

ABSTRACT
Although live-attenuated measles virus (MV) vaccines have been used successfully for over 50 years, the target cells that sustain virus replication in vivo are still unknown. We generated a reverse genetics system for the live-attenuated MV vaccine strain Edmonston-Zagreb (EZ), allowing recovery of recombinant (r)MVEZ. Three recombinant viruses were generated that contained the open reading frame encoding enhanced green fluorescent protein (EGFP) within an additional transcriptional unit (ATU) at various positions within the genome. rMVEZEGFP(1), rMVEZEGFP(3), and rMVEZEGFP(6) contained the ATU upstream of the N gene, following the P gene, and following the H gene, respectively. The viruses were compared in vitro by growth curves, which indicated that rMVEZEGFP(1) was overattenuated. Intratracheal infection of cynomolgus macaques with these recombinant viruses revealed differences in immunogenicity. rMVEZEGFP(1) and rMVEZEGFP(6) did not induce satisfactory serum antibody responses, whereas both in vitro and in vivo rMVEZEGFP(3) was functionally equivalent to the commercial MVEZ-containing vaccine. Intramuscular vaccination of macaques with rMVEZEGFP(3) resulted in the identification of EGFP+ cells in the muscle at days 3, 5, and 7 postvaccination. Phenotypic characterization of these cells demonstrated that muscle cells were not infected and that dendritic cells and macrophages were the predominant target cells of live-attenuated MV.
IMPORTANCE Even though MV strain Edmonston-Zagreb has long been used as a live-attenuated vaccine (LAV) to protect against measles, nothing is known about the primary cells in which the virus replicates in vivo. This is vital information given the push to move toward needle-free routes of vaccination, since vaccine virus replication is essential for vaccination efficacy. We have generated a number of recombinant MV strains expressing enhanced green fluorescent protein. The virus that best mimicked the nonrecombinant vaccine virus was formulated according to protocols for production of commercial vaccine virus batches, and was subsequently used to assess viral tropism in nonhuman primates. The virus primarily replicated in professional antigen-presenting cells, which may explain why this LAV is so immunogenic and efficacious.
INTRODUCTION
The causative agent of measles is a negative-sense, single-stranded enveloped virus with an RNA genome. Disease caused by infection with the virus is characterized by a latent period of 10 to 14 days, a short prodromal phase of fever, cough, coryza, and conjunctivitis, followed by the appearance of a characteristic maculopapular skin rash (1). Rash onset coincides with the development of immune responses and initiation of virus clearance. Infection with wild-type virus provides lifelong immunity to disease (2). Much of the more detailed knowledge of pathogenesis and sites of viral replication has come from studies in nonhuman primates (3). Monkeys experimentally infected with wild-type strains of measles virus (MV) develop a similar disease to humans (4, 5). Significant progress has been made in understanding viral pathogenesis by harnessing wild-type recombinant MVs (rMVs) expressing fluorescent proteins and the measles monkey model (6–9).
MV was first isolated in cell culture in 1954 (10) from a boy named David Edmonston. This wild-type MV strain was passaged multiple times in primary human kidney and amnion cells and adapted to embryonated hen's eggs and chicken embryo fibroblasts to produce the live-attenuated Edmonston-B vaccine virus (11). This was later replaced by further attenuated MV strains, including Edmonston-Zagreb (EZ) (12). These vaccines were shown to be safe and effective, and two-dose regimens combined with high coverage have successfully interrupted endemic MV transmission in large geographic areas (13).
In the developed world, vaccination has led to a dramatic decrease in the number of measles cases and a concomitant reduction in associated morbidity and mortality. However, it is still the leading vaccine-preventable disease, causing an estimated 122,000 deaths in 2012 (14). The immunosuppression associated with MV infection increases susceptibility to opportunistic infections exacerbating morbidity and mortality (9). Deaths mainly occur in Africa, India, and South East Asia, where vaccination rates are lower; contributing factors include the need to maintain the cold chain and the requirement to vaccinate >95% of susceptible individuals due to the highly infectious nature of MV (15).
Despite the huge accomplishments achieved by using live-attenuated measles vaccines, surprisingly little is known about what makes them so effective. Although previous studies have attempted to identify an “attenuation signature,” a definitive mechanism has not been elucidated. In order to induce protective immune responses, vaccine virus replication in the host is pivotal. However, nothing is known about the cells in which the vaccine virus replicates in vivo. With renewed interest in developing alternative vaccination routes, particularly aerosol vaccination (16, 17), this information is of crucial importance.
To address this gap in knowledge, we set out to identify the cells targeted by MV vaccine after injection. To this end, we generated a reverse genetics system for the World Health Organization (WHO)-approved EZ vaccine, which allowed us to generate and subsequently assess viruses containing an additional transcription unit (ATU) encoding enhanced green fluorescent protein (EGFP). The presence of EGFP was necessary to guide sample collection to the small number of virus-infected cells. Viruses expressing EGFP from different positions in the MVEZ genome were assessed, identifying rMVEZEGFP(3) as the one which retained all of the characteristics of the nonrecombinant parent. Virus stocks were produced in the human diploid cell line MRC-5 and formulated with excipients based on protocols provided by the Serum Institute of India (SII), the largest MV vaccine producer in the world. Intramuscular (i.m.) vaccination of cynomolgus macaques with rMVEZEGFP(3) revealed macrophages and dendritic cells (DCs) as the predominant target cells for MV vaccine virus replication in vivo.
MATERIALS AND METHODS
Cells.
MRC-5 and Vero-hCD150 cells were kind gifts from SII and Yusuke Yanagi, respectively. Both were grown in Advanced minimal essential medium, supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 1% (vol/vol) GlutaMAX (Invitrogen). The Vero-hCD150 cells were periodically passaged in the presence of Geneticin (400 μg/ml) to ensure all cells contained the expression plasmid encoding hCD150.
Generation of recombinant vaccine MV expressing EGFP.
A vial of M-VAC vaccine was supplied by SII. The freeze-dried powder was reconstituted in 0.5 ml of sterile water according to the manufacturer's instructions and used directly to isolate viral RNA. The complete consensus genomic sequence was determined following reverse transcription-PCR (RT-PCR) and RACE (rapid amplification of cDNA ends). A full-length cDNA which expressed the MVEZ anti-genome (pMVEZ) was constructed based on a modified pBluescript vector. Replacement of NotI/SacII, SacII/PacI, or PacI/PspXI fragments was used to generate pMVEZEGFP(1), pMVEZEGFP(3), and pMVEZEGFP(6), respectively. The ATU consisted of the EGFP open reading frame (ORF) and MV gene end and gene start specific sequences to allow expression from the viral genome. Plasmid sequences are available on request. Recombinant viruses were recovered from MVA-T7-infected Vero-hCD150 cells transfected with the full-length plasmids along with plasmids expressing MV N, P, and L. Virus stocks were grown in MRC-5 cells and tested negative for contamination with Mycoplasma species. Virus titers were determined by endpoint titration in Vero-hCD150 cells, and expressed in 50% tissue culture infectious dose (TCID50) units.
Multistep growth analysis.
MRC-5 or Vero-hCD150 cells in suspension were infected with each virus in triplicate at a multiplicity of infection (MOI) of 0.02 for 2 h at 37°C. The cells were spun out of the inoculum at 700 × g for 5 min, the pellet was resuspended, and the cell suspension was divided into aliquots in 36-mm-diameter wells (5 × 105 cells/well). For MRC-5 cells, at each indicated time point, the medium was removed from the well and replaced with 1 ml of medium. For Vero-hCD150 cells, at each indicated time point the cells and medium were combined into a tube and subjected to one freeze-thaw cycle to release total virus. Virus present in the sample for each time point was determined by endpoint titration in Vero-hCD150 cells, and quantities are expressed in TCID50 units.
Growth and formulation of vaccine viruses.
Protocols for the growth and formulation of recombinant vaccine viruses were kindly provided by SII. Briefly, MRC-5 cells were seeded into roller bottles along with recombinant virus at an MOI of 0.01. The cells were observed for up to 8 days, and viruses were harvested when the cytopathic effect was maximal. To formulate the stock, stabilizers were added, and the preparation was filtered and subsequently frozen in aliquots at −80°C. At this point the formulation of the recombinant viruses was comparable to the reconstituted M-VAC vaccine as licensed by SII.
Ethics statement.
Animal experiments were conducted in compliance with European guidelines (EU directive on animal testing 86/609/EEC) and Dutch legislation (Experiments on Animals Act, 1997). The protocols (EMC2218 and EMC2646) were approved by the independent animal experimentation ethical review committee DCC in Driebergen, The Netherlands. Animals were housed in groups prior to MV vaccination, received standard primate feed and fresh fruit on a daily basis, and had access to water ad libitum. In addition, their cages contained several sources of environmental enrichment, for example, hiding places, hanging ropes, tires, and other toys. During the vaccination study animals were housed in HEPA-filtered, negatively pressurized BSL-3 isolator cages. Animal welfare was observed on a daily basis, and all animal handling was performed under light anesthesia using ketamine and medetomidine (50/50 [vol/vol], 0.2 ml per kg of body weight [bw], i.m. injection). After handling, atipamezole was administered to antagonize the effect of medetomidine (0.05 ml per kg bw, i.m. injection).
Animal study design.
For each of the evaluation studies, 12 subadult male, MV-seronegative cynomolgus macaques (Macaca fascicularis) were infected via intratracheal (i.t.) inoculation with 104 TCID50 of M-VAC (reformulated according to the manufacturer's instructions), rMVEZ, rMVEZEGFP(1), rMVEZEGFP(3), or rMVEZEGFP(6). For the virus tropism study, six macaques (as described above) were vaccinated via i.m. injection with 104 TCID50 of rMVEZEGFP(3). The injection site was marked to allow collection of the necessary tissue during necropsy on 3 (n = 2 animals), 5 (n = 2 animals), or 7 (n = 2 animals) days postvaccination (d.p.v.).
Samples.
Small-volume EDTA blood samples were collected in Vacuette tubes containing K3EDTA as an anticoagulant 0, 3, 6, 9, 13, 17, 24, 35, 45, and 85 d.p.i. Plasma was separated from the blood by centrifugation, heat inactivated (30 min; 56°C), and stored at −20°C. Peripheral blood mononuclear cells (PBMC) were isolated from EDTA blood 0, 3, 6, 9, and 13 d.p.i. by density gradient centrifugation, resuspended in complete RPMI 1640 medium (Gibco Invitrogen, Carlsbad, CA) supplemented with l-glutamine (2 mM), 10% (vol/vol) FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml), and used for virus isolation. A BAL was performed 3, 6 and 9 d.p.i. by i.t. infusion of 10 ml of phosphate-buffered saline (PBS) through a flexible catheter. Bronchoalveolar lavage (BAL) cells were resuspended in culture medium with supplements as described above and used directly for virus isolation or virus detection by real-time RT-PCR (18). Throat and nose swabs were collected 0, 3, 6, and 9 d.p.i. for virus detection by real-time RT-PCR. For the virus tropism study, animals were euthanized by exsanguination under deep anesthesia using ketamine and medetomidine. After necropsy, injection site skin and muscle were collected in PBS, directly processed and screened for the presence of EGFP by UV microscopy. EGFP+ samples were either transferred to 4% (wt/vol) paraformaldehyde in PBS (to preserve EGFP autofluorescence) for direct imaging or to 10% neutral buffered formalin for further processing. Direct imaging was performed by confocal laser scanning microscopy with a LSM700 system fitted on an Axio Observer Z1 inverted microscope (Zeiss), and images were generated using Zen software.
Virus detection.
Isolation of MV was performed on Vero cells using an infectious-center test as previously described (19, 20). Virus isolations were monitored for cytopathic effect and/or fluorescence on day 6 after isolation. Real-time RT-PCR for BAL samples, and throat and nose swabs were performed as described previously (18). The results are expressed as “+”, indicating virus/genome was isolated/detected (regardless of load), or “–”, indicating no virus was isolated/detected.
MV IgG ELISA.
An in-house indirect enzyme-linked immunosorbent assay (ELISA) was used to measure levels of MV IgG in macaque serum samples. Plates were coated with β-propiolactone-inactivated MV as antigen and then incubated with 1:100-diluted plasma samples. Bound antibodies were detected using anti-human IgG-peroxidase conjugate, and the results are expressed as optical density at 450 nm (OD450) measurements (means of duplicate measurements).
MV neutralizing antibody quantification.
Serial dilutions (2−2 to 2−9) of heat-inactivated plasma samples were incubated with 102 TCID50 of MV for 1 h at 37°C. Vero cells were added, and the plates were cultured for 7 days at 37°C. Complete inhibition of infection, due to the presence of virus-neutralizing (VN) antibody in the plasma samples, was scored. The results are expressed in international units (IU) per ml, based on the WHO second international standard serum for measles provided by National Institute for Biological Standards and Control, Potters Bar, United Kingdom. The detection limit was 0.06 IU/ml.
MV glycoprotein-specific immunofluorescence.
Plasma antibody levels to MV-F or MV-H glycoproteins were detected by using a fluorescence-activated cell sorting (FACS)-measured immunofluorescence assay as previously described (21). Cells were examined using a FACSCanto flow cytometer, and results were determined by measuring the median fluorescence signal of the fluorescence-1 histogram. The data points for each animal are the means of duplicate measurements, expressed in arbitrary fluorescence units (AFU).
Immunohistochemical and immunofluorescence analyses of formalin-fixed tissues.
Samples were treated as previously described (7). Dual-labeling indirect immunofluorescence was performed using polyclonal rabbit anti-EGFP (Invitrogen) or rabbit anti-MV (Novus Biologicals) antibodies and monoclonal mouse antibodies to the muscle marker desmin (Novocastra; cloneDER11), the myeloid (macrophage/DC) marker CD11c (Novocastra; clone 5D11), the T-lymphocyte marker CD3 (Dako; clone F7.2.38), the B-lymphocyte marker CD20 (Dako; clone L26), the macrophage marker CD68 (Dako; clone KP1), or the DC marker DC-SIGN (R&D Systems; clone 120507). Antigen binding sites were detected with a mixture of anti-mouse Alexa 568 and anti-rabbit Alexa 488 (Invitrogen). Sections were counterstained with DAPI hard-set mounting medium (Vector). Fluorescently stained slides were examined at ×100, ×200, ×400, and ×1,000 magnifications on a fluorescence imaging microscope (Leica Microsystems).
RESULTS
Generation and characterization of recombinant viruses.
A vial of the commercial M-VAC vaccine (SII) containing the Edmonston-Zagreb (MVEZ) strain of MV was reconstituted according to the manufacturer's instructions. RNA was extracted and used to obtain a consensus sequence of the complete viral genome, including the 3′ and 5′ ends, which were sequenced following RACE. The sequence was identical to the previously published MVEZ master and working seed stock sequences (GenBank accession numbers AY486083.1 and AY486084.1 (22). Subsequently, a full-length anti-genomic plasmid (pMVEZ) was constructed. The plasmid was modified by the addition of an ATU encoding EGFP at the promoter-proximal position [pMVEZEGFP(1)], after the P gene [pMVEZEGFP(3)], or after the H gene [pMVEZEGFP(6)]. Recombinant viruses, rMVEZ (Fig. 1A), rMVEZEGFP(1) (Fig. 1B), rMVEZEGFP(3) (Fig. 1C), and rMVEZEGFP(6) (Fig. 1D) were recovered after transfection of Vero-hCD150 cells (Fig. 1E to H) and passaged exclusively on human diploid MRC-5 cells (Fig. 1I to L), the same cells used to produce the commercial vaccine product. Examination by fluorescence microscopy revealed that rMVEZEGFP(1) and rMVEZEGFP(3) produced a high level of EGFP expression associated with single infected cells and multinucleated syncytia, while rMVEZEGFP(6) produced a lower level, consistent with the promoter-distal positioning of the EGFP-containing ATU (23). Growth analysis of rMVEZ, rMVEZEGFP(1), rMVEZEGFP(3), and rMVEZEGFP(6) in MRC-5 cells over a period of 3 days, simulating the commercial procedure used to generate the M-VAC vaccine product, demonstrated that the viruses replicated to similar maximum titers by 60 h postinfection (Fig. 1M). There was a small lag in rMVEZEGFP(1) growth at early time points. Further growth analysis of the viruses in Vero-hCD150 cells, which cannot produce interferon (24), demonstrated that rMVEZEGFP(1) was potentially impaired in replication and also that rMVEZEGFP(6) may have a slight replicative advantage in these cells (Fig. 1N). rMVEZEGFP(3) replicated similarly to unmodified rMVEZ in both cell types. Large pools of each virus were prepared, formulated, and divided into aliquots for use in the animal vaccination studies.
FIG 1.

A vial of vaccine containing MVEZ was obtained from the commercial producer SII. The vial was reconstituted, the viral RNA was extracted, and a full-length clone was generated. (A) Virus rescued from this clone is rMVEZ. An ATU encoding EGFP was inserted at three positions within the full-length clone of rMVEZ to generate the recombinant viruses rMVEZEGFP(1) (B), rMVEZEGFP(3) (C), and rMVEZEGFP(6) (D). Genomes are drawn to scale. (E to L) Phase (E and I) or EGFP fluorescence (F to H and J to L) images in monolayers of Vero-hCD150 (E to H; primary syncytia at 6 days posttransfection) and MRC-5 (I to L; at 36 h postinfection at an MOI of 0.02) cells infected with rMVEZ (E and I), rMVEZEGFP(1) (F and J), rMVEZEGFP(3) (G and K), and rMVEZEGFP(6) (H and L). Scale bars: 100 μm, E to H; 200 μm, I to L. (M and N) Multistep growth analysis of rMVEZ, rMVEZEGFP(1), rMVEZEGFP(3), and rMVEZEGFP(6) in MRC-5 (M) or Vero-hCD150 (N) cells over 72 h. Error bars represent one standard error of the mean for all curves (n = 3 for each virus). Symbols for curves in panel N are identical to those in panel M.
rMVEZEGFP(3) is functionally equivalent to M-VAC.
We evaluated the immune responses (MV-specific IgG and virus-neutralizing antibody responses) and viral load (genome detection from throat swabs, nose swabs, and BAL samples and virus isolation from BAL and PBMC samples) after i.t. administration of the different recombinant viruses to macaques. This evaluation was carried out in two parts (studies 1 and 2). Both studies were performed in the same way; male, subadult, MV-seronegative cynomolgus macaques were infected with 104 TCID50 of the respective viruses by i.t. inoculation.
In the first study animals were infected with M-VAC, rMVEZ, rMVEZEGFP(1), or rMVEZEGFP(3) (n = 3 animals per group). All animals mounted an MV-specific IgG response by 17 days postinfection (d.p.i.), which peaked by 35 d.p.i. (Fig. 2A). However, the animals infected with rMVEZEGFP(1) showed a delayed response. With the exception of one animal infected with M-VAC (animal EZ02), the genome could be detected in the BAL samples collected from all animals at 3 and 6 d.p.i. (Table 1). Animal EZ02 did not contain detectable genome at any time point examined, but did mount robust MV-specific IgG and virus neutralizing antibody responses by 17 and 35 d.p.i., respectively. MV genome could not be detected in any animal at any time point in nose or throat swabs. Even though genome was detected in the BAL samples of all animals infected with rMVEZEGFP(1) at 3 and 6 d.p.i, virus could not be isolated from any of those samples. This was in contrast to the animals infected with the other three viruses where virus could be isolated from the BAL samples from at least two animals at 3 d.p.i. (Table 1). Virus could not be isolated from PBMC samples from any animal at any time point. Recombinant, unmodified rMVEZ yielded equivalent data to M-VAC for all parameters assessed.
FIG 2.

Measles virus specific immune responses in macaques after i.t. inoculation of M-VAC, rMVEZ, or one of the rMVEZEGFP derivative viruses. Levels of MV-specific IgG (expressed as OD450 readings, y axes) detected 0, 9, 13, 17, 24, 35, 45, and 85 d.p.i. (x axes) with viruses in study 1 (A) or study 2 (B). Levels of MV-specific IgG (expressed as OD450 readings, y axis [C]), virus-neutralizing (VN) antibody (expressed as IU/ml, y axis [D]), MV-F-specific IgG (expressed as AFU, y axis [E]), and MV-H-specific IgG (expressed as AFU, y axis [F]) detected 0, 9, 13, 17, 24, 35, 45, and 85 d.p.i. (x axes) with M-VAC or rMVEZEGFP(3), when all data from studies 1 and 2 are combined (n = 6 animals for each virus). Error bars represent one standard error of the mean for all curves. Symbols for curves in panels C to F are identical to those in panels A and B.
TABLE 1.
Overview of cynomolgus macaques in studies 1 and 2a
| Virus | Macaque | Result (3/6/9 d.p.i.)a |
|
|---|---|---|---|
| Virus isolated | Genome detected | ||
| Study 1 | |||
| M-VAC | EZ01 | +/+/– | +/+/+ |
| EZ02 | –/–/– | –/–/– | |
| EZ03 | +/+/– | +/+/+ | |
| rMVEZ | EZ04 | +/–/– | +/+/+ |
| EZ05 | +/+/– | +/+/+ | |
| EZ06 | +/–/– | +/+/+ | |
| rMVEZEGFP(1) | EZ07 | –/–/– | +/+/+ |
| EZ08 | –/–/– | +/+/– | |
| EZ09 | –/–/– | +/+/+ | |
| rMVEZEGFP(3) | EZ10 | +/–/– | +/+/+ |
| EZ11 | +/–/– | +/+/+ | |
| EZ12 | –/–/– | +/+/+ | |
| Study 2 | |||
| M-VAC | EZ13 | +/–/– | +/+/+ |
| EZ14 | +/–/– | +/+/+ | |
| EZ15 | +/+/– | +/+/+ | |
| rMVEZEGFP(3) | EZ16 | +/–/– | +/+/+ |
| EZ17 | +/–/– | +/+/– | |
| EZ18 | +/–/– | +/+/+ | |
| rMVEZEGFP(6) | EZ19 | –/–/– | +/+/– |
| EZ20 | +/–/– | +/+/– | |
| EZ21 | +/–/– | +/+/+ | |
| EZ22 | +/–/– | –/–/+ | |
| EZ23 | +/+/– | +/+/+ | |
| EZ24 | –/–/– | +/+/+ | |
BAL samples were tested at various times postinfection. d.p.i., days postinfection.
rMVEZEGFP(3) induced more similar responses to the unmodified M-VAC than rMVEZEGFP(1), so it was compared to rMVEZEGFP(6) in the second study. As a further control for rMVEZEGFP(3), the virus was re-rescued, and a new stock was generated and formulated as before. Three macaques were i.t. infected with M-VAC or the new stock of rMVEZEGFP(3), and six macaques were i.t. infected with rMVEZEGFP(6), bringing the total number of animals infected with these viruses to six per group. All animals infected with M-VAC and rMVEZEGFP(3) mounted a MV-specific IgG response by 13 d.p.i., which peaked by 24 d.p.i. (Fig. 2B). Although all animals infected with rMVEZEGFP(6) mounted an IgG response, it was lower and delayed. Apart from one animal infected with rMVEZEGFP(6) (EZ22), genome was detected in the BAL samples collected from all animals on 3 and 6 d.p.i. (Table 1). Genome could be detected in the BAL sample from animal EZ22 by 9 d.p.i. MV genome could not be detected in any animal at any time point in nose or throat swabs. Virus could be isolated from BAL samples for all animals infected with M-VAC and rMVEZEGFP(3) at 3 d.p.i. but from only two-thirds of the animals infected with rMVEZEGFP(6) (Table 1). Virus could not be isolated from PBMC samples from any animal at any time point.
rMVEZEGFP(1) and rMVEZEGFP(6) were impaired in their ability to induce satisfactory MV-specific IgG responses and comparable viral loads in infected animals. Since the only fluorescent protein-expressing, recombinant virus to behave comparably to M-VAC was rMVEZEGFP(3), the data from both studies were combined for these two viruses (n = 6 animals per virus). The levels of MV-specific IgG (Fig. 2C) and neutralizing antibodies (Fig. 2D) generated in response to the viruses were not significantly different at any of the time points sampled. Further, all animals infected with either virus produced MV-specific anti-F and anti-H antibodies (Fig. 2E and F). Genome could be detected in the BAL samples from all animals at equivalent levels (except animal EZ02, as noted above), and virus could be isolated from BAL samples for five of the six animals infected with each virus at 3 d.p.i. (Table 1). All the data combined indicate that rMVEZEGFP(3) induced equivalent responses to the M-VAC vaccine in vivo.
DCs and macrophages are target cells for rMVEZ after i.m. vaccination.
To identify the target cells infected by the EZ vaccine strain of MV, we vaccinated six MV-seronegative cynomolgus macaques with 104 TCID50 of rMVEZEGFP(3) by i.m. injection. At 3, 5, and 7 d.p.v., two animals were euthanized, and a relatively large piece of muscle tissue was collected from the injection site (10 by 10 by 20 mm). The skin was removed, and the muscle was processed into very thin slices (10 by 3 by 1 mm), which were each screened for fluorescence. The removed skin was also screened separately.
EGFP+ cells were detected in the muscle at the injection site of all animals and in the dermis and/or subcutus of the skin of four of the six animals (one at 3 and 5 d.p.v. and two at 7 d.p.v.) (Table 2) by direct confocal scanning laser microscopy visualization (Fig. 3A and B) and immunohistochemistry (Fig. 3C and D). Some of these had the morphology of DCs. Areas could be identified containing large numbers of EGFP+ cells (Fig. 3B to D), in close association with the striated muscle cells, and many of the cells had clearly visible, long processes (Fig. 3A). Dual immunofluorescence in formalin-fixed sections indicated that the EGFP+ cells were not desmin+ muscle cells, and they localized in the perimysium outside the fascicles (Fig. 4A and B). Staining for MV antigen indicated that the cells were productively infected with MV and were not just EGFP+ (Fig. 4C and D). MV+ cells were not desmin+ and localized outside the fascicles.
TABLE 2.
Overview of cynomolgus macaques in study 3 following vaccination with rMVEZEGFP(3)
| Macaque (d.p.v.)a | Result |
|
|---|---|---|
| EGFP+ cells in skin | EGFP+ cells in muscle | |
| EZ25 (3) | – | + |
| EZ26 (3) | + | + |
| EZ27 (5) | – | + |
| EZ28 (5) | + | + |
| EZ29 (7) | + | + |
| EZ30 (7) | + | + |
Fluorescence was screened at various times postvaccination. d.p.v., days postvaccination.
FIG 3.
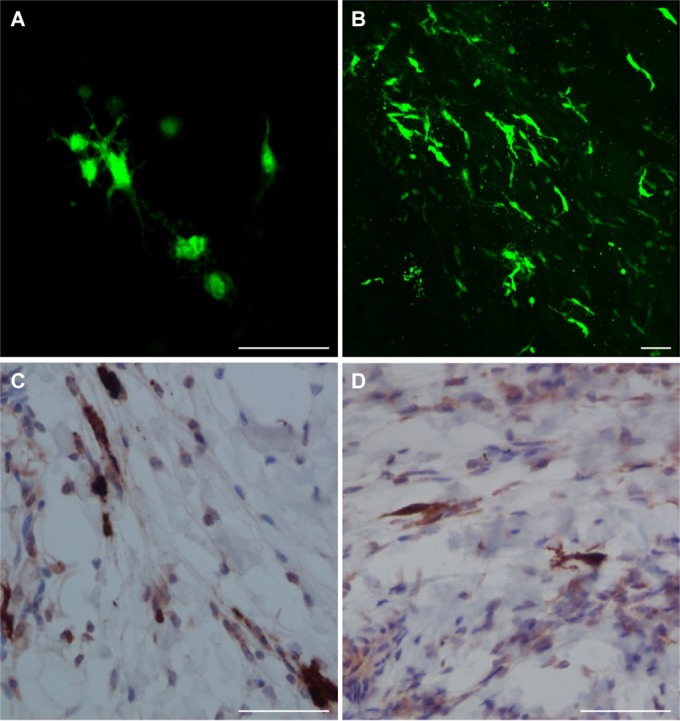
Target cells of rMVEZEGFP(3). EGFP distribution in macaque tissues after administration of rMVEZEGFP(3), detected directly by EGFP fluorescence (green) and confocal microscopy (A, skin from animal EZ26 at 3 d.p.v.; B, muscle tissue from animal EZ28 at 5 d.p.v.) and indirectly by immunodetection of EGFP protein (brown) in formalin fixed muscle tissue sections (C, animal EZ26 at 3 d.p.v.; D, animal EZ30 at 7 d.p.v.). Scale bar, 50 μm (for all images).
FIG 4.

Target cells of rMVEZEGFP(3). Dual indirect immunofluorescence on formalin-fixed muscle tissue sections from a representative macaque infected with rMVEZEGFP(3) (animal EZ26 at 3 d.p.i.). Green staining represents EGFP (A and B and E to H) or MV (C and D). Red staining represents desmin, a marker of muscle cells (A to D), CD68, a marker of macrophages (E), CD11c, a marker of myeloid cells (F), or DC-SIGN, a marker of DCs (G and H). An inset representing the red channel is included (E to H), with arrows marking the same representative double positive cells in the inset and main image. The marked area in panel A is shown at a higher magnification in panel B. An asterisk marks a blood vessel in panel B. Scale bars: 100 μm, A; 50 μm, B to H.
Further dual immunofluorescence indicated that EGFP did not colocalize with the CD3+ T cells or CD20+ B cells present in this peripheral tissue (data not shown). However, a number of the EGFP+ cells were found to be CD68+ (Fig. 4E), CD11c+ (Fig. 4F), or DC-SIGN+ (Fig. 4G and H). Collectively, the panel of markers demonstrated vaccine virus replication in macrophages and DCs, whereas muscle cells were not productively infected.
DISCUSSION
In the present study, we evaluated the immunogenicity of recombinant MVEZ-based measles vaccine viruses compared to the biological parental virus in the macaque model and identified the in vivo target cells of the vaccine virus after i.m. injection. To our knowledge, this is the first study to examine the in vivo tropism of live-attenuated measles vaccine virus.
Although MV vaccination has been used for nearly 50 years (11), little is known about the attenuation profile of the vaccine virus or what cells it targets upon vaccination. It is also not clear whether those target cells are specific to the route of administration, even though regulatory authorities license a vaccine and its route of administration as a single entity (3). In 2002, the WHO established the Measles Aerosol Project, which aimed to achieve licensure for respiratory delivery of currently licensed measles vaccines. The EZ strain of MV is one such licensed MV vaccine and is the most widely used strain in developing countries (25). Currently, the EZ vaccine is predominantly administered by subcutaneous injection. We sought here to identify the target cells of the EZ vaccine when administered by injection with a view to comparison with cells targeted after respiratory administration.
We used our experience from previous studies of MV tropism and pathogenesis, wherein we used viruses expressing EGFP to guide sample collection to regions where the virus was present at early time points when there are relatively small numbers of infected cells present in the animals (6). This necessitated the generation of a reverse genetics system for MVEZ that could be modified to produce a virus encoding EGFP.
We needed to achieve the correct balance of immunogenicity and detection sensitivity. The ATU had to be placed such that it would not perturb replication of the virus and yet produce enough EGFP for sensitive detection of infected cells in vivo. The level of EGFP produced from a particular morbillivirus is dependent on the position of the EGFP-containing ATU within the genome. Most EGFP is produced by promoter-proximal insertion, with less EGFP being produced as the ATU is moved further down the genome distal from the single promoter for gene transcription (23). However, based on observations from canine distemper viruses in the ferret model, it is commonly assumed that a promoter-proximal ATU will have a more attenuating effect on a recombinant virus than a promoter-distal ATU (26). We decided to test three ATU insertion positions. Insertion upstream of the N ORF has historically been used since it leads to high expression levels of the fluorescent protein from the ATU (27, 28). The rationale for insertion after the P gene was that the N and P proteins are predicted to be required in large amounts, while this location can retain balanced levels of envelope associated M, F, and H proteins. Insertion after the H gene has been successful in previous morbillivirus studies (26, 29, 30). We designed the constructs so that the ATUs were identical in rMVEZEGFP(3) and rMVEZEGFP(6). It was not possible to use the identical ATU for rMVEZEGFP(1), since the region upstream of the N ORF contains replication signals [the B box and (GN5)3 motif] which must be conserved (31, 32).
The overattenuation of rMVEZEGFP(6) was a surprise, given that equivalent viruses are apparently unperturbed in other systems (26, 29, 30). Also, in a previous study where IFN-α/β receptor knockout mice expressing human CD46 were injected intraperitoneally with 5 × 106 TCID50 of Schwarz-based rMVs containing an ATU, the position 3 and 6 viruses induced equivalent immune responses at 28 d.p.i. (33). Similarly, subcutaneous vaccination of macaques with 104 infectious units of Moraten-based rMVs found that positions 3 and 6 induced equivalent immune responses for the parameters examined (34). However, another recent study showed that insertion of an ATU into positions 3 and 6 of a human parainfluenza virus 3 vaccine candidate caused a temperature-sensitive phenotype, resulting in attenuation of the virus in vivo (35). The lack of ability of rMVEZEGFP(1) to induce comparable immune responses is in contrast to the situation with wild-type MVs, which tolerate insertion of an ATU in the same position upstream of the N ORF (6–8) and are pathogenic in macaques. This suggests that the vaccine virus is on a narrow “fitness peak” and as such is limited in the genome changes that it can tolerate. Although intriguing, elucidation of the mechanisms of debilitation for these viruses is beyond the scope of the present study, especially since we were able to fulfill the objective of generating a virus which induced equivalent responses to the unmodified biological vaccine with rMVEZEGFP(3).
We were successful in using the fluorescence generated by rMVEZEGFP(3) to locate small numbers of infected cells in the muscle of all six macaques after i.m. vaccination. The standard route of MV vaccination in humans is subcutaneous injection, but the vaccine can also effectively be administered i.m. (36–38). The skin of macaques is much more loosely connected to the subcutus than the skin of humans, and so to avoid redistribution of the vaccine over a large surface area, which would restrict finding and characterizing the target cells, we chose to use i.m. rather than subcutaneous injection in this animal study.
The EGFP+ cells observed 3, 5, and 7 d.p.v. were predominantly macrophages and DCs. We were unable to detect MV-infected muscle cells, even though the infected myeloid cells were in close association with them. Vaccine strains of MV, in contrast to wild-type strains, are able to use CD46 as an entry receptor in vitro (39). CD46 is ubiquitously expressed on nucleated cells (40) and, as such, would be available on muscle cells to allow entry of the vaccine virus. This lack of infection of non-CD150-expressing cells is in keeping with our previous study of attenuated rMVrEdt infection of macaques by aerosol and i.t. routes (7), where the attenuated virus targeted the same CD150-expressing cells as the wild type, and infection of CD46-expressing cells was observed at a very low level. Another study where the H glycoprotein of a wild-type strain of MV was replaced with that from an attenuated strain, which could use CD46 as an entry receptor in vitro, found that replacement of the H gene did not change the tropism of the virus in macaques, where mainly CD150+ infected cells were detected after infection (41). It is also in accordance with our previous studies in macaques infected with the wild-type Khartoum-Sudan (KS) strain of MV (6), where we identified alveolar macrophages and DCs as the early target cells of wild-type MV when administered via the aerosol route.
An interesting question is how long the fluorescent cells persist at the site of injection. The cells also stain positive for MV antigen, indicating that they are productively infected with MV. No gross differences were noted in the numbers of fluorescent cells observed at 3, 5, or 7 d.p.v. Further questions are whether the cells that become infected are resident in the muscle at the time of vaccination or whether they migrate there in response to a stimulus, and where the infected cells are trafficked to; some cells were located close to blood vessels (Fig. 4B), or they could be traveling to lymph nodes draining the site of inoculation, recently shown to be a site of virus replication for a rabies candidate vaccine after i.m. vaccination (42). However, despite significant efforts, we were unable to find EGFP+ cells in draining lymph nodes, suggesting limited migration of these MV-infected DCs. The identification of DCs and macrophages as target cells may explain why subcutaneous injection is such an effective route for the administration of live-attenuated MV vaccine, since these cells are found in high concentrations in subcutaneous tissues, allowing the high seroconversion rates observed in humans immunized under optimal conditions. Identification of the target cells of MV vaccine after injection will now allow a comparative assessment with cells targeted after vaccination by the respiratory route.
ACKNOWLEDGMENTS
This study was funded by the Foundation for the National Institutes of Health through the Bill and Melinda Gates Foundation Grand Challenges in Global Health initiative (grant DUPREX09GCGH0).
We thank R. Dhere and Vivek Vaidya (SII), Connor Bamford, Carlos Hillson, and all members of the Scientific Advisory Board for their time, support, and invaluable feedback.
REFERENCES
- 1.Griffin DE. 2013. Measles virus, p 1042–1069 InKnipe DM, Howley PM (ed), Fields virology, 6th ed, vol 1 Lippincott/Williams & Wilkins Co, Philadelphia, PA. [Google Scholar]
- 2.Muller CP, Huiss S, Schneider F. 1996. Secondary immune responses in parents of children with recent measles. Lancet 348:1379–1380. doi: 10.1016/S0140-6736(05)65440-2. [DOI] [PubMed] [Google Scholar]
- 3.de Swart RL. 2009. Measles studies in the macaque model. Curr Top Microbiol Immunol 330:55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Binnendijk RS, van der Heijden RW, Osterhaus AD. 1995. Monkeys in measles research. Curr Top Microbiol Immunol 191:135–148. [DOI] [PubMed] [Google Scholar]
- 5.Kobune F, Takahashi H, Terao K, Ohkawa T, Ami Y, Suzaki Y, Nagata N, Sakata H, Yamanouchi K, Kai C. 1996. Nonhuman primate models of measles. Lab Anim Sci 46:315–320. [PubMed] [Google Scholar]
- 6.Lemon K, de Vries RD, Mesman AW, McQuaid S, van Amerongen G, Yuksel S, Ludlow M, Rennick LJ, Kuiken T, Rima BK, Geijtenbeek TB, Osterhaus AD, Duprex WP, de Swart RL. 2011. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog 7:e1001263. doi: 10.1371/journal.ppat.1001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Vries RD, Lemon K, Ludlow M, McQuaid S, Yuksel S, van Amerongen G, Rennick LJ, Rima BK, Osterhaus AD, de Swart RL, Duprex WP. 2010. In vivo tropism of attenuated and pathogenic measles virus expressing green fluorescent protein in macaques. J Virol 84:4714–4724. doi: 10.1128/JVI.02633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Swart RL, Ludlow M, de Witte L, Yanagi Y, van Amerongen G, McQuaid S, Yuksel S, Geijtenbeek TB, Duprex WP, Osterhaus AD. 2007. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog 3:e178. doi: 10.1371/journal.ppat.0030178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Vries RD, McQuaid S, van Amerongen G, Yuksel S, Verburgh RJ, Osterhaus AD, Duprex WP, de Swart RL. 2012. Measles immune suppression: lessons from the macaque model. PLoS Pathog 8:e1002885. doi: 10.1371/journal.ppat.1002885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enders JF, Peebles TC. 1954. Propagation in tissue cultures of cytopathogenic agents from patients with measles. Proc Soc Exp Biol Med 86:277–286. doi: 10.3181/00379727-86-21073. [DOI] [PubMed] [Google Scholar]
- 11.Enders JF, Katz SL, Milovanovic MV, Holloway A. 1960. Studies on an attenuated measles-virus vaccine. I. Development and preparation of the vaccine: techniques for assay of effects of vaccination. N Engl J Med 263:153–159. [DOI] [PubMed] [Google Scholar]
- 12.Schwarz AJ. 1962. Preliminary tests of a highly attenuated measles vaccine. Am J Dis Child 103216–219. [DOI] [PubMed] [Google Scholar]
- 13.de Quadros CA, Izurieta H, Carrasco P, Brana M, Tambini G. 2003. Progress toward measles eradication in the region of the Americas. J Infect Dis 187(Suppl 1):S102–S110. doi: 10.1086/368032. [DOI] [PubMed] [Google Scholar]
- 14.Perry RT, Gacic-Dobo M, Dabbagh A, Mulders MN, Strebel PM, Okwo-Bele JM, Rota PA, Goodson JL. 2014. Global control and regional elimination of measles, 2000-2012. MMWR Morbid Mortal Wkly Rep 63:103–107. [PMC free article] [PubMed] [Google Scholar]
- 15.Griffin DE, Pan CH, Moss WJ. 2008. Measles vaccines. Front Biosci 13:1352–1370. doi: 10.2741/2767. [DOI] [PubMed] [Google Scholar]
- 16.Valdespino-Gomez JL, Lourdes Garcia-Garcia M, Fernandez-de Castro J, Henao-Restrepo AM, Bennett J, Sepulveda-Amor J. 2006. Measles aerosol vaccination. Curr Top Microbiol Immunol 304:165–193. [DOI] [PubMed] [Google Scholar]
- 17.Sabin AB, Fernandez de Castro J, Flores Arechiga A, Sever JL, Madden DL, Shekarchi I. 1982. Clinical trials of inhaled aerosol of human diploid and chick embryo measles vaccine. Lancet 320:604. doi: 10.1016/S0140-6736(82)90673-0. [DOI] [PubMed] [Google Scholar]
- 18.El Mubarak HS, de Swart RL, Osterhaus AD, Schutten M. 2005. Development of a semi-quantitative real-time RT-PCR for the detection of measles virus. J Clin Virol 32:313–317. doi: 10.1016/j.jcv.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 19.El Mubarak HS, Yuksel S, van Amerongen G, Mulder PG, Mukhtar MM, Osterhaus AD, de Swart RL. 2007. Infection of cynomolgus macaques (Macaca fascicularis) and rhesus macaques (Macaca mulatta) with different wild-type measles viruses. J Gen Virol 88:2028–2034. doi: 10.1099/vir.0.82804-0. [DOI] [PubMed] [Google Scholar]
- 20.Stittelaar KJ, Wyatt LS, de Swart RL, Vos HW, Groen J, van Amerongen G, van Binnendijk RS, Rozenblatt S, Moss B, Osterhaus AD. 2000. Protective immunity in macaques vaccinated with a modified vaccinia virus Ankara-based measles virus vaccine in the presence of passively acquired antibodies. J Virol 74:4236–4243. doi: 10.1128/JVI.74.9.4236-4243.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Swart RL, Vos HW, UytdeHaag FG, Osterhaus AD, van Binnendijk RS. 1998. Measles virus fusion protein- and hemagglutinin-transfected cell lines are a sensitive tool for the detection of specific antibodies by a FACS-measured immunofluorescence assay. J Virol Methods 71:35–44. doi: 10.1016/S0166-0934(97)00188-2. [DOI] [PubMed] [Google Scholar]
- 22.Baricevic M, Forcic D, Gulija TK, Jug R, Mazuran R. 2005. Determination of the coding and non-coding nucleotide sequences of genuine Edmonston-Zagreb master seed and current working seed lot. Vaccine 23:1072–1078. doi: 10.1016/j.vaccine.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 23.Cattaneo R, Rebmann G, Schmid A, Baczko K, ter Meulen V, Billeter MA. 1987. Altered transcription of a defective measles virus genome derived from a diseased human brain. EMBO J 6:681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosca JD, Pitha PM. 1986. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol Cell Biol 6:2279–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bankamp B, Takeda M, Zhang Y, Xu W, Rota PA. 2011. Genetic characterization of measles vaccine strains. J Infect Dis 204(Suppl 1):S533–S548. doi: 10.1093/infdis/jir097. [DOI] [PubMed] [Google Scholar]
- 26.von Messling V, Milosevic D, Cattaneo R. 2004. Tropism illuminated: lymphocyte-based pathways blazed by lethal morbillivirus through the host immune system. Proc Natl Acad Sci U S A 101:14216–14421. doi: 10.1073/pnas.0403597101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duprex WP, McQuaid S, Hangartner L, Billeter MA, Rima BK. 1999. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J Virol 73:9568–9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hashimoto K, Ono N, Tatsuo H, Minagawa H, Takeda M, Takeuchi K, Yanagi Y. 2002. SLAM (CD150)-independent measles virus entry as revealed by recombinant virus expressing green fluorescent protein. J Virol 76:6743–6749. doi: 10.1128/JVI.76.13.6743-6749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ludlow M, Nguyen DT, Silin D, Lyubomska O, de Vries RD, von Messling V, McQuaid S, De Swart RL, Duprex WP. 2012. Recombinant canine distemper virus strain Snyder Hill expressing green or red fluorescent proteins causes meningoencephalitis in the ferret. J Virol 86:7508–7519. doi: 10.1128/JVI.06725-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Vries RD, Ludlow M, Verburgh RJ, van Amerongen G, Yuksel S, Nguyen DT, McQuaid S, Osterhaus AD, Duprex WP, de Swart RL. 2014. Measles vaccination of nonhuman primates provides partial protection against infection with canine distemper virus. J Virol 88:4423–4433. doi: 10.1128/JVI.03676-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walpita P. 2004. An internal element of the measles virus antigenome promoter modulates replication efficiency. Virus Res 100:199–211. doi: 10.1016/j.virusres.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 32.Rennick LJ, Duprex WP, Rima BK. 2007. Measles virus minigenomes encoding two autofluorescent proteins reveal cell-to-cell variation in reporter expression dependent on viral sequences between the transcription units. J Gen Virol 88:2710–2718. doi: 10.1099/vir.0.83106-0. [DOI] [PubMed] [Google Scholar]
- 33.Lorin C, Mollet L, Delebecque F, Combredet C, Hurtrel B, Charneau P, Brahic M, Tangy F. 2004. A single injection of recombinant measles virus vaccines expressing human immunodeficiency virus (HIV) type 1 clade B envelope glycoproteins induces neutralizing antibodies and cellular immune responses to HIV. J Virol 78:146–157. doi: 10.1128/JVI.78.1.146-157.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.del Valle JR, Devaux P, Hodge G, Wegner NJ, McChesney MB, Cattaneo R. 2007. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. J Virol 81:10597–10605. doi: 10.1128/JVI.00923-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang B, Munir S, Amaro-Carambot E, Surman S, Mackow N, Yang L, Buchholz UJ, Collins PL, Schaap-Nutt A. 2014. Chimeric bovine/human parainfluenza virus type 3 expressing respiratory syncytial virus (RSV) F glycoprotein: effect of insert position on expression, replication, immunogenicity, stability, and protection against RSV infection. J Virol 88:4237–4250. doi: 10.1128/JVI.03481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffin DE, Pan C-H. 2009. Measles: old vaccines, new vaccines. Curr Top Microbiol Immunol 330:191–212. [DOI] [PubMed] [Google Scholar]
- 37.World Health Organization. 2009. Measles vaccines: WHO position paper. Wkly Epidemiol Rec 84:349–360. [PubMed] [Google Scholar]
- 38.Gillet Y, Habermehl P, Thomas S, Eymin C, Fiquet A. 2009. Immunogenicity and safety of concomitant administration of a measles, mumps and rubella vaccine (M-M-RvaxPro) and a varicella vaccine (VARIVAX) by intramuscular or subcutaneous routes at separate injection sites: a randomised clinical trial. BMC Med 7:16. doi: 10.1186/1741-7015-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buckland R, Wild TF. 1997. Is CD46 the cellular receptor for measles virus? Virus Res 48:1–9. doi: 10.1016/S0168-1702(96)01421-9. [DOI] [PubMed] [Google Scholar]
- 40.Liszewski MK, Atkinson JP. 1992. Membrane cofactor protein. Curr Top Microbiol Immunol 178:45–60. [DOI] [PubMed] [Google Scholar]
- 41.Takeuchi K, Nagata N, Kato SI, Ami Y, Suzaki Y, Suzuki T, Sato Y, Tsunetsugu-Yokota Y, Mori K, Van Nguyen N, Kimura H, Nagata K. 2012. Wild-type measles virus with the hemagglutinin protein of the edmonston vaccine strain retains wild-type tropism in macaques. J Virol 86:3027–3037. doi: 10.1128/JVI.06517-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schutsky K, Curtis D, Bongiorno EK, Barkhouse DA, Kean RB, Dietzschold B, Hooper DC, Faber M. 2013. Intramuscular inoculation of mice with the live-attenuated recombinant rabies virus TriGAS results in a transient infection of the draining lymph nodes and a robust, long-lasting protective immune response against rabies. J Virol 87:1834–1841. doi: 10.1128/JVI.02589-12. [DOI] [PMC free article] [PubMed] [Google Scholar]