

ABSTRACT
Regulatory T (Treg) cells are important in the maintenance of self-tolerance, and the depletion of Treg cells correlates with autoimmune development. It has been shown that type I interferon (IFN) responses induced early in the infection of mice can drive memory (CD44hi) CD8 and CD4 T cells into apoptosis, and we questioned here whether the apoptosis of CD44-expressing Treg cells might be involved in the infection-associated autoimmune development. Instead, we found that Treg cells were much more resistant to apoptosis than CD44hi CD8 and CD4 T cells at days 2 to 3 after lymphocytic choriomeningitis virus infection, when type I IFN levels are high. The infection caused a downregulation of the interleukin-7 (IL-7) receptor, needed for survival of conventional T cells, while increasing on Treg cells the expression of the high-affinity IL-2 receptor, needed for STAT5-dependent survival of Treg cells. The stably maintained Treg cells early during infection may explain the relatively low incidence of autoimmune manifestations among infected patients.
IMPORTANCE Autoimmune diseases are controlled in part by regulatory T cells (Treg) and are thought to sometimes be initiated by viral infections. We tested the hypothesis that Treg may die off at early stages of infection, when virus-induced factors kill other lymphocyte types. Instead, we found that Treg resisted this cell death, perhaps reducing the tendency of viral infections to cause immune dysfunction and induce autoimmunity.
INTRODUCTION
Regulatory T (Treg) cells play an important role in maintaining self-tolerance and limiting an overactive immune response. Immunosuppressive “natural” Treg cells develop in the thymus and are marked by the expression of CD4 and a Forkhead/winged-helix family member, forkhead box P3 (Foxp3; Scurfin), which is an activator and a repressor of transcriptional control (1). Scurfy mice, which have a 2-bp insertion in their exon 8 of the X-linked Foxp3 gene (2), develop a lymphoproliferative disorder and perivascular infiltration of hematopoietic cells in multiple peripheral organs (3). Depletion of the Foxp3-expressing population in mice can lead to autoimmune disorders (4–6). Under the transcriptional control of Foxp3, Treg cells express a high level of interleukin-2 (IL-2) receptor α (CD25) and a low level of IL-7 receptor α (CD127) on the cell surface (7). Treg cells do not produce IL-2 or tumor necrosis factor (TNF) but instead depend on common γ chain cytokines from other cells for survival (7, 8).
CD8 and CD4 T cells, particularly those of the memory phenotype, undergo apoptosis and decline in number early (days 2 to 4) during viral (9–11) and some bacterial (12) infections. This attrition is mediated in part by type I interferon (IFN) and occurs after infection with type I IFN-inducing pathogens or after treatment of mice with the type I IFN-inducer poly(I·C). This attrition and apoptosis can be blocked by antibody to type I IFN (13) and is dramatically reduced in type I IFN receptor (IFNAR) knockout (KO) mice (9, 14). Memory CD8 and CD4 T cells express high levels of CD44, and these CD44hi cells show the highest levels of apoptotic loss at the early stage of infection, perhaps making room for a new immune response to rapidly develop (14).
Infection has been implicated as a potential trigger in autoimmune diseases (15–18), but underlying mechanisms have yet to be clarified. One of the theories is that infection may disrupt the balance of immune regulation, perhaps through Treg cells, in the susceptible organs (18, 19). Foxp3+ CD4+ Treg cells exhibit a partial memory phenotype with an intermediate to high expression of the activation and memory marker CD44 (20). Therefore, we questioned whether Treg cells may behave like the CD44hi CD4 and CD44hi CD8 T cells and get driven into apoptosis and decline in number during early stages of infection. If so, their demise might contribute to the phenomenon of virus-induced autoimmunity.
MATERIALS AND METHODS
Mice and virus stocks.
Foxp3-GFP knock-in mice, a kind gift from Vijay K. Kuchroo (21), were bred and maintained in a specific-pathogen-free facility at the University of Massachusetts Medical School (UMMS) (Worcester, MA). C57BL/6 mice between 4 to 6 weeks of age were purchased from the Jackson Laboratory. Experiments were done when mice reached at least 6 to 7 weeks of age.
Lymphocytic choriomeningitis virus (LCMV) strain Armstrong was propagated in baby hamster kidney BHK21 cells (22, 23). Mice were inoculated intraperitoneally (i.p.) with 5 × 104 PFU of LCMV stock. Injection with supernatant from uninfected BHK21 cell cultures (BHK) was used as a sham control in some experiments. Experiments were done in compliance with the Animal Welfare Act and the National Institutes of Health guidelines for the ethical care and use of animals in biomedical research, according to protocols approved by the Institutional Animal Care and Use Committee of UMMS.
Surface staining.
Splenocytes in suspension were washed in staining buffer (1% fetal calf serum [FCS] in phosphate-buffered saline [PBS]), blocked with anti-CD16/32 monoclonal antibody (MAb; clone 2.4G2; Fc block), stained with anti-CD4 (clone RM4-5), anti-CD8α (clone 53-6.7), anti-CD8β (clone YTS156.7.7), anti-CD44 (clone IM7), anti-CD25 (clone PC61.5), anti-CD127 (clone A7R34), anti-CD122 (clone TM-β1), anti-CD132 (clone TUGm2), and anti-Thy1.2 (clone 30-H12) antibodies, and Live/Dead Fixable Aqua (Invitrogen) for 20 min at 4°C, and fixed with CytoFix (BD Biosciences) for 5 min at 4°C. Samples were resuspended in staining buffer until reading by an LSRII flow cytometer (BD Biosciences). Collected data were analyzed by FlowJo software (Tree Star).
TUNEL assay.
To evaluate DNA fragmentation, cells were rested in medium (10% FCS in RPMI 1640 with 2 mM penicillin-streptomycin and 2 mM l-glutamine; Gibco) supplemented with 10 mM HEPES in a 48-well plate for 5 h at 37°C and 5% CO2, Fc blocked, surface stained, and subjected to CytoFix for 15 min at 4°C. After permeabilization with 70% (vol/vol) ethanol for 3.5 days at −20°C, terminal deoxynucleotidyl transferase dUTP-biotin nick end labeling (TUNEL) was performed using the Apo-BrdU-Red In Situ DNA Fragmentation Assay kit (BioVision) according to the manufacturer's instructions. Samples were analyzed within 3 h by LSRII.
Annexin V staining.
Annexin V staining was done directly ex vivo. After the Fc block, surface staining, and one wash with annexin V binding buffer (eBioscience), cells were stained with annexin V (eBioscience) at room temperature for 15 min. Samples were analyzed immediately by LSRII.
PhosFlow staining.
Splenocytes in suspension were stimulated with IL-2 (5 ng/ml; BD Biosciences) for 15 min at 37°C, 5% CO2. After a quick wash in staining buffer, cells were immediately fixed with CytoFix for 15 min at 4°C, Fc blocked, and surface stained as described above. Cells were then permeabilized with PhosFlow Perm buffer III (BD Biosciences) for 30 min on ice and stained with anti-STAT5 (clone 3H7) or anti-pSTAT5 (clone pY694) for 60 min at room temperature in the dark. Samples were resuspended in staining buffer until analysis by LSRII.
Statistical analysis.
Student's t test was calculated using Excel or Prism software. Data are presented as means and standard errors of the means (SEM).
RESULTS
Insignificant reduction of Foxp3+ CD4+ Treg cells after LCMV infection.
As an initial screen to determine if there was a loss in Treg cells during viral infection, C57BL/6 mice were infected intraperitoneally with strain Armstrong of LCMV and examined for CD8, CD4, and Treg cell phenotype and number. In a time course study, mice were examined every 4 days for 2 weeks and then every 7 days for 2 more weeks to determine the stability of the Treg cell population. Figure 1 depicts the frequency (Fig. 1a) and total number (Fig. 1b) of Foxp3+ CD4+ Treg cells as well as CD8 and non-Treg CD4 T cells in the spleen at these different time points. It shows the well-established dramatic rise of the CD8 T cells versus the more stable CD4 T cell number, reflecting the reversal of the CD4 to CD8 T cell ratio during an acute viral infection. The Treg population, however, remained relatively stable during this 4-week time period. This stability of the Treg population was also seen when Treg numbers were separately examined at 0, 2, and 3 days postinfection in many additional experiments as described below.
FIG 1.
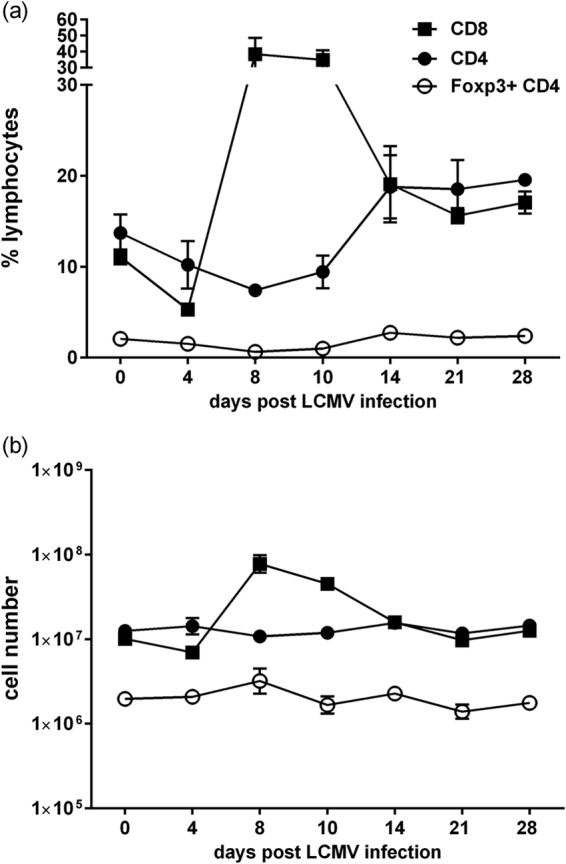
Relatively stable Foxp3+ CD4+ Treg cell numbers during LCMV infection. C57BL/6 mice were infected i.p. with 5 × 104 PFU of LCMV strain Armstrong, and the spleen was examined every 4 days over 4 weeks for the frequency (a) and total number (b) of CD8 T, Foxp3+ CD4+ Treg, and Foxp3-negative CD4 T cells. Each time point represents 3 to 14 mice.
Normally, at early stages (days 2 to 3) of viral infection, there is a substantial type 1 IFN-induced apoptosis-driven loss in CD8 and CD4 T cells expressing the CD44hi memory phenotype. To examine whether Treg cells, which express intermediate to high levels of CD44, are resistant to apoptosis at these very early time points, we examined their apoptosis and attrition in more detail. LCMV induces type I IFN, which peaks at day 2 postinfection (24). Splenocytes from infected Foxp3-GFP knock-in mice and age-matched naive controls were harvested at this type I IFN peak 2 days postinfection and analyzed by flow cytometry (Fig. 2). While the frequency of Foxp3-negative CD44hi CD4 and CD44hi CD8 T cells among lymphocytes decreased considerably after LCMV infection, the frequency of Foxp3+ CD4 T cells was only slightly reduced (Fig. 2b), and the frequency of Foxp3+ CD4 T cells among CD4 T cells before and after LCMV infection was not significantly different (11.6% ± 0.75% versus 10.7% ± 2.1%, P = 0.24). The actual number of T cells in the spleen was then calculated for each individual mouse using their total splenocyte counts separately. As expected, CD44hi CD4 and CD44hi CD8 T cells declined significantly in number (Fig. 2c). However, the total number of Foxp3+ CD4 T cells did not significantly change after infection (Fig. 2c). At day 3 following LCMV infection, a small but consistent decline in the frequency of Foxp3+ CD4 T cells was observed in the spleen (Fig. 2d), but the total number of Foxp3+ CD4 T cells in the spleen of infected animals was not significantly different from that of naive animals (Fig. 2e). Therefore, the presence of Foxp3+ CD4 T cells in the spleen is relatively stable early during LCMV infection.
FIG 2.

Foxp3+ CD4+ Treg cells resist loss after LCMV infection in comparison to CD44hi CD8 and CD44hi Foxp3-negative CD4 T cells. Foxp3-GFP mice were infected i.p. with 5 × 104 PFU LCMV. Two (b and c) or 3 (d and e) days later, surface-stained splenocytes from infected and uninfected (naive) mice were analyzed by flow cytometry, with gating as shown in panel a. (b and d) The frequencies of CD8 T cells and Foxp3+ CD4+ Treg cells were obtained by direct gating, while the frequency of Foxp3-negative CD4 T cells was calculated by Boolean gating in FlowJo. (c and e) The absolute numbers of the different populations were then calculated from the total live splenocyte counts. (b and c) Combined data from 3 separate experiments (n = 9). (d and e) Representative data from seven separate experiments (n = 3). *, statistical significance (P < 0.05) between naive and LCMV-infected groups.
Infection can cause Foxp3+ CD4 T cells to traffic between the secondary lymphoid organs. Therefore, the Treg cells from inguinal, axillary/brachial, and mesenteric lymph nodes were examined after LCMV infection, and we found no significant changes in the number and frequency of Treg cells at days 2 and 3 postinfection (data not shown).
Foxp3+ CD4 T cells are resistant to apoptosis during LCMV infection.
Cell death by apoptosis is a coordinated process that ultimately leads to DNA fragmentation. The 3′-OH end of the fragmented or nicked DNA can be visualized by TUNEL assay, which covalently attaches a dUTP to the 3′-OH end. Subsequent labeling with a fluorescence-conjugated anti-dUTP antibody allows quantitative assessment of DNA fragmentation by flow cytometry. A previous study showed that both CD44hi CD8 and CD44hi CD4 T cells in the spleen were more TUNEL positive after poly(I·C) treatment (11). Infection with LCMV also increases the frequency of TUNEL-positive CD44hi CD8 T cells in the spleen early after infection (11), and tissue sections have shown two peaks of TUNEL-positive splenocytes after LCMV infection: one at day 3, closely following the peak of the type I IFN response, which we examined here, and the other at day 11, as the response enters the contraction phase (25).
To test if Treg cells were sensitive to virus infection-induced apoptosis, splenocytes were harvested from Foxp3-GFP mice at day 2 after LCMV infection, rested in medium at 37°C for 5 h, and stained with the TUNEL assay (Fig. 3). Without infection, CD44hi Foxp3-negative CD4 T cells and Foxp3+ CD4+ Treg cells appeared to be more TUNEL positive than CD8 T cells (Fig. 3a). Infection with LCMV increased the percentage of TUNEL-positive CD4 and CD8 T cells but had no effect on Foxp3+ CD4 T cells. As expected, the increase in apoptotic events among CD44hi T cells was more dramatic than that among CD44lo T cells after LCMV infection (Fig. 3b). Although Foxp3+ CD4+ Treg cells from naive mice had a measurable TUNEL background, their levels of apoptosis were similar to those of the relatively resistant CD44lo CD4 and CD44lo CD8 T cells at day 2 after LCMV infection, and they were much less apoptotic than CD44hi CD4 and CD44hi CD8 T cells.
FIG 3.

Foxp3+ CD4+ Treg cells are less TUNEL positive than CD44hi Foxp3-negative CD4 and CD44hi CD8 T cells in the spleen at day 2 following LCMV infection. Foxp3-GFP mice were infected i.p. with 5 × 104 PFU LCMV. Splenocytes harvested at 2 days postinfection were stained with TUNEL after incubating in medium at 37°C for 5 h. (a) Histograms of TUNEL staining intensity of CD4, CD8, and Treg cells from a naive animal and a day 2 LCMV-infected animal in a representative experiment. (b) Combined data from 3 separate experiments (total n = 9). *, statistical significance (P < 0.05) between naïve mice and mice at day 2 following LCMV infection. (c) A representative panel of dot plots showing TUNEL reactivity versus CD44 staining among CD4 T cells, CD8 T cells, and Treg cells.
Activated caspase disturbs the asymmetry of the plasma membrane, causing exposure of phosphatidylserine on the cell surface (26). Therefore, apoptosis of splenocytes from LCMV-infected Foxp3-GFP mice was also examined ex vivo with amine-reactive live/dead stain and annexin V, which binds to exposed phosphatidylserine. Similar to the data from the TUNEL assay, Foxp3-negative CD44hi CD4 T cells were highly annexin V positive, even among cells from naive spleens (Fig. 4). Annexin V reactivity among CD4 and CD8 T cells increased after LCMV infection but remained low among Foxp3+ CD4+ Treg cells.
FIG 4.

Foxp3+ CD4+ Treg cells are less annexin V positive than CD8 and Foxp3-negative CD4 T cells in the spleen at day 2 following LCMV infection. Foxp3-GFP mice were infected i.p. with 5 × 104 PFU LCMV. Splenocytes harvested at 2 days postinfection were stained ex vivo with Live/Dead Fixable stain and annexin V. (a) Histograms of annexin V staining intensity of live CD4, CD8, and Treg cells from a naive animal and a day 2 LCMV-infected animal in a representative experiment. (b) Data are from a representative of three separate experiments (n = 3). *, statistical significance (P < 0.05) between naive cells and cells at day 2 following LCMV infection. (c) Representative panel of dot plots showing annexin V reactivity versus CD44 staining among CD4 and CD8 T cells and Treg cells.
Similar results were observed at day 3 after LCMV infection. Foxp3+ CD4 T cells from naive animals displayed some TUNEL and annexin V background reactivities, but their staining intensity was not increased by LCMV infection, while CD8- and Foxp3-negative CD4 T cells, especially their CD44hi subsets, became much more TUNEL and annexin V positive after infection (Fig. 5). Both the TUNEL and annexin V data therefore indicate that Foxp3+ CD4+ Treg cells are much more resistant to virus infection-induced apoptosis than CD44hi CD4 and CD44hi CD8 T cells in the spleen.
FIG 5.

Foxp3+ CD4+ Treg cells are less apoptotic than CD44hi Foxp3-negative CD4 and CD8 T cells in spleen at day 3 following LCMV infection. Foxp3-GFP mice were infected i.p. with 5 × 104 PFU LCMV. Splenocytes harvested at 3 days postinfection were stained with TUNEL after incubating in medium at 37°C for 5 h (a) or ex vivo with Live/Dead Fixable stain and annexin V (b). Data are representative of at least three separate experiments (n = 3). *, statistical significance (P < 0.05) between naive cells and cells at day 3 following LCMV infection.
Survival factors regulating T cell apoptosis.
Cellular apoptosis is generally regulated by a balance between intracellular pro- and antiapoptotic proteins and the availability of survival cytokines and their receptors. The complete mechanism behind the type I IFN-induced apoptosis of memory T cells during viral infection remains unclear, and it is also not certain if the mechanism of apoptosis for the CD44hi CD8 T cells is the same as that for the CD44hi CD4 T cells. However, it has been shown that the apoptosis of these T cells requires the proapoptotic molecule Bim and that it occurs in T cells lacking CD127, the receptor for the T cell survival factor IL-7 (11, 27). Treg cells are reported to express very high levels of CD25 (7), and their survival is correlated with IL-2-mediated signaling of STAT5 (28, 29). We therefore examined each of these factors in uninfected and day 2 LCMV-activated T cell populations and determined how they correlated with apoptosis.
Although our previous study had shown that the proapoptotic Bim molecule was required for apoptosis of memory T cell populations (11), examination by flow cytometry found no correlation between proapoptotic Bim or BAK expression and the apoptotic cell populations within the uninfected and LCMV-inoculated naive (CD44lo) and memory (CD44hi) CD4 and CD8 T cell populations and Treg populations. There also was no correlation between expression of the antiapoptotic proteins Bcl-2, Bcl-xL, or Mcl-1 or of the ratio between the pro- and antiapoptotic proteins throughout these five gated cell populations (data not shown).
Failing to find illuminating results by examining pro- and antiapoptotic molecule expression, we focused on the expression of cytokine receptors. It has been shown that IL-7 signaling is critical for the survival of naive (30, 31) and memory (32, 33) T cells in the periphery, whereas IL-2 is important for Treg cell maintenance (34). Therefore, we examined the surface expression of CD127, CD25, IL-2 receptor β (CD122), and the common cytokine receptor γ chain (CD132) on T cells in naive and LCMV-infected Foxp3-GFP mice (where GFP is green fluorescent protein) at day 2 postinfection (Fig. 6). As published previously (35), surface expression of CD127 on Foxp3+ CD4 T cells was much lower than that on Foxp3-negative CD4 T cells in the spleen. CD127 expression on CD44hi Foxp3-negative CD4 T cells was lower than that on CD44hi CD8 T cells (Fig. 6b). After LCMV infection, CD127 was downregulated in all CD8 and CD4 T cells regardless of Foxp3 or CD44 expression status. This downregulation of receptors for this T cell survival cytokine after viral infection may therefore have made conventional CD4 and CD8 T cells more susceptible to apoptosis. Expression of CD25 was high on Foxp3+ CD4+ Treg cells but was negligible on all other CD4 and CD8 T cells (Fig. 6c). LCMV infection, in contrast to its lowering of expression of CD127, upregulated CD25 expression on Treg cells substantially but did not affect the low levels of expression of CD25 on other CD4 or CD8 T cells at day 2 postinfection. As published previously (36), expression of CD122 was the highest on CD44hi CD8 T cells but very low on CD44lo CD4 T cells. We found that Foxp3+ CD4+ Treg cells expressed higher levels of CD122 than Foxp3-negative CD4 T cells and CD44lo CD8 T cells (P < 0.02), and CD122 expression was not changed much by LCMV infection (Fig. 6d). All CD4 and CD8 T cells and Treg cells expressed CD132 (Fig. 6e). Although the expression of CD132 was elevated on Foxp3-negative CD4 and CD8 T cells after LCMV infection, it was unchanged on Treg cells. The pattern of receptor expression indicates that although LCMV infection downregulates the IL-7 receptor on all T cells, the high-affinity IL-2 receptor remains intact or is more highly expressed on Treg cells. This may provide a survival advantage to the Treg cells.
FIG 6.

The IL-7 receptor is downregulated on T cells, while the high-affinity IL-2 receptor remains on Foxp3+ CD4+ Treg cells after LCMV infection. Foxp3-GFP mice were infected i.p. with 5 × 104 PFU LCMV. Splenocytes harvested at 2 days postinfection were stained ex vivo for CD127 (b), CD25 (c), CD122 (d), and CD132 (e), and the T cell populations were analyzed by flow cytometry (a). Data are representative of at least three separate experiments showing geometric mean fluorescence intensity between naive mice and mice at day 2 following LCMV infection (n = 3). *, statistical significance (P < 0.05) between naive mice and mice at day 2 following LCMV infection (n = 3). Histograms of a representative animal from each group were overlaid for visual comparison.
STAT5A and STAT5B mediate IL-2 and IL-7 signaling (28, 37, 38). Phosphorylated STAT5 is involved not only in sustaining Foxp3 expression (39) but also in regulating CD8 T cell homeostasis (40, 41). PhosFlow staining was used to examine the intracellular STAT5 and its phosphorylated form. The percentages of CD4 and CD8 T cells were similar to those obtained by surface staining alone. When the expression levels of STAT5 before and after LCMV infection were compared, we found that the total expression of STAT5 was the highest among Foxp3+ CD4 T cells in naive animals and was marginally increased after LCMV infection (Fig. 7a). In contrast, the total STAT5 expression was slightly reduced among CD8 T cells (both CD44hi and CD44lo) and CD44lo Foxp3-negative CD4 T cells after infection. Consistent with the expression pattern of CD25 (Fig. 6b), only Treg cells were able to respond to IL-2 stimulation in vitro and phosphorylate STAT5 (Fig. 7b). A time course experiment also revealed that STAT5 in Foxp3+ CD4 T cells could be phosphorylated in response to IL-2 within seconds of encounter (data not shown). Therefore, the ability of Foxp3+ CD4+ Treg cells to receive survival signals through IL-2 receptor and STAT5 was not diminished early during LCMV infection.
FIG 7.
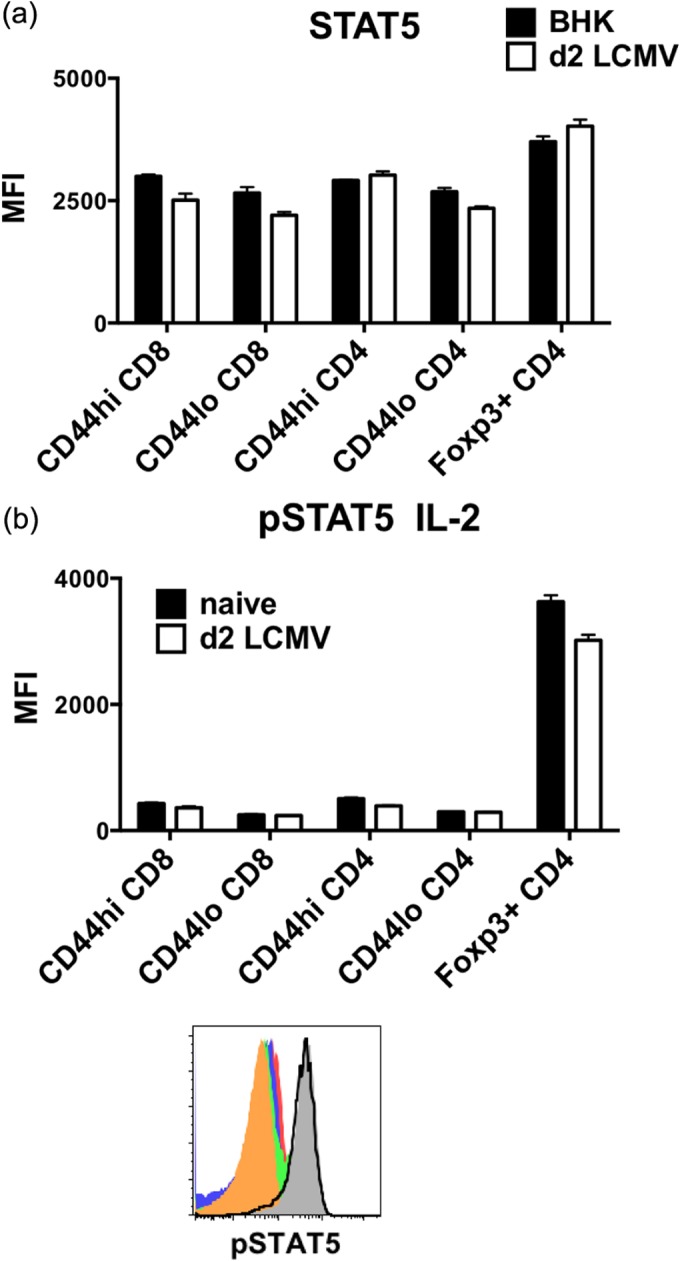
Foxp3+ CD4+ Treg cells display the highest level of STAT5 expression and respond well to IL-2 stimulation in vitro even after LCMV infection. Foxp3-GFP mice were infected i.p. with 5 × 104 PFU LCMV. (a) Splenocytes harvested at 2 days postinfection were stained ex vivo for STAT5. (b) Phosphorylation of STAT5 (pSTAT5) was assessed among CD44hi CD8 (solid red), CD44lo CD8 (solid blue), CD44hi Foxp3-negative CD4 (solid green), CD44lo Foxp3-negative CD4 (solid orange), and Foxp3+ CD4 (solid gray) T cells from a representative naive animal and Foxp3+ CD4 T cells (black line) from a day 2 LCMV-infected animal after stimulating splenocytes with 5 ng/ml IL-2 for 15 min at 37°C. Data are representative of two separate experiments showing geometric mean fluorescence intensity between BHK sham control or naive versus day 2 LCMV-infected groups of 2 or 3 mice.
DISCUSSION
Previous work had established that CD44hi CD8 and CD44hi CD4 T cells are driven to apoptosis by virus infection- and poly(I·C)-induced type I IFN (9, 11, 13, 14). The current study extends our knowledge to the Foxp3+ CD4+ natural Treg cells. Treg cells are major regulators of self-reactive T cells and are strong suppressors of autoimmunity. There are several experimental models of virus-induced autoimmunity in animals, and there are epidemiological correlates linking viral infections to the induction or exacerbations of a variety of autoimmune diseases, including diabetes, multiple sclerosis, rheumatoid arthritis, myocarditis, etc. (15, 42). A possible mechanism for virus-induced autoimmunity could be a consequence of the infection-induced apoptosis of Treg cells, because a transient loss of Treg cells is sufficient to initiate an autoimmune disorder (34). However, most of the time viruses do not induce autoimmunity. It is not uncommon to see some self-reactive antibodies generated during infection (43–45), but most of the time the host survives a viral infection without serious autoimmune manifestations. It was, therefore, of interest to determine whether Foxp3+ CD4+ natural Treg cells, present at the beginning of viral infections, were resistant or sensitive to the T cell apoptosis that occurs early during infection. Human Treg cells are more resistant to apoptosis induced by irradiation or a topoisomerase inhibitor than non-Treg CD4 T cells (46), and both murine and human Treg cells are resistant to activation-induced cell death (47, 48). Here we show that Treg cells were substantially more resistant to viral infection-induced apoptosis than were the CD44hi CD4 and CD8 memory T cells with which they coexist. This resistance of Treg cells to apoptosis could be part of the explanation why autoimmunity, which can occur after a virus infection, is relatively rare.
Treg cells might be more resistant to virus infection-induced apoptosis because of their ability to receive signals from IL-2. Both IL-7 and IL-2 have been shown to affect Treg population size in the periphery through regulating Foxp3 expression (49, 50). Foxp3 as a transcription factor can downregulate the expression of CD127 but upregulate CD25 expression (7), resulting in mature Treg cells being CD127lo and CD25hi. Recent work has also identified a CD127hi Foxp3+ population in bone marrow and in skin (35), but these were not examined in this study. Upon LCMV infection, CD127 expression was further reduced on Foxp3+ CD4+ Treg cells, but the expression of CD25, CD122, and CD132 was either increased or unchanged. The ability to continue to receive survival signals through the IL-2/STAT5 pathway may allow Treg cells to survive and maintain functionality during infection. A high dose (1 mg/mouse) of IL-2-neutralizing antibody has been shown to deplete Foxp3+ CD25+ CD4+ Treg cells over 3 days in vivo (34), and the presence of IL-2 (0.1 to 10 ng/ml) has been shown to protect CD25+ CD4+ Treg cells but not CD25− conventional CD4 T cells from spontaneous and dexamethasone-induced cell death in a 12-h in vitro assay (51). Our preliminary attempt to induce Treg cell death by adding a depleting anti-IL-2 antibody similarly supported the role of IL-2 in Treg cell survival, though more tests are needed to confirm the observation.
The downregulation of CD127 might have made T cells more sensitive to apoptosis. CD127 is thought to be downregulated in response to T cell activation, but CD127 expression on wild-type (WT) donor CD8 T cells is much higher in IFNAR KO hosts than in WT hosts after acute LCMV infection (52). It is noteworthy that the early apoptosis of memory T cells does not occur in LCMV-infected IFNAR KO mice. Thus, type I IFN probably indirectly downregulates CD127 on T cells as it induces their apoptosis. While the expression of CD127 is important for naive and memory CD8 T cell survival, signaling through the IL-7 receptor is not required during the initial proliferative response to an infection or immunization (32). Decreasing the sensitivity of T cells to IL-7 through downregulation of CD127 may be a way to allow the selection and programming of responding T cells by other cytokines to generate specific responses to newly encountered antigens while simultaneously eliminating some preexisting memory T cells.
Two recent reports have examined the effects of type I IFN on Treg cells under inflammatory conditions. One concludes that type I IFN may actually act to maintain Foxp3 expression and Treg activity, a result wholly consistent with our observations of Treg cell resistance to IFN-induced apoptosis (53). A second paper using the LCMV system notes that type I IFN serves to inhibit the proliferation of Treg cells at later stages of infection (54). Clearly, IFN can have many effects on Treg and other types of immune cells, but the early apoptosis of Treg cells does not seem to be one of them.
Previous attempts to decipher the mechanism of memory CD44hi CD8 T cell apoptosis and early attrition induced by type I IFN had not established any connection to NK cells, gamma interferon receptor (IFN-γR), perforin, FasL, granzyme B, TNF-related apoptosis-inducing ligand (TRAIL), TNF receptor 1 (TNFR1), or TNFR2 (9) (K. Bahl and R. M. Welsh, unpublished data). However, Bim is instrumental to the process. In Bim KO mice, the frequency of CD44hi CD8 T cells is relatively stable after LCMV infection, and the CD44hi CD8 T cells are substantially less TUNEL positive at day 3 postinfection with LCMV (11). Bim can physically interact with Bcl-2, Bcl-xL, and Mcl-1 to initiate apoptosis (55, 56). Since Treg cells are more resistant to virus infection-induced early apoptosis, it was expected that the relative abundance of antiapoptotic and proapoptotic molecules might be different between Treg cells and the CD44hi CD8 and CD4 T cells. However, the mean fluorescence intensity (MFI) ratios of Bim to antiapoptotic molecule did not increase with the increased occurrences of apoptosis observed in the CD44hi CD8 and CD4 T cells after LCMV infection (data not shown).
The functions of Treg cells may make them useful to be preserved during infections and yet inevitably problematic in some circumstances. Treg cells play a critical role of regulating effector T cells, NK cells, neutrophils, monocytes, and macrophages early during viral infections, mitigating the pathology incurred by the immune response and prolonging survival of the infected mice (57–60). The presence of virus-specific memory Treg cells can preserve lung function by limiting CD8 T cell response and reducing pathology during homologous influenza rechallenge (61). However, during heterologous infections, the nature of their persistence may also pose a problem, as demonstrated in the influenza A virus-immune mice during intranasal LCMV challenge, in which the higher number of Treg cells contribute to more-severe immunopathology in the immune mice by preventing exhaustion of responding CD8 T cells and supporting migration of CD8 T cells into the lungs (62).
The association between infection, IFN therapy, and autoimmunity (63–65) had us questioning whether virus-induced type I IFN might drive Treg cells into apoptosis and reduction in number, thereby allowing activation of autoreactive cells. Our study, however, indicates that the Foxp3+ CD4+ natural Treg population is relatively stable early during LCMV infection. Similarly, our preliminary studies with mice treated with type I IFN inducer poly(I·C) for 1 day also showed more stable Foxp3+ Treg cells by TUNEL assay (data not shown), suggesting that type I IFN may not compromise the survival of natural Treg cells. This may explain why the autoimmunity that develops in IFN-treated or infected patients is at a relatively low frequency. Many other factors are involved in the development of autoimmunity, and further investigation will be needed to dissect the specific process for each autoimmune disease.
ACKNOWLEDGMENTS
We thank Kapil Bahl, Varun Kapoor, and Stina Urban for helpful discussions on the staining protocols.
This work was funded by U.S. National Institutes of Health Training grant T32 AI007349 and research grants R37 AI017672, PO1 AI046620, AI081675, and AI-46578.
The views expressed are those of the authors and not necessarily those of NIH.
REFERENCES
- 1.Zheng Y, Josefowicz SZ, Kas A, Chu T-T, Gavin MA, Rudensky AY. 2007. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 2.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko S-A, Wilkinson JE, Galas D, Ramsdell SFZF. 2001. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 3.Godfrey VL, Wilkinson JE, Russell LB. 1991. X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am J Pathol 138:1379–1387. [PMC free article] [PubMed] [Google Scholar]
- 4.Feuerer M, Shen Y, Littman DR, Benoist C, Mathis D. 2009. How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity 31:654–664. doi: 10.1016/j.immuni.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. 2007. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med 204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JM, Rasmussen JP, Rudensky AY. 2007. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 7.Williams LM, Rudensky AY. 2007. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol 8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 8.Pandiyan P, Lenardo MJ. 2008. The control of CD4+CD25+Foxp3+ regulatory T cell survival. Biol Direct 3:6. doi: 10.1186/1745-6150-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNally JM, Zarozinski CC, Lin M-Y, Brehm MA, Chen HD, Welsh RM. 2001. Attrition of bystander CD8 T cells during virus-induced T-cell and interferon responses. J Virol 75:5965–5976. doi: 10.1128/JVI.75.13.5965-5976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang J, Anaraki F, Blank KJ, Murasko DM. 2003. Cuttine edge: T cells from aged mice are resistant to depletion early during virus infection. J Immunol 171:3353–3357. doi: 10.4049/jimmunol.171.7.3353. [DOI] [PubMed] [Google Scholar]
- 11.Bahl K, Hüebner A, Davis RJ, Welsh RM. 2010. Analysis of apoptosis of memory T cells and dendritic cells during the early stages of viral infection or exposure to toll-like receptor agonists. J Virol 84:4866–4877. doi: 10.1128/JVI.02571-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang J, Lau LL, Shen H. 2003. Selective depletion of nonspecific T cells during the early stage of immune responses to infection. J Immunol 171:4352–4358. doi: 10.4049/jimmunol.171.8.4352. [DOI] [PubMed] [Google Scholar]
- 13.Jiang J, Gross D, Nogusa S, Elbaum P, Murasko DM. 2005. Depletion of T cells by type I interferon: differences between young and aged mice. J Immunol 175:1820–1826. doi: 10.4049/jimmunol.175.3.1820. [DOI] [PubMed] [Google Scholar]
- 14.Bahl K, Kim S-K, Calcagno C, Ghersi D, Puzone R, Celada F, Selin LK, Welsh RM. 2006. IFN-induced attrition of CD8 T cells in the presence or absence of cognate antigen during the early stages of viral infections. J Immunol 176:4284–4295. doi: 10.4049/jimmunol.176.7.4284. [DOI] [PubMed] [Google Scholar]
- 15.van der Werf N, Kroese FGM, Rozing J, Hillebrands J-L. 2007. Viral infections as potential triggers of type 1 diabetes. Diabetes Metab Res Rev 23:169–183. doi: 10.1002/dmrr.695. [DOI] [PubMed] [Google Scholar]
- 16.Huber SA. 2006. Autoimmunity in coxsackievirus B3 induced myocarditis. Autoimmunity 39:55–61. doi: 10.1080/08916930500484906. [DOI] [PubMed] [Google Scholar]
- 17.Sherbet G. 2009. Bacterial infections and the pathogenesis of autoimmune conditions. Br J Med Practitioners 2:6–13. [Google Scholar]
- 18.Zipris D, Hillebrands J-L, Welsh RM, Rozing J, Xie JX, Mordes JP, Greiner DL, Rossini AA. 2003. Infections that induce autoimmune diabetes in BBDR rats modulate CD4+CD25+ T cell populations. J Immunol 170:3592–3602. doi: 10.4049/jimmunol.170.7.3592. [DOI] [PubMed] [Google Scholar]
- 19.Jun H-S, Yoon J-W. 2003. A new look at viruses in type 1 diabetes. Diabetes Metab Res Rev 19:8–31. doi: 10.1002/dmrr.337. [DOI] [PubMed] [Google Scholar]
- 20.Min B, Thornton A, Caucheteux SM, Younes S-A, Oh K, Hu-Li J, Paul WE. 2007. Gut flora antigens are not important in the maintenance of regulatory T cell heterogeneity and homeostasis. Eur J Immunol 37:1916–1923. doi: 10.1002/eji.200737236. [DOI] [PubMed] [Google Scholar]
- 21.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 22.Welsh RM, Seedhom MO. 2008. Lymphocytic choriomeningitis virus (LCMV): propagation, quantitation, and storage. Curr Protoc Microbiol Chapter 15:Unit 15A.1. doi: 10.1002/9780471729259.mc15a01s8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welsh RM, Lampert PW, Burner PA, Oldstone MBA. 1976. Antibody-complement interactions with purified lymphocytic choriomeningitis virus. Virology 73:59–71. doi: 10.1016/0042-6822(76)90060-X. [DOI] [PubMed] [Google Scholar]
- 24.Welsh RM. 1978. Cytotoxic cells induced during lymphocytic choriomeningitis virus infection of mice. I. Characterization of natural killer cell induction. J Exp Med 148:163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Razvi ES, Jiang Z, Woda BA, Welsh RM. 1995. Lymphocyte apoptosis during the silencing of the immune response to acute viral infections in normal, lpr, and Bcl-2-transgenic mice. Am J Pathol 147:79–91. [PMC free article] [PubMed] [Google Scholar]
- 26.Fink SL, Cookson BT. 2005. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wojciechowski S, Jordan MB, Zhu Y, White J, Zajac AJ, Hildeman DA. 2006. Bim mediates apoptosis of CD127lo effector T cells and limits T cell memory. Eur J Immunol 36:1694–1706. doi: 10.1002/eji.200635897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wuest TY, Willette-Brown J, Durum SK, Hurwitz AA. 2008. The influence of IL-2 family cytokines on activation and function of naturally occurring regulatory T cells. J Leukoc Biol 84:973–980. doi: 10.1189/jlb.1107778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antov A, Yang L, Vig M, Baltimore D, Van Parijs L. 2003. Essential role for STAT5 signaling in CD25+CD4+ regulatory T cell homeostasis and the maintenance of self-tolerance. J Immunol 171:3435–3441. doi: 10.4049/jimmunol.171.7.3435. [DOI] [PubMed] [Google Scholar]
- 30.Boursalian TE, Bottomly K. 1999. Survival of naive CD4 T cells: roles of restricting versus selecting MHC class II and cytokine milieu. J Immunol 162:3795–3801. [PubMed] [Google Scholar]
- 31.Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, Surh CD. 2001. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A 98:8732–8737. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. 2000. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat Immunol 1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 33.Kondrack RM, Harbertson J, Tan JT, McBreen ME, Surh CD, Bradley LM. 2003. Interleukin 7 regulates the survival and generation of memory CD4 cells. J Exp Med 198:1797–1806. doi: 10.1084/jem.20030735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. 2005. Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med 201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simonetta F, Chiali A, Cordier C, Urrutia A, Girault I, Bloquet S, Tanchot C, Bourgeois C. 2010. Increased CD127 expression on activated Foxp3+CD4+ regulatory T cells. Eur J Immunol 40:2528–2538. doi: 10.1002/eji.201040531. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Sun S, Hwang I, Tough DF, Sprent J. 1998. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 8:591–599. doi: 10.1016/S1074-7613(00)80564-6. [DOI] [PubMed] [Google Scholar]
- 37.Xue H-H, Fink DW, Zhang X, Qin J, Turck CW, Leonard WJ. 2002. Serine phosphorylation of Stat5 proteins in lymphocytes stimulated with IL-2. Int Immunol 14:1263–1271. doi: 10.1093/intimm/dxf101. [DOI] [PubMed] [Google Scholar]
- 38.Tripathi P, Kurtulus S, Wojciechowski S, Sholl A, Hoebe K, Morris SC, Finkelman FD, Grimes HL, Hildeman DA. 2010. STAT5 is critical to maintain effector CD8+ T cell responses. J Immunol 185:2116–2124. doi: 10.4049/jimmunol.1000842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio C-WJ, Vegoe AL, Hsieh C-S, Jenkins MK, Farrar MA. 2008. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity 28:112–121. doi: 10.1016/j.immuni.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly J, Spolski R, Imada K, Bollenbacher J, Lee S, Leonard WJ. 2003. A role for Stat5 in CD8+ T cell homeostasis. J Immunol 170:210–217. doi: 10.4049/jimmunol.170.1.210. [DOI] [PubMed] [Google Scholar]
- 41.Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, Liu Y, Kaech SM. 2010. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci U S A 107:16601–16606. doi: 10.1073/pnas.1003457107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim B, Kaistha SD, Rouse BT. 2006. Viruses and autoimmunity. Autoimmunity 39:71–77. doi: 10.1080/08916930500484708. [DOI] [PubMed] [Google Scholar]
- 43.Sjöwall C, Cardell K, Boström EA, Bokarewa MI, Enocsson H, Ekstedt M, Lindvall L, Frydén A, Almer S. 2012. High prevalence of autoantibodies to C-reactive protein in patients with chronic hepatitis C infection: association with liver fibrosis and portal inflammation. Hum Immunol 73:382–388. doi: 10.1016/j.humimm.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Sane J, Kurkela S, Lokki M-L, Miettinen A, Helve T, Vaheri A, Vapalahti O. 2012. Clinical Sindbis Alphavirus infection is associated with HLA-DRB1*01 allele and production of autoantibodies. Clin Infect Dis 55:358–363. doi: 10.1093/cid/cis405. [DOI] [PubMed] [Google Scholar]
- 45.Berlin T, Zandman-Goddard G, Blank M, Matthias T, Pfeiffer S, Weis I, Toubi E, Singh S, Asherson R, Fraser A, Gilburd B, Sapir T, Levy Y, Lukac J, Rozman B, Kveder T, Shoenfeld Y. 2007. Autoantibodies in nonautoimmune individuals during infections. Ann N Y Acad Sci 1108:584–593. doi: 10.1196/annals.1422.061. [DOI] [PubMed] [Google Scholar]
- 46.Winzler C, Fantinato M, Giordan M, Calore E, Basso G, Messina C. 2011. CD4(+) T regulatory cells are more resistant to DNA damage compared to CD4(+) T effector cells as revealed by flow cytometric analysis. Cytometry A 79:903–911. doi: 10.1002/cyto.a.21132. [DOI] [PubMed] [Google Scholar]
- 47.Fritzsching B, Oberle N, Eberhardt N, Quick S, Haas J, Wildemann B, Krammer PH, Suri-Payer E. 2005. In contrast to effector T cells, CD4+CD25+Foxp3+ regulatory T cells are highly susceptible to CD95 ligand- but not to TCR-mediated cell death. J Immunol 175:32–36. doi: 10.4049/jimmunol.175.1.32. [DOI] [PubMed] [Google Scholar]
- 48.Banz A, Pontoux C, Papiernik M. 2002. Modulation of Fas-dependent apoptosis: a dynamic process controlling both the persistence and death of CD4 regulatory T cells and effector T cells. J Immunol 169:750–757. doi: 10.4049/jimmunol.169.2.750. [DOI] [PubMed] [Google Scholar]
- 49.Kim GY, Ligons DL, Hong C, Luckey MA, Keller HR, Tai X, Lucas PJ, Gress RE, Park J-H. 2012. An in vivo IL-7 requirement for peripheral Foxp3+ regulatory T cell homeostasis. J Immunol 188:5859–5866. doi: 10.4049/jimmunol.1102328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, Frank DA, Ritz J. 2006. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 108:1571–1579. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen X, Murakami T, Oppenheim JJ, Howard OMZ. 2004. Differential response of murine CD4+CD25+ and CD4+CD25− T cells to dexamethasone-induced cell death. Eur J Immunol 34:859–869. doi: 10.1002/eji.200324506. [DOI] [PubMed] [Google Scholar]
- 52.Nakayama Y, Plisch EH, Sullivan J, Thomas C, Czuprynski CJ, Williams BRG, Suresh M. 2010. Role of PKR and Type I IFNs in viral control during primary and secondary infection. PLoS Pathog 6:e1000966. doi: 10.1371/journal.ppat.1000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee SE, Li X, Kim JCK, Lee J, González-Navajas JM, Hong SH, Park I-K, Rhee JH, Raz E. 2012. Type I interferons maintain Foxp3 expression and T-regulatory cell functions under inflammatory conditions in mice. Gastroenterology 143:145–154. doi: 10.1053/j.gastro.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srivastava S, Koch MA, Pepper M, Campbell DJ. 2014. Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J Exp Med 160:521. doi: 10.1084/jem.20131556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hübner A, Barrett T, Flavell RA, Davis RJ. 2008. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell 30:415–425. doi: 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DCS. 2007. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 57.Lund JM, Hsing L, Pham TT, Rudensky AY. 2008. Coordination of early protective immunity to viral infection by regulatory T cells. Science 320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fulton RB, Meyerholz DK, Varga SM. 2010. Foxp3+ CD4 regulatory T cells limit pulmonary immunopathology by modulating the CD8 T cell response during respiratory syncytial virus infection. J Immunol 185:2382–2392. doi: 10.4049/jimmunol.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Veiga-Parga T, Suryawanshi A, Mulik S, Gimenez F, Sharma S, Sparwasser T, Rouse BT. 2012. On the role of regulatory T cells during viral-induced inflammatory lesions. J Immunol 189:5924–5933. doi: 10.4049/jimmunol.1202322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bedoya F, Cheng GS, Leibow A, Zakhary N, Weissler K, Garcia V, Aitken M, Kropf E, Garlick DS, Wherry EJ, Erikson J, Caton AJ. 2013. Viral antigen induces differentiation of Foxp3+ natural regulatory T cells in influenza virus-infected mice. J Immunol 190:6115–6125. doi: 10.4049/jimmunol.1203302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brincks EL, Roberts AD, Cookenham T, Sell S, Kohlmeier JE, Blackman MA, Woodland DL. 2013. Antigen-specific memory regulatory CD4+Foxp3+ T cells control memory responses to influenza virus infection. J Immunol 190:3438–3446. doi: 10.4049/jimmunol.1203140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kraft ARM, Wlodarczyk MF, Kenney LL, Selin LK. 2013. PC61 (anti-CD25) treatment inhibits influenza A virus-expanded regulatory T cells and severe lung pathology during a subsequent heterologous lymphocytic choriomeningitis virus infection. J Virol 87:12636–12647. doi: 10.1128/JVI.00936-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rönnblom LE, Alm GV, Oberg KE. 1991. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann Intern Med 115:178–183. doi: 10.7326/0003-4819-115-3-178. [DOI] [PubMed] [Google Scholar]
- 64.Fattovich G, Giustina G, Favarato S, Ruol A. 1996. A survey of adverse events in 11,241 patients with chronic viral hepatitis treated with alfa interferon. J Hepatol 24:38–47. [DOI] [PubMed] [Google Scholar]
- 65.Dumoulin FL, Leifeld L, Sauerbruch T, Spengler U. 1999. Autoimmunity induced by interferon alpha therapy for chronic viral hepatitis. Biomed Pharmacother 53:242–254. doi: 10.1016/S0753-3322(99)80095-X. [DOI] [PubMed] [Google Scholar]