

ABSTRACT
Nuclear delivery of the adenoviral genome requires that the capsid cross the limiting membrane of the endocytic compartment and traverse the cytosol to reach the nucleus. This endosomal escape is initiated upon internalization and involves a highly coordinated process of partial disassembly of the entering capsid to release the membrane lytic internal capsid protein VI. Using wild-type and protein VI-mutated human adenovirus serotype 5 (HAdV-C5), we show that capsid stability and membrane rupture are major determinants of entry-related sorting of incoming adenovirus virions. Furthermore, by using electron cryomicroscopy, as well as penton- and protein VI-specific antibodies, we show that the amphipathic helix of protein VI contributes to capsid stability by preventing premature disassembly and deployment of pentons and protein VI. Thus, the helix has a dual function in maintaining the metastable state of the capsid by preventing premature disassembly and mediating efficient membrane lysis to evade lysosomal targeting. Based on these findings and structural data from cryo-electron microscopy, we suggest a refined disassembly mechanism upon entry.
IMPORTANCE In this study, we show the intricate connection of adenovirus particle stability and the entry-dependent release of the membrane-lytic capsid protein VI required for endosomal escape. We show that the amphipathic helix of the adenovirus internal protein VI is required to stabilize pentons in the particle while coinciding with penton release upon entry and that release of protein VI mediates membrane lysis, thereby preventing lysosomal sorting. We suggest that this dual functionality of protein VI ensures an optimal disassembly process by balancing the metastable state of the mature adenovirus particle.
INTRODUCTION
Adenoviruses (AdVs) are nonenveloped, double-stranded DNA viruses that assemble in the nuclei of productively infected cells and are released at the end of the infection cycle into the extracellular milieu. Productive infection of new cells requires that the capsid follow a stepwise disassembly process upon entry that, if perfectly executed, results in highly efficient genome transfer to the nucleus (1, 2).
The AdV virion is composed of 13 different polypeptides that form an icosahedral capsid encompassing the genome-containing viral core. The capsid is mainly composed of trimeric hexons building up the facets of the capsid. Pentons and fibers are located at each of the 12 vertices, where they form a pentameric penton base from which the trimeric fiber molecule elongates (3–5). In addition the capsid is stabilized via the cement proteins IIIa, VI, VIII, and IX. The capsid encloses the viral core, with the viral genome organized into chromatin through association with the major core protein VII and proteins V, X, TP, and IVa2 (3–5). Following (or concomitant with) capsid assembly, several of the virion proteins undergo proteolytic processing by the virion-incorporated adenoviral proteinase (AVP) (6). This process of virus maturation is essential to render newly assembled particles infectious (7). The assembly process itself does not require capsid maturation and incorporation of the AVP into the particle (6, 8). The temperature-sensitive human adenovirus serotype 2 mutant HAdV-C2-PRO-P137L (also termed ts1) was initially discovered in a chemical mutagenesis screen for growth-arrested viruses at the restrictive temperature of 38.5°C compared to the permissive temperature of 33°C (9). Subsequent analysis showed that the phenotype relies on a single point mutation in the AVP (P137L) that prevents virion incorporation of the AVP (8, 10). Genome-containing PRO-P137L capsids show a lack of proteolytic maturation, resulting in hyperstable capsids, presumably due to nonprocessed interactions between cement proteins and the viral core (11, 12). PRO-P137L particles can be produced and purified to the same extent as their mature counterparts. In contrast to mature viruses, PRO-P137L particles show severely reduced infectivity. While binding and endocytosis are efficient, postentry sorting is different from that of mature viruses, and PRO-P137L particles are targeted into lysosomes for degradation (13).
Several studies, mainly performed on human adenovirus serotype 5 (HAdV-C5), show that mature virions exit the endocytic compartment during or shortly after uptake to traverse the cytosol for efficient nuclear delivery (14–16). This process involves a capsid-associated membrane-lytic activity absent from PRO-P137L particles. Initially believed to be a penton, subsequent in vitro analysis and in vivo observations have identified the internal capsid protein VI as the membrane lytic factor of AdV (17, 18). Unprocessed, i.e., precursor protein VI (pVI) is involved in virus assembly by translocating the hexon into the nucleus during progeny virion production (19). Upon maturation, protein VI is processed by the AVP at both its N and C termini (20). The cleaved C-terminal peptide serves as a coactivator of the viral protease (20, 21). The N-terminal region of processed protein VI forms an amphipathic helix capable of membrane association and lysis in vitro and in vivo (18, 22, 23). Prior to assembly and in the assembled virion, the helix is shielded through association with hexons (12, 24). A recent random-mutagenesis approach to the amphipathic helix performed in the viral backbone of HAdV-C5 showed that it is indispensable for the viability of the virus (25). It was reported that partially breaking the N-terminal helix by introducing the mutation PVI-L40Q reduces membrane lysis and viral infectivity without affecting thermally induced capsid destabilization. Upon entry, HAdV-C5–PVI-L40Q virions were found to associate with the endosomal compartment for prolonged times compared to wild-type (wt) virions (25). It was shown that in intact virions, protein VI remains inaccessible to protein VI-specific antibodies (26). Following uptake into cells, protein VI is liberated and thus becomes accessible to antibody staining, providing direct evidence for entry-dependent disassembly and protein VI release (26). A recent study using cellular markers of membrane damage provided the first experimental system to show in vivo visualization of the membrane lysis step of AdV entry, demonstrating that membrane damage coincided with protein VI deployment from the AdV capsid (16, 22, 23). The work showed that membrane lysis and parting of the remaining capsid from the damaged membrane compartment are separated in time and subcellular space, functionally discriminating between the two events. The entry-associated sorting of endocytosed capsids and the exact compartment where and mechanisms by which disassembly, protein VI release, and membrane lysis and/or separation from the membrane compartment occur remain controversial, and it was suggested that differences in the cell types or the genotypes of the viruses used in different studies may explain the inconsistencies (27–29).
Cell-based assays suggested that disassembly starts at the cell surface by mechanical forces executed through differential receptor binding, releasing fiber and inducing conformational changes in pentons through integrin binding, ultimately allowing protein VI release, although it remains unclear how disassembly and protein VI release are linked (1, 2, 30, 31).
Protein VI release from the capsid can be triggered in vitro through mild heat treatment of purified capsids, which releases the whole vertex region, including peripentonal hexons, leaving the remaining capsid intact (18). The combination of crystallography and image reconstruction using cryo-electron microscopy (cryo-EM), has provided a detailed picture of the structural organization of the viral capsid. A recent detailed mass spectroscopic (MS) analysis of the AdV capsid confirmed previous estimations of ∼360 copies of protein VI per virion (32). Most tentative allocations based on high-resolution structures place protein VI at the inner capsid surface under the penton/vertex region, with the N termini of protein VI contacting peripentonal hexons (5, 33), or in the internal cavity of the hexon trimer (11, 34, 35). Recently, a large part of the structure of protein VI was assigned using a full-particle crystallography approach (36). However, the assigned location of protein VI is in disagreement with a cryo-EM-based high-resolution model that assigns the same density to protein IIIa (34). Based on in vitro disassembly studies and the provisional structure model, two hypothetical modes of capsid disassembly and protein VI release have been proposed: starting with penton and fiber release (as a unit or sequentially), followed by release of the peripentonal hexons, thus liberating protein VI (12, 37); alternatively, initial fiber and penton release also release protein VI, while capsid disintegration and peripentonal hexon release occur only after translocation to the nuclear pore (11, 38).
In this study, we use AdVs with a mutated amphipathic helix or AdVs that lack protein VI release and show that membrane lysis, in addition to protein VI release, is required to save viruses from lysosomal targeting. Furthermore, our data identify a role for the amphipathic helix in maintaining the metastability of the AdV capsid by preventing premature penton release and protein VI exposure. Based on our observations, we suggest a refined model for the structural changes taking place upon AdV entry.
MATERIALS AND METHODS
Cell culture and transfection.
U2OS cells (ATCC HTB-96), HEK293 cells (ATCC CRL-1573), and A549 cells (ATCC CCL-185) were grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal calf serum (FCS) (Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) in a 5% CO2 atmosphere at 37°C. Transfections were done using Lipofectamine 2000 and OptiMEM (Life Technologies) according to the manufacturer's instructions.
BAC mutagenesis and virus production and labeling.
Bacterial artificial chromosome (BAC) mutagenesis was used to generate recombinant adenoviruses following a protocol described previously (26, 39), with the following modifications: bp 1 to 3513 of the HAdV-C5 genome, including the left inverted terminal repeat (ITR) and the whole E1 region, was replaced by a flippase recognition target (FRT) site and maintained as a BAC to perform lambda red recombination essentially as described in reference 39. Briefly, BAC mutagenesis is achieved by first replacing the target sequence in the viral genome with a selectable marker (GalK/Amp) through homologous recombination. In the second step, the mutated genomic region is recombined into the viral genome, replacing the marker. Following this principle, the L40Q mutation was introduced into the protein VI open reading frame in pBA5-FRT (39), and the P137L mutation was inserted into the viral PRO open reading frame separately. To reconstitute the left ITR, as well as to introduce a cytomegalovirus (CMV) promoter-driven green fluorescent protein (GFP) expression cassette, Flp recombination was used to recombine plasmids pO6-Ad5-CMV-GFP (encoding the left ITR and CMV-GFP) and modified pBA5-FRT and its mutant derivatives (carrying most of the genome and the right ITR) to reconstitute GFP expressing HAdV-C5 BAC with E1 and E3 deleted (BxAd5GFP) and mutants BxAd5GFP-VI-wt and -L40Q and BxAd5GFP-P137L, respectively. Vectors BxAd5GFP, BxAd5GFP-VI-L40Q, and BxAd5GFP-P137L BAC were digested with a PacI restriction enzyme to release the viral genome and transfected into HEK293 cells to reconstitute the recombinant viruses. The viruses were amplified in HEK293 cells and purified by double banding on CsCl gradients, extensively dialyzed against phosphate-buffered saline (PBS)-10% glycerol, and stored in aliquots at −80°C. The viruses were quantified as physical particles per microliter following a method described previously (40). The concentrated and dialyzed viruses were labeled with Alexa 488 dye using a small-scale protein-labeling kit (Invitrogen) as described previously (16).
Western blotting and Coomassie gels.
SDS-PAGE was done using 11% polyacrylamide (40% acrylamide and bis-acrylamide solution; 37.5:1; Bio-Rad) gels and either stained with Coomassie blue or transferred to nitrocellulose membranes. The membranes were blocked in Tris-buffered saline (TBS)-0.1% Tween 20 containing 10% dry milk, followed by overnight detection of antigens using primary antibodies diluted in TBS-0.1% Tween 20 containing 10% dry milk. Primary antibodies were detected using species-specific horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma) at a dilution of 1:5,000. Specific signals were revealed using the enhanced-chemiluminescence (ECL) detection system (Pierce).
Immunofluorescence.
Cells grown on coverslips were synchronously infected with virus by prebinding the virus at 4°C for 30 min, followed by a shift to 37°C. At the indicated time points, the cells were rinsed in PBS, fixed for 20 min with 4% paraformaldehyde (electron microscopy grade; Electron Microscopy Sciences) in PBS, and blocked/permeabilized for 5 min with IF buffer (10% FCS in PBS and 0.2% saponin). Primary and secondary antibodies where applied to the coverslip in IF buffer for 1 h each. The cells were mounted in mounting medium (Fluorescence Mounting Medium; Dako) containing 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole dihydrochloride; Sigma) and analyzed by confocal microscopy. The antibodies used in this study were monoclonal antibody (MAb) 9F10 against protein VI (see below), rabbit anti-penton (kindly provided by G. Nemerow), rat anti-protein VII (kindly provided by K. Nagata), rabbit anti-Ad5 (kindly provided by R. Iggo), MAb 3C2-2A3 against galectin 3 (Gal-3) (Sigma), and MAb H4B4 against lamp-2 (Santa Cruz). The secondary antibodies Alexa 546 (Molecular Probes) and Atto647 (Sigma) were used as recommended by the supplier. Image analysis and colocalization studies were done in batch analysis using an automated macro in ImageJ. Briefly, individual cells were imaged, and for each channel, a particle analysis was performed and used to create a mask identifying objects (e.g., virus particles or stain for a cellular marker) by using the size/intensity threshold followed by automatic quantification of colocalization between the channels. Over 500 particles were analyzed for each time point in at least two independent experiments.
Monoclonal antibody production.
Monoclonal antibody generation against capsid protein VI from HAdV-C5 was done as previously described (41). Briefly, 20 μg of recombinant purified protein VI was emulsified with complete Freund's adjuvant and injected into each footpad of the back legs of BALB/c mice (n = 3). Twelve days later, the mice were boosted with incomplete Freund's adjuvant, and at day 15, popliteal lymph nodes were collected and B cells were extracted and fused with the myeloma partner P3U1 in polyethylene glycol (PEG) 1500 (Roche Diagnostic). Hybridomas were grown until confluent and screened by indirect enzyme-linked immunosorbent assay (ELISA). Nunc MaxiSorp flat 96-well plates were coated with 0.5 μg of recombinant protein VI in carbonate buffer and saturated with 0.2% gelatin. After PBS washes, 50 μl of hybridoma supernatants was added and incubated for 1 h at 37°C, washed, and detected with HRP-conjugated secondary anti-mouse IgG and IgM antibodies (Jackson ImmunoResearch; 1:5,000). Signals were revealed using 0.015% ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)[-H2O2, and absorbance was measured at 405 nm after the PBS washes. Hybridomas, which secreted high levels of specific antibody to protein VI, were cloned by limiting dilutions and screened by immunofluorescence on infected cells. MAb 9F10-B2 (IgG1) was selected for its highly specific signal and used as described above. The antibody recognizes mature protein VI and the precursor of protein VI equally well.
FACS assay.
For fluorescence-activated cell sorter (FACS) analysis, 5 × 104 U2OS cells were seeded per well of a 24-well plate, transduced with the GFP-expressing AdV constructs at 100 physical particles per cell (pp/c), and infected for 3 h at 37°C, followed by removal of viruses that were not endocytosed by the cell. Cells were harvested 24 h later and analyzed without fixation and additional staining in PBS containing 2% FCS, and GFP-specific fluorescence was recorded with a Canto II cytometer (BD Biosciences). Acquisitions were controlled and FACS data were processed and analyzed with FACS DIVA software (BD Biosciences) according to the manufacturer's instructions.
Sarcin assay.
Sarcin assays were performed as described previously (26) with some minor modifications. A549 cells were seeded at 5 × 104 in 48-well plates (Corning) the day before the experiment. Viruses diluted in 0.2 mg/ml of the translational inhibitor α-sarcin (Sigma) containing RPMIst (Roswell Park Memorial Institute starvation medium; Sigma), 10 mM HEPES, pH 7.4, and l-glutamine were preadsorbed for 30 min at room temperature, followed by 90 min at 37°C. Fresh RPMIst containing 0.4 μCi [35S]methionine/cysteine (l-[35S]methionine/cysteine, 0.10 mCi; Hartmann Analytic) was added for another 1 h at 37°C. The cells were washed with PBS; 200 μl lysis buffer containing 1% Triton X-100, 150 mM NaCl, 20 mM Tris-HCl (pH 8) supplemented with Complete protease inhibitor cocktail (Roche) was added to the cells; and the plate was stored at −20°C overnight. The lysates were harvested the following day and clarified by centrifugation at 20,000 × g for 15 min at 4°C. To remove the residual free l-[35S]methionine, 100 μl cleared lysates was further purified with Zeba Desalt Spin Columns (Thermo Scientific). l-[35S]methionine/cysteine incorporation was measured as counts per minute by liquid scintillation (200 μl OptiPhase Supermix Cocktail plus 50 μl purified lysate) using a MicroBeta counter (PerkinElmer).
Disassembly assay and DMF treatment.
Snap-frozen aliquots of CsCl-purified viruses were subject to 3 or 4 consecutive freeze-thaw cycles by freezing the viruses at −80°C (freeze) followed by thawing on ice for ∼20 min (thaw). Cells were treated with dimethylformamide (DMF) for 3 h, followed by infection with virus diluted in medium containing either no DMF or 130 mM or 260 mM DMF for 3 h and then replaced with fresh medium without DMF. Virus infectivity was scored using FACS analysis 24 h postinfection.
Cryo-electron tomography.
Cryo-electron tomography (cryo-ET) was performed at 300 keV, using a TF30 Polara electron microscope (FEI). Low-dose, zero-loss images were acquired with SerialEM (42) in energy-filtered transmission electron microscopy (EFTEM) mode with a Quantum 964 (Gatan) postcolumn energy filter operated with a 20-eV-wide energy-selecting slit. Images of wt and PVI-L40Q purified particles were recorded at a nominal magnification of ×34,000, sampling the specimens at a calibrated pixel size of 1.16 nm after binning at a factor of 2. Tomographic tilt series were collected in 3° increments covering an angular range from −50° to +54° and −6-μm to −8-μm defocus using SerialEM (42). The total electron doses for the tilt series were kept between 75 and 85 electrons/Å2. Tomographic reconstructions were calculated in IMOD (43) using weighted back projection (41). The visualization of reconstructed tomograms was done in Amira 5.4 (Visage Imaging).
Virus particle picking and subtomogram averaging.
One hundred ten wt and 100 PVI-L40Q virus particles were picked from the tomograms for subtomogram averaging using the 3dmod program from IMOD (41, 43). Subtomogram averaging was then performed using PEET version 1.9 (44), with one particle from each data set as the initial reference for particle alignment. No symmetry was applied at that stage. Four iterations of alignment were used to create the initial nonsymmetrized map. Icosahedral symmetry was then applied using Bsoft (45, 46). Finally, an inner mask was applied to the average, resulting in the final three-dimensional (3D) reconstruction maps corresponding to the capsid shell. The resolution of each averaged structure was determined by Fourier shell correlation (FSC), calculated between two maps generated from two independent half-sets of the data using a threshold of 0.143 (47), at 4.6-nm and 4.3-nm resolution for the wt and PVI-L40Q structures, respectively.
Difference map calculation and atomic-model fitting.
3D reconstructions of the PVI-L40Q and wt final symmetrized maps were normalized for difference map analysis using Bsoft (45, 46). Difference mapping was performed with Chimera (48) and visualized with a threshold set at 1.9σ and 1.6σ above the mean density for main and subtle difference analysis, respectively. The atomic model of the Ad5-wt capsid proteins (Protein Data Bank [PDB] IDs 3IYN and 4CWU) was fitted into the wt EM density map using Chimera.
Statistical analysis.
Unless otherwise indicated, experiments were done in triplicate, and the error bars represent standard deviations (SD). P values were calculated using Student's t test.
Ethics statement.
All animal experiments were performed in accordance with institutional guidelines as determined by the Service Commun des Animaleries de l'Université Bordeaux (approval number A33-063-916), which specifically approved the study.
RESULTS
A recombinant virus with a mutated amphipathic helix of protein VI (PVI-L40Q) or a mutated proteinase (PRO-P137L) has reduced transduction efficiency and is impaired in membrane lysis.
Adenovirus membrane penetration and escape from the endocytic compartment are required for cytosolic accumulation and nuclear transport of the capsid. A mutagenesis screen within the amphipathic helix of membrane-lytic internal capsid protein VI determined that the mutation PVI-L40Q functionally impairs membrane lysis and impairs infectivity (25). We confirmed a reduced membrane association for PVI-L40Q compared to the wild type by transfection assays using traceable fusion proteins (unpublished observation). To further analyze the effect of the PVI-L40Q mutation in the viral context, the mutation was next introduced into the protein VI open reading frame in the context of a first-generation HAdV-C5 GFP-expressing vector using recombineering (39). For comparison, we also constructed a virus that lacks protein VI release upon entry. For that purpose, we introduced the P137L mutation into the viral proteinase gene, which was described as the PRO-P137L-associated mutation in HAdV-C2 (8, 10). The resulting GFP-expressing mutant was called HAdV-C5-GFP-PRO-P137L. Both mutant viruses could be reconstituted following PacI release from the BAC and transfection into HEK293 cells at 37°C and 33°C, respectively. CsCl-purified particles, propagated at 37°C or at 38.5°C for HAdV-C5-GFP-PRO-P137L, were analyzed by Coomassie gel analysis and anti-protein VI Western blotting (Fig. 1A, left). This analysis showed the presence of all the capsid proteins and confirmed the temperature-dependent maturation defect of capsid precursor proteins pIIIA, pVI, pVII, and pVIII for the newly generated HAdV-C5-GFP-PRO-P137L when grown at the nonpermissive temperature (Fig. 1A, right). Next, the relative infection efficiencies of the three viruses were determined using GFP expression in transduced cells as the readout. Cells were transduced at 100 pp/c. The transduction efficiency was determined by FACS analysis and normalized for the wt virus (Fig. 1B). This analysis revealed an ∼5-fold-reduced transduction efficiency for HAdV-C5-GFP-PVI-L40Q and over 70-fold-reduced transduction efficiency for HAdV-C5-GFP-PRO-P137L grown at the nonpermissive temperature. Next, the membrane-lytic capacity upon entry of the three viruses was determined. Cells were incubated with 150, 1,500, or 15,000 pp/c in the presence of the non-cell-permeable translation inhibitor α-sarcin (18, 26). The release of α-sarcin into the cytosol following endocytic uptake is entirely dependent on the membrane-lytic activity of the virus. Reduction of translational efficiency in the cells by α-sarcin was measured by the level of 35S incorporation (Fig. 1C). The results showed that HAdV-C5-GFP-wt particles inhibited translation in a dose-dependent manner, while the PRO-P137L control virus lacked translation inhibition even at the highest particle concentration. In contrast, at 1,500 pp/c, HAdV-C5-GFP-PVI-L40Q showed ∼4-fold-diminished membrane lysis capacity compared to the wt virus, which is in agreement with the observed 5-fold-reduced infectivity. This analysis confirmed that membrane lysis is the principal defect associated with the PVI-L40Q mutant virus, as previously suggested (25). In summary, the data showed that efficient membrane lysis is linked to transduction efficiency and that the mutation responsible for the PRO-P137L phenotype can be transferred to the HAdV-C5 background, and this also prevented entry-associated membrane lysis.
FIG 1.

Transduction efficiencies and membrane lysis of adenovirus mutants. (A) (Left, top) Coomassie gel of purified GFP-expressing adenoviruses as indicated above the lanes. The corresponding viral proteins are identified on the right. (Left, bottom) Western blot using antibodies against protein VI of the same viruses. Note the size shift of the VI signal for the PRO-P137L virus. (Right) Side-by-side comparison of wt virus and PRO-P137L (grown at the nonpermissive temperature) showing the selective migration differences of unprocessed precursor capsid proteins. Unprocessed precursor capsid proteins are labeled and indicated by arrows in gray and mature capsid proteins in black. (B) Relative transduction efficiency of the wt virus compared to the PRO-P137L and PVI-L40Q mutant virus. Cells were transduced with GFP-expressing viruses at 100 pp/c, and the transduction efficiency was normalized to 1 for the wt virus for comparison. (C) Membrane lysis assay. For each virus, cells were incubated with three different particle amounts (the x axis represents pp/c) in the presence of the membrane-impermeable translation inhibitor α-sarcin. 35S incorporation was measured and plotted against the incorporation efficiency in the presence of α-sarcin alone, arbitrarily set as a 100% incorporation rate. The shorter bars correspond to translational inhibition due to α-sarcin uptake following membrane lysis. The error bars correspond to SD for the results of two individual experiments, each performed in triplicate.
HAdV-C5-GFP-PVI-L40Q virions are targeted to lysosomes.
We previously showed that protein VI release upon entry into cells can be visualized by antibody staining, because the antibody epitope remains hidden prior to capsid disassembly (26). Likewise, we showed that cytoplasmic Gal-3, a galactose binding lectin that binds galactosyl residues exposed to the cytoplasm upon membrane damage (49), can be used to monitor AdV-induced membrane damage in living and fixed cells via Gal-3 accumulation at damaged membranes (16, 23). Using these approaches, we quantified protein VI release and induced membrane damage over time for all three viruses. Cells were infected with Alexa 488-labeled wt, PRO-P137L, or PVI-L40Q viruses and fixed at different time points postinfection, followed by staining for protein VI (Fig. 2A, top) or Gal-3 (Fig. 2A, middle), respectively, and analyzed using quantitative image analysis. The results were normalized by background subtraction and are depicted in Fig. 2A. Release of protein VI from wt viruses was fast and efficient, with a maximum at 20 min postinfection. In contrast, no VI release was detected for the PRO-P137L virus. Protein VI release from the PVI-L40Q mutant virus followed the wt kinetics, although with somewhat reduced efficiency (Fig. 2A, top). Detection of Gal-3 punctae in fixed cells relies on the preservation of membrane damage, which was technically difficult. The analysis showed a rapid association of wt virus with the Gal-3 signal essentially following the kinetics of protein VI release. In contrast, PRO-P137L viruses remained Gal-3 negative, coinciding with the absence of protein VI release. The PVI-L40Q virus showed reduced and delayed formation of Gal-3 punctae compared to the wt virus, which is in sharp contrast to its protein VI release kinetics (Fig. 2A, middle). We confirmed the data obtained for membrane lysis using in vivo imaging analysis of A488 virus-infected galectin 3-mCherry-expressing cells, as previously described (50). Previous reports showed that HAdV-C2-PRO-P137L is sorted into lysosomes (13). Therefore, we also quantified the association of the different viruses with the lysosomal marker lamp-2 (51). Quantification of the association of the different viruses with the lysosome marker lamp-2 showed rapid association of PRO-P137L and PVI-L40Q viruses with lamp-2 (Fig. 2A, bottom). Wt viruses, in contrast, did not efficiently associate with lamp-2, suggesting early and rapid escape prior to sorting into lysosomes.
FIG 2.

Quantitative analysis of protein VI release, membrane lysis, and lysosome association during virus entry. (A) (Top) Quantification of protein VI release over time. Cells were infected with Alexa 488-labeled wt virus (black circles), PVI-L40Q virus (open circles), or PRO-P137L virus (black triangles), and protein VI-positive viruses were identified by antibody staining at the indicated time points. The error bars indicate cell-to-cell variations (n ≥ 100 particles). (Middle) Quantification of galectin 3 association of viruses over time. Cells were infected with Alexa 488-labeled viruses as for the top graph, fixed, and stained for endogenous galectin 3 at the indicated time points. The error bars indicate cell-to-cell variations (n ≥ 100 particles). (Bottom) Quantification of virus association with lamp-2. Cells were infected with Alexa 488-labeled virus as for the top graph, and association of virus with lamp-2 was quantified at each time point. The error bars indicate cell-to-cell variations (n ≥ 100 particles). (B) Immunofluorescence analysis of virus association with lamp-2. Cells were infected with Alexa 488-labeled wt virus (top), PRO-P137L virus (middle), and PVI-L40Q virus (bottom) for 1 h and stained for lamp-2 association. Viruses are depicted in green, lamp-2 in red, and the DAPI signal in gray. Enlargements of the dashed boxes are depicted on the right. Clusters of virus colocalizing with lamp-2 appear yellow and are indicated by arrows. Scale bars, 10 μm.
The analysis also confirmed that several clusters of PRO-P137L and PVI-L40Q viruses, which we observed in fixed cells (see below) or in vivo (data not shown), colocalized with lysosomes. Representative images of all three viruses at 60 min post-temperature shift are shown in Fig. 2B. While the wt viruses are largely separated from the lamp-2 stain (Fig. 2B, top), the PRO-P137L virus fully (Fig. 2B, middle) and the PVI-L40Q virus partly (Fig. 2B, bottom) colocalize with the lamp-2 stain. In summary, the data suggested that membrane lysis via the amphipathic helix of protein VI is an important step to avoid capsid sorting into lysosomes or to escape from the lysosomal compartment, while protein VI release alone is not sufficient to rescue the virus from lysosomal sorting.
Premature protein VI release contributes to HAdV-C5-GFP-PVI-L40Q lysosome targeting.
Our analysis showed that the PVI-L40Q mutant virus, like the PRO-P137L mutant virus, was sorted into lysosomes. In contrast, the PVI-L40Q virus had only 5-fold-reduced infectivity compared to ∼70-fold for the PRO-P137L virus. We next asked whether this increased infectivity is because some PVI-L40Q virus escapes from lysosomes or if a subpopulation escapes from the endosomal compartment prior to lysosomal sorting. We switched to antibody detection of unlabeled viruses as the detection method to confirm cluster formation with labeled virus and the association with lysosomes. Using this method, we observed that virus-specific signals were either small, likely representing single viral particles, or larger, likely reflecting damaged particles or clusters of several viruses (Fig. 3A). We noticed that large signals were more common in cells infected with the PVI-L40Q virus and also at earlier time points. We next quantified the virus signals over time in infected cells, classifying virus signals by using a size cutoff at 1 μm (<1 μm, small; >1 μm, large). Larger virus signals appeared more often and earlier in PVI-L40Q-infected cells and were present even at 5 min post-temperature shift. In contrast, in PRO-P137L- or wt-infected cells only, they appeared at later time points and to a lesser extent (Fig. 3B). We next repeated the kinetic analysis of lysosome accumulation by separately analyzing PVI-L40Q large and small signals (Fig. 3C). This analysis showed that large PVI-L40Q signals rapidly accumulated in lysosomes, similar to those of PRO-P137L, while small signals more closely followed the kinetics of the wt particles. We then repeated the experiment and stained for protein VI. The analysis shown in Fig. 3D indicated that the vast majority of large signals were positive for protein VI even at the earliest time point measured (5 min postinfection). In contrast, small signals showed maximum protein VI release at 20 min postinfection, similar to the wild type but without the subsequent drop. These data suggested that premature protein VI exposure contributes to lysosomal sorting. Whether the large, protein VI-positive signals represent clusters of viral particles or damaged particles displaying additional epitopes for antibody recognition remains to be determined. In contrast, small PVI-L40Q signals represent particles that release protein VI with wt kinetics and did not associate with lysosomes. This suggests that a subpopulation of PVI-L40Q virus may escape from the endosomal compartment with wt kinetics, which could explain the remaining infectivity. We tested this hypothesis, assuming that similar endosomal-escape kinetics between wt and PVI-L40Q virus should result in similar nuclear arrival of the genome, differing only in the overall amount, while escape from the lysosomal compartment would delay nuclear genome arrival. We next quantified protein VII dots in the nucleus over time (Fig. 3E). Protein VII is a genome-bound viral histone-like protein that becomes accessible to antibody staininig only once the genome has reached the nucleus. The results showed that wt and PVI-L40Q genomes arrived in the nucleus at similar times (Fig. 3E), while no PRO-P137L genomes could be detected (data not shown), suggesting early endosomal escape for some PVI-L40Q virus.
FIG 3.

Differential sorting of PVI-L40Q mutant virus into lysosomes. (A) Sorting of PVI-L40Q mutant virus into lysosomes. Cells were infected with unlabeled PVI-L40Q mutant virus and fixed at 60 min postinfection, followed by antibody detection of virus particles. Enlarged details from the dashed box are shown on the right, with the virus signal (top; green), antibody detection of lamp-2 (middle; red), and merged image (bottom). The white arrows indicate lamp-2-free viruses (small particles), and the orange arrows indicate virus associated with lamp-2 (large particles). (B) Particle detection by size. Cells were infected with unlabeled wt, PVI-L40Q, or PRO-P137L virus and fixed at the indicated times, and particles were detected using anti-Ad5 antibodies. The signals were quantified and scored according to size using the indicated cutoffs and are displayed as percentages of the total. The error bars indicate cell-to-cell variation. (C) PVI-L40Q mutant virus lamp-2 association by size. Infections were carried out as for panel B. Virus was detected using antibodies against Ad5 and lamp-2, and lamp-2-positive viruses were plotted over time. The black circles represent wt and the black triangles PRO-P137L virus. The open circles represent PVI-L40Q mutant viruses below the size cutoff (as in panel B) and the open triangles viruses above the size cutoff. (D) PVI-L40Q mutant virus protein VI association by size. The experiment was performed as for panel C. Virus was detected using antibodies against Ad5 and protein VI, and protein VI-positive viruses were plotted over time. The symbols are as in panel C. (E) Nuclear accumulation of protein VII dots. Cells were infected as for panel B, fixed at the indicated times, and stained with antibodies against protein VII. The distribution of protein VII signals per nucleus is shown as box-and-whisker plots (minimum to maximum). Shown are cells infected with the wt virus (left) and with the PVI-L40Q mutant virus (right).
The PVI-L40Q mutation, in contrast to the PRO-P137L mutation, destabilizes the AdV capsid.
The exact location of protein VI in the entering particle and the spatiotemporal organization of disassembly events necessary for entry and endosomal escape are currently under debate. Premature disassembly and exposure of protein VI, e.g., through mild heat treatment of the particle, render particles noninfectious, as we have observed for some PVI-L40Q viruses. When we performed transduction experiments with the GFP-expressing PVI-L40Q virus, we noticed that the infectivity of the virus preparation was sensitive to repeated freeze-thaw cycles, unlike preparations containing wt or PRO-P137L virus (Fig. 4A). This sensitivity was more pronounced upon long-term storage of the virus preparations at −80°C, because freshly made PVI-L40Q virus was not sensitive (Fig. 4A). Likewise, when we tried to test virus infectivity in the presence of drugs, we noticed that the PVI-L40Q virus was sensitive to the addition of the vehicle DMF compared to the wt or PRO-P137L counterparts, preventing such an approach (Fig. 4B). We reasoned that DMF destabilizes hydrophobic interactions and thus could interfere with molecular interactions of the amphipathic helix inside the viral capsid. In turn, this suggested a role for the amphipathic helix in capsid stabilization prior to deployment. To investigate this hypothesis, we decided to determine capsid integrity by staining Alexa 488-labeled viruses bound to cells at 4°C for penton association. Pentons are located at the vertices of the AdV capsid, and vertex removal is thought to occur during partial disassembly and protein VI exposure (18). Neither the wt nor the PRO-P137L virus showed a difference in penton association with the capsid after one or three freeze-thaw cycles when bound to cells in the cold (Fig. 4C). On the other hand, the VI-L40Q mutant virus (after extended storage at −80°C) lost significant amounts of penton association between one and three freeze-thaw cycles when bound to cells at 4°C (Fig. 4C). This result was supported by our observation that fewer particles of the latter virus bound to the cell after three freeze-thaw cycles (presumably because fiber is also removed), with more penton-free capsids, as well as capsid-free penton signals, associated with the cells (Fig. 4D). Using unlabeled viruses and staining for protein VI as a marker for premature disassembly, we saw that a single thaw of PVI-L40Q viruses (but none of the other viruses) was sufficient to render some particles positive for protein VI, confirming that the virus had decreased stability and could already expose protein VI upon cell surface binding at 4°C (Fig. 4E). In summary the data suggest that pentons are less tightly associated with the viral capsid in PVI-L40Q viruses, e.g., compared to the wt virus, which can be revealed by exposing the mutant virus to repeated freeze-thaw cycles and by sensitivity to DMF treatment.
FIG 4.

Altered capsid stability for Ad-PVI-L40Q. (A) Freeze-thaw (FT) sensitivity. Aliquoted virus was used in transduction assays and refrozen as indicated below the bars. Transduction efficiency was normalized to the efficiency after the first thaw (100%). Two independent virus preparations were used. Only the PVI-L40Q preparation from 2012, which had been in storage for 2 years at −80°C, was sensitive to repeated freeze-thaw cycles. Error bars represent the mean for one representative experiment performed in triplicate. (B) Solvent sensitivity. Viruses, as indicated below the bars, were incubated with DMF at the indicated concentrations, and the relative transduction efficiency was compared to that of the wt virus in DMEM only (100%). Note that both PVI-L40Q preparations were sensitive to DMF treatment. Error bars represent the mean for one representative experiment performed in duplicate. (C) Penton association upon repeated freeze-thaw cycles. Viruses, as indicated below the bars, were subjected to one or three repeated freeze-thaw cycles and incubated with cells at 4°C for 30 min, followed by fixation and antibody staining for pentons. The percentages of penton-positive particles are indicated. Note the loss of penton association for the PVI-L40Q virus upon repeated freeze-thaw treatment. Error bars represent cell-to-cell variation. (D) As for C, comparing 2012 PVI-L40Q particles bound to cells after one (top) or three (bottom) freeze-thaw cycles. Details of the boxed areas, with viruses in green, penton antibody staining in red, and the overlaid images, are shown on the right. Penton-positive particles are indicated by solid arrows, and particles lacking penton staining are indicated by open arrows. (E) Premature protein VI exposure. Cells were incubated with unlabeled PVI-L40Q mutant virus (1 thaw) at 4°C and fixed, followed by immunofluorescence detection of virus particles and protein VI. Enlarged details from the dashed box are shown on the right, with the virus signal (top; green), immunofluorescence of protein VI (middle; red), and merged images (bottom). Viruses positive for protein VI appear yellow. Similar results were obtained for both PVI-L40Q virus preparations. Scale bars, 10 µm.
Freeze-thaw treatment of Alexa 488-labeled PVI-L40Q virus removes most pentons, but not entire vertices.
Our data implied a destabilizing effect on the AdV capsid if the amphipathic helix of protein VI was weakened, resulting in premature penton, protein VI, and/or vertex separation from the capsid. To further understand the structural differences between the wt and the PVI-L40Q viruses, we performed cryo-ET on the same batch of purified Alexa 488-labeled particles used in the infection assays after freeze-thaw cycle treatment. Initial observations of the purified particles in cryo-ET revealed that the PVI-L40Q particles were not disassembled (Fig. 5A and B). We then calculated a 3D average of the virus capsid by computationally extracting and averaging individual capsids from the tomograms, a process termed subvolume averaging (52) (Fig. 5). Icosahedral symmetry was applied to the two capsid averages to create the final reconstructions of wt and PVI-L40Q capsids. This allowed us to analyze the main differences between the two virus capsid types (Fig. 5C and E versus D and F). The PVI-L40Q density map clearly showed that the pentons of the capsids were missing from the capsid shell (Fig. 5D and F). Interestingly, only the penton bases of the capsid were missing; all peripentonal hexons were still present. In addition, weak densities on the 5-fold axes of the PVI-L40Q virus were detectable. This is likely caused by a minority of pentons remaining in the capsids that contributed to the average structure (Fig. 5F, insets). This result is consistent with the decrease in infectivity of the mutant and the loss of pentons in the cell binding studies (Fig. 4). However, we noted that the freeze-thaw treatment did not quantitatively release the pentons from unlabeled PVI-L40Q viruses, suggesting that the labeling procedure might introduce an additional destabilizing force that facilitates penton release.
FIG 5.
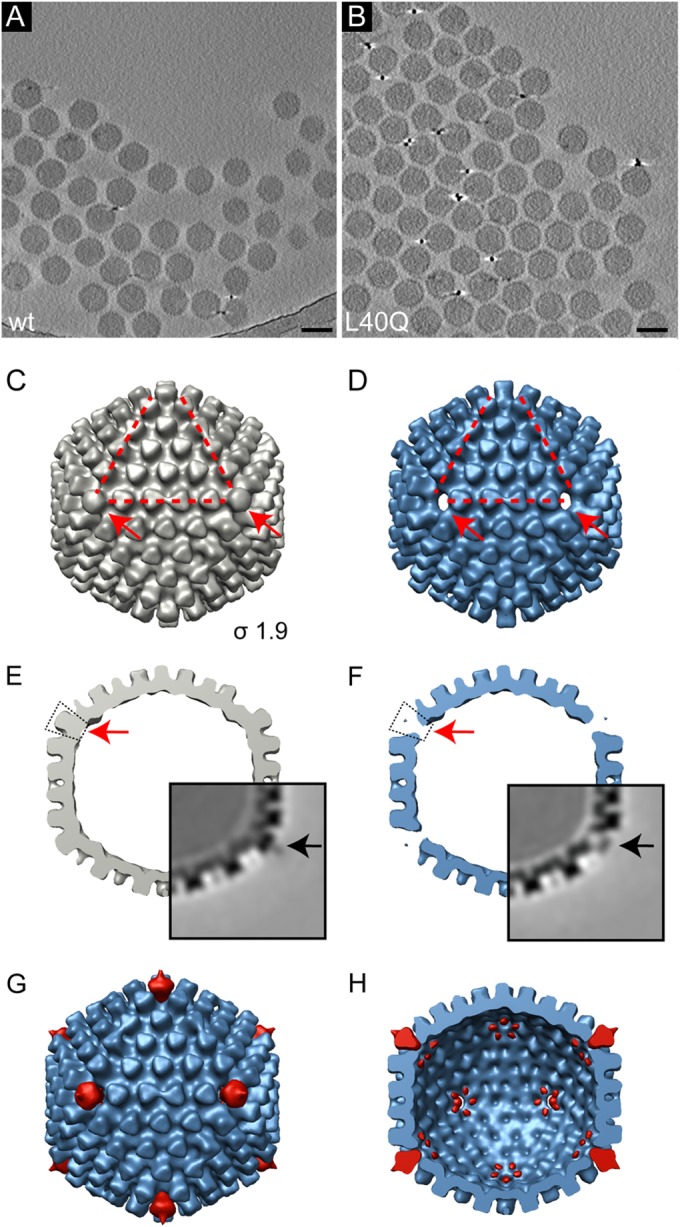
Cryo-ET and 3D reconstructions from subtomogram averaging of freeze-thaw-treated wt and PVI-L40Q mutant adenoviruses reveals loss of pentons. (A and B) Computational slices through a tomogram of frozen, hydrated preparations of freeze-thaw-treated (three times) Alexa 488-labeled wt viruses (A) and PVI-L40Q mutant viruses (B). Particles are intact, showing no apparent disruption. Scale bar, 100 nm. (C and D) Subtomogram averages of wt (C) (gray) and PVI-L40Q (D) (blue). The isosurface threshold was set 1.9σ above the mean density. The corners of the red triangles indicate the positions of 3 of the 12 vertices, which are also indicated by the arrows. (E and F) Same reconstruction as in panels C and D, but viewed as a central section. The red arrows indicate the vertex positions of the capsid, revealing the absence of penton bases in the mutant capsid (F). The insets show the central slice of the subvolume-averaged density map in grayscale. Higher density is shown in black. The electron density in light gray indicates the presence of fiber proteins in the wt average (E, black arrow) and few remaining penton bases in the PVI-L40Q virus (F, black arrow). (G and H) Difference maps (red) calculated between the wt and PVI-L40Q average superimposed on the PVI-L40Q average (blue) as seen from the outside (G) and as a cut open view into the inside of the capsid (H). The capsid reconstructions and difference maps are shown at the same threshold (1.9σ). The main difference is the loss of the penton base proteins, with starfish-shaped rearrangements of the peripentonal proteins located on the inner side of the capsid shell.
To further analyze the structural differences between wt and PVI-L40Q viruses, we calculated a difference map (Fig. 5G and H) between the respective subtomogram-averaged structures (Fig. 5C and D). This confirmed that the differences are limited to the vertex positions. The comparison indicates that there are also missing densities in the PVI-L40Q capsids in the peripentonal area on the inside of the capsid shell. At a lower surface-rendering threshold level (1.6 σ), the missing densities are visible as a starfish-shaped density. At a higher threshold level (1.9 σ), these missing densities appear as isolated densities. Next, we fitted both the previously published Ad5-wt atomic structure (34) (Fig. 6A to D) and the newly assigned structure of the adenovirus capsid cement protein from a recent X-ray crystallography study (36) (Fig. 6E to H) into the wt map and overlaid it with the difference map. The location of the starfish-shaped missing densities matches the inner cavity of the peripentonal hexons (Fig. 6C and G). Various suggestions for the location of VI have been put forward in recent years (11, 34, 53, 54), and there remains ambiguity in the locations of at least the recently unassigned parts of protein VI (residues 158 to 250) (55, 56). The difference density was observed between the location of the previously described protein IIIa (Fig. 6A to D, green), making it a possible location of VI, as VI had not been assigned in this atomic model (34). This location in the difference map is also close to the location where Reddy and Nemerow (36) assigned parts of proteins VI (called IIIa in reference 34) (Fig. 6H, blue) and V (Fig. 6G, yellow). Thus, the position of the density lost in the PVI-L40Q capsids could correspond to the remaining unmapped residues of VI and/or a rearrangement of the peripentonal cement proteins upon penton removal.
FIG 6.

Difference maps between wt and PVI-L40Q mutant adenoviruses identify structural differences in the vertex region. Shown are difference maps (red) between the wt and the PVI-L40Q mutant superimposed onto the wt average (light gray). The isosurface threshold was set 1.9σ above the mean density. The two most recent, and contradictory, atomic models of wt capsid proteins (PDB 3IYN and 4CWU) were fitted into the wt subtomogram average map (A to D and E to H, respectively). Only penton proteins (red) and peripentonal cement proteins located inside the virion are displayed as ribbons: IIIa (green), V (yellow), and VI (blue). The positions of the peripentonal hexons are indicated by the numbers 1 to 5 in panel A. The superimposed maps are viewed from the outside, with a transparent surface rendering (A and E) or sliced through (B and F) or inside (C and G) the capsid vertex region, as indicated by the pictogram scheme in the middle of each column. (C and G) The difference map shown at a lower threshold of 1.6σ (lighter red) indicates that the structural differences between the wt and PVI-L40Q affects the whole peripentonal area (starfish-like density), with a maximum difference in the peripentonal hexons inner cavity (arrow). (D and H) Direct comparison between the locations of the atomic models and the difference map shows that the difference map includes part of V and is tightly associated with VI (H).
Penton release coincides with protein VI exposure in vivo.
Our results suggested that in the in vivo situation, penton release is linked to protein VI. When we avoided repeated freeze-thaw cycles and compared the dynamic dissociation of pentons from Alexa 488-labeled capsids following synchronized infections, we observed a time-dependent efficient separation of pentons from entering wt capsids (Fig. 7), which coincided with the appearance of protein VI (see below). In contrast, pentons from PRO-P137L and PVI-L40Q viruses remained largely associated with the capsid upon entry, likely reflecting lack of disassembly (PRO-P137L) and absence or delayed membrane lysis (PVI-L40Q) (Fig. 7A and B shows the quantification).
FIG 7.

Entry-dependent penton release. (A) Cells were infected with Alexa 488-labeled VI-L40Q viruses (top row), PRO-P137L viruses (middle row), and wt viruses (bottom row); fixed after 5 min (left column) or 20 min (right column); and stained for pentons using specific antibodies. Viruses are depicted in green and penton antibody stain in red. The dashed boxes in each panel are enlarged on the right, depicting virus, pentons, and a merged image. Note that separation of the penton and virus signal for the wt virus occurs at 20 min, but not for the PRO-P137L or the L40Q virus. (B) The experiment was performed as for panel A. Cells were infected with Alexa 488-labeled wt virus (black circles), PVI-L40Q virus (open circles), or PRO-P137L virus (black triangles). Antibody staining was quantified at 5 min, 20 min, and 60 min postinfection, and penton-positive virions were plotted for each virus. The error bars are cell-to-cell variations (n > 100 virus particles). Scale bars, 10 μm.
To investigate the observed penton deployment difference in vivo, we used antibody detection to identify penton and protein VI association with incoming Alexa 488-labeled viral particles. The data in Fig. 8 depict a representative cell at 20 min postinfection with wt (left) and PVI-L40Q (right) virus. As shown in Fig. 8, bottom, at 20 min postinfection, most wt particles appeared positive for either protein VI or pentons. However, we also observed several particles that were double positive for pentons and protein VI, suggesting codeployment of both proteins. Penton/VI-positive particles were more abundant but not restricted to PVI-L40Q virus-infected cells, confirming that membrane lysis is needed for separation of the penton and protein VI (Fig. 8, bottom). In addition, we observed occasional capsid-free penton/VI staining for both viruses, suggesting that the penton and protein VI can form a complex that exists independently of viral capsids upon productive entry, which would be in agreement with codeployment from the capsid (Fig. 8, bottom).
FIG 8.

Analysis of codeployment of pentons and protein VI from entering virus particles. Shown is immunofluorescence analysis of virus association with capsid protein VI and pentons. Cells were infected with Alexa 488-labeled wt virus (left) and PVI-L40Q virus (right) for 20 min and stained for capsid protein VI and penton association. Viruses are depicted in green, pentons in red, protein VI in light blue, and the DAPI signal in gray. Enlargements of the dashed boxes showing pairs of associations are depicted below. Viruses positive for pentons are indicated by small arrows, and viruses positive for protein VI are indicated by large arrows. In the bottom images, double-positive viruses are indicated with two white stars, protein VI-positive viruses with one white star, and penton/protein VI signal without viruses by two yellow stars. Scale bars, 10 μm.
DISCUSSION
To reach the cytosolic compartment following endosomal uptake, AdVs follow a set of coordinated entry-associated sorting steps until virus-induced membrane lysis and endosomal escape end the sorting process. To accomplish the sorting, the AdV capsid has to undergo sequential disassembly steps culminating in the deployment of the membrane-lytic internal capsid protein VI. In an excellent recent review, Leopold and Crystal pointed out that AdV postentry sorting steps can be influenced by several factors, such as the viral genotype or the metabolic state and cell type (15). Thus, the complexity of experimental systems described in the literature make general assumptions about AdV sorting difficult. Here, we followed a different approach by keeping the same cellular context and the same genetic background of the virion (HAdV-C5-GFP). We compared parameters of stability and endosomolysis by using a hyperstable mutant virus (PRO-P137L) and a mutant virus with reduced membrane-lytic activity (PVI-L40Q) in comparison to the wild type. We used time course analysis to study the fates of the capsid proteins VI and penton using specific antibodies. Our analysis identified capsid stability and membrane lysis as two essential factors that affect postentry sorting of AdVs. We showed that wt virions rapidly deploy protein VI; lyse the membranes, as shown by galectin 3 recruitment; and prevent association with lysosomes, presumably via efficient endosomal escape, supported by in vivo imaging (16, 23). In contrast, the hyperstable PRO-P137L virions do not deploy protein VI and consequently do not induce membrane lysis and were rapidly and efficiently sorted into lysosomes, preventing the virions from being infectious. The PVI-L40Q virions, on the other hand, are less stable, and a fraction deployed protein VI prematurely, thereby also failing to escape from the endocytic compartment. In their initial study of the PVI-L40Q mutant virus, Moyer et al. (25) observed a prolonged association of the virions with the endosomal compartment but did not study their destiny. Surprisingly, we observed that part of the PVI-L40Q virions associate with lysosomal markers with initial kinetics similar to those of the PRO-P137L virions, suggesting rapid lysosomal sorting despite efficient disassembly and protein VI release. In contrast, we also detected a fraction of viruses that escape from lysosome association, presumably also escaping from the endosomal compartment. Consequently, the PVI-L40Q virions retained infectivity at ∼20% compared to the wt, unlike the PRO-P137L virions, which do not disassemble. In living cells, PVI-L40Q virions appear in large clusters reminiscent of PRO-P137L lysosome clusters, but galectin 3-positive structures are also present, suggesting some membrane lysis has occurred (unpublished observation). How the PVI-L40Q virus is capable of partially escaping from the endosomal compartment remains to be determined. One possibility is that accumulations of PVI-L40Q result in high local concentrations to overcome the reduced lytic activity. A mechanism of escape from lysosomes instead of early endosomal structures, as suggested for subgroup B viruses (27, 28), appears less likely, as we show that genomes from PVI-L40Q viruses are delivered to the nucleus with kinetics similar to those of the wt virus, although at reduced efficiency.
Our analysis revealed a second property of the PVI-L40Q mutant that may provide insight into how protein VI is released during entry. Repeated freeze-thawing of highly purified virions stored for extended periods at −80°C reduced virus infectivity, which did not occur with either wt or PRO-P137L virions. Subsequent cryo-ET analysis of Alexa 488-labeled PVI-L40Q virions showed that they had quantitatively lost pentons and potentially other vertex-associated proteins upon freeze-thaw (see below). Previous studies have not observed any stability differences between wt and PVI-L40Q virions when the particles were exposed to sequential temperature rises (25). This discrepancy may be explained by the fact that, in contrast to temperature-induced disassembly, freeze-thaw cycles in combination with chemical labeling via primary amines induce dehydration of and mechanical stress on the virion particle, which can render virion components more sensitive and weaken hydrophobic interactions (57). However, mild physical stress was shown to also be able to sequentially remove pentons from wild-type virions without affecting the peripentonal hexons (38).
In addition to the density assigned to the penton in the freeze-thaw-treated PVI-L40Q capsid, additional densities are missing below the penton compared to the wt. These densities form a starfish-shaped radial density most likely indicative of a rearrangement of the peripentonal-hexon cement proteins. It may also indicate the location of full-length protein VI inside the capsid. Though the cryo-ET averages are informative in respect to the locations of the densities, the comparatively low resolution does not allow an exact description (e.g., specific secondary-structure elements) of the volume difference. Protein VI has been recently quantified using mass spectrometry and was estimated to be present at ∼360 copies per virion (32). This count is compatible with the possibility that the starfish-shaped density is protein VI, assuming 30 units per vertex organized into five bundles. Additionally, the cleaved N terminus of protein VI has been mapped to associate with the five peripentonal hexons, suggesting that the radial symmetry could initially extend to the peripentonal hexons stabilizing the capsid (53). Processing by the adenoviral proteinase would then weaken the interaction between protein VI and the peripentonal hexons and create a deployable penton-protein VI entity. This entity would sit at the vertex, similar to a “champagne cork,” where the wider base of the cork would be protein VI, holding the structure in place by interactions of the amphipathic helix with peripentonal hexons. Release of the putative penton-protein VI entity (possibly containing additional capsid proteins, such as IIIa) would follow the principle of opening a champagne bottle, leaving the vertex uncovered. In fact, we observed such penton-protein VI entities upon infection of cells using dual-antibody stain, confirming previous reports of sequential penton separation from the capsid (1). In such a scenario, the introduction of the PVI-L40Q mutation would weaken the interactions that hold the entity in place, opening it to structural alterations (e.g., via freeze-thaw or labeling procedures) that result in premature penton-protein VI release. Such a scenario would be consistent with all our accumulated data and most of the literature, including the recent partial structure assignment of protein VI inside the capsid. It would provide an elegant method of protein VI deployment by assigning dual functionality to the amphipathic helix in maintaining metastability of the capsid and providing membrane-lytic activity upon deployment. A role for the penton, in addition to protein VI, in the membrane lysis step is also suggested in the literature. Long before the discovery of protein VI as the AdV membrane-lytic factor, it was clear that AdVs possessed a membrane-destabilizing activity (58, 59) that could be blocked by mild heat treatment of AdV particles at 45°C (60), reminiscent of premature vertex dissociation and protein VI exposure. Antibody inhibition studies suggested that the penton provides the membrane-lytic function following uptake into the endocytic compartment (17). This was corroborated by in vitro studies exposing AdV to mild acidic treatment, showing association of the penton and/or the vertex region with Triton X114, suggesting a pH-dependent deployment of an amphiphilic virion property believed to be the penton (61). Interestingly, the EM images provided in the study do not suggest removal of peripentonal hexons and, rather, are in agreement with our champagne cork model (61). Selective antibody depletion experiments and in vitro permeabilization studies showed that the membrane-lytic activity is in fact encoded by the N-terminal amphipathic helix of protein VI rather than the penton (18, 62) (22, 25). Our direct observations confirm that the kinetics of protein VI deployment from wt virions coincides with galectin 3 recruitment to virions, reminiscent of membrane lysis. Moreover, the observation that pentons and protein VI can be found either in close association with virions or independent of viral particles suggests that both proteins could be released in a joint manner in the context of AdV entry.
In summary, our data suggest that the timed release of a penton-protein VI entity, in combination with membrane lysis, determines the postentry sorting of the entering virions and prevents their lysosomal degradation. Thus, differences in capsid stability may be the primary determinant of entry-associated sorting for all AdVs, allowing endosomal escape in very early endocytic compartments (as observed for species C AdVs) or after prolonged sorting from the late endosomal/lysosomal compartment, as was suggested for species B viruses (28, 63). In contrast, premature protein VI release (e.g., PVI-L40Q) or failure to release protein VI at all (PRO-P137L) would result in noninfectious virions. Changing the entry-associated sorting of AdVs is of great pharmacological interest, e.g., to increase the efficiency of vaccines or to decrease the toxicity of gene therapy vectors. While previous studies have assessed changing genotypes or producing hybrid vectors to optimize sorting for entry of recombinant particles, our data provide compelling evidence that modulating the sorting of incoming AdV particles can be achieved within the same AdV genotype by altering its stability based on structure-function information. We conclude that AdVs tolerate a range of different states of metastability that are compatible with infectivity but would result in differential sorting and, hence, the compartment from which the bulk of the virions escape or where they end up, which may provide additional means to increase the utility of clinically relevant AdV vectors currently in use.
ACKNOWLEDGMENTS
This work was supported in part by an Equipe FRM grant (DEQ 20110421299 to H.W.), a Wellcome Trust Senior Research Fellowship (to K.G.), a Wellcome Trust JIF award (060208/Z/00/Z) and a Wellcome Trust equipment grant (093305/Z/10/Z) to the Oxford Particle Imaging Centre, and a Wellcome Trust core award 090532/Z/09/Z to the Wellcome Trust Centre for Human Genetics. S.A. is supported through a department of defense (DGA) fellowship. H.W. is an Inserm fellow. Z.R. is supported by the ForBIMed program of the Bavarian Research Council (BFS).
We thank Sigrid Seelmair for excellent technical assistance.
REFERENCES
- 1.Greber UF, Willetts M, Webster P, Helenius A. 1993. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 75:477–486. doi: 10.1016/0092-8674(93)90382-Z. [DOI] [PubMed] [Google Scholar]
- 2.Suomalainen M, Greber UF. 2013. Uncoating of non-enveloped viruses. Curr Opin Virol 3:27–33. doi: 10.1016/j.coviro.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Smith JG, Wiethoff CM, Stewart PL, Nemerow GR. 2010. Adenovirus. Curr Top Microbiol Immunol 343:195–224. doi: 10.1007/82_2010_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemerow GR, Stewart PL, Reddy VS. 2012. Structure of human adenovirus. Curr Opin Virol 2:115–121. doi: 10.1016/j.coviro.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart PL, Burnett RM. 1995. Adenovirus structure by X-ray crystallography and electron microscopy. Curr Top Microbiol Immunol 199:25–38. [DOI] [PubMed] [Google Scholar]
- 6.Yeh-Kai L, Akusjarvi G, Alestrom P, Pettersson U, Tremblay M, Weber J. 1983. Genetic identification of an endoproteinase encoded by the adenovirus genome. J Mol Biol 167:217–222. doi: 10.1016/S0022-2836(83)80044-8. [DOI] [PubMed] [Google Scholar]
- 7.Cotten M, Weber JM. 1995. The adenovirus protease is required for virus entry into host cells. Virology 213:494–502. doi: 10.1006/viro.1995.0022. [DOI] [PubMed] [Google Scholar]
- 8.Rancourt C, Keyvani-Amineh H, Sircar S, Labrecque P, Weber JM. 1995. Proline 137 is critical for adenovirus protease encapsidation and activation but not enzyme activity. Virology 209:167–173. doi: 10.1006/viro.1995.1240. [DOI] [PubMed] [Google Scholar]
- 9.Weber J. 1976. Genetic analysis of adenovirus type 2. III. Temperature sensitivity of processing viral proteins. J Virol 17:462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imelli N, Ruzsics Z, Puntener D, Gastaldelli M, Greber UF. 2009. Genetic reconstitution of the human adenovirus type 2 temperature-sensitive 1 mutant defective in endosomal escape. Virol J 6:174. doi: 10.1186/1743-422X-6-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Berna AJ, Marabini R, Scheres SH, Menendez-Conejero R, Dmitriev IP, Curiel DT, Mangel WF, Flint SJ, San Martin C. 2009. Structure and uncoating of immature adenovirus. J Mol Biol 392:547–557. doi: 10.1016/j.jmb.2009.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silvestry M, Lindert S, Smith JG, Maier O, Wiethoff CM, Nemerow GR, Stewart PL. 2009. Cryo-electron microscopy structure of adenovirus type 2 temperature-sensitive mutant 1 reveals insight into the cell entry defect. J Virol 83:7375–7383. doi: 10.1128/JVI.00331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gastaldelli M, Imelli N, Boucke K, Amstutz B, Meier O, Greber UF. 2008. Infectious adenovirus type 2 transport through early but not late endosomes. Traffic 9:2265–2278. doi: 10.1111/j.1600-0854.2008.00835.x. [DOI] [PubMed] [Google Scholar]
- 14.Leopold PL, Kreitzer G, Miyazawa N, Rempel S, Pfister KK, Rodriguez-Boulan E, Crystal RG. 2000. Dynein- and microtubule-mediated translocation of adenovirus serotype 5 occurs after endosomal lysis. Hum Gene Ther 11:151–165. doi: 10.1089/10430340050016238. [DOI] [PubMed] [Google Scholar]
- 15.Leopold PL, Crystal RG. 2007. Intracellular trafficking of adenovirus: many means to many ends. Adv Drug Deliv Rev 59:810–821. doi: 10.1016/j.addr.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Martinez R, Burrage AM, Wiethoff CM, Wodrich H. 2013. High temporal resolution imaging reveals endosomal membrane penetration and escape of adenoviruses in real time. Methods Mol Biol 1064:211–226. doi: 10.1007/978-1-62703-601-6_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seth P, Fitzgerald D, Ginsberg H, Willingham M, Pastan I. 1984. Evidence that the penton base of adenovirus is involved in potentiation of toxicity of Pseudomonas exotoxin conjugated to epidermal growth factor. Mol Cell Biol 4:1528–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. 2005. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J Virol 79:1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wodrich H, Guan T, Cingolani G, Von Seggern D, Nemerow G, Gerace L. 2003. Switch from capsid protein import to adenovirus assembly by cleavage of nuclear transport signals. EMBO J 22:6245–6255. doi: 10.1093/emboj/cdg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sung MT, Cao TM, Lischwe MA, Coleman RT. 1983. Molecular processing of adenovirus proteins. J Biol Chem 258:8266–8272. [PubMed] [Google Scholar]
- 21.Webster A, Hay RT, Kemp G. 1993. The adenovirus protease is activated by a virus-coded disulphide-linked peptide. Cell 72:97–104. doi: 10.1016/0092-8674(93)90053-S. [DOI] [PubMed] [Google Scholar]
- 22.Maier O, Galan DL, Wodrich H, Wiethoff CM. 2010. An N-terminal domain of adenovirus protein VI fragments membranes by inducing positive membrane curvature. Virology 402:11–19. doi: 10.1016/j.virol.2010.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maier O, Marvin SA, Wodrich H, Campbell EM, Wiethoff CM. 2012. Spatiotemporal dynamics of adenovirus membrane rupture and endosomal escape. J Virol 86:10821–10828. doi: 10.1128/JVI.01428-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthews DA, Russell WC. 1994. Adenovirus protein-protein interactions: hexon and protein VI. J Gen Virol 75:3365–3374. doi: 10.1099/0022-1317-75-12-3365. [DOI] [PubMed] [Google Scholar]
- 25.Moyer CL, Wiethoff CM, Maier O, Smith JG, Nemerow GR. 2011. Functional genetic and biophysical analyses of membrane disruption by human adenovirus. J Virol 85:2631–2641. doi: 10.1128/JVI.02321-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wodrich H, Henaff D, Jammart B, Segura-Morales C, Seelmeir S, Coux O, Ruzsics Z, Wiethoff CM, Kremer EJ. 2010. A capsid-encoded PPxY-motif facilitates adenovirus entry. PLoS Pathog 6:e1000808. doi: 10.1371/journal.ppat.1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyazawa N, Leopold PL, Hackett NR, Ferris B, Worgall S, Falck-Pedersen E, Crystal RG. 1999. Fiber swap between adenovirus subgroups B and C alters intracellular trafficking of adenovirus gene transfer vectors. J Virol 73:6056–6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyazawa N, Crystal RG, Leopold PL. 2001. Adenovirus serotype 7 retention in a late endosomal compartment prior to cytosol escape is modulated by fiber protein. J Virol 75:1387–1400. doi: 10.1128/JVI.75.3.1387-1400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carey B, Staudt MK, Bonaminio D, van der Loo JC, Trapnell BC. 2007. PU.1 redirects adenovirus to lysosomes in alveolar macrophages, uncoupling internalization from infection. J Immunol 178:2440–2447. doi: 10.4049/jimmunol.178.4.2440. [DOI] [PubMed] [Google Scholar]
- 30.Nakano MY, Boucke K, Suomalainen M, Stidwill RP, Greber UF. 2000. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J Virol 74:7085–7095. doi: 10.1128/JVI.74.15.7085-7095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burckhardt CJ, Suomalainen M, Schoenenberger P, Boucke K, Hemmi S, Greber UF. 2011. Drifting motions of the adenovirus receptor CAR and immobile integrins initiate virus uncoating and membrane lytic protein exposure. Cell Host Microbe 10:105–117. doi: 10.1016/j.chom.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 32.Benevento M, Di Palma S, Snijder J, Moyer CL, Reddy VS, Nemerow GR, Heck AJ. 2014. Adenovirus composition, proteolysis, and disassembly studied by in-depth qualitative and quantitative proteomics. J Biol Chem 289:11421–11430. doi: 10.1074/jbc.M113.537498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart PL, Fuller SD, Burnett RM. 1993. Difference imaging of adenovirus: bridging the resolution gap between X-ray crystallography and electron microscopy. EMBO J 12:2589–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu H, Jin L, Koh SB, Atanasov I, Schein S, Wu L, Zhou ZH. 2010. Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 329:1038–1043. doi: 10.1126/science.1187433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddy VS, Natchiar SK, Stewart PL, Nemerow GR. 2010. Crystal structure of human adenovirus at 3.5 A resolution. Science 329:1071–1075. doi: 10.1126/science.1187292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy VS, Nemerow GR. 2014. Structures and organization of adenovirus cement proteins provide insights into the role of capsid maturation in virus entry and infection. Proc Natl Acad Sci U S A 111:11715–11720. doi: 10.1073/pnas.1408462111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Wu L, Zhou ZH. 2011. Model of the trimeric fiber and its interactions with the pentameric penton base of human adenovirus by cryo-electron microscopy. J Mol Biol 406:764–774. doi: 10.1016/j.jmb.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Berna AJ, Ortega-Esteban A, Menendez-Conejero R, Winkler DC, Menendez M, Steven AC, Flint SJ, de Pablo PJ, San Martin C. 2012. The role of capsid maturation on adenovirus priming for sequential uncoating. J Biol Chem 287:31582–31595. doi: 10.1074/jbc.M112.389957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruzsics Z, Lemnitzer F, Thirion C. 2014. Engineering adenovirus genome by bacterial artificial chromosome (BAC) technology. Methods Mol Biol 1089:143–158. doi: 10.1007/978-1-62703-679-5_11. [DOI] [PubMed] [Google Scholar]
- 40.Mittereder N, March KL, Trapnell BC. 1996. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol 70:7498–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sandberg K, Mastronarde DN, Beylkin G. 2003. A fast reconstruction algorithm for electron microscope tomography. J Struct Biol 144:61–72. doi: 10.1016/j.jsb.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 42.Mastronarde DN. 2005. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 43.Kremer JR, Mastronarde DN, McIntosh JR. 1996. Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 44.Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR. 2006. The molecular architecture of axonemes revealed by cryoelectron tomography. Science 313:944–948. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- 45.Heymann JB, Belnap DM. 2007. Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol 157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 46.Heymann JB, Cardone G, Winkler DC, Steven AC. 2008. Computational resources for cryo-electron tomography in Bsoft. J Struct Biol 161:232–242. doi: 10.1016/j.jsb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenthal PB, Henderson R. 2003. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 48.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 49.Paz I, Sachse M, Dupont N, Mounier J, Cederfur C, Enninga J, Leffler H, Poirier F, Prevost MC, Lafont F, Sansonetti P. 2010. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol 12:530–544. doi: 10.1111/j.1462-5822.2009.01415.x. [DOI] [PubMed] [Google Scholar]
- 50.Bosse JB, Ragues J, Wodrich H. 2013. Fast generation of stable cell lines expressing fluorescent marker molecules to study pathogen induced processes. Methods Mol Biol 1064:153–169. doi: 10.1007/978-1-62703-601-6_11. [DOI] [PubMed] [Google Scholar]
- 51.Eskelinen EL. 2006. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med 27:495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 52.Briggs JA. 2013. Structural biology in situ—the potential of subtomogram averaging. Curr Opin Struct Biol 23:261–267. doi: 10.1016/j.sbi.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 53.Snijder J, Benevento M, Moyer CL, Reddy V, Nemerow GR, Heck AJ. 2014. The cleaved N-terminus of pVI binds peripentonal hexons in mature adenovirus. J Mol Biol 426:1971–1979. doi: 10.1016/j.jmb.2014.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saban SD, Silvestry M, Nemerow GR, Stewart PL. 2006. Visualization of alpha-helices in a 6-angstrom resolution cryoelectron microscopy structure of adenovirus allows refinement of capsid protein assignments. J Virol 80:12049–12059. doi: 10.1128/JVI.01652-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reddy VS, Nemerow GR. 2014. Reply to Campos. Revised structures of adenovirus cement proteins represent a consensus model for understanding virus assembly and disassembly. Proc Natl Acad Sci U S A 111:E4544–E4545. doi: 10.1073/pnas.1417014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campos SK. 2014. New structural model of adenoviral cement proteins is not yet concrete. Proc Natl Acad Sci U S A 111:E4542–E4543. doi: 10.1073/pnas.1415364111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katouzian-Safadi M, Favre A, Haenni AL. 1980. Effect of freezing and thawing on the structure of turnip yellow mosaic virus. Eur J Biochem 112:479–486. [DOI] [PubMed] [Google Scholar]
- 58.FitzGerald DJ, Padmanabhan R, Pastan I, Willingham MC. 1983. Adenovirus-induced release of epidermal growth factor and pseudomonas toxin into the cytosol of KB cells during receptor-mediated endocytosis. Cell 32:607–617. doi: 10.1016/0092-8674(83)90480-4. [DOI] [PubMed] [Google Scholar]
- 59.FitzGerald DJ, Trowbridge IS, Pastan I, Willingham MC. 1983. Enhancement of toxicity of antitransferrin receptor antibody-Pseudomonas exotoxin conjugates by adenovirus. Proc Natl Acad Sci U S A 80:4134–4138. doi: 10.1073/pnas.80.13.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seth P, Pastan I, Willingham MC. 1985. Adenovirus-dependent increase in cell membrane permeability. J Biol Chem 260:9598–9602. [PubMed] [Google Scholar]
- 61.Seth P, Willingham MC, Pastan I. 1985. Binding of adenovirus and its external proteins to Triton X-114. Dependence on pH. J Biol Chem 260:14431–14434. [PubMed] [Google Scholar]
- 62.Smith JG, Nemerow GR. 2008. Mechanism of adenovirus neutralization by human alpha-defensins. Cell Host Microbe 3:11–19. doi: 10.1016/j.chom.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 63.Teigler JE, Kagan JC, Barouch DH. 2014. Late endosomal trafficking of alternative serotype adenovirus vaccine vectors augments antiviral innate immunity. J Virol 88:10354–10363. doi: 10.1128/JVI.00936-14. [DOI] [PMC free article] [PubMed] [Google Scholar]