

ABSTRACT
Phosphoinositides and phosphoinositide binding proteins play a critical role in membrane and protein trafficking in eukaryotes. Their critical role in replication of cytoplasmic viruses has just begun to be understood. Poxviruses, a family of large cytoplasmic DNA viruses, rely on the intracellular membranes to develop their envelope, and poxvirus morphogenesis requires enzymes from the cellular phosphoinositide metabolic pathway. However, the role of phosphoinositides in poxvirus replication remains unclear, and no poxvirus proteins show any homology to eukaryotic phosphoinositide binding domains. Recently, a group of poxvirus proteins, termed viral membrane assembly proteins (VMAPs), were identified as essential for poxvirus membrane biogenesis. A key component of VMAPs is the H7 protein. Here we report the crystal structure of the H7 protein from vaccinia virus. The H7 structure displays a novel fold comprised of seven α-helices and a highly curved three-stranded antiparallel β-sheet. We identified a phosphoinositide binding site in H7, comprised of basic residues on a surface patch and the flexible C-terminal tail. These residues were found to be essential for viral replication and for binding of H7 to phosphatidylinositol-3-phosphate (PI3P) and phosphatidylinositol-4-phosphate (PI4P). Our studies suggest that phosphoinositide binding by H7 plays an essential role in poxvirus membrane biogenesis.
IMPORTANCE Poxvirus viral membrane assembly proteins (VMAPs) were recently shown to be essential for poxvirus membrane biogenesis. One of the key components of VMAPs is the H7 protein. However, no known structural motifs could be identified from its sequence, and there are no homologs of H7 outside the poxvirus family to suggest a biochemical function. We have determined the crystal structure of the vaccinia virus (VACV) H7 protein. The structure displays a novel fold with a distinct and positively charged surface. Our data demonstrate that H7 binds phosphatidylinositol-3-phosphate and phosphatidylinositol-4-phosphate and that the basic surface patch is indeed required for phosphoinositide binding. In addition, mutation of positively charged residues required for lipid binding disrupted VACV replication. Phosphoinositides and phosphoinositide binding proteins play critical roles in membrane and protein trafficking in eukaryotes. Our study demonstrates that VACV H7 displays a novel fold for phosphoinositide binding, which is essential for poxvirus replication.
INTRODUCTION
Phosphoinositides are lipids derived from reversible phosphorylation of phosphatidylinositol at positions 3, 4, and 5 of the inositol head group (1). They include three monophosphorylated (phosphatidylinositol-3-phosphate [PI3P], PI4P, and PI5P), three bisphosphorylated [PI(3,4)P2, PI(3,5)P2, and PI(4,5)P2], and one trisphosphorylated [PI(3,4,5)P3] isoform. Cellular organelles are partially defined by their phosphoinositide compositions, and specific recognition of phosphoinositides on different organelles is crucial for protein sorting and membrane trafficking (1, 2). Proteins that specifically bind phosphoinositides include phox homology (PX), pleckstrin homology (PH), FYVE (Fab1p/YOTB/Vac1p/EEA1), and ENTN (epsin N-terminal homology) domain-containing proteins (3, 4). The PX domain-containing proteins include a diverse family of sorting nexins that play various roles in membrane trafficking and remodeling, protein sorting, and actin cytoskeletal organization (5). Their binding to phosphoinositides typically involves electrostatic interactions with the negative charge of the phosphate(s) on the inositol ring (5).
A critical role of phosphoinositides in viral replication has just begun to be understood (6, 7). The replication of many cytoplasmic viruses involves elaborate strategies of remodeling the intracellular membranes (8). Positive-sense, single-stranded RNA viruses, for example, modify cytoplasmic membranes to establish a specialized organelle which concentrates replication complexes and facilitates viral genome replication (8). Specifically, enteroviruses recruit phosphatidylinositol 4-kinase IIIb (PI4KIIIb), which catalyzes the production of PI4P lipids (9). PI4P lipids in turn bind several viral proteins, including the RNA polymerase, and recruit these viral proteins to the replication organelle. Viral RNA synthesis is disrupted when PI4P is depleted from cells (9). Poxviruses (10), a family of cytoplasmic DNA viruses, also rely on intracellular membranes for their replication. Poxvirus intracellular mature virions (MVs) acquire their envelope from the endoplasmic reticulum (ER), through a poorly understood process (11, 12). MVs are enriched in phosphatidylinositol (12, 13), and inhibition of host phosphatidylinositol 3-kinase (PI3K) affects multiple steps of poxvirus morphogenesis, including the formation of MVs (14). However, the exact role of phosphoinositides in poxvirus morphogenesis is unclear, and no poxvirus proteins have any homology to eukaryotic phosphoinositide binding domains.
Poxvirus virion morphogenesis is a complex process involving a series of intermediate stages discernible by electron microscopy (reviewed in reference 15). The electron-dense viroplasms, comprised of viral core proteins, appear first. This is followed by the development of crescent-shaped membranes at the periphery of viroplasms. The crescent membranes appear to be open membrane structures (12, 16), which is quite unusual for membranes in cells. Next, the crescent membranes engulf part of the viroplasm to form the spherical immature virions (IVs). IVs subsequently undergo additional transformations to become infectious MVs. The origin and biogenesis of the crescent membranes are among the least understood aspects of poxvirus biology, but several viral proteins that are involved in crescent formation were recently identified in vaccinia virus (VACV), the prototypical poxvirus. These proteins, including A11 (17), H7 (18), L2 (19), A6 (20), and A30.5 (21), are collectively referred to as the viral membrane assembly proteins (VMAPs) (21). VMAPs were thought to work together to generate breaks in the ER membrane, to stabilize the open-ended crescent membranes, and to elongate the crescents by membrane fusion (21). However, the functions of the individual VMAPs and how VMAPs work together are unknown. In this study, we determined the crystal structure of the VACV H7 protein and found that H7 binds to phosphoinositides. Mutations of the putative phosphoinositide binding site on H7 disrupted viral replication, suggesting that phosphoinositide binding by H7 plays an essential role in poxvirus membrane biogenesis.
MATERIALS AND METHODS
Protein purification and crystallization.
The vaccinia virus H7 protein was cloned into a modified pET vector as a SUMO fusion with an N-terminal 6×His tag. The protein was expressed in Escherichia coli BL21(DE3) Gold cells (Stratagene). All H7 mutants and the truncated H7 protein [H7-Δ(119-146)] were cloned and expressed in the same way as the wild-type (WT) protein. The individual proteins were purified using a double Ni-nitrilotriacetic acid (Ni-NTA) procedure similar to that described previously (22). Briefly, the His-tagged fusion proteins were first purified from the cell lysate by use of a Ni-NTA affinity column (Qiagen) and then codialyzed with the ULP1 protease to remove the SUMO moiety. The cleaved protein mixtures were subsequently passed through a second Ni-NTA column and further purified by size-exclusion chromatography on a Superdex s200 column. The pooled peak fractions were further purified by ion-exchange chromatography using a Q Sepharose column (GE Healthcare). The purified H7 proteins were concentrated to 11 mg/ml. The selenomethionine (SeMet)-substituted H7 protein was expressed in M9 minimal medium supplemented with amino acids as described previously (23) and then purified similarly to the native protein, using the procedures described above. For optimal reproducibility of crystallization, all purified proteins were flash frozen and stored at −80°C until usage (24). The full-length and truncated [H7-Δ(119-146)] H7 derivatives (C89S) crystallized under conditions including 0.08 M bis-tris phosphate-citric acid, pH 8.8, and 18% (wt/vol) polyethylene glycol 3350 (PEG 3350). Twenty percent glycerol was added to the mother liquid as the cryoprotectant.
Structure determination.
A set of data was collected from an SeMet-substituted full-length H7 protein (C89S) crystal at beamline 19-ID at the Advanced Photon Source (APS), Argonne National Laboratory. The structure was solved by the single-wavelength anomalous dispersion method, using the program HKL3000 (25). A nearly complete model was constructed from the experimental phases obtained from the SeMet crystal data. This model was used to solve the native structure by the molecular replacement method, using the program phaser (26). The PHENIX program (27) was used for refinement, and Coot (28) was used for iterative manual model building. The full-length H7 crystal structure was refined to a 2.7-Å resolution, with crystallographic Rwork and Rfree values of 25.6% and 30.3%, respectively. The C-terminal 29 residues (amino acids [aa] 118 to 146) were disordered and could not be modeled. H7-Δ(119-146) protein crystals diffracted to a 2.0-Å resolution at APS. The structure of H7-Δ(119-146) was solved by the molecular replacement method as described above, and the final structure was refined to a 2.0-Å resolution, with crystallographic Rwork and Rfree values of 19.6% and 24.7%, respectively. The final model has 97.8% of all residues residing in the most-favored region of the Ramachandran plot and 2.2% of residues in additionally allowed regions, as calculated by the Molprobity server (29). All molecular graphic figures were generated with PYMOL (30). The current model has excellent geometry and refinement statistics (Table 1) and was validated by wwpdb validation servers (31).
TABLE 1.
X-ray crystallographic data and refinement statistics
| Parameter | Value or descriptiona |
||
|---|---|---|---|
| Full-length H7 with SeMet (WT) | Full-length H7 (native) | H7-Δ(119-146) | |
| Crystallographic parameters | |||
| Beamline | 19-ID, APS | 19-ID, APS | 19-ID, APS |
| Wavelength (Å) | 0.97937 | 0.97937 | 0.97937 |
| Space group | P3221 | P21212 | C2 |
| Cell parameters | |||
| a, b, c (Å) | 64.4, 67.4, 144.1 | 118.7, 68.0, 81.8 | 112.8, 34.2, 67.2 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 | 90, 99.5, 90 |
| Resolution (Å) | 50–2.9 | 50–2.7 | 50–2.0 |
| Total no. of reflections | 85,860 | 127,772 | 77,807 |
| No. of unique reflections | 8,812 | 18,495 | 16,966 |
| Redundancy | 9.7 | 6.9 | 4.6 |
| Completeness (%) | 99.1 (92.6) | 98.4 (88.7) | 97.6 (87.5) |
| I/σ | 21.9 (2.1) | 19.5 (1.9) | 17.0 (4.6) |
| Rsymb (%) | 11.4 (61.7) | 9.5 (53.7) | 9.0 (27.6) |
| Refinement statistics | |||
| Resolution range used (Å) | 48.0–2.7 | 39.5–2.0 | |
| No. of reflections used | 18,426 | 16,276 | |
| Rwork/Rfreeb | 25.6/30.3 | 19.6/24.7 | |
| RMSD | |||
| Bond lengths (Å) | 0.009 | 0.007 | |
| Bond angles (°) | 1.170 | 0.957 | |
| No. of atoms | |||
| Protein | 3,756 | 1,899 | |
| Water | 19 | 168 | |
| Ramachandran value (%) | |||
| Preferred regions | 92.1 | 97.8 | |
| Allowed regions | 7.9 | 2.2 | |
Values in parentheses are for the highest-resolution shell: 3.0 to 2.9 Å (full-length H7 with SeMet), 2.80 to 2.70 Å (full-length H7 [native]), or 2.07 to 2.00 Å [H7-Δ(119-146)].
Rsym = Σ |Iobs − Iavg|/Σ Iavg. Rwork = Σ ||Fobs| − |Fcalc||/Σ Fobs. Rfree was calculated using 10% of the data.
Lipid overlay assay.
Nitrocellulose membranes spotted with 100-pmol aliquots of phospholipids (PIP strips; Echelon Biosciences) were blocked in 3% fat-free bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 h and then incubated with 1 μg/ml recombinant H7 proteins in 3% BSA for 1 h. The membranes were washed with TBS-T and incubated with mouse polyclonal anti-H7 antibodies (32) for 1 h. The membranes were washed again with TBS-T and incubated with an anti-mouse secondary antibody for 1 h. The H7 proteins that bound to the membrane were detected by chemiluminescence. Membranes incubated with different proteins were washed for the same length of time and detected with the same exposure time in each experiment.
Fluorescence polarization-based lipid binding assay.
Experiments were performed on a Beacon 2000 fluorescence polarization system (Life Technologies). The phosphoinositides used in this assay possess C6-acyl chains and are water soluble. The lipids are covalently linked to a fluorescent moiety, BODIPY, at the sn-1 position, through an amide linkage. Experiments were conducted by adding the indicated concentration of H7-WT or H7-Δ(119-146) to a solution containing 0.2 nM BODIPY-PI3P or BODIPY-PI4P (Echelon Biosciences) in binding buffer (20 mM Tris and 10 mM NaCl; pH 8.0), using a 100-μl final reaction volume. Protein-ligand samples were incubated for 30 s inside the chamber at 25°C prior to measuring the millipolarization (ΔmP). Data were analyzed using a two-tailed t test. Polarization filters were selected that matched the excitation and emission spectra (503 nm and 513 nm, respectively) for the BODIPY fluorescence label. All steps were performed under reduced light. Error bars represent standard errors of the means. The bar graph was generated via GraphPad Prism v.5 software.
Cells and viruses.
BSC-1 (BS-C-1; ATCC CCL-26) and BSC-H7 (a cell line derived from BS-C-1 cells expressing H7) (32) cells were cultured in minimum essential medium with Earle's balanced salts (Invitrogen) supplemented with 10% fetal bovine serum (FBS). The ΔH7 virus and all H7R VACV mutants were propagated on BSC-H7 cells, and crude viral stocks were prepared by lysing the infected cells via three freeze-thaw cycles and sonication in a cup horn sonicator, essentially as described previously (33). The viruses were titrated on BSC-H7 cells.
Construction of H7R mutant VACVs.
A PCR fragment containing the left and right flanking sequences of H7R and the coding sequences for H7 and green fluorescent protein (GFP) under the control of the VACV late-stage promoter P11 was assembled by recombinant PCR, essentially as described previously (34). The PCR fragment was TA cloned into the pGEM-T vector (Promega) to produce pWX22. The plasmids for making H7R mutant VACVs were all derived from pWX22 by use of recombinant PCR techniques. For each H7 mutation, a pair of mutagenesis primers with complementary sequences was used together with either a 5′ primer or 3′ primer in two separate PCRs. The resulting PCR products were then assembled by recombinant PCR and substituted for the H7R region of pWX22. VACVs expressing WT or various mutant H7 proteins were constructed by homologous recombination of the ΔH7 mutant with pWX22 or derivatives of pWX22, using standard methods (35). Briefly, pWX22 or its derivatives were transfected into BSC-H7 cells that were infected with the ΔH7 virus. Recombinant viruses encoding GFP and H7 were picked under a fluorescence microscope and purified by four rounds of plaque purification on BSC-H7 cells.
Plaque formation and growth curve analysis of H7R mutants.
BSC-1 or BSC-H7 cells in 12-well plates were incubated with different H7R mutants for 2 h at room temperature. Following adsorption, the cells were washed twice with phosphate-buffered saline (PBS) and moved to a 37°C incubator. For comparison of plaque morphologies, medium containing 0.5% methylcellulose was added, and plaques were visualized at 48 h postinfection (hpi), after staining with crystal violet. For growth curve analysis, infected cells were harvested at 0, 12, and 48 hpi. The viral titers in the cell lysates were determined by plaque assays on BSC-H7 cells. The results were confirmed by repeating the same experiment at least one time.
Western blot analysis.
BSC-H7 cells infected with various H7R mutants were harvested at 8 hpi. The cleared cell lysates were solubilized in sodium dodecyl sulfate (SDS) sample buffer, resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and blocked with Tris-buffered saline supplemented with 5% nonfat dried milk and 0.05% Tween 20 for 1 h at room temperature. Subsequently, the membranes were incubated with a mouse polyclonal antibody against H7 (32) or a monoclonal antibody against E3 (unpublished data), washed, incubated with horseradish peroxidase-conjugated secondary antibodies (Amersham), and analyzed with chemiluminescence reagents (Pierce).
Protein structure accession numbers.
The structure factors and atomic coordinates for full-length H7-C89S and H7-Δ(119-146) have been deposited in the Protein Data Bank under accession numbers 4W60 and 4W5X, respectively.
RESULTS
Crystal structure of H7.
H7 has no recognizable sequence motifs and no homologs outside the poxvirus family. To gain insight into the mechanism by which H7 functions, we set out to determine the structure of the H7 protein. Recombinant H7 protein was expressed and purified from E. coli. The wild-type (WT) H7 protein formed crystals that were of low quality and did not give good diffraction. However, an H7 protein with a cysteine-to-serine substitution at residue 89 (H7-C89S) yielded crystals of higher quality, which gave rise to a 2.7-Å structure. The structure includes all but the C-terminal 29 amino acids (residues 118 to 146). SDS-PAGE analysis of the crystal used for data collection showed a band identical to that for purified, full-length H7, excluding the possibility that the C terminus was degraded during crystallization (data not shown). We also found that there is a large unoccupied space at the C-terminal end of the H7 structure in the crystal packing lattices, which may accommodate the disordered C-terminal 29 residues. In addition, deletion of the C-terminal tail further improved the quality of the crystals, which diffracted to 2.0 Å (see Materials and Methods) (Table 1). The two structures are essentially identical, with a root mean square deviation (RMSD) of 0.55 Å over 116 aligned C-α atoms. Therefore, it appears that the C-terminal tail has a flexible conformation, which may be involved in dynamic associations with the core of H7.
H7 displays an L-shaped monomeric structure, comprised of seven α-helices (α1 to α7) and a highly curved three-stranded antiparallel β-sheet (β1, β2, and a very short β3) (Fig. 1). Helices α1, α2, α3, α6, and α7 form a compact core. The long helix α5 runs parallel to helix α2 and perpendicular to the short helices α4 and α6. The β-sheet packs tightly against helices α2 and α5 and is embraced by helices α3 and α4 and the intervening loop.
FIG 1.
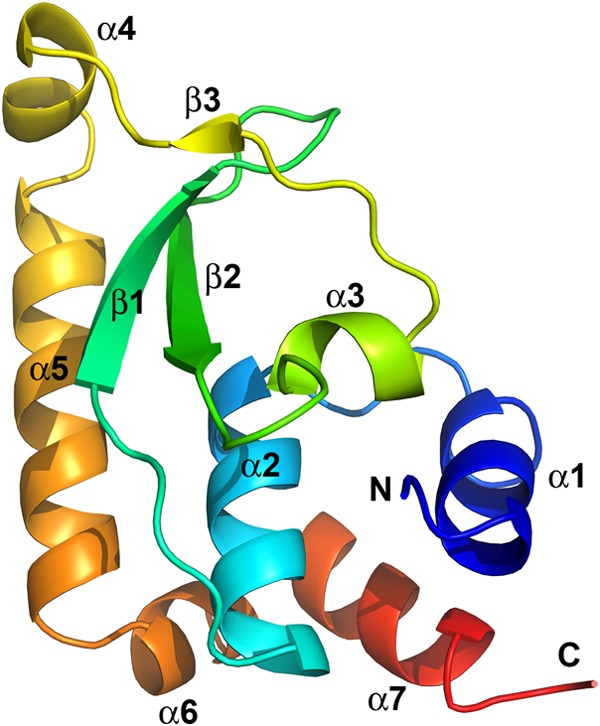
The structure of vaccinia virus H7 protein displays a novel fold. The secondary structures are shown as ribbon models and were colored in rainbow colors and labeled. The N and C termini of H7 are indicated. The C-terminal 29 residues (aa 118 to 146) are disordered and are not visible in this structure.
A structure similarity search using the SSM server (36) and the DALI server (37) did not yield any close matches to the H7 structure, suggesting that H7 adopts a novel fold. Lowering the threshold for acceptable similarity to below 50% for the aligned structures, however, yielded some hits. Among the top 20 hits, 5 were structures of PX domains, which have a globular fold of approximately 110 residues in length, composed of three β-strands followed by three α-helices (5). Comparison of the structures of one of the hits (the PX domain of the human p40 subunit of NADPH oxidase [PDB entry 1H6H] [38]) and H7 indeed showed substantial overall differences (Fig. 2A and B). Only 22% of the secondary structures could be aligned (RMSD of 3.17 Å over 53 C-α atoms that could be aligned). PX domains are found in cellular proteins that are often involved in intracellular protein trafficking, and each has an electropositive pocket that binds negatively charged phosphate groups of phosphoinositides (Fig. 2C). Despite the very limited structural similarity with PX domains, the structure of H7 displays a prominent surface patch comprised of basic residues, including K108, R109, and K112 (Fig. 2D). The basic surface patch, however, is present at a location on the protein that is different from that of the electropositive pocket in PX domains. Specifically, the phosphoinositide binding site on the PX domain of p40 is located at a basic cleft that is formed between two helices and the edge of the β-sheet. In contrast, the basic surface patch on H7 is located predominantly on the C-terminal helix α7 and away from the β-sheet (Fig. 2).
FIG 2.

Comparison of the structures of H7 and the PX domain. (A) The structure of the PX domain of the human p40 subunit of NADPH oxidase (PDB entry 1H6H) is shown as a ribbon. The bound PI3P is shown as a ball-and-stick model. (B) H7 is shown as a ribbon, and the basic surface residues (K108, R109, and K112) on helix α7 are shown as sticks, with dotted envelopes indicating the van der Waals radius. The color scheme is the same as that for Fig. 1. (C and D) Electropotential surfaces of the PX domain (PDB entry 1H6H) (C) and H7 (D). The PX domain uses a large basic pocket for lipid binding. Similarly, H7 also displays a prominent basic surface patch centered on a cluster of positively charged residues, including K108, R109, and K112.
VACV H7 binds PI3P and PI4P.
The structural features of H7 prompted us to examine whether H7 could bind phosphoinositides and whether the observed basic surface patch could be the binding site. A lipid overlay assay was first used to determine the specificities of lipid binding by the H7 protein (Fig. 3A). Purified recombinant H7 proteins (Fig. 3A, right panel) were incubated with a hydrophobic membrane spotted with various lipids. H7 proteins that bound to lipids after extensive washes were detected with the anti-H7 antibody. Full-length wild-type H7 protein bound PI3P and PI4P (Fig. 3A, left panel). In contrast, an H7 protein with K108E/R109E/K112E substitutions did not bind any lipids. In additional lipid overlay assays, H7-C89S also bound PI3P and PI4P, while the truncated H7 protein lacking the C-terminal tail [H7-Δ(119-146)] failed to bind the lipids (data not shown).
FIG 3.

H7 binds PI3P and PI4P. (A) Lipid overlay assay. Nitrocellulose strips containing 100-pmol spots of 15 different lipids were incubated with 1 μg/ml of the purified H7 protein, and the bound protein was detected by Western blotting with anti-H7 antibody. The numbers on the blots mark the positions of the lipids, and their names are given below the blots. The H7 proteins (WT or K108E/R109E/K112E mutant) used are indicated above the blots. Coomassie staining of the purified H7 protein used for lipid overlay is shown on the right. The sizes of molecular mass markers are indicated in kilodaltons. (B) Fluorescence polarization assay. A sample containing 0.2 nM BODIPY-labeled PI3P (top) or PI4P (bottom) was mixed with increasing concentrations of either H7-WT or H7-Δ(119-146). Binding was determined by measuring the change in millipolarization (ΔmP). (C) Binding experiments were conducted at a fixed concentration (20 μM) of either H7-WT or H7-Δ(119-146). Error bars represent standard errors of the means. Significance was found for differences between H7-WT and H7-Δ(119-146) (P < 0.01 by the two-tailed t test).
We also performed a fluorescence polarization-based lipid binding assay to provide a more quantitative comparison of PI3P and PI4P binding. For this purpose, BODIPY-labeled water-soluble PI3P and PI4P lipids were utilized. We observed an increase in polarization for wild-type H7 (Fig. 3B and C), which is indicative of binding. In contrast, H7-Δ(119-146) did not bind to the lipids. Due to the low affinity of these interactions, we were unable to saturate the binding with either phosphoinositide or protein. For H7-WT, millipolarization (ΔmP) as a function of protein concentration remained linear at 20 μM, consistent with an at least 5- to 10-fold higher equilibrium dissociation constant (Kd).
These data indicated that the H7 protein binds to PI3P and PI4P and that the binding requires the C-terminal tail (residues 119 to 146) and a positively charged surface patch centered on K108/R109/K112.
The putative lipid binding site on H7 is essential for viral replication.
To determine whether binding to phosphoinositides is essential for H7 function in viral replication, we performed site-directed mutagenesis of H7 and tested the effects of the mutations on viral replication. H7R (the gene that encodes H7) mutant VACVs were constructed by introducing mutated H7R into a previously constructed H7R deletion VACV (ΔH7) with a complementing cell line that stably expresses H7 (BSC-H7) (32). As expected, the ΔH7 virus and all H7R mutant VACVs replicated similarly to WT VACV on the complementing cell line (Fig. 4 and 5). The effects of the H7 mutations on viral replication were then determined by examining the plaque morphologies and growth curves for the individual mutant viruses on a regular cell line (BSC-1) (Fig. 4 and 5B). The results are summarized in Table 2.
FIG 4.

Plaque morphologies of H7 mutant VACVs. BSC-1 (top rows) and BSC-H7 (bottom rows) cells in 12-well plates were infected with the indicated H7R mutant VACVs in semisolid medium for 48 h. The cells were then stained with crystal violet to reveal plaques. For each mutant, the same amount of virus was used to infect BSC-1 and BSC-H7 cells. The mutants were studied in groups in four separate experiments (A to D), each with a WT control. Thus, the plaque sizes of different mutants can be compared within groups but not between groups.
FIG 5.

Growth curves of H7 mutant VACVs. BSC-1 (A) and BSC-H7 (B) cells in 12-well plates were infected with the indicated viruses at a multiplicity of infection (MOI) of 5 PFU/cell. Viral titers at 0, 24, and 48 h postinfection (hpi) were determined by plaque assay on BSC-H7 cells. The mutants were studied in groups in three separate experiments, each with a WT control.
TABLE 2.
Summary of phenotypes of H7 mutant VACVs
| H7 mutation(s)a | Plaque sizeb | One-step growthc |
|---|---|---|
| WT | ++++ | ++++ |
| K108E/R109E/K112E | − | − |
| K108E | − | + |
| R109E | +++ | +++ |
| K112E | + | ++ |
| K102E | ++++ | ND |
| K113E | ++++ | ++++ |
| R115E | ++++ | ND |
| F32A | ++++ | ND |
| K33E | ++++ | ND |
| C89S | ++++ | ++++ |
| Δ(119-146) | − | − |
| R121E | ++++ | ++++ |
| K128E | − | − |
| K130E | ++++ | ++++ |
| K135E | +++ | +++ |
| D138K | ++++ | ++++ |
| E140K | ++++ | ++++ |
| K143E | − | − |
| D144K | ++++ | ++++ |
Underlining indicates H7 mutants with impaired viral replication.
−, no plaques; ++++, WT plaque size.
−, no growth after 48 hpi; ++++, growth rate of WT virus; ND, not determined.
Since the H7 protein used to obtain the crystal structure had the C89S substitution, we evaluated the effect of H7-C89S on viral replication. VACV with the H7-C89S mutation formed plaques of a size similar to that for VACV with H7-WT on BSC-1 cells (Fig. 4C), indicating that the C89S substitution did not affect H7 function in viral replication. Indeed, the one-step growth curve analysis showed that the mutant virus was amplified to a level (>100-fold increase in titer) similar to that of WT VACV (Fig. 5A). In contrast, VACVs with H7 basic patch mutations (K108E/R109E/K112E) (Fig. 4A) or a C-terminal deletion [H7-Δ(119-146)] (Fig. 4C) failed to form plaques on BSC-1 cells, and their titers decreased in the one-step growth curve analysis (Fig. 5A). These data indicate that these mutations disrupt H7 function.
To pinpoint the residues that are essential for H7 function, we constructed mutant viruses with single amino acid substitutions in H7. The K108E, R109E, and K112E substitutions all negatively affected H7 function in viral replication, but to different degrees (Fig. 4A and 5A). The K108E mutation had the greatest effect, as VACV with the H7-K108E substitution did not form plaques on BSC-1 cells, and its titer did not increase after 48 h of infection on BSC-1 cells. The R109E mutation had a relatively minor effect, as VACV with the H7-R109E substitution formed plaques on BSC-1 cells, but the plaque size was slightly reduced compared to that of WT VACV. Accordingly, after 48 h of infection on BSC-1 cells, the titer of the mutant virus increased ∼70-fold, instead of ∼150-fold for WT VACV. The titer difference, however, did not reach statistical significance (Student's t test; P = 0.3). The K112E mutation had an intermediate effect, as VACV with the H7-K112E mutation formed very small plaques on BSC-1 cells, and its titer increased only ∼10-fold after 48 h of infection. Several charged or hydrophobic residues located near the K108/R109/K112 residues were also mutated, but none of these single amino acid substitutions (F32A, K33E, K102E, K113E, and R115E) had any effect on H7 function in viral replication (Fig. 4B).
We performed similar studies on all charged residues present in the C-terminal tail of H7. While the R121E, K130E, D138K, E140K, and D144K single amino acid substitutions had no negative effect on H7 function, the K128E and K143E mutations completely disrupted the function of H7 in viral replication (Fig. 4D and 5A). VACV with either the H7-K128E or H7-K143E substitution did not form plaques on BSC-1 cells, and the titer did not increase after 48 h of infection of BSC-1 cells. The K135E mutation may have a minor effect on H7 function, as the titer of VACV with H7-K135E increased ∼85-fold, instead of >150-fold for WT VACV. The titer difference, however, did not reach statistical significance (Student's t test; P = 0.2).
To assess whether the H7 mutations had any significant impact on H7 protein stability, the levels of H7 protein expressed by the mutant VACVs in BSC-H7 cells were determined by Western blotting. In the same Western blot, the level of an unrelated viral protein, E3, was also determined as an infection and loading control (Fig. 6). The Western blot indicated that none of the H7 mutations that disrupted H7 function had any negative effects on the stability of the H7 protein. Together, these data show that specific positively charged residues around K108 and in the C-terminal tail are essential for viral replication, correlating with a role of these residues in phosphoinositide binding.
FIG 6.

Western blots of viral proteins expressed by H7 mutant VACVs. BSC-H7 cells were infected with the indicated viruses at an MOI of 5 PFU/cell. The levels of H7 and E3 proteins at 8 hpi were determined by Western blotting with antibodies against H7 and E3, respectively. E3 is a viral protein that is unrelated to H7 and served as a control for infectivity and gel loading. The sizes of molecular mass markers are shown in kilodaltons. The mutants were studied in groups in four separate experiments, each with a WT control.
DISCUSSION
In this study, we determined the crystal structure of VACV H7, which is one of the poxvirus viral membrane assembly proteins (VMAPs) (21). VMAPs are proteins that were recently discovered, through viral genetics, to be essential for poxvirus membrane biogenesis. VACV mutants lacking any one of the VMAPs have very similar defects (11, 17, 19, 20, 32), with viral assembly arrested at similar stages right before the formation of crescent membranes, the precursors of the viral envelope. There are at least 5 proteins in this group, varying in size from 5 to 20 kDa for A30.5, L2, and H7 and around 40 kDa for A11 and A6. A6 and H7 are localized predominantly in the cytosol (18, 32, 39), while L2 and A30.5 are localized in the ER (19, 21), and A11 associates with viral membranes (40, 41). How these diverse proteins function together to facilitate acquisition of membranes from the ER is an intriguing question. However, a further mechanistic understanding of this process is greatly hindered by a lack of understanding of any biochemical functions of the VMAPs. As VMAPs lack any sequence homology to proteins outside the poxvirus family, structural studies of VMAPs represent an avenue for gaining novel insights into the process of poxvirus membrane biogenesis.
Protein engineering played a critical role in our successful determination of the H7 crystal structure. Specifically, the C89S mutation drastically improved the quality of the crystal, leading to the determination of the structure. The three Cys residues in the H7 protein (C76, C89, and C146) are too far from each other to form any intramolecular disulfide bonds. However, C89 is surface exposed and may potentially form intermolecular disulfide bonds with Cys residues in another H7 molecule. Since the H7 protein is predominantly monomeric in solution, as determined by size-exclusion chromatography (data not shown), any intermolecular disulfide bonds are likely nonspecific. The C89S mutation probably eliminates the nonspecific intermolecular disulfide bond and consequently improves protein homogeneity and protein crystal quality. The C89S substitution had no negative effect on H7 function in viral replication, further confirming that any intermolecular disulfide bond involving C89 is biologically irrelevant.
Consistent with the fact that H7 has no sequence homology outside the poxvirus family, the structure of H7 is rather novel and dissimilar to any existing structures in the Protein Data Bank (PDB). Its secondary structure and surface properties, however, have some similarity to those of the PX domains, providing some clues to H7 function. Notably, H7 has a basic surface patch that appears to be suitable for binding phosphoinositides. We used two biochemical assays to test whether H7 indeed binds to phosphoinositides. The lipid overlay assay showed that H7 bound to PI3P and PI4P immobilized on a nitrocellulose membrane. The fluorescence polarization experiments confirmed the binding of H7 with PI3P and PI4P in solution and indicated that the affinity is on the order of 100 to 200 μM. Importantly, reversing the charges of three positively charged residues or truncating the C-terminal tail disrupted lipid binding. The affinity of H7 for phosphoinositides is rather modest, but it is comparable to the typical lipid binding affinities of the PX domains, which vary from nanomolar to millimolar values depending on the PX domain studied (5).
The recent development of an H7 deletion VACV mutant and a complementary cell line greatly facilitated the structure-function analysis of this essential protein (32). Previously, the functionality of mutant alleles of essential poxvirus genes could be assessed only by a transient-complementation assay (42), in which mutant alleles were transfected into cells to transiently complement a virus that was repressed for expression of the endogenous gene. With the H7 complementary cell line, we could now generate mutant H7R VACVs with lethal phenotypes and test the mutants without any ambiguity caused by the presence of the wild-type H7 allele. Through structure-guided mutagenesis and analysis of H7 mutant viruses, we uncovered two regions of H7 that are essential for viral replication. One region is a basic surface patch centered on K108, and the other region is the flexible C-terminal tail, specifically two basic residues on the C-terminal tail (K128 and K143). Our in vitro lipid binding assays further demonstrated that both regions are required for the binding of H7 to PI3P and PI4P. These data suggested that phosphoinositide binding by H7 is essential for viral replication. The structure of H7 implied that the C-terminal tail of H7 may be dynamic in solution. Our cumulative data suggested a model in which the two regions of H7 together form a positively charged composite pocket for binding of phosphoinositides, and the C-terminal tail of H7 might serve as a switch for binding and release of phosphoinositides. As the C-terminal tail was not visible in the H7 structure, a de novo model of the tail was constructed and combined with the H7 structure to create a model of the full-length H7 structure (Fig. 7). This model shows a large, positively charged composite pocket including the basic patch centered at K108/R109/K112 and additional basic residues (K128 and K135) from the C-terminal tail (Fig. 7). It will be interesting to determine the structure of full-length H7 in complex with phosphoinositides to provide more insights into the detailed mechanism of phosphoinositide binding and regulation by H7.
FIG 7.
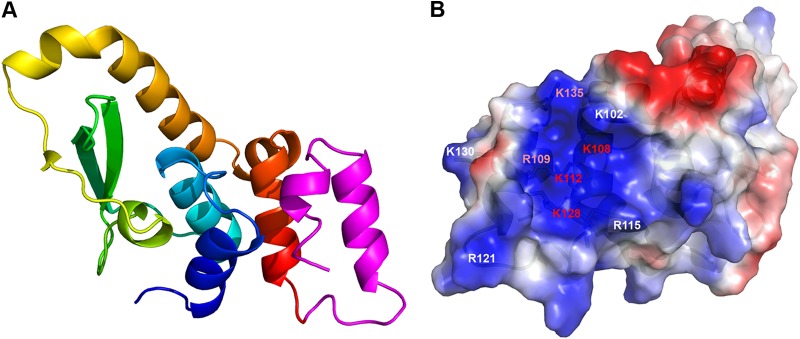
Modeled structure of full-length H7. The structure of full-length H7 was modeled with the I-TASSER server (43) by using the crystal structure of H7(1-118) as the template. The model with the highest confidence score (C score) as suggested by I-TASSER was selected. (A) The modeled full-length H7 structure is shown as a ribbon. The part of H7 observed from the crystal structure is colored as in Fig. 2B. The modeled C terminus (residues 119 to 146) is shown in magenta. (B) Electropotential surface of the modeled full-length H7 protein. The basic residues at the putative lipid binding pocket are indicated and labeled in color according to their importance for H7 function, as determined by the experiments shown in Fig. 5 and 6. Red, white, and salmon represent essential, nonessential, and modestly important regions, respectively.
PI3P and PI4P are present primarily in the Golgi apparatus and early endosomes, respectively. Although the H7 protein binds to PI3P and PI4P, the H7 protein in infected cells does not localize to any particular cellular organelle and appears to localize predominantly in the cytosol (18, 32). This is not surprising, as the specificity for membrane attachment may require other protein partners in vivo. Most of the mammalian PX domain-containing proteins harbor additional domains with diverse functions (5). Apart from lipid binding by the PX domains, these additional domains also independently bind membrane lipids or other membrane-associated proteins, driving and enhancing membrane attachment through “coincident detection” (1). Therefore, it is possible that H7 associates with other VMAPs and that together they specifically bind to certain membrane structures.
Although our data suggest that phosphoinositide binding by H7 is essential for viral replication, its exact role in poxvirus membrane biogenesis remains unclear. Poxvirus crescent membranes are unusual membrane structures in cells, as they appear to be open membrane structures that may have derived from breaking of the cellular membranes (16, 21). One possible function for H7 may be to target VMAPs to cellular membranes to generate breaks. Alternatively, H7 may bind to lipids at the ends of crescent membranes to prevent fusion of the ends. As all cellular membranes are closed, lipid-binding viral proteins, such as H7, may be required to stabilize the open-ended crescent membranes. Further studies of H7 and other VMAPs will be necessary to elucidate the mechanisms used by this class of poxviral assembly proteins.
ACKNOWLEDGMENTS
We thank the staff of beamline 19-ID at the Advanced Photon Source for their generous support.
This work was supported by NIH grants AI113539 (J.D.), AI079217 (Y.X.), and AI053531 (C.E.C.) and by the Oklahoma Agricultural Experiment Station at Oklahoma State University, under project OKL02848 (J.D.).
We declare that we have no competing interests.
REFERENCES
- 1.Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 2.Balla T. 2013. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93:1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemmon MA. 2008. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol 9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- 4.Kutateladze TG. 2010. Translation of the phosphoinositide code by PI effectors. Nat Chem Biol 6:507–513. doi: 10.1038/nchembio.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teasdale RD, Collins BM. 2012. Insights into the PX (phox-homology) domain and SNX (sorting nexin) protein families: structures, functions and roles in disease. Biochem J 441:39–59. doi: 10.1042/BJ20111226. [DOI] [PubMed] [Google Scholar]
- 6.Delang L, Paeshuyse J, Neyts J. 2012. The role of phosphatidylinositol 4-kinases and phosphatidylinositol 4-phosphate during viral replication. Biochem Pharmacol 84:1400–1408. doi: 10.1016/j.bcp.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishe B, Syed G, Siddiqui A. 2012. Phosphoinositides in the hepatitis C virus life cycle. Viruses 4:2340–2358. doi: 10.3390/v4102340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moss B. 2007. Poxviridae: the viruses and their replication, p 2905–2946 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 11.Maruri-Avidal L, Weisberg AS, Bisht H, Moss B. 2013. Analysis of viral membranes formed in cells infected by a vaccinia virus L2-deletion mutant suggest their origin from the endoplasmic reticulum. J Virol 87:1861–1871. doi: 10.1128/JVI.02779-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krijnse Locker J, Chlanda P, Sachsenheimer T, Brugger B. 2013. Poxvirus membrane biogenesis: rupture not disruption. Cell Microbiol 15:190–199. doi: 10.1111/cmi.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sodeik B, Doms RW, Ericsson M, Hiller G, Machamer CE, van't Hof W, van Meer G, Moss B, Griffiths G. 1993. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J Cell Biol 121:521–541. doi: 10.1083/jcb.121.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNulty S, Bornmann W, Schriewer J, Werner C, Smith SK, Olson VA, Damon IK, Buller RM, Heuser J, Kalman D. 2010. Multiple phosphatidylinositol 3-kinases regulate vaccinia virus morphogenesis. PLoS One 5:e10884. doi: 10.1371/journal.pone.0010884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res 66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- 16.Chlanda P, Carbajal MA, Cyrklaff M, Griffiths G, Krijnse-Locker J. 2009. Membrane rupture generates single open membrane sheets during vaccinia virus assembly. Cell Host Microbe 6:81–90. doi: 10.1016/j.chom.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 17.Resch W, Weisberg AS, Moss B. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J Virol 79:6598–6609. doi: 10.1128/JVI.79.11.6598-6609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satheshkumar PS, Weisberg A, Moss B. 2009. Vaccinia virus H7 protein contributes to the formation of crescent membrane precursors of immature virions. J Virol 83:8439–8450. doi: 10.1128/JVI.00877-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maruri-Avidal L, Domi A, Weisberg AS, Moss B. 2011. Participation of vaccinia virus L2 protein in the formation of crescent membranes and immature virions. J Virol 85:2504–2511. doi: 10.1128/JVI.02505-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng X, Embry A, Rose L, Yan B, Xu C, Xiang Y. 2012. Vaccinia virus A6 is essential for virion membrane biogenesis and localization of virion membrane proteins to sites of virion assembly. J Virol 86:5603–5613. doi: 10.1128/JVI.00330-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maruri-Avidal L, Weisberg AS, Moss B. 2013. Direct formation of vaccinia virus membranes from the endoplasmic reticulum in the absence of the newly characterized L2-interacting protein A30.5. J Virol 87:12313–12326. doi: 10.1128/JVI.02137-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krumm B, Meng X, Li Y, Xiang Y, Deng J. 2008. Structural basis for antagonism of human interleukin 18 by poxvirus interleukin 18-binding protein. Proc Natl Acad Sci U S A 105:20711–20715. doi: 10.1073/pnas.0809086106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. 1993. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol 229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 24.Deng J, Davies DR, Wisedchaisri G, Wu M, Hol WG, Mehlin C. 2004. An improved protocol for rapid freezing of protein samples for long-term storage. Acta Crystallogr D Biol Crystallogr 60:203–204. doi: 10.1107/S0907444903024491. [DOI] [PubMed] [Google Scholar]
- 25.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. 2006. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 26.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. 2004. Coot: model building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeLano WL. 2002. The PyMOL molecular graphics system. http://www.pymol.org.
- 31.Berman H, Henrick K, Nakamura H. 2003. Announcing the worldwide Protein Data Bank. Nat Struct Biol 10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 32.Meng X, Wu X, Yan B, Deng J, Xiang Y. 2013. Analysis of the role of vaccinia virus H7 in virion membrane biogenesis with an H7-deletion mutant. J Virol 87:8247–8253. doi: 10.1128/JVI.00845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Earl PL, Cooper N, Wyatt LS, Moss B, Carroll MW. 2001. Preparation of cell cultures and vaccinia virus stocks. Curr Protoc Mol Biol Chapter 16:Unit16.16. doi: 10.1002/0471142727.mb1616s43. [DOI] [PubMed] [Google Scholar]
- 34.Meng X, Xiang Y. 2006. Vaccinia virus K1L protein supports viral replication in human and rabbit cells through a cell-type-specific set of its ankyrin repeat residues that are distinct from its binding site for ACAP2. Virology 353:220–233. doi: 10.1016/j.virol.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 35.Moss B, Earl PL. 2002. Overview of the vaccinia virus expression system. Curr Protoc Mol Biol Chapter 16:Unit16.15. doi: 10.1002/0471142727.mb1615s60. [DOI] [PubMed] [Google Scholar]
- 36.Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 37.Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bravo J, Karathanassis D, Pacold CM, Pacold ME, Ellson CD, Anderson KE, Butler PJ, Lavenir I, Perisic O, Hawkins PT, Stephens L, Williams RL. 2001. The crystal structure of the PX domain from p40(phox) bound to phosphatidylinositol 3-phosphate. Mol Cell 8:829–839. doi: 10.1016/S1097-2765(01)00372-0. [DOI] [PubMed] [Google Scholar]
- 39.Meng X, Embry A, Sochia D, Xiang Y. 2007. Vaccinia virus A6L encodes a virion core protein required for formation of mature virion. J Virol 81:1433–1443. doi: 10.1128/JVI.02206-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu X, Meng X, Yan B, Rose L, Deng J, Xiang Y. 2012. Vaccinia virus virion membrane biogenesis protein A11 associates with viral membranes in a manner that requires the expression of another membrane biogenesis protein, A6. J Virol 86:11276–11286. doi: 10.1128/JVI.01502-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maruri-Avidal L, Weisberg AS, Moss B. 2013. Association of the vaccinia virus A11 protein with the endoplasmic reticulum and crescent precursors of immature virions. J Virol 87:10195–10206. doi: 10.1128/JVI.01601-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mercer J, Traktman P. 2003. Investigation of structural and functional motifs within the vaccinia virus A14 phosphoprotein, an essential component of the virion membrane. J Virol 77:8857–8871. doi: 10.1128/JVI.77.16.8857-8871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]