhibitor of acyl-CoA:cholesterol acyltransferase (ACAT) that catalyzes the biosynthesis of cholesteryl esters from cholesterol and fatty acyl-CoA, on HCV production. A metabolic labeling experiment with [3H]acetate verified that treatment with 30 μM Sandoz 58-035 inhibits cholesteryl ester synthesis but not cholesterol and triglyceride syntheses in Huh-7.5.1-8 cells grown in serum-free medium (Fig. 8A). When Huh-7.5.1-8 cells were infected with HCV JFH-1 and then treated with either 30 μM Sandoz 58-035 or DMSO (control) in serum-free medium, virus secretion from the drug-treated cells was similar to that from the control cells (Fig. 8B). Taken together with the results shown in Fig. 3 and 4, these results suggest that biosynthesis of cholesterol, but not that of cholesteryl esters, is important for HCV production.
FIG 8.
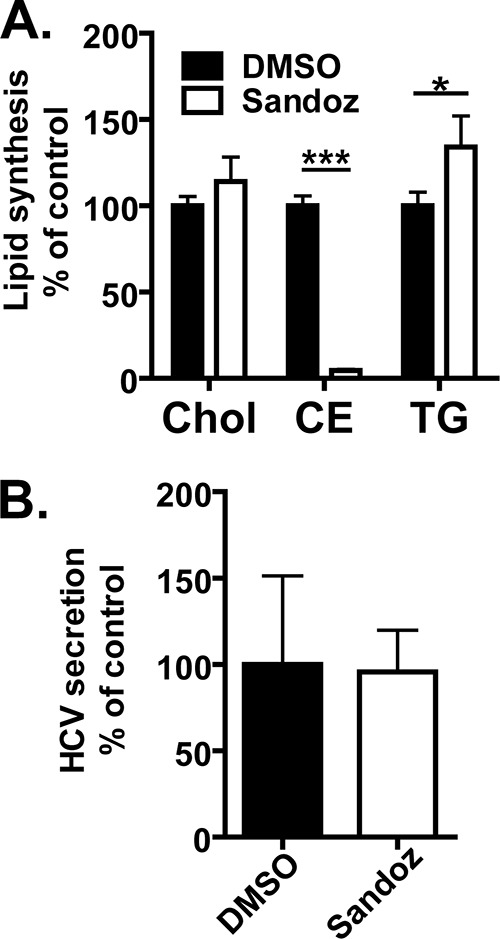
An ACAT inhibitor, Sandoz 58-035, does not exhibit an anti-HCV effect. (A) Huh-7.5.1-8 cells were pretreated with 30 μM Sandoz 58-035 or its vehicle, DMSO, in serum-free medium for 24 h. The cells were subsequently labeled using [3H]acetate in the same medium as for the pretreatment for 18 h. The lipid fractions were extracted from cells and separated by TLC. The incorporation of [3H]acetate into cholesterol (Chol), cholesteryl esters (CE), and triglycerides (TG) was quantified and expressed as a percentage of the control value. (B) Huh-7.5.1-8 cells were infected with HCV JFH-1 and then treated with Sandoz 58-035 or DMSO (control) under the same conditions as described above. The culture supernatants were harvested on the fifth day postinfection. The amount of secreted viral particles in each culture supernatant was determined by ELISA for the core protein and is expressed as a percentage of the control value. Data in each graph are means ± SD for triplicate samples from one representative experiment. Similar results were obtained in two independent experiments. *, P < 0.05; ***, P < 0.001.
YM-53601 inhibits RNA replication of HCV JFH-1.
To investigate which stages of the HCV life cycle are targeted by YM-53601, we conducted a transient-replication assay using a subgenomic replicon, SGR-JFH1/Luc (45). When cells are transfected with this replicon RNA, the self-encoded viral RNA replicase (NS3-NS5B) is expressed under the control of the EMCV IRES and then amplifies the replicon in the cells. The replicon also encodes luciferase translated under HCV IRES control, thereby allowing quantitation of viral RNA replication and translation activities via luciferase expression. Parallel transfection with a replication-incompetent mutant replicon, SGR-JFH1/Luc-GND (45), enables estimation of the level of replication-independent luciferase expression from the input replicon. As shown in Fig. 9A, luciferase activity in the wild-type replicon-transfected cells reached its peak at 47 h posttransfection and then declined. In the presence of YM-53601, the peak activity was decreased to approximately half of that in the control cells. The mutant replicon yielded very low luciferase activity irrespective of the drug treatment, confirming that the activity yielded by the wild-type replicon at 23 to 71 h posttransfection was dependent on viral RNA replication. Multiple experiments showed that the drug treatment lowered the luciferase activity at 46 to 50 h posttransfection to 52.6% ± 11.3% (mean ± standard error of the mean [SEM]; n = 5) of the control activity.
FIG 9.

YM-53601 inhibits HCV RNA replication of HCV JFH-1. (A) Transient-replication assay using JFH-1 subgenomic replicons. Huh-7.5.1-8 cells were transfected with SGR-JFH1/Luc (closed symbols) or SGR-JFH1/Luc-GND (open symbols) RNAs by electroporation and then placed in serum-free medium. At 5 h posttransfection, YM-53601 (final concentration, 1.5 μM) (squares and dashed line) or DMSO (circles and solid line) was added to the medium. The cells were harvested at the indicated time points (posttransfection) and assayed for luciferase activity. (B and C) Huh-7.5.1-8 cells were pretreated with 1.5 μM YM-53601 or DMSO in serum-free medium for 42 h and then transfected with SGR-JFH1/Luc-GND RNA by lipofection. After transfection, the cells were further treated in the same medium and harvested at the indicated time points. (B) The cells were lysed and assayed for luciferase activity. (C) An equal amount of protein (10 μg/lane) in each cell lysate was subjected to immunoblotting for NS3 and GAPDH proteins, and each protein band was quantified. The relative amount of NS3 protein was calculated by dividing its intensity by that of GAPDH protein in the same lane. Data are expressed as a percentage of the relative amount at 9 h posttransfection. The value at 9 h posttransfection was not significantly different between the drug-treated and untreated cells (data not shown). Data in each graph are means ± SD for triplicate samples from one representative experiment. Similar results were obtained in at least two independent experiments. Statistical analysis was performed between drug-treated and control cells harboring the same replicon. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To test whether YM-53601 inhibits HCV IRES-dependent translation, we transfected drug-pretreated Huh-7.5.1-8 cells with the replication-incompetent mutant replicon SGR-JFH1/Luc-GND and then monitored luciferase expression in the presence of the drug for up to 21 h. Because the replicon cannot be replicated, luciferase activity yielded by the mutant replicon is attributable exclusively to HCV IRES-dependent translation and reflects the residual amount of the input replicon RNA. As shown in Fig. 9B, luciferase activity in the drug-treated cells and untreated control cells reached its peak at 3 h posttransfection and then declined. During the time course, the activity in the drug-treated cells was not lower, but rather was higher, than the activity in the control cells. Furthermore, the level of NS3 protein that was expressed from the mutant replicon changed similarly in the drug-treated and control cells, reaching its peak at 3 to 6 h posttransfection (Fig. 9C). Thus, it appears unlikely that YM-53601 impairs HCV IRES-dependent translation or viral RNA and NS protein stability.
Taken together, these results suggest that YM-53601 inhibits the RNA replication of HCV JFH-1.
YM-53601 does not affect the cellular distribution of NS4B protein.
It is possible that YM-53601 alters the formation of the HCV-specific ultrastructure termed the membranous web, which serves as a scaffold for the viral RNA replication complex (55, 56), thereby inhibiting viral RNA replication. To test this possibility, we treated Huh-7.5.1-8 cells stably expressing HA-tagged NS4B protein with YM-53601 for 3 days in serum-free medium. It has been shown that the membranous web is induced by NS4B protein alone (55, 57) and appears as NS4B-accumulating foci or dots under fluorescence microscopy (58, 59). We found small intense foci that were detected with an anti-HA antibody in non-drug-treated cells (Fig. 10C) and are similar to the NS4B foci previously reported (58, 60, 61). The foci were not detected in Huh-7.5.1-8 cells transfected with a backbone plasmid (Fig. 10A and B). Drug treatment resulted in no apparent alteration in NS4B foci (Fig. 10D) or the expression level of NS4B protein (data not shown), suggesting that the drug does not grossly alter the formation of the membranous web by NS4B protein.
FIG 10.

YM-53601 does not affect NS4B foci in Huh-7.5.1-8 cells. Huh-7.5.1-8 cells that were stably transfected with modified pCXN2 (A and B) or pCXN2/HA-TEV-NS4B (C and D) were grown on coverslips and treated with 1.5 μM YM-53601 (B and D) or DMSO (A and C) in serum-free medium for 3 days. The cells were fixed and subjected to immunofluorescence analysis using confocal microscopy. HA-tagged NS4B protein was detected with a rat anti-HA antibody followed by an Alexa Fluor 488-conjugated anti-rat antibody (green), and the nucleus was stained with DAPI (blue). Scale bars represent 20 μm.
RNA replication of HCV genotype 1b is not inhibited by YM-53601.
To examine whether YM-53601 is able to inhibit viral RNA replication of HCV strains other than the JFH-1 strain (genotype 2a), we performed a transient-replication assay using a subgenomic replicon of the Con-1 strain (genotype 1b), FK-I389Luci/NS3-3′/NK5.1 (46), and its replication-incompetent mutant, FK-I389/Luci/NS3-3′/NK5.1/ΔGDD. Consistent with the previous report (46), time-dependent luciferase expression in the Con-1 replicon-transfected cells exhibited a downward-sloping pattern: luciferase activity at early time points (2.5 to 7 h posttransfection) was higher than the activity at later time points (Fig. 11A). At the early time points, the activity in the wild-type replicon-transfected cells was lower than the activity in the mutant replicon-transfected cells, indicating that RNA replication is too low to be detected at these early points. Afterwards, the activity in the wild-type replicon-transfected cells stayed higher than the activity in the mutant replicon-transfected cells, indicating that the difference between these activities was attributed to viral RNA replication. Unlike in the case of the JFH-1 replicon, treatment with YM-53601 did not lower RNA replication-dependent luciferase expression but rather enhanced it. From multiple experiments, the luciferase activity in the drug-treated cells at 46 to 50 h posttransfection was 284% ± 62% (mean ± SEM; n = 4) of the control activity.
FIG 11.
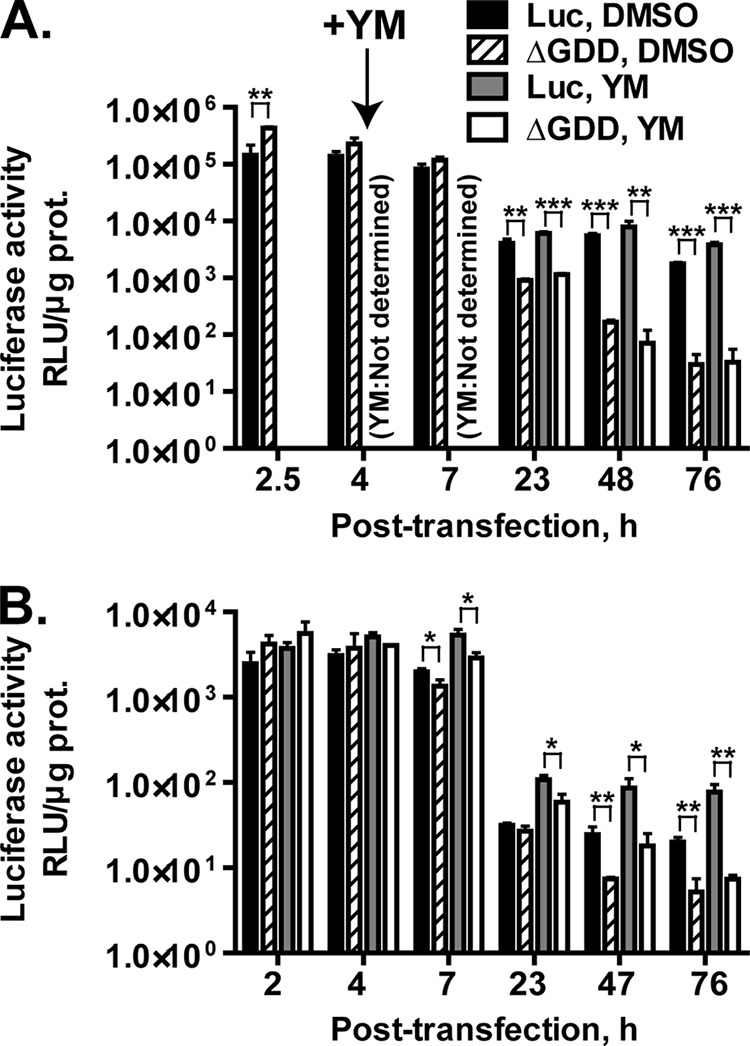
YM-53601 does not inhibit transient replication of Con-1 subgenomic replicons. (A) Huh-7.5.1-8 cells were transfected with FK-I389Luci/NS3-3′/NK5.1 (Luc) (black and gray bars) or FK-I389/Luci/NS3-3′/NK5.1/ΔGDD (ΔGDD) (hatched and white bars) RNAs by electroporation and then placed in serum-free medium. At 4 h posttransfection, YM-53601 (YM) (final concentration, 1.5 μM) (gray and white bars) or DMSO (black and hatched bars) was added to the medium. The cells were harvested at the indicated time points (posttransfection) and assayed for luciferase activity. (B) Huh-7.5.1-8 cells were pretreated with 1.5 μM YM-53601 or DMSO in serum-free medium for 47 h. The cells were transfected with FK-I389Luci/NS3-3′/NK5.1 or FK-I389/Luci/NS3-3′/NK5.1/ΔGDD RNAs and then further treated in the same medium. The cells were harvested at the indicated time points (posttransfection) and assayed for luciferase activity. Bars are as described for panel A. Data in each graph are means ± SD for triplicate samples from one representative experiment and are presented on a logarithmic scale because of large range of values. Some error bars are not visible due to their small sizes. Similar results were obtained in at least two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To increase the impact of YM-53601, we performed a similar transient-replication assay using Huh-7.5.1-8 cells pretreated with the drug in serum-free medium for 2 days. Unexpectedly, serum-free preculture before transfection led to an overall decrease of two orders of magnitude in luciferase expression (Fig. 11B). In untreated control cells, RNA replication-dependent luciferase expression (i.e., the difference between the activity yielded by the wild-type replicon and that yielded by the mutant replicon) was not clearly found until 47 h posttransfection (compare black bars with hatched bars). However, RNA replication-dependent luciferase expression in drug-treated cells was found at and after 7 h posttransfection (compare gray bars with white bars) and was slightly higher than that in the untreated cells. Thus, the RNA replication-dependent luciferase expression does not appear to be inhibited by even a prolonged drug treatment.
Taken together, these results suggest that RNA replication of the Con-1 strain is not inhibited by YM-53601.
Entry of HCVpp of genotype 2a but not genotype 1b is blocked by YM-53601.
To further investigate how YM-53601 blocks HCV production, we conducted an entry assay for HCV pseudoparticles (HCVpp), which enter cells by using HCV envelope protein but replicate via a retroviral system (52). Although YM-53601 was added to cells after infection (Fig. 3 and 7), a block at the step of entry of progeny virus is possible because more than one round of infection can occur under our experimental conditions. Huh-7.5.1-8 cells were preincubated with YM-53601 in serum-free medium for 2 days and then infected in the presence of the drug with HCVpp harboring envelope glycoproteins from the JFH-1 strain. The cells were thereafter incubated in the absence of the drug for 3 days, and luciferase activity, reflecting the degree of HCVpp entry into host cells, was measured. Treatment with YM-53601 reduced luciferase activity to less than 50% of the activity in untreated cells (Fig. 12, left two bars). Infection with mock HCVpp prepared without envelope glycoproteins did not yield luciferase activity (<3 relative light units [RLU]/μg protein), confirming that luciferase expression is dependent on the envelope glycoproteins (data not shown). When the drug was added only at HCVpp infection, no reduction in the luciferase expression was found (Fig. 12, third bar from left), suggesting that the drug targets cells but not HCVpp. These results are consistent with the previous report showing partial cholesterol dependency of HCV entry (14). Similarly, we tested the effect of the drug on HCVpp harboring envelope glycoproteins from genotype 1b HCV (strain TH). Drug treatment before and during infection or only during infection did not significantly alter luciferase expression (Fig. 12, right three bars). Taken together, these results suggest that YM-53601 blocks entry of HCV genotype 2a but not that of genotype 1b.
FIG 12.
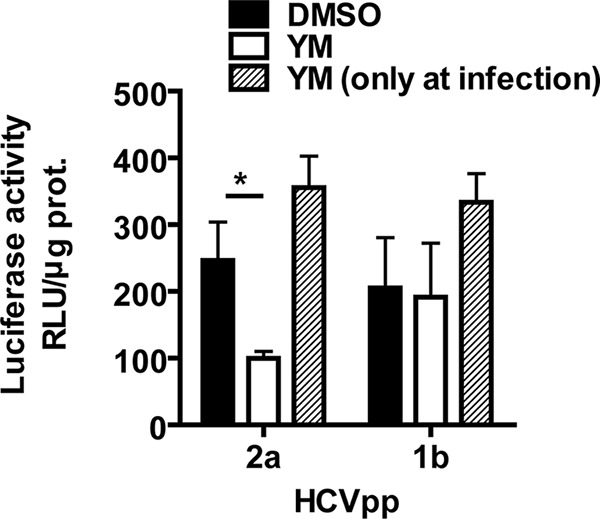
YM-53601 blocks entry of genotype 2a, but not genotype 1b, HCVpp. Huh-7.5.1-8 cells were grown in serum-free medium that contained 1.5 μM YM-53601 (white bars) or DMSO (black and hatched bars) for 2 days and then infected with HCVpp in the presence (white and hatched bars) or absence (black bars) of the drug. The cells were further grown in complete medium without the drug for 3 days and assayed for luciferase activity. Data are means ± SD for triplicate samples from one representative experiment. Similar results were obtained in two independent experiments. *, P < 0.05.
DISCUSSION
The main aim of this study was to elucidate the importance of the committed pathway of cholesterol biosynthesis in the HCV life cycle. We have shown that three types of SQS inhibitor, YM-53601 (Fig. 3), zaragozic acid A (Fig. 5), and siSQS (Fig. 6), inhibited HCV JFH-1 production in Huh-7.5.1-8 cells in a similar manner. In particular, YM-53601 exerted an antiviral effect without remarkable cell toxicity. The antiviral effect of SQS inhibition was reversed by the addition of LDL (Fig. 6 and 7), indicating that the effect is attributable to cellular cholesterol and/or cholesteryl ester deficiencies (Fig. 4 and 6). Unlike YM-53601, no antiviral effect was observed with the ACAT inhibitor Sandoz 58-035 (Fig. 8), suggesting that synthesis of cholesterol rather than that of cholesteryl esters is important for HCV production. From these findings, we conclude that the committed pathway of cholesterol biosynthesis that begins with squalene synthesis (Fig. 1) plays an important role in the HCV life cycle. This conclusion is consistent with recent studies showing that inhibition of oxidosqualene cyclase, lanosterol C14-demethylase, 24-dehydrocholesterol reductase, 7-dehydrocholesterol reductase, and SQS (discussed below) leads to decreased HCV production (31–33, 62). Furthermore, we propose that SQS is a potential target for anti-HCV strategies because all the SQS inhibitors tested in this study exerted anti-HCV effects. It has been reported that the peak plasma concentration of YM-53601 is 0.92 μg/ml (approximately 2.5 μM) after oral administration in rats at a dose with a cholesterol-lowering effect (38, 63). This concentration is roughly close to the IC50 of YM-53601 for HCV production in the presence of serum. Thus, YM-53601 might exert an anti-HCV effect in vivo.
Using a transient-replication assay (Fig. 9A) and the HCVpp system (Fig. 12), we found that suppression of HCV RNA replication and entry is involved in the antiviral mechanism of YM-53601 against JFH-1 virus. However, the degrees of suppression of these processes were at most approximately 50% in our assays. Accordingly, these mechanisms alone may not explain the more severe inhibition of HCV production observed in the HCV cell culture system (Fig. 3). Possibly, some steps in the HCV life cycle other than RNA replication and entry might be sensitive to the drug. Alternatively, some steps which are not reproduced in the subgenomic replicon and HCVpp systems might be more sensitive to the drug.
YM-53601 inhibited transient RNA replication of the subgenomic reporter replicon from the JFH-1 strain (genotype 2a) (Fig. 9A) but somewhat enhanced that of the subgenomic replicon from the Con-1 strain (genotype 1b) (Fig. 11). Similarly, the drug inhibited entry of genotype 2a, but not genotype 1b, HCVpp (Fig. 12). These findings raise the possibility that the cholesterol requirement for HCV RNA replication and entry varies among virus genotypes. Consistent with our results, previous studies have shown that SQS inhibition by zaragozic acid A leads to an enhancement of genotype 1b RNA replication (28, 31). This effect appears to be caused by an increase in geranylgeranyl pyrophosphate, which is required for geranylgeranylation of a viral host factor, and elevated expression of HMG-CoA reductase (31). In the case of genotype 2a, the effect might be overwhelmed by antiviral effect caused by cholesterol depletion. Interestingly, genotype-specific inhibition of HCV RNA replication was also observed with inhibitors of sphingomyelin biosynthesis (19, 64, 65). Thus, major components of lipid rafts, i.e., cholesterol and sphingomyelin, appear to be similar in that they both contribute to HCV RNA replication in a genotype-dependent manner.
During preparation of this paper, Park et al. reported that siRNAs against farnesyl-diphosphate farnesyltransferase 1 (another name for SQS) and YM-53601 impair propagation of the HCV Jc1 strain (genotype 2a) in Huh-7.5 cells (62). They suggested that these agents target viral RNA replication by using a luciferase-encoding full genomic replicon of the JFH-1 strain and genotype 2a subgenomic replicon cells. These findings are consistent with our results. However, their finding that the viral RNA level in genotype 1b subgenomic replicon cells is decreased by SQS knockdown appears to argue against our results, as we could not find any antiviral effect of YM-53601 on genotype 1b RNA replication (Fig. 11). Although the reason for this discrepancy is currently unknown, differences in the culture conditions (serum-containing medium versus serum-free medium), replication assay (RT-qPCR versus reporter), methods of SQS inhibition (siRNA versus drug), and origin of the subgenomic replicon might be involved. In any case, we should evaluate the effects of SQS inhibitors on the complete life cycle of HCV genotype 1b when cell culture systems capable of supporting its growth are developed.
Our data suggest that biosynthesis of cholesterol, rather than that of cholesteryl esters, is important for HCV production (Fig. 8). Treatment with YM-53601 led to only a slight reduction in cholesterol levels (Fig. 4D) but severely impaired HCV production, implying that the drug selectively decreases relatively minor but specific pools of cellular cholesterol that are important for HCV production. Given that lipid rafts may serve as sites for viral RNA replication (15–17), assembly (19, 20), and virus entry (14, 19, 66), one scenario is that YM-53601 might selectively decrease lipid raft-associated cholesterol, thereby perturbing these processes. Consistent with this proposition, inhibition of SQS in prostate cancer cells results in a decrease of raft-associated cholesterol rather than nonraft cholesterol (67). On the other hand, a recent study has shown that purified double-membrane vesicles containing active HCV RNA replication complexes are highly enriched with cholesterol (68), although they originate from the ER, which is poor in cholesterol (69). It has also been shown that cholesterol depletion from the double-membrane vesicles decreases viral RNA levels associated with them, suggesting that cholesterol is an important structural component of HCV RNA replication complexes. Cholesterol biosynthesis (70) and HCV RNA replication (71, 72) both occur in the ER, and some cholesterol biosynthetic enzymes, including SQS, are partially copurified with components of HCV RNA replication complexes (73), implying that the cholesterol biosynthetic machinery might be closely associated with HCV RNA replication complexes in the ER. Thus, another scenario is that YM-53601 might decrease newly synthesized ER cholesterol pools, which might be preferentially used for structural components of membrane-bound viral RNA replication complexes. Preferential use of newly synthesized cholesterol in the formation of envelope membranes of human immunodeficiency virus has been found (74). Note that we could not detect any impact of YM-53601 on the morphology of NS4B-induced foci, which are considered scaffolds of viral RNA replication complexes, under fluorescence microscopy (Fig. 10). Thus, alteration in the structure of RNA replication complexes caused by YM-53601, if any, might be found at the ultrastructural level.
Our data provide evidence that the committed pathway of cholesterol biosynthesis is important for HCV production, consistent with recent studies (31–33, 62). Moreover, we found that biosynthesis of cholesterol, but not of cholesteryl esters, is important for this process. The identity of the cholesterol pools required for HCV production and the molecular mechanisms underlying the cholesterol requirement should be elucidated in future studies. Our data also provide concrete evidence that SQS is a potential anti-HCV target. Further studies are required to ascertain the anti-HCV activity of SQS inhibitors in vivo. SQS inhibitors are expected to exert fewer adverse effects on human cells than statins because SQS inhibitors lower cholesterol without depleting nonsterol isoprenoids (75, 76). For this reason, many compounds targeting SQS have been developed in the past by the pharmaceutical industry as potential cholesterol-lowering drugs for hypercholesterolemia. Thus, reevaluation of these compounds for potential anti-HCV activity might offer a time-saving and cost-effective approach for developing anti-HCV drugs.
ACKNOWLEDGMENTS
The pFK-I389Luci/NS3-3′/NK5.1 and pFK-I389neo/NS3-3′/NK5.1/ΔGDD plasmids were kind gifts from Ralf Bartenschlager. We thank Kiyoshi Kawasaki (Doshisha Woman's College) for useful suggestions and Toshiyuki Yamaji for technical assistance.
This study was supported by the National Cancer Center Research and Development Fund (grant no. 7 to M.F.) from the National Cancer Center of Japan, by Health and Labor Sciences research grants for research on hepatitis from the Ministry of Health, Labor and Welfare of Japan, and by Grants-in-Aid for Scientific Research (C) JSPS KAKENHI (grant no. 21590085 to K.S. and 23590104 to M.F.) from the Japan Society for the Promotion of Science.
REFERENCES
- 1.World Health Organization. 2014. Hepatitis C fact sheet no. 164. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs164/en/. [Google Scholar]
- 2.Ghany MG, Strader DB, Thomas DL, Seeff LB. 2009. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 49:1335–1374. doi: 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexopoulou A, Papatheodoridis GV. 2012. Current progress in the treatment of chronic hepatitis C. World J Gastroenterol 18:6060–6069. doi: 10.3748/wjg.v18.i42.6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doyle JS, Aspinall E, Liew D, Thompson AJ, Hellard ME. 2013. Current and emerging antiviral treatments for hepatitis C infection. Br J Clin Pharmacol 75:931–943. doi: 10.1111/j.1365-2125.2012.04419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawitz E, Sulkowski MS, Ghalib R, Rodriguez-Torres M, Younossi ZM, Corregidor A, DeJesus E, Pearlman B, Rabinovitz M, Gitlin N, Lim JK, Pockros PJ, Scott JD, Fevery B, Lambrecht T, Ouwerkerk-Mahadevan S, Callewaert K, Symonds WT, Picchio G, Lindsay KL, Beumont M, Jacobson IM. 2014. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet 384:1756–1765. doi: 10.1016/S0140-6736(14)61036-9. [DOI] [PubMed] [Google Scholar]
- 6.Schneider MD, Sarrazin C. 2014. Antiviral therapy of hepatitis C in 2014: do we need resistance testing? Antiviral Res 105:64–71. doi: 10.1016/j.antiviral.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Fraser CS, Doudna JA. 2007. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat Rev Microbiol 5:29–38. doi: 10.1038/nrmicro1558. [DOI] [PubMed] [Google Scholar]
- 8.Grakoui A, Wychowski C, Lin C, Feinstone SM, Rice CM. 1993. Expression and identification of hepatitis C virus polyprotein cleavage products. J Virol 67:1385–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H. 1993. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J Virol 67:3835–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hijikata M, Mizushima H, Tanji Y, Komoda Y, Hirowatari Y, Akagi T, Kato N, Kimura K, Shimotohno K. 1993. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc Natl Acad Sci U S A 90:10773–10777. doi: 10.1073/pnas.90.22.10773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 12.Moradpour D, Gosert R, Egger D, Penin F, Blum HE, Bienz K. 2003. Membrane association of hepatitis C virus nonstructural proteins and identification of the membrane alteration that harbors the viral replication complex. Antiviral Res 60:103–109. doi: 10.1016/j.antiviral.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 13.Felmlee DJ, Hafirassou ML, Lefevre M, Baumert TF, Schuster C. 2013. Hepatitis C virus, cholesterol and lipoproteins—impact for the viral life cycle and pathogenesis of liver disease. Viruses 5:1292–1324. doi: 10.3390/v5051292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. 2007. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol 81:374–383. doi: 10.1128/JVI.01134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aizaki H, Lee KJ, Sung VM, Ishiko H, Lai MM. 2004. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324:450–461. doi: 10.1016/j.virol.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 16.Shi ST, Lee KJ, Aizaki H, Hwang SB, Lai MM. 2003. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J Virol 77:4160–4168. doi: 10.1128/JVI.77.7.4160-4168.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao L, Aizaki H, He JW, Lai MM. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J Virol 78:3480–3488. doi: 10.1128/JVI.78.7.3480-3488.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lingwood D, Simons K. 2010. Lipid rafts as a membrane-organizing principle. Science 327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 19.Aizaki H, Morikawa K, Fukasawa M, Hara H, Inoue Y, Tani H, Saito K, Nishijima M, Hanada K, Matsuura Y, Lai MM, Miyamura T, Wakita T, Suzuki T. 2008. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J Virol 82:5715–5724. doi: 10.1128/JVI.02530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matto M, Rice CM, Aroeti B, Glenn JS. 2004. Hepatitis C virus core protein associates with detergent-resistant membranes distinct from classical plasma membrane rafts. J Virol 78:12047–12053. doi: 10.1128/JVI.78.21.12047-12053.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 22.Bartenschlager R, Penin F, Lohmann V, Andre P. 2011. Assembly of infectious hepatitis C virus particles. Trends Microbiol 19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Hishiki T, Shimizu Y, Tobita R, Sugiyama K, Ogawa K, Funami K, Ohsaki Y, Fujimoto T, Takaku H, Wakita T, Baumert TF, Miyanari Y, Shimotohno K. 2010. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J Virol 84:12048–12057. doi: 10.1128/JVI.01063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem 286:3018–3032. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein JL, Brown MS. 1990. Regulation of the mevalonate pathway. Nature 343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 26.Schachter M. 2005. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol 19:117–125. doi: 10.1111/j.1472-8206.2004.00299.x. [DOI] [PubMed] [Google Scholar]
- 27.Ye J, Wang C, Sumpter R Jr, Brown MS, Goldstein JL, Gale M Jr. 2003. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci U S A 100:15865–15870. doi: 10.1073/pnas.2237238100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kapadia SB, Chisari FV. 2005. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci U S A 102:2561–2566. doi: 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikeda M, Abe K, Yamada M, Dansako H, Naka K, Kato N. 2006. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology 44:117–125. doi: 10.1002/hep.21232. [DOI] [PubMed] [Google Scholar]
- 30.Brautbar A, Ballantyne CM. 2011. Pharmacological strategies for lowering LDL cholesterol: statins and beyond. Nat Rev Cardiol 8:253–265. doi: 10.1038/nrcardio.2011.2. [DOI] [PubMed] [Google Scholar]
- 31.Owens CM, Mawhinney C, Grenier JM, Altmeyer R, Lee MS, Borisy AA, Lehar J, Johansen LM. 2010. Chemical combinations elucidate pathway interactions and regulation relevant to hepatitis C replication. Mol Syst Biol 6:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodgers MA, Villareal VA, Schaefer EA, Peng LF, Corey KE, Chung RT, Yang PL. 2012. Lipid metabolite profiling identifies desmosterol metabolism as a new antiviral target for hepatitis C virus. J Am Chem Soc 134:6896–6899. doi: 10.1021/ja207391q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takano T, Tsukiyama-Kohara K, Hayashi M, Hirata Y, Satoh M, Tokunaga Y, Tateno C, Hayashi Y, Hishima T, Funata N, Sudoh M, Kohara M. 2011. Augmentation of DHCR24 expression by hepatitis C virus infection facilitates viral replication in hepatocytes. J Hepatol 55:512–521. doi: 10.1016/j.jhep.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Tansey TR, Shechter I. 2000. Structure and regulation of mammalian squalene synthase. Biochim Biophys Acta 1529:49–62. doi: 10.1016/S1388-1981(00)00137-2. [DOI] [PubMed] [Google Scholar]
- 35.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shirasago Y, Sekizuka T, Saito K, Suzuki T, Wakita T, Hanada K, Kuroda M, Abe R, Fukasawa M. 25November2014. Isolation and characterization of a Huh.7.5.1-derived cell clone highly permissive to hepatitis C virus. Jpn J Infect Dis doi: 10.7883/yoken.JJID.2014.231. [DOI] [PubMed] [Google Scholar]
- 38.Ugawa T, Kakuta H, Moritani H, Matsuda K, Ishihara T, Yamaguchi M, Naganuma S, Iizumi Y, Shikama H. 2000. YM-53601, a novel squalene synthase inhibitor, reduces plasma cholesterol and triglyceride levels in several animal species. Br J Pharmacol 131:63–70. doi: 10.1038/sj.bjp.0703545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergstrom JD, Kurtz MM, Rew DJ, Amend AM, Karkas JD, Bostedor RG, Bansal VS, Dufresne C, VanMiddlesworth FL, Hensens OD, Liesch JM, Zink DL, Wilson KE, Onishi J, Milligan JA, Bills G, Kaplan L, Nallin Omstead M, Jenkins RG, Huang L, Meinz MS, Quinn L, Burg RW, Kong YL, Mochales S, Mojena M, Martin I, Pelaez F, Diez MT, Alberts AW. 1993. Zaragozic acids: a family of fungal metabolites that are picomolar competitive inhibitors of squalene synthase. Proc Natl Acad Sci U S A 90:80–84. doi: 10.1073/pnas.90.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross AC, Go KJ, Heider JG, Rothblat GH. 1984. Selective inhibition of acyl coenzyme A:cholesterol acyltransferase by compound 58-035. J Biol Chem 259:815–819. [PubMed] [Google Scholar]
- 41.Hanada K, Nishijima M, Kiso M, Hasegawa A, Fujita S, Ogawa T, Akamatsu Y. 1992. Sphingolipids are essential for the growth of Chinese hamster ovary cells. Restoration of the growth of a mutant defective in sphingoid base biosynthesis by exogenous sphingolipids. J Biol Chem 267:23527–23533. [PubMed] [Google Scholar]
- 42.Kato T, Matsumura T, Heller T, Saito S, Sapp RK, Murthy K, Wakita T, Liang TJ. 2007. Production of infectious hepatitis C virus of various genotypes in cell cultures. J Virol 81:4405–4411. doi: 10.1128/JVI.02334-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 44.Mizoguchi T, Edano T, Koshi T. 2004. A method of direct measurement for the enzymatic determination of cholesteryl esters. J Lipid Res 45:396–401. doi: 10.1194/jlr.D300024-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Kato T, Date T, Miyamoto M, Sugiyama M, Tanaka Y, Orito E, Ohno T, Sugihara K, Hasegawa I, Fujiwara K, Ito K, Ozasa A, Mizokami M, Wakita T. 2005. Detection of anti-hepatitis C virus effects of interferon and ribavirin by a sensitive replicon system. J Clin Microbiol 43:5679–5684. doi: 10.1128/JCM.43.11.5679-5684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krieger N, Lohmann V, Bartenschlager R. 2001. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J Virol 75:4614–4624. doi: 10.1128/JVI.75.10.4614-4624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 48.Inoue Y, Murakami K, Hmwe SS, Aizaki H, Suzuki T. 2007. Transcriptomic comparison of human hepatoma Huh-7 cell clones with different hepatitis C virus replication efficiencies. Jpn J Infect Dis 60:173–178 http://www0.nih.go.jp/JJID/60/173.pdf. [PubMed] [Google Scholar]
- 49.Yamaji T, Nishikawa K, Hanada K. 2010. Transmembrane BAX inhibitor motif containing (TMBIM) family proteins perturbs a trans-Golgi network enzyme, Gb3 synthase, and reduces Gb3 biosynthesis. J Biol Chem 285:35505–35518. doi: 10.1074/jbc.M110.154229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 51.Yamaji T, Mitsuki M, Teranishi T, Hashimoto Y. 2005. Characterization of inhibitory signaling motifs of the natural killer cell receptor Siglec-7: attenuated recruitment of phosphatases by the receptor is attributed to two amino acids in the motifs. Glycobiology 15:667–676. doi: 10.1093/glycob/cwi048. [DOI] [PubMed] [Google Scholar]
- 52.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murakami Y, Fukasawa M, Kaneko Y, Suzuki T, Wakita T, Fukazawa H. 2013. Selective estrogen receptor modulators inhibit hepatitis C virus infection at multiple steps of the virus life cycle. Microbes Infect 15:45–55. doi: 10.1016/j.micinf.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 54.Wakita T, Wands JR. 1994. Specific inhibition of hepatitis C virus expression by antisense oligodeoxynucleotides. In vitro model for selection of target sequence. J Biol Chem 269:14205–14210. [PubMed] [Google Scholar]
- 55.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 77:5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Konan KV, Giddings TH Jr, Ikeda M, Li K, Lemon SM, Kirkegaard K. 2003. Nonstructural protein precursor NS4A/B from hepatitis C virus alters function and ultrastructure of host secretory apparatus. J Virol 77:7843–7855. doi: 10.1128/JVI.77.14.7843-7855.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lundin M, Monne M, Widell A, Von Heijne G, Persson MA. 2003. Topology of the membrane-associated hepatitis C virus protein NS4B. J Virol 77:5428–5438. doi: 10.1128/JVI.77.9.5428-5438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gretton SN, Taylor AI, McLauchlan J. 2005. Mobility of the hepatitis C virus NS4B protein on the endoplasmic reticulum membrane and membrane-associated foci. J Gen Virol 86:1415–1421. doi: 10.1099/vir.0.80768-0. [DOI] [PubMed] [Google Scholar]
- 60.Manna D, Aligo J, Xu C, Park WS, Koc H, Heo WD, Konan KV. 2010. Endocytic Rab proteins are required for hepatitis C virus replication complex formation. Virology 398:21–37. doi: 10.1016/j.virol.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han Q, Aligo J, Manna D, Belton K, Chintapalli SV, Hong Y, Patterson RL, van Rossum DB, Konan KV. 2011. Conserved GXXXG- and S/T-like motifs in the transmembrane domains of NS4B protein are required for hepatitis C virus replication. J Virol 85:6464–6479. doi: 10.1128/JVI.02298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park EM, Nguyen LN, Lim YS, Hwang SB. 2014. Farnesyl-diphosphate farnesyltransferase 1 regulates hepatitis C virus propagation. FEBS Lett 588:1813–1820. doi: 10.1016/j.febslet.2014.03.043. [DOI] [PubMed] [Google Scholar]
- 63.Ishihara T, Kakuta H, Moritani H, Ugawa T, Sakamoto S, Tsukamoto S-I, Yanagisawa I. 2003. Syntheses of 3-ethylidenequinuclidine derivatives as squalene synthase inhibitors. Part 2: Enzyme inhibition and effects on plasma lipid levels. Bioorg Med Chem 11:3735–3745. doi: 10.1016/S0968-0896(03)00336-5. [DOI] [PubMed] [Google Scholar]
- 64.Sakamoto H, Okamoto K, Aoki M, Kato H, Katsume A, Ohta A, Tsukuda T, Shimma N, Aoki Y, Arisawa M, Kohara M, Sudoh M. 2005. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat Chem Biol 1:333–337. doi: 10.1038/nchembio742. [DOI] [PubMed] [Google Scholar]
- 65.Weng L, Hirata Y, Arai M, Kohara M, Wakita T, Watashi K, Shimotohno K, He Y, Zhong J, Toyoda T. 2010. Sphingomyelin activates hepatitis C virus RNA polymerase in a genotype-specific manner. J Virol 84:11761–11770. doi: 10.1128/JVI.00638-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Voisset C, Lavie M, Helle F, Op De Beeck A, Bilheu A, Bertrand-Michel J, Terce F, Cocquerel L, Wychowski C, Vu-Dac N, Dubuisson J. 2008. Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell Microbiol 10:606–617. doi: 10.1111/j.1462-5822.2007.01070.x. [DOI] [PubMed] [Google Scholar]
- 67.Brusselmans K, Timmermans L, Van de Sande T, Van Veldhoven PP, Guan G, Shechter I, Claessens F, Verhoeven G, Swinnen JV. 2007. Squalene synthase, a determinant of raft-associated cholesterol and modulator of cancer cell proliferation. J Biol Chem 282:18777–18785. doi: 10.1074/jbc.M611763200. [DOI] [PubMed] [Google Scholar]
- 68.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. 2013. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J Virol 87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lange Y. 1991. Disposition of intracellular cholesterol in human fibroblasts. J Lipid Res 32:329–339. [PubMed] [Google Scholar]
- 70.Ikonen E. 2008. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- 71.Wolk B, Buchele B, Moradpour D, Rice CM. 2008. A dynamic view of hepatitis C virus replication complexes. J Virol 82:10519–10531. doi: 10.1128/JVI.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Targett-Adams P, Boulant S, McLauchlan J. 2008. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J Virol 82:2182–2195. doi: 10.1128/JVI.01565-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M Jr, Ye J. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci U S A 104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng YH, Plemenitas A, Fielding CJ, Peterlin BM. 2003. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc Natl Acad Sci U S A 100:8460–8465. doi: 10.1073/pnas.1437453100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Charlton-Menys V, Durrington PN. 2008. Human cholesterol metabolism and therapeutic molecules. Exp Physiol 93:27–42. doi: 10.1113/expphysiol.2007.035147. [DOI] [PubMed] [Google Scholar]
- 76.Trapani L, Segatto M, Ascenzi P, Pallottini V. 2011. Potential role of nonstatin cholesterol lowering agents. IUBMB Life 63:964–971. doi: 10.1002/iub.522. [DOI] [PubMed] [Google Scholar]