

ABSTRACT
Replication of (+)RNA viruses depends on several co-opted host proteins but is also under the control of cell-intrinsic restriction factors (CIRFs). By using tombusviruses, small model viruses of plants, we dissect the mechanism of inhibition of viral replication by cellular WW-domain-containing proteins, which act as CIRFs. By using fusion proteins between the WW domain and the p33 replication protein, we show that the WW domain inhibits the ability of p33 to bind to the viral RNA and to other p33 and p92 replication proteins leading to inhibition of viral replication in yeast and in a cell extract. Overexpression of WW-domain protein in yeast also leads to reduction of several co-opted host factors in the viral replicase complex (VRC). These host proteins, such as eEF1A, Cdc34 E2 ubiquitin-conjugating enzyme, and ESCRT proteins (Bro1p and Vps4p), are known to be involved in VRC assembly. Simultaneous coexpression of proviral cellular factors with WW-domain protein partly neutralizes the inhibitory effect of the WW-domain protein. We propose that cellular WW-domain proteins act as CIRFs and also as regulators of tombusvirus replication by inhibiting the assembly of new membrane-bound VRCs at the late stage of infection. We suggest that tombusviruses could sense the status of the infected cells via the availability of cellular susceptibility factors versus WW-domain proteins for binding to p33 replication protein that ultimately controls the formation of new VRCs. This regulatory mechanism might explain how tombusviruses could adjust the efficiency of RNA replication to the limiting resources of the host cells during infections.
IMPORTANCE Replication of positive-stranded RNA viruses, which are major pathogens of plants, animals, and humans, is inhibited by several cell-intrinsic restriction factors (CIRFs) in infected cells. We define here the inhibitory roles of the cellular Rsp5 ubiquitin ligase and its WW domain in plant-infecting tombusvirus replication in yeast cells and in vitro using purified components. The WW domain of Rsp5 binds to the viral RNA-binding sites of p33 and p92 replication proteins and blocks the ability of these viral proteins to use the viral RNA for replication. The WW domain also interferes with the interaction (oligomerization) of p33 and p92 that is needed for the assembly of the viral replicase. Moreover, WW domain also inhibits the subversion of several cellular proteins into the viral replicase, which otherwise play proviral roles in replication. Altogether, Rsp5 is a CIRF against a tombusvirus, and it possibly has a regulatory function during viral replication in infected cells.
INTRODUCTION
Plus-stranded (+)RNA viruses, which are widespread and emerging pathogens, replicate in the cytosol of infected cells by assembling membrane-bound viral replicase complexes (VRCs). The VRCs consist of the viral RNA and viral proteins, as well as co-opted host-coded proteins (1–8). Rapid progress has recently been made in understanding the functions of the viral replication proteins, including the viral RNA-dependent RNA polymerase (RdRp) and auxiliary replication proteins, and yet the functions of many subverted host proteins in VRC assembly are less well characterized (9, 10). The growing list of subverted host proteins contributing to VRC assembly includes translation factors, protein chaperones, RNA-modifying enzymes, and cellular proteins involved in lipid biosynthesis (11–15). The cellular ESCRT proteins, reticulons, and amphiphysins could be involved in membrane deformation occurring during VRC assembly (4, 16, 17). Altogether, it seems that the VRC assembly is a rather complex process driven by many factors; thus, it is likely regulated by viral and host factors for optimal replication in infected cells.
In addition to the subverted cellular proteins helping viral replication as susceptibility factors, many host proteins have been identified, which act as cell-intrinsic restriction factors (CIRFs) (11–15, 18–22). These factors might be components of the innate immune responses and used by the host for antiviral defense (23–26) or utilized by viruses as regulatory factors to keep the replication process under control (27).
Tomato bushy stunt virus (TBSV) is a small (+)RNA virus of plants. TBSV is used to study virus-host interactions using yeast (Saccharomyces cerevisiae) as a model host (5, 28–31). The auxiliary p33 replication protein, which is an RNA chaperone, recruits the TBSV (+)RNA to the site of replication, which occurs at the cytosolic surface of peroxisomal membranes (27, 32–36). The interaction between the RdRp protein p92pol and the p33 replication protein is required for assembling the functional VRCs (30, 34, 37–39).
A dozen systematic genome-wide screens and global proteomics approaches in yeast or in vitro have led to the identification of ∼500 host proteins/genes involved in TBSV replication. The host proteins interacted with the viral replication proteins and viral RNA or affected TBSV replication and recombination when deleted/downregulated or overexpressed in host cells (11, 13, 40–48). Cataloging of the host factors affecting TBSV replication is one of the most complete among pathogens at a single cell level, thus facilitating mechanistic studies.
Several co-opted host factors are known to be involved in the assembly of the membrane-bound VRCs of tombusviruses. These proteins include the host heat shock protein 70 (Hsp70), the eukaryotic elongation factor 1A (eEF1A), Vps23p ESCRT (endosomal sorting complexes required for transport) protein, Bro1p ESCRT-associated protein, and Vps4p AAA+ ATPase (39, 46, 47, 49–59). Cdc34p E2 ubiquitin-conjugating enzyme binds to p33, and it functions as a permanent member of the viral VRC, affecting the activity of the VRC (47).
Pex19p peroxisomal transport protein binds to p33 and promotes the recruitment of p33 to the peroxisomes (32, 36, 60). Interestingly, the Pex19p-p33 interaction is not essential for TBSV replication and, in the absence of Pex19p, p33 is recruited to the ER via another unidentified host protein/pathway (32, 60).
Other subverted cellular proteins are involved in viral RNA synthesis. The list includes eEF1A, the eukaryotic elongation factor 1Bγ (eEF1Bγ), the DDX3-, DDX5-, and eIF4AIII-like DEAD box helicases, and Tdh2p (GAPDH [glyceraldehyde-3-phosphate dehydrogenase]), all of which facilitate RNA replication (51, 53, 59, 61–63).
To test the possible regulatory functions of recruited cellular proteins, we chose the WW-domain-containing cellular proteins (64). The yeast Nedd4-type Rsp5p E3 ubiquitin ligase carrying WW domain was identified in several genome-wide screens previously (13, 42, 48). In addition to Rsp5p, several cellular WW-domain proteins, including Wwm1p, Prp40p, and plant AtDrh1, AtFCA, and AtPrp40c, bind to the tombusvirus replication proteins and inhibit their functions (64, 65). Accordingly, binding of Rsp5p and other WW-domain proteins to the p92pol replication protein leads to the degradation of p92pol (64, 65).
The WW domain is a simple and highly conserved protein domain involved in protein-protein interactions (66, 67). The sequences of WW domains are highly variable (except from the conserved residues), which likely affect substrate specificity (66). The canonical WW domain contains two signature tryptophan residues and a conserved proline residue, which are part of a globular fold with three beta-sheets. WW-domain proteins, which are represented by multiple proteins in various organisms, including humans, bind to ligands usually carrying proline-rich sequences (66).
In the present study, we dissected the detailed function of the WW-domain proteins in viral replication. We show that the expression of the WW-domain protein interfered with complex formation between the p33 replication protein and several cellular proteins that act as susceptibility factors during TBSV replication. In addition, the WW domain inhibited the binding of p33 to the viral RNA and p33-p33 self-interaction in yeast. We also show that the WW domain can efficiently block tombusvirus replication in yeast or in a cell-free replication assay. We propose models on the CIRF activity and regulatory role of the WW-domain proteins in controlling TBSV replication via inhibition of VRC assembly at the late stage of replication.
MATERIALS AND METHODS
Yeast strains and plasmids.
The Saccharomyces cerevisiae strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) was obtained from Open Biosystems. Yeast strains expressing C-terminal hemagglutinin (HA)-tagged proteins were made by homologous recombination using BY4741 as a parental strain. PCRs for 3×HA tagging of SSA1, TEF1, and TDH2 were performed with the primer pairs 5075/2947, 5076/5077, and 5078/5079, respectively, using plasmid pYM-24 (Euroscarf) as the template. PCRs for 6×HA tagging of CDC34, PEX19, and VPS4 were performed with the primer pairs 5080/5081, 5180/5181, and 3258/3259, respectively, using plasmids pYM-14 (Euroscarf) as the template for CDC34 and PEX19 and pYM-16 for VPS4. The obtained PCR products were transformed into BY4741 strain. Recombinant yeasts were selected on YPD plates supplemented with hygromycin or with G418-Geneticin. The BRO1-6×HA strain has been described before (4).
To create the yeast strain GAL1-CDC34, containing an extra copy of CDC34, whose expression driven by the GAL1 promoter integrated at retrotransposon sites, CDC34 sequence was PCR amplified with the primer pair 1846/1847. The PCR product was digested with BglII and XhoI and ligated into BamHI/XhoI-digested pESC-HIS (Agilent Technologies). The cassette comprising GAL1::CDC34-CYCt was amplified with the primer pair 3874/3654. The resulting product was digested with BglII and ligated to BglII-digested pFA6-hphNT1 (Euroscarf). The ligation was used as the template for PCR with the primers 3653 and 3654. The resulting PCR product was transformed into yeast strain BY4741. Recombinant yeast was selected on yeast extract-peptone-dextrose plates supplemented with hygromycin.
Plasmids pGBK-Hisp33-CUP1/DI72-GAL1, pGAD-His92-CUP1, pGAD-Flag92-CUP1, and pGBK-His33-CUP1 have been described before (47, 64, 68). To create plasmid pGAD-CFP-p92-CUP1, the 6×His-CFP-p92 cassette was amplified by PCR with the primers 807 and 952 using plasmid pGAD-CFP-p92 as the template (34). The PCR product was digested with NcoI and XhoI and ligated to pGAD-His92-CUP1 previously digested with NcoI and XhoI. To create pGBK-CFP-p33-CUP1, the 6×His-CFP-p33 cassette was PCR amplified with the primers 807 and 992B from plasmid pGBK-His-CFP-p33 (34). The product was digested with NcoI and PstI and ligated into pGBK-His33-CUP1 previously digested with NcoI and PstI.
To make plasmid pGAD-WW-p92-CUP1, the RSP5 WW region was PCR amplified from plasmid pYES-Rsp5 (64) with the primers 3045 and 2800 and then digested with BglII and NheI, while the CNV p92 open reading frame (ORF) was PCR amplified with the primers 2261 and 952 and digested with SpeI. The two PCR products were ligated together with pYES2/NT/C digested with BamHI. The ligation product was used as the template for PCR with the primers 807 and 952. The resulting product was digested with NcoI and XhoI and ligated into NcoI/XhoI-digested pGAD-His92-CUP1. To make pGBK-WW-p33-CUP1, a similar strategy was used except that CNV p33 was PCR amplified with the primers 2261 and 992B. After ligation with pYES2/NT/C and RSP5-WW, the cassette was amplified with the primers 807 and 992B digested with NcoI and PstI and ligated into NcoI/PstI-digested pGBK-His33-CUP1.
The plasmid pESC-HisYFP-p33-GAL1/DI-72-GAL10 has been described (32). To create pESC-WW-YFP-p33-GAL1/DI-72-GAL10, the WW region of RSP5 was PCR amplified with the primers 4937 and 4608 and then digested with BglII. YFP was PCR amplified with the primers 1291 and 4938 and then digested with BamHI. The two digested PCR products were ligated and reamplified by PCR with the primers 4937 and 4938, digested with NcoI and BglII, and ligated into NcoI/BamHI-digested pESC-His-p33-GAL1/DI-72-GAL10 (60). Plasmid pGAD-pex13-CFP has been described elsewhere (34).
pMAL-p33, expressing the maltose-binding protein (MBP) fused with TBSV p33, was made as follows. The TBSV p33 ORF was PCR amplified with the primers 788 and 2744. The PCR product was digested with BamHI and PstI and ligated into BamHI/PstI-digested pMALc2x (New England Biolabs). To make pMAL-p92, TBSV p92 was PCR amplified with the primers 4688 and 826, digested with EcoRI and XhoI, and ligated into EcoRI/SalI-digested pMALc2x. To make pMAL-WW-p33, the WW region of RSP5 was PCR amplified with the primers 2805 and 4608, digested with EcoRI and BglII, and ligated into EcoRI/BamHI-digested pMAL-p33. To make pMAL-WW-p92, the WW region of RSP5 was PCR amplified with the primers 2805 and 4608 and digested with BglII. TBSV p92 was PCR amplified with the primers 4619 and 826 and digested with BglII. The two PCR products were ligated together and reamplified with PCR using the primers 2805 and 826. The product was digested with EcoRI and XhoI and ligated into EcoRI/XhoI-digested pMALc2x.
pMAL constructs expressing truncated versions of TBSV p33 fused to MBP have been described before (69). pMAL-p33c-ΔRPR was made as follows. TBSV p33 (amino acids [aa] 168 to 209) was PCR amplified with the primers 48 and 1134 and digested with XbaI. TBSV p33 (aa 221 to 296) was PCR amplified with the primers 3564 and 10 and digested with NheI. The two PCR products were ligated and reamplified with PCR using primers 48 and 10. The obtained PCR product was digested with BamHI and XbaI and ligated into pMALc2x digested with BamHI and XbaI. pMAL–p33-ΔRPR was made using a similar strategy. TBSV p33 (aa 1 to 209) was PCR amplified with the primers 788 and 1134, digested with XbaI, and ligated with NheI-digested TBSV p33 (aa 221 to 296) product. The ligation products were used for PCR with the primers 788 and 10. The generated PCR product was digested with EcoRI and XbaI and ligated into EcoRI/XbaI-digested pMALc2x. The plasmid pGEX-His-Rsp5, for expression of GST (glutathione S-transferase)-His6-Rsp5p in Escherichia coli, has been described before (64).
The plasmid pESC-URA (Agilent Technologies) was modified to replace the GAL10 promoter with the ADH1 promoter. To do so, the GAL1 promoter was amplified by PCR with the primers 1147 and 5002. The ADH1 promoter was amplified with the primers 953 and 5003, using pGAD-His92 as the template (30). The resulting PCR products were digested with Bsp1407I, ligated together, and reamplified with PCR using the primers 5002 and 5003. The PCR product was digested with BamHI and EcoRI and ligated into BamHI/EcoRI-digested pESC-URA.
To create pESC-His-WW-GAL1/ADH1, a cassette comprising the GAL1 promoter and the His6-tagged WW region of RSP5 was obtained by PCR using pYES-Rsp5 (64) with the primers 1147 and 2800. The ADH1 promoter was PCR-amplified with the primers 953 and 5003 as described above. The two PCR products were digested with Bsp1407I, ligated together, and reamplified with PCR using the primers 2800 and 5003. The generated PCR product was digested with XhoI and NotI and ligated into XhoI/NotI-digested pESC-URA. The His6-tagged YFP, SSA1, TEF1, and TDH2 were PCR amplified with the primers 5035/5036, 4960/4961, 4958/4959, and 5090/5005, respectively. The obtained PCR products were digested with NotI and SacI and ligated into NotI/SacI-digested pESC-GAL1/ADH1 or into pESC-HisWW-GAL1/ADH1.
For the YTH-based studies, we created plasmid pGBK-p33C carrying the C-terminal half (aa 168 to 296) of TBSV p33 ORF. The cDNA of TBSV p33 ORF was PCR amplified with the primers 183 and 1593, digested with EcoRI and XhoI, and ligated into EcoRI/SalI-digested pGBK-T7 (Clontech). Similarly, the EcoRI/XhoI-digested p33C PCR product was ligated into EcoRI/XhoI-digested pGAD-T7 (Clontech) to generate pGAD-p33C. To create pGAD-WW-p33C, the WW region of RSP5 was PCR amplified with the primers 2805/4608 and digested with BglII. The TBSV p33C (aa 168 to 296) was PCR amplified with the primers 633/1593 and digested with BamHI. The two PCR products were ligated together and reamplified with PCR using the primers 2805 and 1593. The resulting PCR product was digested with EcoRI and XhoI and ligated into EcoRI/XhoI-digested pGAD-T7.
To generate plasmid pYES-CypA, expressing the human His6-tagged cyclophilin A (CypA) protein from the GAL1 promoter, a PCR was performed with the primers 5031 and 5032 to amplify CypA sequences. The PCR product was digested with BamHI and XhoI and ligated into similarly digested pYES2/NT/C. The tetratricopeptide repeat (TPR) domain of the yeast CPR7 was amplified with the primers 3195 and 3196, digested with BamHI and XhoI, and ligated into equally digested pESC-GAL1/ADH1 to generate pESC-His-TPR-GAL1/ADH1.
Analysis of TBSV replication and p33 and p92 expression in yeast.
Yeast strain BY4741 was transformed with plasmids pGBK-His33-CUP1/DI72-GAL1 and pGAD-His92-CUP1 or combinations of CFP-tagged p33/p92 or WW-tagged p33/p92. Transformed yeasts were cultured in SC medium supplemented with 2% galactose for 16 h at 29°C. CuSO4 was then added to a final concentration of 50 μM, and the cultures were incubated for 24 h at 29°C as previously described (64). The total RNA was extracted, and the accumulation of DI-72 (+)replicon RNA [(+)repRNA] was analyzed by Northern blotting. The level of 18S rRNA accumulation was used for normalization (30). Alternatively, BY4741 was transformed with plasmids pESC-hisYFP-p33-GAL1/DI-72-GAL10 or pESC-WW-YFP-p33-GAL1/DI-72-GAL10 plus pGAD-FLAGp92-CUP1; the transformed yeasts were then grown as described above, and the accumulation of DI-72 (+)repRNA was analyzed by Northern blotting (30).
For the analysis of TBSV repRNA replication in yeast overexpressing WW and other host proteins, BY4741 was transformed with plasmids pGBK-His33-CUP1/DI72-GAL1, pGAD-His92-CUP1, and pESC-GAL1/ADH1 plasmids coexpressing His6-tagged WW and other host proteins. Transformed yeasts were pregrown in liquid medium supplemented with 2% glucose for 16 h at 29°C, washed in 2% galactose medium, and used to inoculate 2% galactose cultures (the starting optical density at 600 nm was 0.3). These cultures were incubated for 8 h at 29°C and then supplemented with 50 μM CuSO4, followed by incubation for an additional 24 h at 29°C.
The accumulation of p33 and p92 viral replication proteins was analyzed by Western blotting. Total proteins were extracted from the aliquots of cultures used to analyze repRNA by using NaOH and SDS-PAGE loading buffer, as described previously (30). Proteins were detected by using anti-His antibody, followed by alkaline phosphatase-conjugated anti-mouse antibody and NBT-BCIP detection (30). p33 and p92 mRNAs were detected by Northern blotting as described previously (64).
In vivo protein-protein interaction assays.
Yeast two-hybrid assays were performed as described previously (70).
Confocal microscopy.
Yeast strain BY4741 was cotransformed with plasmids pESC-HisYFP-p33-GAL1/DI-72-GAL10 and pGAD-pex13-CFP or plasmids pESC-WW-YFP-p33-GAL1/DI-72-GAL10 and pGAD-pex13-CFP (32). Transformed yeast colonies were grown in SC minimal medium supplemented with 2% galactose at 23°C for 24 h. Confocal laser microscopy was performed as previously described (32).
Copurification of selected host factors with the tombusvirus replicase from yeast.
Yeast strains expressing HA-tagged host proteins from their chromosomal locations were transformed with plasmids pGBK-Hisp33-CUP1/DI72-GAL1 or pGBK-FLAGp33-CUP1/DI72-GAL1, plus pGAD-His92-CUP1 and pYES2/NT/C or pYES-Rsp5-WW1-3 (64). Transformed yeasts were pregrown for 16 h at 29°C in SC minimal medium containing 2% glucose and 100 μM Bathocuproine disulfonate (BCS; Acros Organics) to chelate the copper ions in the media. Cultured yeasts were transferred to SC medium supplemented with 2% galactose and 100 μM BCS, followed by incubation for 8 h at 29°C. The yeasts were then transferred to SC medium supplemented with 2% galactose and 5 μM CuSO4, followed by incubation for an additional 24 h at 29°C. The cultures were centrifuged, washed with phosphate-buffered saline (PBS), and incubated in PBS plus 1% formaldehyde for 1 h on ice to cross-link proteins. Formaldehyde was quenched by addition of glycine (0.1 M final concentration), and the yeast was recovered by centrifugation. The viral replicase complex was purified as described previously (47) based on FLAG-tagged p33 replication protein using anti-FLAG M2 agarose. Purified p33 was analyzed by Western blotting with anti-FLAG antibody, followed by anti-mouse antibody conjugated to alkaline phosphatase. Copurified HA-tagged host proteins were analyzed with anti-HA antibody, followed by alkaline phosphatase-conjugated anti-rabbit antibody and detection with NBT-BCIP as described previously (4, 30).
TBSV replication in yeast cell extracts.
Cell-free extracts (CFE) from yeast strain BY4741 were prepared as described previously (56). The MBP-tagged proteins were purified from E. coli as described previously (69). The in vitro CFE assays were carried out with 0.1 μg of each purified protein, 0.5 μg of in vitro-transcribed DI-72 (+)repRNA, and 2 μl of CFE in a 20-μl final volume. The mixtures were incubated at 25°C for 3 h, and the amount of newly synthesized 32P-labeled repRNA was analyzed in denaturing polyacrylamide/urea gels as described previously (56).
In another set of experiments, affinity-purified GST or GST-tagged WW-domain, Rsp5p, and Prp40p proteins (5 pmol, each) were added directly to CFE assay, or these proteins were preincubated with MBP-p33 for 10 min at room temperature; then, all other components (not including MBP-p33) were added. The mixtures were incubated at 25°C for 3 h, and the amount of newly synthesized 32P-labeled repRNA was analyzed in denaturing polyacrylamide/urea gels as described previously (56). Each experiment was repeated three times.
Analysis of membrane association of p33 replication protein.
Membrane fractionation was performed according to the same procedure for membrane-enriched fractions described elsewhere (64). Briefly, BY4741 yeast transformed with plasmids pESC-hisYFP-p33-GAL1/DI-72-GAL10 or pESC-WW-YFP-p33-GAL1/DI-72-GAL10 plus pGAD-Hisp92 was grown in 2% glucose minimum medium for 16 h at 29°C, transferred to 2% galactose medium, and then grown for 24 h at 29°C. Yeasts were collected by centrifugation, resuspended in buffer E, and broken with glass beads. Unbroken cells were removed by low-speed centrifugation (100 × g for 5 min). Membrane fractions were pelleted by centrifugation at 15,000 × g for 20 min. Both the membrane fractions and the supernatant were used to analyze His6-YFP-p33 and WW-YFP-p33 accumulation by SDS-PAGE and Western blotting with anti-His antibody.
In vitro protein-protein interaction assays.
Pulldown assays were performed as previously described (64). Briefly, the MBP-tagged TBSV p33, expressed in E. coli, was bound to amylose columns. Lysates of E. coli expressing recombinant GST-His6-Rsp5p were then passed through the columns. After washing, MBP-tagged proteins were eluted with maltose. The amount of GST-His6-Rsp5p bound to MBP-tagged p33 was analyzed by SDS-PAGE and Western blotting with anti-His antibody as described previously (64).
In vitro RNA-protein binding assays.
Electrophoresis mobility shift assays (EMSA) were performed as described previously (69). Briefly, reaction mixtures contained 5 ng of 32P-labeled DI-72 (+)RNA plus 50, 200, or 500 ng of purified MBP-tagged proteins. After incubation for 15 min at 25°C, samples were placed on ice and run on nondenaturing 5% polyacrylamide gels.
In another set of EMSA, we used GST (10 pmol) or GST-tagged Prp40p, Rsp5p, and WW-domain proteins (two values for each, 5 and 10 pmol), which were preincubated for 10 min at room temperature with MBPp33C (5 pmol), and then all other components were added [including ∼0.1 pmol of 32P-labeled (−)DI-72]. After incubation for 15 min at 25°C, the samples were placed on ice and run on nondenaturing 5% polyacrylamide gels. Each experiment was repeated twice.
In vitro replicase assay.
Yeast strains R1158 (wild type [wt] for yTHC library series) (41) and Δ3WW (wwm1Δ/Tet::RSP5/Tet::PRP40) (65) were cotransformed with plasmids pGBK-CUP1-Flag-p33/GAL1-DI72 and pGAD-Cup-Flag-p92, and colonies were selected using SC-LH− plates. After growing in 50 ml of SC-LH− medium supplemented with 2% glucose and 1 mg/ml doxycycline for 24 h at 29°C, yeasts were pelleted, washed with SC-LH− medium supplemented with 2% galactose, and resuspended in 50 ml of SC-LH− medium supplemented with 2% galactose, 1 mg/ml doxycycline, and 50 μM CuSO4. Yeast cells were grown for 16 or 64 h at 29°C and then pelleted. About 200 mg of pellet was used to isolate tombusvirus replicase (based on Flag-p33 and Flag-p92) with anti-Flag M2-agarose as described previously (49). Replicase preparations isolated from different yeast strains were balanced and their activities were measured in vitro using (−)R1/3 template in the standard RdRp assay (30). The presence of host factors was detected by using the following primary antibodies: anti-eEF1A, anti-eEF1Bγ, anti-CDC34 and by using alkaline phosphatase-conjugated anti-rabbit antibody (Sigma) and NBT-BCIP detection (30).
Kinetic measurements with surface plasmon resonance.
Kinetic measurements were done using a BLITZ instrument (ForteBio). Briefly, the GST-tagged yeast proteins (0.2 μM) were loaded onto the GST-chip-based biosensor for 5 min with shaking (1,000 rpm). Binding to chip was measured in MBP-elution buffer (30 mM HEPES-KOH [pH 7.4], 25 mM NaCl, 1 mM EDTA) at room temperature as follows. MBP-tagged proteins were diluted with MBP elution buffer to 0.1 to 12 μM, and 4 μl of protein was interacted with GST-tagged proteins bound to the biosensor. Kinetic data were obtained as recommended: 30 s for baseline (buffer), 2 min for association (MBP-tagged protein), 2 min for dissociation (buffer). Association rate constant (ka), dissociation rate constant (kd), and interaction affinity constant (KD) values were calculated using the BLITZ software. The negative control was purified MBP binding to the immobilized GST-WW protein or GST-Cdc34 on the GST-chip-based biosensor.
RESULTS
Inhibition of the RNA-binding and protein-interaction functions of tombusvirus replication proteins by the WW-domain protein.
Previous studies revealed that the yeast Rsp5p E3 ubiquitin ligase acts as a CIRF through binding via its WW domain to tombusvirus p33 and p92pol replication proteins and inhibits TBSV replication in yeast and in vitro, while several WW proteins inhibit TBSV infection in plants (64, 65). To unravel the mechanism of WW-domain-protein-driven inhibition on TBSV replication, we first defined the binding site for the WW domain of Rsp5p in the p33 replication protein using a set of truncated proteins in a pulldown assay (Fig. 1A). These experiments defined that the arginine-proline-rich (RPR) motif in p33 involved in viral RNA binding is the preferred binding site for Rsp5p WW-domain protein (Fig. 1B and C).
FIG 1.

Defining the sequence within the TBSV p33 protein needed for binding to the Rsp5p WW-domain protein in vitro. (A) Schematic representation of the TBSV p33 and its truncated derivatives used in the affinity-binding assay. The various domains include the following: TMD, transmembrane domain; RPR, arginine-proline-rich RNA-binding domain; P; phosphorylated serine and threonine; S1 and S2 subdomains involved in p33–p33/p92 interaction. (B) Affinity binding (pulldown) assay to detect interaction between GST-His6-tagged Rsp5p and the MBP-tagged viral p33 protein derivatives. The MBP-tagged viral proteins produced in E. coli were immobilized on amylose-affinity columns. Then, GST-His6-Rsp5p expressed in E. coli was passed through the amylose-affinity columns with immobilized MBP-tagged proteins. The affinity-bound proteins were eluted with maltose from the columns. (Top) The eluted proteins were analyzed by Western blotting with anti-His antibody to detect the amount of GST-His6-Rsp5p specifically bound to MBP-tagged viral proteins. (Bottom) SDS-PAGE analysis of the purified MBP-p33 and its derivatives. (C) Pulldown assay to detect interaction between GST-His6-tagged Rsp5p and the MBP-tagged p33CΔRPR protein. See further details in panel B.
To test whether Rsp5p WW-domain protein affects the ability of p33 to bind to the viral RNA, we first expressed and purified the WW domain of Rsp5p, the full-length Rsp5p, and Prp40, another yeast protein with WW domain, which has moderate CIRF activity against tombusviruses (65). Using affinity-purified proteins and 32P-labeled viral RNA, we performed an electrophoretic mobility shift assay (EMSA). These experiments revealed that both WW-domain and full-length Rsp5p completely inhibited the RNA-binding function of p33 in vitro (Fig. 2A, lanes 7 to 10 versus lanes 1 and 2). Prp40 was less efficient inhibitor of p33 binding to RNA.
FIG 2.

WW-domain inhibits viral RNA binding by p33 in vitro. (A) For the the top image, EMSA was performed with 5 pmol of purified MBP-tagged p33C (wt, carrying 151 to 296 aa). The 32P-labeled RNA probe was wt DI-72 (−)RNA. Samples contained 10 pmol of GST (lanes 2 and 4) and 10 or 5 pmol of GST-Prp40 (lanes 5 and 6), GST-Rsp5 (lanes 7 and 8), or GST-WW domain (lanes 9 and 10), respectively. Arrows depict the bound and unbound RNA probes. For the bottom image, SDS-PAGE, followed by Coomassie blue staining of the purified recombinant proteins used in panel A, was performed. (B) Schematic representation of the fusion protein used for expression in E. coli. The WW domain of Rsp5p containing three WW repeats and the C-terminal region of p33 (termed p33C) containing the known RNA-binding site is shown. (C) For the top image, EMSA was performed with 1.2, 5, or 12 pmol of purified MBP, MBP-tagged p33C, or MBP-WW-p33C. The 32P-labeled RNA probe was wt DI-72 (+)RNA. Arrows depict the bound and unbound RNA probes. For the bottom image, SDS-PAGE, followed by Coomassie blue staining of the purified recombinant proteins used in panel C, was performed. (D) Interaction between two p33 replication proteins is inhibited by the WW domain in yeast. A yeast two-hybrid assay was performed to test binding between p33C and the shown prey proteins. The empty prey vector was used as a negative control. (E) Reduced copurification of His6-p33 with the viral replicase from yeast coexpressing His6-WW-domain protein. The viral replicase was purified through FLAG-tagged p33 from yeast extracts by using a FLAG-affinity column. Western blot analysis of copurified His6-p33 using anti-His antibody. Each experiment was repeated three times.
In case of the second approach, we made a fusion protein containing the WW domain of Rsp5p, and the C-terminal portion of p33, termed p33C, separated by a linker sequence (Fig. 2B). The fusion protein strategy is used to guarantee that each p33 sequence can efficiently interact with the WW domain. Another advantage of the fusion strategy is that the WW domain is known to fold efficiently in the absence of cofactors (67, 71, 72). Using affinity-purified proteins and 32P-labeled viral RNA in EMSA, we demonstrated that the WW domain completely inhibited the RNA-binding function of p33 in vitro (Fig. 2C, lanes 10 to 12 versus lanes 6 to 8).
Using a similar strategy with the fusion protein, we also showed that the WW domain inhibited the self-interaction between p33 molecules in a yeast two-hybrid assay (Fig. 2D). Similar to the observation with the above fusion strategy, copurification of His6-tagged p33 with the FLAG-p33 from membranes was inhibited by the separate coexpression of the WW-domain protein in yeast (Fig. 2E, lane 3 versus lane 2). Altogether, these data suggest that the interaction of the WW domain with the C-terminal region of p33 inhibits both the RNA-binding and p33-p33 interaction functions of the viral replication protein. Because p92pol shares the same sequence with p33 in its N-terminal region, it is likely that the previously shown interaction between Rsp5p and p92pol (64, 65) also leads to the inhibition of the ability of p92pol to interact with p33 and the viral RNA.
The WW domain inactivates the replication function of both p33 and p92 in the CFE-based replication assay.
To test the inhibitory function of the WW domain (derived from Rsp5p and contains three WW motifs) under defined conditions in vitro, we used the purified recombinant WW-domain protein in a CFE-based TBSV replication assay (Fig. 3A). The TBSV repRNA can go through a single full cycle of replication (producing double-stranded RNA intermediate on added plus-stranded template and excess amount of new plus-stranded RNAs) in yeast CFE when purified recombinant p33 and p92pol replication proteins are provided (Fig. 3B, lane 1) (37, 56, 73). When WW-domain protein or Rsp5p were added to the CFE assay, then TBSV replication was ∼10% of the control assay containing purified GST protein (Fig. 3B, lane 2 versus lane 1). Preincubation of the WW-domain protein and p33 replication protein did not increase further the inhibitory effect of the WW-domain protein (Fig. 3B, lane 6).
FIG 3.

WW domain does not show dominant-negative effect on TBSV replication in a yeast cell extract. (A) Scheme of the CFE-based TBSV replication assay with purified recombinant proteins and added TBSV DI-72 (+)repRNA. All of the recombinant proteins were added simultaneously to the CFE. (B) Denaturing PAGE analysis shows the level of repRNA accumulation in the CFE-based replication assay. The yeast cell extract (CFE) containing host factors and cellular membranes required for TBSV replication were programmed with in vitro-synthesized DI-72 (+)repRNA and purified MBP-p33, MBP-p92, and the shown GST-tagged yeast proteins, all of which were expressed in E. coli. The reactions included [32P]UTP to detect newly synthesized DI-72 repRNA. Note that the CFE is capable of supporting full cycle of TBSV replication, leading to asymmetrical (+)RNA and (−)RNA synthesis on the added (+)repRNA. For the samples in lanes 5 to 8, the GST-tagged yeast proteins were preincubated with MBP-p33 for 10 min prior to the CFE-based replication assay. (C) For the top panel, denaturing PAGE analysis shows the level of repRNA accumulation in the CFE-based replication assay. The CFE assays were performed as in panel B, except that purified MBP-p33 and MBP-p92 or the shown fusion proteins, all of which were expressed in E. coli, were used. For the bottom panel, SDS-PAGE, followed by Coomassie blue staining of the purified recombinant proteins used in panel C, was performed. (D) The lack of dominant-negative effect of WW domain in the fusion protein on the activities of p33 and p92 replication proteins in a CFE-based replication assay. See further details in panels B and C.
However, the above-described experiments did not define whether the WW-domain protein or Rsp5p inhibited the function of p33, p92pol, or both. Therefore, we used the fusion protein approach in the CFE-based replication assay (Fig. 3C). When WW-p33 was provided with p92pol in the CFE assay, then TBSV replication was undetectable (Fig. 3C, lanes 3 and 4). The WW-p92 in combination with p33 supported a low level of TBSV RNA replication (Fig. 3C, lanes 5 and 6), suggesting that the replication function of p92pol is also inhibited by the fusion with the WW domain. Altogether, these data confirmed that the WW domain efficiently inhibits the replication functions of both p33 and p92pol in vitro.
To test whether the WW domain makes p33 molecules dominant negative (i.e., also inhibiting wt p33 or p92pol molecules that are part of the VRCs), we added the functional p33 and p92pol, together with WW-p33, to the CFE assay. Interestingly, we observed no inhibitory effect by the WW-p33 in the CFE assay, suggesting that WW-p33 had no dominant-negative effect on TBSV replication (Fig. 3D, lanes 3 and 4 versus lanes 1 and 2). This is in contrast with the dominant-negative effect of the p33 mutant lacking the RPR region responsible for RNA binding (mutant p33ΔRPR, lanes 5 and 6, Fig. 3D) (74). The lack of dominant-negative effect of WW domain in WW-p33 on the viral replicase activity could be important during regulation of tombusvirus replication in infected cells (see Discussion).
Inhibition of tombusvirus replication by the WW-domain protein in yeast.
Using the fusion protein approach, we tested the effect of the WW domain on TBSV replication to define if the WW-domain protein inhibited the function of p33, p92pol or both in yeast cells (Fig. 4A). Expression of WW-p33 with His6-p92 or His6-p33 with WW-p92 completely blocked TBSV repRNA accumulation (Fig. 4B, lanes 7 to 12 versus lanes 1 to 6). Western blot analysis showed that WW-p33 accumulated in yeast (Fig. 4C, lanes 5 and 6), suggesting that the lack of TBSV replication in the presence of WW-p33 was likely due to the inactivation of p33 functions by the WW domain. On the other hand, WW-p92 did not accumulate at a detectable level in yeast cells (Fig. 4C, lanes 7 and 8), suggesting that p92pol was degraded in the presence of the WW domain, as shown previously with separately expressed proteins (64, 65). Indeed, we were able to detect mRNA expression for WW-p92 (Fig. 4B, bottom panel). In contrast, either His6-p33 and His6-p92 or His6-CFP-p33 and His6-CFP-p92 supported TBSV RNA accumulation efficiently (Fig. 4B, lanes 1 to 6), suggesting that the N-terminal tags attached to p33/p92pol, which do not interact with the p33 and p92pol sequences, do not inhibit TBSV replication.
FIG 4.

WW domain of Rsp5p blocks TBSV RNA replication in yeast. (A) Schematic representation of the fusion proteins used for expression in yeast. The WW domain of Rsp5p containing three WW repeats and the p33 sequence were fused as shown. The functional CFP-p33 fusion protein was chosen as control. (B) For the top panel, Northern blot analysis to detect DI-72(+) repRNA accumulation in yeast coexpressing the shown combination of p33 and p92 fusion proteins was performed. The accumulation level of DI-72(+) repRNA was normalized based on 18S rRNA. For the bottom panel, Northern blot analysis of p33 and p92 mRNA levels in yeast was performed. (C) Western blot analysis of total protein extracts with anti-His antibody. (D) For the top panel, Northern blot analysis to detect DI-72(+) repRNA accumulation in yeast coexpressing the shown combination of p33 and p92 fusion proteins was performed. For the middle and bottom panels, Western blot analysis of total protein extracts with anti-His or anti-Flag antibodies was performed. See further details in panels B and C. (E) Membrane association of the various fusion proteins in yeast. Broken yeast cells were fractionated to obtain supernatant (S, soluble fraction) and membrane fraction (P, pellet). Note that the yeast membrane fraction was washed with 1 M NaCl to remove peripheral membrane proteins. Lanes 9 and 10 represent the total, not fractionated, proteins as standards. (F) The WW-domain–p33 fusion protein shows mostly peroxisomal localization in yeast. Confocal laser microscopy images show the subcellular localization of WW-YFP-p33 fusion protein expressed from GAL1 promoter in the BY4741 yeast strain. The peroxisomes were visualized with Pex13p-CFP marker. The merged images show the colocalization of WW-YFP-p33 and Pex13p-CFP marker. Differential interference contrast (DIC) images are shown on the right. Each row represents a separate yeast cell. (G) Peroxisomal localization of YFP-p33 fusion protein. Yeast was grown under similar conditions and images were taken as in panel F. Each experiment was repeated two to three times.
We also used a different fusion protein, in which the YFP sequence separated the WW domain from the p33 sequence (construct WW-YFP-p33, Fig. 4A). This protein, when coexpressed with p92pol, could not support TBSV repRNA accumulation in yeast (Fig. 4D, lanes 3 to 4), confirming that the WW domain interferes with the functions of p33 replication protein. Similar to wt p33 (34, 75) and the YFP-p33, the WW-YFP-p33 is associated with membranes (was inserted into the lipid bilayer) in yeast based on fractionation and washing with 1 M NaCl, which could remove peripheral membrane proteins (Fig. 4E). Also, confocal microscopic analysis showed the mostly peroxisomal localization of WW-YFP-p33 (Fig. 4F), similar to YFP-p33 (Fig. 4G) (32, 34, 60). Thus, the YFP sequence is correctly folded within the WW-YFP-p33 fusion protein and the peroxisomal targeting sequence and membrane association of p33 have also remained functional. These data are in agreement with the prediction that the WW domain precisely inactivates p33 functions, such as the RNA-binding function, p33-p33 interaction, and p33-host protein interactions, when present in the fusion protein.
Expression of the WW-domain protein decreases the amount of copurified cellular proteins in the tombusvirus replicase.
To test the possible regulatory role of the WW-domain proteins in TBSV replication, we expressed the WW domain of Rsp5p in yeast. We chose to express only the WW domain and not the full-length protein, since the WW domain is the functionally relevant portion of Rsp5p during TBSV replication (65), and the presence of the E3 ubiquitin ligase domain of Rsp5p might affect the functions of numerous client cellular proteins when overexpressed. The same yeast cells also coexpressed p33 and p92pol replication proteins and the DI-72 replicon (rep)RNA and selected HA-tagged cellular proteins, those which are known to function as susceptibility factors for TBSV, from chromosomal locations. After affinity purification of the membrane-bound tombusvirus VRCs (via purification of p33 and p92pol), we analyzed the amount of copurified host proteins.
Interestingly, the amounts of six subverted VRC-associated host proteins decreased by 45 to 90% in the VRC preparations purified from yeast coexpressing the WW-domain protein (Fig. 5B to G), while copurification of one host factor, the Ssa1p Hsp70, was not affected (Fig. 5A). The largest decrease in copurification was observed with the cellular ESCRT proteins (i.e., Vps4p and Bro1p) and Cdc34p E2 ubiquitin-conjugating enzyme, all of which are known to affect the assembly of the tombusvirus VRCs (4, 47, 51). The extent of inhibition of the additional copurified host proteins was also significant, but somewhat less pronounced for (i) Tef1p (eEF1A) translation elongation factor known to affect many viral functions, including the stability of p33, the recruitment of the viral RNA, and the assembly of the VRCs and (−)RNA synthesis (46, 51, 53); (ii) Tdh2p (GAPDH) involved in (+)RNA synthesis (62, 63); and (iii) Pex19p cytosolic shuttle protein that targets p33 and p92pol to the peroxisomes (60). Altogether, the inhibition of recruitment of multiple host factors to the VRCs by WW-domain protein suggests a regulatory function, possibly via competition of the WW-domain protein with these cellular proteins for binding to p33 and p92pol. It is possible that some of the proviral host proteins compete, while the rest of them just being disturbed by the active binding of the ectopically expressed WW domain to p33.
FIG 5.

Reduced copurification of selected co-opted host proteins with the viral replicase from yeast coexpressing WW-domain protein. The viral replicase was purified through FLAG-tagged p33 from yeast extracts using a FLAG-affinity column. (A to G) For the top panel, Western blot analysis of copurified HA-tagged host protein expressed from the chromosomal location with anti-HA antibody was performed. For the middle panel, Western blot analysis of the purified Flag-with with anti-FLAG antibody was performed. For the bottom panel, Western blot analysis of total protein extract with anti-HA antibody to detect the total amount of the HA-tagged host protein expressed from the chromosomal location in each sample was performed. Lane 1, negative control based on yeast expressing His6-tagged p33 and the HA-tagged host protein; lane 2, yeast coexpressing Flag-p33 and the HA-tagged host protein; lane 3, yeast coexpressing Flag-p33 and the HA-tagged host protein in combination with WW-domain protein. All yeast strains actively replicate the TBSV repRNA (not shown). Each experiment was repeated two to four times.
Overexpression of selected cellular proteins decreases the inhibitory effect of the WW-domain protein on TBSV replication.
If competition for binding to p33 replication proteins exists among the WW-domain proteins and the subverted stimulatory host factors, then overexpression of selected stimulatory host proteins is expected to neutralize the inhibitory effect of the WW-domain proteins on TBSV replication in yeast cells. To test this model, we individually overexpressed four stimulatory host proteins in yeast, also coexpressing the WW-domain protein.
The increase of TBSV replication was ∼4-fold in yeast overexpressing both Cdc34p and the WW-domain proteins in comparison with the overexpression of the WW-domain protein only (Fig. 6A, lanes 7 and 8 versus lanes 3 and 4). Thus, overexpression of Cdc34p completely neutralized the strong inhibitory effect of the overexpressed WW-domain protein. This neutralization effect by Cdc34p likely due to efficient recruitment of the overexpressed Cdc34p by the p33 replication protein, because Cdc34p does not interact with Rsp5p (76). Overexpression of Ssa1p or Tef1p slightly increased TBSV replication in wt yeast (Fig. 6B, lanes 5 and 6 and 9 and 10), while both proteins partly neutralized the inhibitory effect of the WW-domain protein (leading to a 2- to 2.5-fold increase) in TBSV replication in yeast cells overexpressing the WW-domain protein (Fig. 6B, lanes 7 and 8 and 11 and 12 versus lanes 3 and 4). The incomplete neutralization of the inhibitory effect by WW-domain protein could be due to the physical interaction between Ssa1p or Tef1p and Rsp5p (76) that might be responsible for the reduced overexpression level of both Ssa1p and Tef1p in yeast coexpressing the WW domain of the Rsp5p protein (Fig. 6B, lanes 4 and 6 versus lanes 3 and 5). Overexpression of the fourth host factor, Tdh2p, had lesser neutralization effect on the inhibitory function of the overexpressed WW-domain protein (Fig. 6B, lanes 15 and 16). However, overexpression of Tdh2p did not increase TBSV repRNA replication in yeast (lanes 13 and 14, Fig. 6B), suggesting that Tdh2p is not a limiting factor in wt yeast under the given experimental conditions.
FIG 6.

Partial recovery of TBSV RNA replication in yeast overexpressing selected subverted proviral host proteins. (A) For the top panel, Northern blot analysis to detect DI-72(+) repRNA accumulation in yeast coexpressing Cdc34p and the WW-domain protein was performed. The accumulation level of DI-72(+) repRNA was normalized based on 18S rRNA. To launch TBSV repRNA replication, we expressed Flag-p33 and His6-p92 from the copper-inducible CUP1 promoter and DI-72(+) repRNA from the galactose-inducible GAL1 promoter. While Cdc34p was expressed from GAL1 promoter from the chromosome, the WW-domain protein (the WW domain of Rsp5p containing three WW repeats) was expressed from a plasmid based on the GAL1 promoter. For the bottom panel, Northern blot analysis of CDC34 mRNA level before launching TBSV repRNA replication was performed. (B) For the top panel, Northern blot analysis was used to detect DI-72(+) repRNA accumulation in yeast coexpressing the shown host factor (or YFP as a control) and the WW-domain protein. The His6-WW-domain protein was expressed from GAL1 promoter from a plasmid, while the shown host susceptibility factor (also His6 tagged) was expressed from the constitutive ADH1 promoter from the same plasmid as the WW-domain protein. See further details in panel A. For the middle and bottom panels, Western blot analysis of total protein extracts with anti-His or anti-FLAG antibodies was performed.
Overexpression of the above host factors in yeast also overexpressing the WW-domain protein was not effective enough to increase p33 or p92pol levels to that observed in wt yeast cells coexpressing YFP (Fig. 6B). These observations suggest that the overexpressed WW-domain protein is a strong competitor against the stimulatory host factors in binding to the viral replication protein. This could be a reason why these stimulatory host proteins only had partial neutralizing effects against the inhibitory effect of the overexpressed WW-domain protein. Altogether, these data suggest that selected stimulatory host factors have neutralizing effects on the inhibitory WW-domain protein during TBSV replication.
Expression of the TPR-domain protein or cyclophilin A did not inhibit the amount of copurified cellular proteins in the tombusvirus replicase.
In addition to the WW-domain-containing host factors, other cellular factors with CIRF functions might also be involved in regulation of TBSV replication (22). To further test the possible regulatory functions of recruited cellular proteins, we chose two additional cellular CIRF factors that inhibit TBSV replication. These were the TPR-domain-containing cellular proteins (77) and cyclophilins (45). The TPR domain from Cyp40-like Cpr7p chaperone and the CypA (homolog of the yeast Cpr1p) cyclophilin have been shown to bind to the tombusvirus replication proteins (45, 65, 78).
Similar to the strategy described above with the WW-domain protein (Fig. 5), we overexpressed either the TPR domain of Cpr7p or CypA cyclophilin in yeast coexpressing p33 and p92pol replication proteins, the TBSV repRNA, and selected HA-tagged stimulatory cellular proteins from chromosomal locations (Fig. 7). After affinity purification of the membrane-bound tombusvirus VRCs, we analyzed the amount of copurified stimulatory host proteins. We found that the copurification of Cdc34p E2 ubiquitin-conjugating enzyme, Ssa1p HSP70 chaperone, and Bro1p ESCRT protein was not affected by the overexpression of the TPR-domain protein or CypA cyclophilin (Fig. 7A to C, lanes 3 and 4 versus lane 2). Moreover, copurification of Pex19 shuttle protein with the tombusviral VRC was increased from yeast expressing the TPR domain and especially CypA protein (Fig. 7D, lanes 3 and 4). Since Cdc34p and Bro1p cellular proteins were among those most affected by overexpression of the WW-domain protein (Fig. 5) and yet their recruitment into the VRCs was not affected by the overexpression of the TPR-domain protein or CypA (Fig. 7), we suggest that the CypA and TPR-domain proteins and the WW-domain proteins have different regulatory roles during TBSV replication in yeast. This is surprising because, similar to the WW domain, the TPR domain of Cpr7p and CypA also bind to the RPR region in p33 responsible for viral RNA binding (77, 78).
FIG 7.

Efficient copurification of selected co-opted host proteins with the viral replicase from yeast coexpressing TPR-domain protein or cyclophilin A (CypA). The viral replicase was purified through FLAG-tagged p33 from yeast extracts using a FLAG-affinity column. (A to D) For the top panel, Western blot analysis of copurified HA-tagged host protein expressed from the chromosomal location with anti-HA antibody was performed. For the middle panel, Western blot analysis of the purified Flag-p33 using anti-FLAG antibody was performed. For the bottom panel, Western blot analysis of total protein extract with anti-HA antibody was used to detect the total amount of the HA-tagged host protein expressed from the chromosomal location in each sample. Lane 1, negative control based on yeast expressing His6-tagged p33 and the HA-tagged host protein; lane 2, yeast coexpressing Flag-p33 and the HA-tagged host protein; lane 3, yeast coexpressing Flag-p33 and the HA-tagged host protein in combination with TPR-domain protein; lane 4, yeast coexpressing Flag-p33 and the HA-tagged host protein in combination with CypA protein. All yeast strains actively replicate the TBSV repRNA (data not shown). Each experiment was repeated. (E) Western blot analysis of total protein extracts from yeasts expressing the shown proteins through anti-His antibody.
The contrasting data on WW-domain protein versus cyclophilins suggest that WW-domain proteins have a unique role during TBSV replication. Nevertheless, these results indicate that WW domain selectively affects the recruitment of stimulatory cellular proteins into VRCs, while overexpression of the TPR-domain protein or CypA do not detectably influence these activities.
Binding kinetics suggest a faster association of proviral cellular factors to p33 than Rsp5p.
In the infected cells, subverted proviral cellular factors and antiviral restriction factors, CIRFs, likely compete with one another for binding to the tombusvirus replication proteins. To test the binding kinetics of selected cellular proteins to the p33 replication protein, we used surface plasmon resonance measurements with purified recombinant proteins, which were separately immobilized on the chip, in analyzing binding constants to the soluble C-terminal portion of p33. Interestingly, five of the known co-opted cellular factors bound with ∼2- to 7-fold-higher ka value to p33 than Rsp5p (Table 1). Among these host proteins Tdh2p and Tef1p are present in large amounts in cells, further increasing the chance that these host proteins bind to p33 first or earlier than Rsp5p. This suggests that p33 likely have a better chance to bind to the co-opted host factors at the early stage of infection when p33 concentration is low and the “free” proviral cellular factors are still abundant.
TABLE 1.
Kinetics of interaction between p33 replication protein and cellular proteinsa
| Protein or control | KD (M) | Mean ka (1/Ms) ± SD | kd (1/s) |
|---|---|---|---|
| Proteins | |||
| Rsp5 | 6.548e–7 | 2,203 ± 96 | 1.442e–3 |
| WW | 5.200e–7 | 7,035 ± 23 | 3.659e–3 |
| Tdh2 | 3.806e–7 | 15,900 ± 457 | 6.052e–3 |
| Vps4 | 1.907e–7 | 11,380 ± 234 | 2.170e–3 |
| Tef1 | 3.633e–7 | 6,977 ± 72 | 2.535e–3 |
| Bro1 | 6.529e–7 | 5,961 ± 151 | 3.892e–3 |
| Cdc34 | 8.756e–7 | 4,549 ± 217 | 3.983e–3 |
| Pex19 | 3.822e–6 | 1,446 + 411 | 5.526e–3 |
| Negative controls | |||
| WW:MBP | 3.746e–6 | 417 | 1.566e–3 |
| Cdc34:MBP | 7.959e–6 | 737 | 5.867e–3 |
KD, affinity of interaction constant; ka, association rate constant; kd, dissociation rate constant.
The tombusvirus replicase purified at late stage of infection is less precise when isolated from cells with depleted WW-domain proteins.
To test whether Rsp5p might have a function in regulation of VRC assembly and whether this function is manifested at the late stage of infection, we purified tombusvirus replicase from yeast with depleted WW-domain proteins (TET::RSP5/TET::PRP40/wwm1Δ) at early and late time points. Then, the purified replicase preparations were tested with a (−)RNA template that is used to measure the precision of initiation events by the RdRp (79, 80). While the replicase preparation from yeast with depleted WW-domain proteins prepared at the early time point (16 h) was ∼2-fold more active than the similar preparation from wt yeast, the ratio of internal initiation and 3′terminal extension (3′-TEX) products versus full-length cRNA synthesis product (this product is required for viral replication) was comparable by these replicases (Fig. 8B, lane 3 versus lane 1). Thus, the replicase from yeast with depleted WW-domain proteins is more active, but comparable in precision with the wt replicase.
FIG 8.
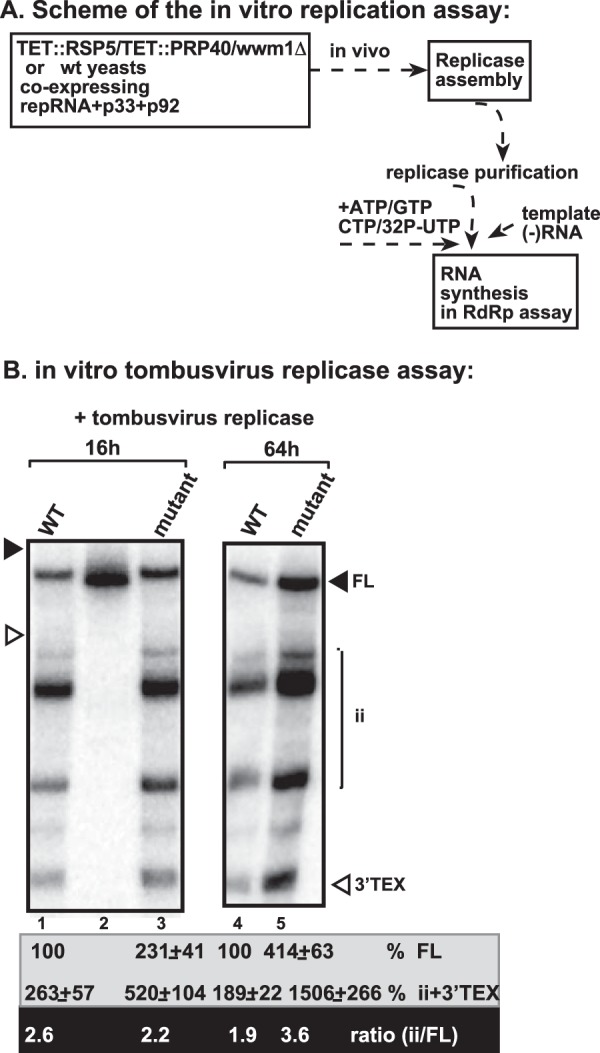
Increased imprecise initiation by the tombusvirus replicase from yeast with depleted WW-domain proteins. (A) Scheme of the tombusvirus replicase assay. wt or mutant (TET::RSP5/TET::PRP40/wwm1Δ to deplete WW-domain proteins) yeasts coexpressing FLAG-p33, FLAG-p92, and DI-72 repRNA were used to purify the tombusvirus replicase after solubilization of the membrane fraction of yeast. Note that yeasts were grown for 16 or 64 h after induction of TBSV replication prior to replicase purification. The replicase preparations were programmed with TBSV RI/III (−)RNA that leads to precise initiation [FL, full-length complementary (+)RNA product] and imprecise initiations (ii, internal initiation from cryptic promoter-like sequences; or 3′-TEX, 3′-terminal extension). (B) Denaturing PAGE analysis of the in vitro replicase products. The ratio of imprecise initiation versus precise initiation was calculated.
Interestingly, the replicase preparation from yeast with depleted WW-domain proteins prepared at the late time point (64 h) was ∼4-fold more active than the similar preparation from wt yeast. However, the replicase with depleted WW-domain proteins was ∼2-fold less precise than the wt replicase, based on the ratio of internal initiation and 3′-TEX versus full-length cRNA synthesis (Fig. 8B, lane 5 versus lane 4). These data suggest that the WW-domain proteins not only inhibit tombusvirus replicase assembly, but surprisingly, they also help making the replicase more precise at the late time points.
DISCUSSION
Rsp5p and WW-domain proteins act as CIRFs against tombusviruses.
(+)RNA viruses replicate the viral RNA efficiently in infected cells via assembling membrane-bound VRCs consisting of viral and subverted host proteins (1–7, 36, 81, 82). However, some cellular factors could act as CIRFs restricting virus replication (22, 83, 84). Among the several CIRFs against tombusviruses, Rsp5p and the WW-domain proteins are strong inhibitors, which are present in both yeast and plant cells (64, 65). In the present study, we show data that support the model that the cellular WW-domain proteins could inhibit assembly of new tombusvirus VRCs. The presented data indicate that binding of the WW domain from Rsp5p to p33 and p92pol (that contains p33 sequence due to the overlapping expression strategy) blocks interaction of the replication proteins with (i) the viral RNA, (ii) the oligomerization with other p33 and p92pol molecules, and (iii) a set of stimulatory (susceptibility) host factors subverted for TBSV replication. All of these interactions are needed for the assembly of new VRCs and the activation of p92pol, which is initially inactive in the cytosol after translation (30, 37, 39, 56). Based on these features involving many of the VRC components, the WW-domain proteins seem to exhibit a complex mechanism as CIRFs.
Interestingly, overexpression of WW-domain protein inhibited the subversion of several host proteins and their recruitment into the VRCs (Fig. 5). Notably, the list of host factors affected by the WW-domain protein includes co-opted host proteins, such as eEF1A, Bro1p, Vps4p, and Cdc34p that play important roles in the assembly of the tombusvirus VRCs in vitro, in yeast and plant cells (4, 46, 47, 51, 53). We propose that overexpression of WW-domain proteins leads to strong competition between the WW-domain proteins and the co-opted susceptibility host factors in binding to the viral replication proteins even at the early stage of replication due to the high abundance of free WW-domain proteins in these cells (Fig. 9C). This should lead to inhibition of the VRC assembly and reduced level of replication. Accordingly, overexpression of Rsp5p and several other WW-domain proteins has resulted in strong inhibition of TBSV RNA replication in yeast and plants (64, 65). Simultaneous overexpression of both WW-domain protein and a co-opted host protein is also expected to lead to strong competition in binding to the viral replication proteins due to the high abundance of free WW-domain protein and the given stimulatory host factor in these cells. Indeed, we observed that overexpression of selected co-opted host proteins increased TBSV replication even in the presence of abundant WW-domain protein (Fig. 6). On the contrary, deletion or downregulation of WW-domain proteins increased TBSV replication in yeast cells (64, 65) or in vitro (Fig. 3 and 8). Thus, our data support that Rsp5p and WW-domain proteins act as CIRFs under certain cellular conditions.
FIG 9.

Models for the role of WW-domain proteins in regulation of tombusvirus replication. We propose that WW-domain proteins inhibit the assembly of new VRCs but do not affect already-assembled VRCs. (A) At the early stage of replication, the tombusvirus replication proteins bind primarily to the abundant cellular susceptibility factors (proviral host factors [HFs]), to other viral replication proteins, and to the viral (+)RNA to recruit the viral (+)RNA to cellular membranes and assemble functional VRCs. Binding to WW-domain proteins by the replication proteins is inefficient under these conditions. (B) At the late stage of replication, the easily accessible host factors are depleted (scarce due to sequestration into previously assembled VRCs) and the new viral replication proteins bind to WW-domain proteins, leading to a blockage in new VRC assembly. (C) Overexpression of the WW-domain proteins facilitates binding to the viral replication proteins to WW-domain proteins (at the expense of interaction between replication proteins and host factors), leading to a blockage in VRC assembly and reduced level of TBSV replication. Thus, WW-domain proteins can also act as CIRFs.
Rsp5p and WW-domain proteins might also function as regulatory factors for TBSV replication.
The VRC assembly is likely highly regulated to prevent the extensive production of incomplete or truncated viral RNAs at the late stage of replication, when one or more cellular components become limiting or even depleted, making them unavailable for new rounds of VRC assembly and virus replication. Our multiple genome-wide screens with TBSV and yeast showed that missing a proviral host factor (due to depletion or gene deletion) does not necessarily prevent the assembly of the VRCs (11, 13, 40–42, 45, 48). Instead, it usually leads to incorrect VRC assembly that changes some of the activities of the viral replicase (such as increased rate of RNA recombination or reduced efficiency of replication). It could be advantageous for the virus to have an active mechanism(s) to regulate the assembly of new VRCs toward the late phase of infection when several subverted proviral host factors might be already depleted during the previous rounds of VRC assembly.
Based on these ideas, we predict that somehow an RNA virus could sense the state of the host cell and decide whether new VRCs should be assembled or not. Based on the features presented in the present study, Rsp5p and WW-domain proteins are likely highly suitable for regulatory functions during TBSV replication. For example, the regulatory role of the WW-domain proteins in TBSV replication is supported by several interesting observations. (i) Rsp5p binds to the p33 replication protein not as rapidly as several proviral cellular factors (based on ka values in Table 1), suggesting that the proviral factors might be favored by TBSV to act early during replication, while Rsp5p might function at a latter stage of infection when some of the proviral factors are mostly depleted. (ii) The binding of a single Rsp5p or WW-domain protein likely inhibits the function of a single p33 or p92pol replication protein but does not have dominant-negative effect on the VRCs, as shown by the use of WW-p33 fusion protein in a CFE-based viral replication assay (Fig. 3). This could be important for tombusviruses, since the previously assembled VRCs should not be blocked or destroyed by the regulatory protein. Instead, only the formation of new VRCs should be inhibited at the late stage of infection. It is also possible that the WW-domain protein cannot access the previously assembled VRCs, because those are closed from cytosolic proteins due to the spherule structure (57) or, alternatively, the p33 and p92 proteins are already oligomerized (via p33-p33 self-interaction or p33-p92 interaction) or bound to proviral host factors. (iii) A regulatory protein is expected to block the interaction between the viral replication proteins and the viral RNA in order to facilitate a nonreplicative use of the viral RNA (e.g., for encapsidation or cell-to-cell movement), instead of keeping the viral RNA trapped in the translation/replication cycle. (iv) It could be useful if the regulatory protein would facilitate the degradation of excess amounts of viral replication proteins to prevent the functional interference of these replication proteins with other, nonreplicative functions. Indeed, p92pol is efficiently degraded when Rsp5p or Wwm1p WW-domain proteins are overexpressed in yeast (65). (v) It is predicted that VRCs assembled at the late stage of infection could be especially error-prone if one or more proviral cellular proteins are not recruited into VRCs due to their depletion in previous rounds of VRC assembly. Accordingly, we detected a higher error rate for incorrect initiation of RNA synthesis with the purified replicase when derived from yeasts with depleted WW-domain proteins at a late stage (Fig. 8). This aberrant feature of the replicase could be due to incorrect assembly of the replicase at the late stage, possibly due to depletion of one or more proviral factors. Interestingly, we have shown that the WW-domain proteins fulfill all of these features during TBSV replication.
Based on these observations, we suggest a new model for the interplay between proviral factors and WW-domain proteins, such as Rsp5p, in the regulation of tombusvirus VRC assembly. We propose that the tombusvirus replication proteins first interact with the host susceptibility factors, which are co-opted for virus replication at the beginning of infection when these susceptibility factors are abundant and/or accessible (Fig. 9A). These events lead to efficient assembly of VRCs and robust viral replication at the early stage of replication. As the amounts of newly produced p33 and p92pol replication proteins increase due to ongoing translation, the cell likely runs out of one or more available susceptibility factors at the late stage of replication. Depletion of the susceptibility factors allows the viral replication proteins to interact with the cellular WW-domain proteins (Fig. 9B). This will then lead to a blockage for the assembly of new VRCs and inhibition of the formation of new p33-viral RNA complexes and to the degradation of p92pol replication protein. Thus, viral replication, especially the formation of new VRCs, will be slowed down at the late stage, and the newly made viral (+)RNAs will be able to leave the translation/replication cycle and can become committed to additional functions, such as encapsidation or cell-to-cell movement. Altogether, we propose that tombusviruses could sense the status of the infected cells via “measuring” the availability of cellular susceptibility factors versus cellular WW-domain proteins, which then determines whether new VRCs are assembled or the VRC assembly process is halted.
Why would an RNA virus select WW-domain proteins for such a regulatory function? We propose that the WW-domain proteins are very suitable for these functions, since they are present in the cytosol of all eukaryotic cells, and they also represent an ancient, very simple motif selected for protein-protein interactions (66, 72, 85). Indeed, we were able to identify several WW-domain proteins in both yeasts and plants (65), which could be used by tombusviruses for such regulatory roles. Interestingly, the unrelated nodaviruses (insect RNA viruses) could also be inhibited by overexpression of yeast Rsp5p and Wwm1p WW-domain proteins (65). The question remains if additional RNA viruses could also take advantage of WW-domain proteins or other cellular proteins for regulatory functions to optimize their replication in various cells and hosts.
Unlike the WW-domain proteins, other inhibitory cellular CIRF proteins, such as the TPR-domain containing Cyp40-like Cpr7p chaperone or CypA cyclophilin (45, 77), do not seem to affect the recruitment of stimulatory host factors into the VRCs (Fig. 7). Thus, these cellular restriction factors are not involved in regulation of tombusvirus replication in a manner similar to WW-domain proteins, or they function utilizing different regulatory mechanisms.
ACKNOWLEDGMENTS
We thank Judit Pogany for critical reading of the manuscript and for very helpful suggestions. We thank Scott D. Emr (Cornell University) for yeast strains and plasmids. We thank Mark Goebl (Indiana University) for anti-Cdc34 antibody and Terry Goss Kinzy (UMDNJ Robert Wood Johnson Medical School) for anti-eEF1A and anti-eEF1Bγ antibodies.
This research was supported by NIH-NIAID (1R21AI109529).
REFERENCES
- 1.Kopek BG, Perkins G, Miller DJ, Ellisman MH, Ahlquist P. 2007. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol 5:e220. doi: 10.1371/journal.pbio.0050220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.den Boon JA, Diaz A, Ahlquist P. 2010. Cytoplasmic viral replication complexes. Cell Host Microbe 8:77–85. doi: 10.1016/j.chom.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barajas D, Jiang Y, Nagy PD. 2009. A unique role for the host ESCRT proteins in replication of Tomato bushy stunt virus. PLoS Pathog 5:e1000705. doi: 10.1371/journal.ppat.1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagy PD, Pogany J. 2012. The dependence of viral RNA replication on co-opted host factors. Nat Rev Microbiol 10:137–149. doi: 10.1038/Nrmicro2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salonen A, Ahola T, Kaariainen L. 2005. Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol 285:139–173. doi: 10.1007/3-540-26764-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartenschlager R, Cosset FL, Lohmann V. 2010. Hepatitis C virus replication cycle. J Hepatol 53:583–585. doi: 10.1016/j.jhep.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 8.de Castro IF, Volonte L, Risco C. 2013. Virus factories: biogenesis and structural design. Cell Microbiol 15:24–34. doi: 10.1111/cmi.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagy PD. 2008. Yeast as a model host to explore plant virus-host interactions. Annu Rev Phytopathol 46:217–242. doi: 10.1146/annurev.phyto.121407.093958. [DOI] [PubMed] [Google Scholar]
- 10.Nagy PD, Pogany J. 2008. Multiple roles of viral replication proteins in plant RNA virus replication. Methods Mol Biol 451:55–68. doi: 10.1007/978-1-59745-102-4_4. [DOI] [PubMed] [Google Scholar]
- 11.Panavas T, Serviene E, Brasher J, Nagy PD. 2005. Yeast genome-wide screen reveals dissimilar sets of host genes affecting replication of RNA viruses. Proc Natl Acad Sci U S A 102:7326–7331. doi: 10.1073/pnas.0502604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cherry S, Doukas T, Armknecht S, Whelan S, Wang H, Sarnow P, Perrimon N. 2005. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev 19:445–452. doi: 10.1101/gad.1267905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang Y, Serviene E, Gal J, Panavas T, Nagy PD. 2006. Identification of essential host factors affecting tombusvirus RNA replication based on the yeast Tet promoters Hughes Collection. J Virol 80:7394–7404. doi: 10.1128/JVI.02686-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kushner DB, Lindenbach BD, Grdzelishvili VZ, Noueiry AO, Paul SM, Ahlquist P. 2003. Systematic, genome-wide identification of host genes affecting replication of a positive-strand RNA virus. Proc Natl Acad Sci U S A 100:15764–15769. doi: 10.1073/pnas.2536857100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, Brass AL, Adametz R, Tsui M, Qian F, Montgomery RR, Lev S, Mason PW, Koski RA, Elledge SJ, Xavier RJ, Agaisse H, Fikrig E. 2008. RNA interference screen for human genes associated with West Nile virus infection. Nature 455:242–245. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diaz A, Wang X, Ahlquist P. 2010. Membrane-shaping host reticulon proteins play crucial roles in viral RNA replication compartment formation and function. Proc Natl Acad Sci U S A 107:16291–16296. doi: 10.1073/pnas.1011105107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neuvonen M, Kazlauskas A, Martikainen M, Hinkkanen A, Ahola T, Saksela K. 2011. SH3 domain-mediated recruitment of host cell amphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathog 7:e1002383. doi: 10.1371/journal.ppat.1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, Elledge SJ. 2009. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A 106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. 2007. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci U S A 104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sessions OM, Barrows NJ, Souza-Neto JA, Robinson TJ, Hershey CL, Rodgers MA, Ramirez JL, Dimopoulos G, Yang PL, Pearson JL, Garcia-Blanco MA. 2009. Discovery of insect and human dengue virus host factors. Nature 458:1047–1050. doi: 10.1038/nature07967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, Chung RT. 2009. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5:298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sasvari Z, Alatriste Gonzalez P, Nagy PD. 2014. Tombusvirus-yeast interactions identify conserved cell-intrinsic viral restriction factors. Front Plant Sci 5:383. doi: 10.3389/fpls.2014.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diamond MS, Gale M Jr. 2012. Cell-intrinsic innate immune control of West Nile virus infection. Trends Immunol 33:522–530. doi: 10.1016/j.it.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aoshi T, Koyama S, Kobiyama K, Akira S, Ishii KJ. 2011. Innate and adaptive immune responses to viral infection and vaccination. Curr Opin Virol 1:226–232. doi: 10.1016/j.coviro.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Jensen S, Thomsen AR. 2012. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86:2900–2910. doi: 10.1128/JVI.05738-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding SW. 2010. RNA-based antiviral immunity. Nat Rev Immunol 10:632–644. doi: 10.1038/nri2824. [DOI] [PubMed] [Google Scholar]
- 27.Nagy PD, Barajas D, Pogany J. 2012. Host factors with regulatory roles in tombusvirus replication. Curr Opin Virol 2:685–692. doi: 10.1016/j.coviro.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Nagy PD, Pogany J. 2006. Yeast as a model host to dissect functions of viral and host factors in tombusvirus replication. Virology 344:211–220. doi: 10.1016/j.virol.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 29.White KA, Nagy PD. 2004. Advances in the molecular biology of tombusviruses: gene expression, genome replication, and recombination. Prog Nucleic Acids Res Mol Biol 78:187–226. doi: 10.1016/S0079-6603(04)78005-8. [DOI] [PubMed] [Google Scholar]
- 30.Panaviene Z, Panavas T, Serva S, Nagy PD. 2004. Purification of the cucumber necrosis virus replicase from yeast cells: role of coexpressed viral RNA in stimulation of replicase activity. J Virol 78:8254–8263. doi: 10.1128/JVI.78.15.8254-8263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panavas T, Nagy PD. 2003. Yeast as a model host to study replication and recombination of defective interfering RNA of Tomato bushy stunt virus. Virology 314:315–325. doi: 10.1016/S0042-6822(03)00436-7. [DOI] [PubMed] [Google Scholar]
- 32.Jonczyk M, Pathak KB, Sharma M, Nagy PD. 2007. Exploiting alternative subcellular location for replication: tombusvirus replication switches to the endoplasmic reticulum in the absence of peroxisomes. Virology 362:320–330. doi: 10.1016/j.virol.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 33.Pogany J, White KA, Nagy PD. 2005. Specific binding of tombusvirus replication protein p33 to an internal replication element in the viral RNA is essential for replication. J Virol 79:4859–4869. doi: 10.1128/JVI.79.8.4859-4869.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panavas T, Hawkins CM, Panaviene Z, Nagy PD. 2005. The role of the p33:p33/p92 interaction domain in RNA replication and intracellular localization of p33 and p92 proteins of Cucumber necrosis tombusvirus. Virology 338:81–95. doi: 10.1016/j.virol.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 35.Stork J, Kovalev N, Sasvari Z, Nagy PD. 2011. RNA chaperone activity of the tombusviral p33 replication protein facilitates initiation of RNA synthesis by the viral RdRp in vitro. Virology 409:338–347. doi: 10.1016/j.virol.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCartney AW, Greenwood JS, Fabian MR, White KA, Mullen RT. 2005. Localization of the Tomato bushy stunt virus replication protein p33 reveals a peroxisome-to-endoplasmic reticulum sorting pathway. Plant Cell 17:3513–3531. doi: 10.1105/tpc.105.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pogany J, Nagy PD. 2008. Authentic replication and recombination of Tomato bushy stunt virus RNA in a cell-free extract from yeast. J Virol 82:5967–5980. doi: 10.1128/JVI.02737-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panaviene Z, Panavas T, Nagy PD. 2005. Role of an internal and two 3′-terminal RNA elements in assembly of tombusvirus replicase. J Virol 79:10608–10618. doi: 10.1128/JVI.79.16.10608-10618.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pogany J, Nagy PD. 2012. p33-independent activation of a truncated p92 RNA-dependent RNA polymerase of Tomato bushy stunt virus in yeast cell-free extract. J Virol 86:12025–12038. doi: 10.1128/JVI.01303-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serviene E, Shapka N, Cheng CP, Panavas T, Phuangrat B, Baker J, Nagy PD. 2005. Genome-wide screen identifies host genes affecting viral RNA recombination. Proc Natl Acad Sci U S A 102:10545–10550. doi: 10.1073/pnas.0504844102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Serviene E, Jiang Y, Cheng CP, Baker J, Nagy PD. 2006. Screening of the yeast yTHC collection identifies essential host factors affecting tombusvirus RNA recombination. J Virol 80:1231–1241. doi: 10.1128/JVI.80.3.1231-1241.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shah Nawaz-Ul-Rehman M, Martinez-Ochoa N, Pascal H, Sasvari Z, Herbst C, Xu K, Baker J, Sharma M, Herbst A, Nagy PD. 2012. Proteome-wide overexpression of host proteins for identification of factors affecting tombusvirus RNA replication: an inhibitory role of protein kinase C. J Virol 86:9384–9395. doi: 10.1128/JVI.00019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagy PD. 2011. The roles of host factors in tombusvirus RNA recombination. Adv Virus Res 81:63–84. doi: 10.1016/B978-0-12-385885-6.00008-0. [DOI] [PubMed] [Google Scholar]
- 44.Nagy PD, Pogany J. 2010. Global genomics and proteomics approaches to identify host factors as targets to induce resistance against Tomato bushy stunt virus. Adv Virus Res 76:123–177. doi: 10.1016/S0065-3527(10)76004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mendu V, Chiu M, Barajas D, Li Z, Nagy PD. 2010. Cpr1 cyclophilin and Ess1 parvulin prolyl isomerases interact with the tombusvirus replication protein and inhibit viral replication in yeast model host. Virology 406:342–351. doi: 10.1016/j.virol.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 46.Li Z, Pogany J, Panavas T, Xu K, Esposito AM, Kinzy TG, Nagy PD. 2009. Translation elongation factor 1A is a component of the tombusvirus replicase complex and affects the stability of the p33 replication cofactor. Virology 385:245–260. doi: 10.1016/j.virol.2008.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Z, Barajas D, Panavas T, Herbst DA, Nagy PD. 2008. Cdc34p ubiquitin-conjugating enzyme is a component of the tombusvirus replicase complex and ubiquitinates p33 replication protein. J Virol 82:6911–6926. doi: 10.1128/JVI.00702-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shah Nawaz-Ul-Rehman M, Reddisiva Prasanth K, Baker J, Nagy PD. 2013. Yeast screens for host factors in positive-strand RNA virus replication based on a library of temperature-sensitive mutants. Methods 59:207–216. doi: 10.1016/j.ymeth.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 49.Serva S, Nagy PD. 2006. Proteomics analysis of the tombusvirus replicase: Hsp70 molecular chaperone is associated with the replicase and enhances viral RNA replication. J Virol 80:2162–2169. doi: 10.1128/JVI.80.5.2162-2169.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma M, Sasvari Z, Nagy PD. 2011. Inhibition of phospholipid biosynthesis decreases the activity of the tombusvirus replicase and alters the subcellular localization of replication proteins. Virology 415:141–152. doi: 10.1016/j.virol.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Z, Nagy PD. 2011. Diverse roles of host RNA binding proteins in RNA virus replication. RNA Biol 8:305–315. doi: 10.4161/rna.8.2.15391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma M, Sasvari Z, Nagy PD. 2010. Inhibition of sterol biosynthesis reduces tombusvirus replication in yeast and plants. J Virol 84:2270–2281. doi: 10.1128/JVI.02003-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Z, Pogany J, Tupman S, Esposito AM, Kinzy TG, Nagy PD. 2010. Translation elongation factor 1A facilitates the assembly of the tombusvirus replicase and stimulates minus-strand synthesis. PLoS Pathog 6:e1001175. doi: 10.1371/journal.ppat.1001175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang RY, Stork J, Pogany J, Nagy PD. 2009. A temperature-sensitive mutant of heat shock protein 70 reveals an essential role during the early steps of tombusvirus replication. Virology 394:28–38. doi: 10.1016/j.virol.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang RY, Stork J, Nagy PD. 2009. A key role for heat shock protein 70 in the localization and insertion of tombusvirus replication proteins to intracellular membranes. J Virol 83:3276–3287. doi: 10.1128/JVI.02313-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pogany J, Stork J, Li Z, Nagy PD. 2008. In vitro assembly of the Tomato bushy stunt virus replicase requires the host Heat shock protein 70. Proc Natl Acad Sci U S A 105:19956–19961. doi: 10.1073/pnas.0810851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barajas D, Martin IF, Pogany J, Risco C, Nagy PD. 2014. Noncanonical role for the host Vps4 AAA+ ATPase ESCRT protein in the formation of Tomato bushy stunt virus replicase. PLoS Pathog 10:e1004087. doi: 10.1371/journal.ppat.1004087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kovalev N, Nagy PD. 2014. The expanding functions of cellular helicases: the tombusvirus RNA replication enhancer co-opts the plant eIF4AIII-like AtRH2 and the DDX5-like AtRH5 DEAD-box RNA helicases to promote viral asymmetric RNA replication. PLoS Pathog 10:e1004051. doi: 10.1371/journal.ppat.1004051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kovalev N, Pogany J, Nagy PD. 2012. A co-opted DEAD-box RNA helicase enhances tombusvirus plus-strand synthesis. PLoS Pathog 8:e1002537. doi: 10.1371/journal.ppat.1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pathak KB, Sasvari Z, Nagy PD. 2008. The host Pex19p plays a role in peroxisomal localization of tombusvirus replication proteins. Virology 379:294–305. doi: 10.1016/j.virol.2008.06.044. [DOI] [PubMed] [Google Scholar]
- 61.Sasvari Z, Izotova L, Kinzy TG, Nagy PD. 2011. Synergistic roles of eukaryotic translation elongation factors 1Bγ and 1A in stimulation of tombusvirus minus-strand synthesis. PLoS Pathog 7:e1002438. doi: 10.1371/journal.ppat.1002438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang TS, Nagy PD. 2011. Direct inhibition of tombusvirus plus-strand RNA synthesis by a dominant-negative mutant of a host metabolic enzyme, glyceraldehyde-3-phosphate dehydrogenase, in yeast and plants. J Virol 85:9090–9102. doi: 10.1128/JVI.00666-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang RY, Nagy PD. 2008. Tomato bushy stunt virus co-opts the RNA-binding function of a host metabolic enzyme for viral genomic RNA synthesis. Cell Host Microbe 3:178–187. doi: 10.1016/j.chom.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 64.Barajas D, Li Z, Nagy PD. 2009. The Nedd4-type Rsp5p ubiquitin ligase inhibits tombusvirus replication by regulating degradation of the p92 replication protein and decreasing the activity of the tombusvirus replicase. J Virol 83:11751–11764. doi: 10.1128/JVI.00789-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qin J, Barajas D, Nagy PD. 2012. An inhibitory function of WW domain-containing host proteins in RNA virus replication. Virology 426:106–119. doi: 10.1016/j.virol.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 66.Hesselberth JR, Miller JP, Golob A, Stajich JE, Michaud GA, Fields S. 2006. Comparative analysis of Saccharomyces cerevisiae WW domains and their interacting proteins. Genome Biol 7:R30. doi: 10.1186/gb-2006-7-4-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Macias MJ, Gervais V, Civera C, Oschkinat H. 2000. Structural analysis of WW domains and design of a WW prototype. Nat Struct Biol 7:375–379. doi: 10.1038/75144. [DOI] [PubMed] [Google Scholar]
- 68.Jaag HM, Stork J, Nagy PD. 2007. Host transcription factor Rpb11p affects tombusvirus replication and recombination via regulating the accumulation of viral replication proteins. Virology 368:388–404. doi: 10.1016/j.virol.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 69.Rajendran KS, Nagy PD. 2003. Characterization of the RNA-binding domains in the replicase proteins of tomato bushy stunt virus. J Virol 77:9244–9258. doi: 10.1128/JVI.77.17.9244-9258.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rajendran KS, Nagy PD. 2004. Interaction between the replicase proteins of Tomato bushy stunt virus in vitro and in vivo. Virology 326:250–261. doi: 10.1016/j.virol.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 71.Wiesner S, Stier G, Sattler M, Macias MJ. 2002. Solution structure and ligand recognition of the WW domain pair of the yeast splicing factor Prp40. J Mol Biol 324:807–822. doi: 10.1016/S0022-2836(02)01145-2. [DOI] [PubMed] [Google Scholar]
- 72.Salah Z, Alian A, Aqeilan RI. 2012. WW domain-containing proteins: retrospectives and the future. Front Biosci 17:331–348. doi: 10.2741/3930. [DOI] [PubMed] [Google Scholar]
- 73.Kovalev N, Pogany J, Nagy PD. 2014. Template role of double-stranded RNA in tombusvirus replication. J Virol 88:5638–5651. doi: 10.1128/JVI.03842-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pathak KB, Jiang Z, Ochanine V, Sharma M, Pogany J, Nagy PD. 2013. Characterization of dominant-negative and temperature-sensitive mutants of tombusvirus replication proteins affecting replicase assembly. Virology 437:48–61. doi: 10.1016/j.virol.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 75.Xu K, Huang TS, Nagy PD. 2012. Authentic in vitro replication of two tombusviruses in isolated mitochondrial and endoplasmic reticulum membranes. J Virol 86:12779–12794. doi: 10.1128/JVI.00973-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gupta R, Kus B, Fladd C, Wasmuth J, Tonikian R, Sidhu S, Krogan NJ, Parkinson J, Rotin D. 2007. Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Mol Syst Biol 3:116. doi: 10.1038/msb4100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin JY, Mendu V, Pogany J, Qin J, Nagy PD. 2012. The TPR domain in the host Cyp40-like cyclophilin binds to the viral replication protein and inhibits the assembly of the tombusviral replicase. PLoS Pathog 8:e1002491. doi: 10.1371/journal.ppat.1002491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kovalev N, Nagy PD. 2013. Cyclophilin a binds to the viral RNA and replication proteins, resulting in inhibition of tombusviral replicase assembly. J Virol 87:13330–13342. doi: 10.1128/JVI.02101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Panavas T, Pogany J, Nagy PD. 2002. Internal initiation by the cucumber necrosis virus RNA-dependent RNA polymerase is facilitated by promoter-like sequences. Virology 296:275–287. doi: 10.1006/viro.2002.1422. [DOI] [PubMed] [Google Scholar]
- 80.Panavas T, Pogany J, Nagy PD. 2002. Analysis of minimal promoter sequences for plus-strand synthesis by the Cucumber necrosis virus RNA-dependent RNA polymerase. Virology 296:263–274. doi: 10.1006/viro.2002.1423. [DOI] [PubMed] [Google Scholar]
- 81.Schwartz M, Chen J, Janda M, Sullivan M, den Boon J, Ahlquist P. 2002. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol Cell 9:505–514. doi: 10.1016/S1097-2765(02)00474-4. [DOI] [PubMed] [Google Scholar]
- 82.Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. 2005. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol Cell 97:147–172. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yasunaga A, Hanna SL, Li J, Cho H, Rose PP, Spiridigliozzi A, Gold B, Diamond MS, Cherry S. 2014. Genome-wide RNAi screen identifies broadly acting host factors that inhibit arbovirus infection. PLoS Pathog 10:e1003914. doi: 10.1371/journal.ppat.1003914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Diamond MS, Farzan M. 2013. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S. 2012. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc Natl Acad Sci U S A 109:16858–16863. doi: 10.1073/pnas.1209751109. [DOI] [PMC free article] [PubMed] [Google Scholar]