

ABSTRACT
The interaction between viruses and immune cells of the host may lead to modulation of intracellular signaling pathways and to subsequent changes in cellular behavior that are of benefit for either virus or host. ERK1/2 (extracellular signal regulated kinase 1/2) signaling represents one of the key cellular signaling axes. Here, using wild-type and gE null virus, recombinant gE, and gE-transfected cells, we show that the gE glycoprotein of the porcine Varicellovirus pseudorabies virus (PRV) triggers ERK1/2 phosphorylation in Jurkat T cells and primary porcine T lymphocytes. PRV-induced ERK1/2 signaling resulted in homotypic T cell aggregation and increased motility of T lymphocytes. Our study reveals a new function of the gE glycoprotein of PRV and suggests that PRV, through activation of ERK1/2 signaling, has a substantial impact on T cell behavior.
IMPORTANCE Herpesviruses are known to be highly successful in evading the immune system of their hosts, subverting signaling pathways of the host to their own advantage. The ERK1/2 signaling pathway, being involved in many cellular processes, represents a particularly attractive target for viral manipulation. Glycoprotein E (gE) is an important virulence factor of alphaherpesviruses, involved in viral spread. In this study, we show that gE has the previously uncharacterized ability to trigger ERK1/2 phosphorylation in T lymphocytes. We also show that virus-induced ERK1/2 signaling leads to increased migratory behavior of T cells and that migratory T cells can spread the infection to susceptible cells. In conclusion, our results point to a novel function for gE and suggest that virus-induced ERK1/2 activation may trigger PRV-carrying T lymphocytes to migrate and infect other cells susceptible to PRV replication.
INTRODUCTION
Alphaherpesviruses constitute the largest subfamily of the herpesviruses. This subfamily contains closely related pathogens, including herpes simplex virus 1 (HSV-1), HSV-2, and varicella-zoster virus (VZV) in humans. Another member of the alphaherpesvirus subfamily is the porcine pseudorabies virus (PRV), which is often used as a model to study general features of alphaherpesvirus biology (1).
PRV encodes 11 glycoproteins (2) incorporated in the viral envelope, which are embedded in different host membranes of the infected cell, including the plasma membrane. One of these glycoproteins is glycoprotein E (gE), which is important for virulence and viral (neuronal) spread (3–10). For both PRV and HSV-1, there are indications that gE may have a signaling function in immune cells, as it drives signaling-dependent processes like cell surface antigen capping (11–13). However, to date, there are no reports that gE indeed triggers any particular signaling pathway.
The extracellular signal-regulated kinase 1/2 (ERK1/2) mitogen-activated protein kinase (MAPK) signaling pathway is an evolutionarily conserved pathway, controlling many fundamental cellular events, such as cell proliferation, survival, differentiation, migration, apoptosis, and metabolism (14–16). It may come as no surprise that many viruses, including alphaherpesviruses, modulate the ERK1/2 signaling pathway (17–21).
Several studies have described alphaherpesvirus modulation of ERK1/2 signaling in fibroblasts and/or epithelial cells, but relatively little is known about such modulation in immune cells. Investigating ERK1/2 modulation in T lymphocytes may be of special interest since this signaling pathway is involved in T cell activation, aggregation, and motility (22–25) and since T lymphocytes may be involved in virus spread and transmission of some alphaherpesviruses. The latter is particularly evident for the Varicellovirus species VZV, whose tropism for T cells contributes to several central aspects of its pathogenesis, including viral dissemination in the body, transmission to skin cells, and spread to new hosts (26–28). Other members of the Varicellovirus genus, like PRV, have also been reported to interact with T lymphocytes (29, 30).
In this report, we describe that PRV activates ERK1/2 signaling in T cells and that PRV gE plays an important role in this process. We also report that PRV-induced ERK1/2 activation leads to cellular aggregation and migration of primary T lymphocytes in vitro. These data provide additional insights in the interaction of PRV with T lymphocytes and reveal a new role for gE during PRV infection, which may contribute to the complex interaction that alphaherpesviruses like PRV have developed with the host immune system.
MATERIALS AND METHODS
Cells and viruses.
Jurkat T cells, clone E6-1, purchased from American Type Culture Collection, were grown in RPMI 1640 (Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Life Technologies), 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Life Technologies). Porcine peripheral blood mononuclear cells (PBMC) were isolated from whole blood obtained from healthy pigs aged 6 to 15 weeks using Lymphoprep density centrifugation. Porcine primary T lymphocytes were isolated from PBMC as described before (31) and cultured overnight prior to stimulation in RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 0.2 mM HEPES (Life Technologies). Swine testicle (ST) cells were grown in Eagle's minimal essential medium (MEM; Life Technologies) supplemented with 10% fetal calf serum (FCS), glutamine (0.3 mg/ml), and antibiotics (100 U/ml penicillin, 0.1 mg/ml streptomycin). Wild-type (WT) PRV strain Becker (here referred to as PRV WT) and PRV isogenic mutants gE-null PRV 91 (PRV ΔgE [32]) and PRV107, lacking the cytoplasmic domain of gE (gEΔcd [33]), were grown and titrated on ST cells and stored at −80°C. Both isogenic mutant viruses were a kind gift from L. Enquist, Princeton University.
Infection and gE stimulation.
Jurkat T cell infection was performed in 24-well plates. Cells (1.5 × 106) were inoculated with either PRV WT or the isogenic mutants PRV ΔgE and PRV gEΔcd at a multiplicity of infection (MOI) of 10 for 24 h (except where mentioned otherwise). For gE stimulation assays, 1 × 106/ml Jurkat T cells or 1 × 107/ml primary T lymphocytes were resuspended in medium containing 10 μg/ml or 100 μg/ml of gE recombinant protein (34), respectively, and incubated for different time points at 37°C. gE recombinant protein was blocked with two different antibodies raised against gE, a rabbit polyclonal antibody (1/100) and a mouse monoclonal antibody (1/100), for 30 min at 37°C. Both gE recombinant protein and polyclonal antibody against gE were a kind gift from K. Bienkowska-Szewczyk, University of Gdansk; monoclonal antibody 5F8 against gE was a kind gift from H. Nauwynck, Ghent University.
Transfection and cocultures.
ST cells were transfected according to the manufacturer's guidelines (Jet Prime; Polyplus Transfection) with a plasmid encoding wild-type gE (pIB2; kind gift from Lynn Enquist [35]) for 48 h. As a control, cells were transfected with either empty vector or a green fluorescent protein (GFP)-expressing plasmid (pcDNA-GFP, made in-house). Rested T lymphocytes (2.5 ×106 cells per well) were added to transfected ST cells and cocultured for different time periods. After coculture, medium containing T lymphocytes was gently collected for analysis via SDS-PAGE and Western blotting. To ensure collection of the majority of T lymphocytes, wells were washed carefully with phosphate-buffered saline (PBS), which was also collected, always ensuring that ST cells remained adherent.
To determine the transmission of infection from T cells to ST cells, primary porcine T lymphocytes were inoculated with PRV WT for 24 h, treated with citrate buffer for 1 min (40 mM Na citrate, 10 mM KCl, 135 mM NaCl, pH 3.0), washed 3 times with PBS–1% FCS, and cocultured with ST cells for another 24 h. In some experiments, virus-neutralizing anti-PRV polyclonal antibodies were added prior to and during coculture, neutralizing possibly remaining virus attached to the T lymphocytes cell surface. ST cells were then fixed with 3% paraformaldehyde and stained with an anti-PRV fluorescein isothiocyanate (FITC)-conjugated antibody.
Western blotting.
After infection or gE stimulation, cells were washed with PBS, resuspended in ice-cold lysis buffer (TNE buffer [consisting of Tris-HCl, NaCl, and EDTA], 10% NP-40, 1 mM Na3VO4, 10 mM NaF, protease inhibitor cocktail), and incubated for 20 min at 4°C. SDS-PAGE and Western blotting were performed as described previously (36). Twenty to 30 μg/sample was used for all experiments. Protein concentration was determined using the bicinchoninic acid (BCA) protein assay reagent (Thermo Scientific) according to the manufacturer's instructions. Blots were blocked in 5% nonfat dry milk in PBS-Tween 20 for 1 h at room temperature and incubated with primary antibodies for 1 h or overnight (according to the manufacturer's instructions). After several washes in 0.1% Tris-buffered saline–Tween 20 (TBS-T), blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature and developed using enhanced chemiluminescence. Phospho-specific ERK1/2 antibody (1/1,000; Cell Signaling) signal was detected with ECL Plus (GE Healthcare). Total ERK1/2 antibody (1/1,000; Cell Signaling) signal and viral protein antibody, i.e., gE (1/500), US3 (1/500), and gB (1/200), signals used as loading and infection controls, respectively, were detected with Pierce ECL (Thermo Scientific).
Homotypic T cell aggregation assay.
Primary T lymphocytes (0.5 × 106) were placed in a 96-well plate and inoculated with either PRV WT or PRV ΔgE (MOI, 100). When assessing the involvement of ERK1/2 signaling, the ERK1/2 signaling inhibitor U0126 (10 μM; Cell Signaling) was used. U0126 was dissolved in dimethyl sulfoxide (DMSO) at 10 mM and diluted 1,000 times in medium to reach the working concentration. Neither U0126 nor the DMSO-based diluent affected cell viability (data not shown). U0126 or DMSO-based diluent was added to the T lymphocytes 30 min prior to inoculation with the virus and at 2 h postinoculation (hpi). At 24 hpi, 20 randomly selected fields per condition were observed under the microscope and pictures were taken. Cell aggregates were counted, and their surface area was measured using ImageJ 1.48. Background aggregation levels observed in mock-infected cells were used as the threshold to distinguish small from large aggregates. This threshold was determined as the surface area at which ≤2% of the identified aggregates in mock-infected samples had a larger surface area. For the other conditions, aggregates with a surface area above this threshold value were considered large aggregates.
Transwell migration assay.
Primary T lymphocytes were inoculated with either PRV WT or PRV ΔgE (MOI, 100). After 24 h of virus inoculation, primary T lymphocytes (0.5 × 106) were placed onto a 5-μm Corning Transwell (24-well plate, Sigma-Aldrich) and incubated further to allow migration. To determine levels of migration, a collagen-coated coverslip was placed at the bottom of each well, so that migrating cells would be able to attach to the coverslip. At 24 h postincubation, Transwells were removed and the adherent cells were counted. When assessing the involvement of ERK1/2 signaling, U0126 was added to the T lymphocytes 30 min prior to virus inoculation and at 2 hpi. As a control, the same experiments were performed in the presence of DMSO-based U0126 diluent.
Statistics.
Statistical analysis was performed on GraphPad Prism 5 (GraphPad Software, Inc.). Data sets (n = 3) were analyzed using one-way analysis of variance (ANOVA) (P < 0.05) combined with Tukey's multiple-comparison test (95% confidence interval).
RESULTS
PRV induces ERK1/2 activation in Jurkat T cells.
We first analyzed whether PRV affects ERK1/2 signaling in T cells. To this end, Jurkat T cells were used, a cell line widely utilized for signaling and functional studies in T cells (37). Cells were either mock inoculated or inoculated with wild-type virus (PRV WT), and ERK1/2 phosphorylation was assessed by Western blotting. Figure 1A indicates that at 24 h postinoculation (hpi), levels of ERK1/2 phosphorylation were substantially increased in infected Jurkat T cells compared to mock-infected cells. A time course assay showed that PRV induces ERK1/2 phosphorylation at a relatively late stage of infection, from 12 hpi onwards (Fig. 1B), suggesting the potential involvement of late/structural viral proteins. The onset of ERK1/2 phosphorylation coincided with expression of the viral gE protein (Fig. 1B).
FIG 1.

PRV infection induces ERK1/2 phosphorylation in Jurkat T cells. (A) Cells were either mock inoculated or inoculated with PRV WT (MOI, 10) and lysed at 24 hpi. ERK1/2 activation was detected via Western blotting using a phospho ERK1/2 (pERK)-specific antibody. Loading and infection controls were performed by detecting for total ERK1/2 and viral proteins gB and US3, respectively. (B) A time course infection was performed, whereby cells were either mock inoculated or inoculated with PRV WT (MOI, 10) and lysed at different time points postinoculation. Phospho ERK1/2 and total ERK1/2 and viral protein gE were detected. m, mock.
Glycoprotein E is required for PRV-induced ERK1/2 activation in Jurkat T cells.
Alphaherpesvirus gE is involved in viral cell-cell spread (7, 9) as well as other functions. It has been suggested that gE of different alphaherpesviruses may affect cell signaling, although gE-mediated modulation of particular signaling axes has not been demonstrated yet (11–13, 38–41). To investigate if gE is involved in PRV-induced ERK1/2 activation, Jurkat T cells were inoculated with either PRV WT or an isogenic PRV mutant that lacks gE expression (PRV ΔgE). ERK1/2 phosphorylation was assessed at 24 hpi (Fig. 2A). Contrary to PRV WT, infection with PRV ΔgE did not cause ERK1/2 phosphorylation, indicating that gE is required for PRV-mediated ERK1/2 activation.
FIG 2.

The gE glycoprotein is required for PRV-induced ERK1/2 phosphorylation in Jurkat T cells. (A, B) Cells were mock inoculated or inoculated with PRV WT (A and B), PRV ΔgE (A), or PRVgEΔcd (B) (MOI, 10) and lysed at 24 hpi. ERK1/2 activation was detected via Western blotting using a phospho ERK1/2 (pERK)-specific antibody. Loading and infection controls were performed by detecting for total ERK1/2 and US3, respectively. Glycoprotein gE was also detected to confirm absence of the entire protein or the cytoplasmic domain.
The cytoplasmic domain of PRV gE contains potential signaling domains, i.e., tyrosine-based YXXΦ motifs (13). We used another isogenic PRV mutant, lacking the cytoplasmic domain of gE (PRV gEΔcd), to study the involvement of putative cytoplasmic signaling domains in ERK1/2 activation (Fig. 2B). Jurkat T cells were inoculated with PRV WT, PRV ΔgE, or PRV gEΔcd, and ERK1/2 phosphorylation was detected at 24 hpi. The absence of the cytoplasmic domain of gE did not affect PRV-mediated ERK1/2 activation, as ERK1/2 phosphorylation levels were similar when cells were infected with either PRV gEΔcd or PRV WT. These results show that ERK1/2 activation upon PRV infection depends on gE but does not require the cytoplasmic domain of gE.
Addition of recombinant gE to Jurkat T cells leads to rapid and transient ERK1/2 activation.
To confirm that gE affects ERK1/2 phosphorylation through its extracellular domain, Jurkat T cells were incubated with recombinant gE protein for several time points. Already at 2 min postincubation, there was a clear increase in ERK1/2 phosphorylation (Fig. 3A), which quickly decreased, reaching background levels at 5 min postincubation. Preincubation of recombinant gE with gE-specific antibodies prior to Jurkat T cells stimulation abrogated its ability to induce ERK1/2 phosphorylation, confirming that the effect is gE dependent (Fig. 3B). Thus, binding of PRV gE to Jurkat T cells causes a rapid and transient activation of ERK1/2.
FIG 3.
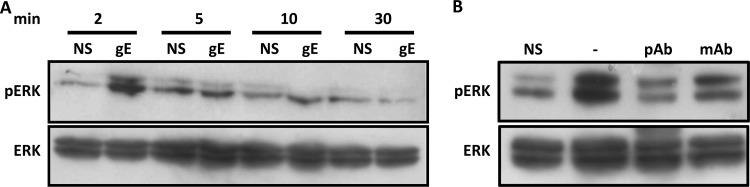
Addition of gE recombinant to Jurkat T cells results in rapid and transient ERK1/2 phosphorylation. (A) Cells were stimulated for different time points with recombinant gE (10 μg/ml) and lysed, and ERK1/2 phosphorylation (pERK) was detected. (B) Recombinant gE was either preincubated for 30 min with two blocking antibodies (mAb and pAb, 1/100) raised against gE or not, prior to stimulation of Jurkat T cells for 2 min at 37°C. NS, nonstimulated Jurkat T cells.
Providing gE in trans leads to ERK1/2 activation in primary porcine T lymphocytes.
Jurkat T cells are widely used for signaling and functional studies in T cells. To investigate whether PRV gE also leads to ERK1/2 activation in primary T cells of its natural host, in a first experimental assay, primary porcine T lymphocytes were incubated with recombinant gE for different time points (Fig. 4A). Similar to our observations in Jurkat T cells, recombinant gE led to a fast increase in ERK1/2 phosphorylation, decreasing back to basal levels at 30 min. Since ERK1/2 activation appeared slightly less pronounced in primary porcine T lymphocytes than in Jurkat T cells, a second experimental assay was used to confirm these findings. ST cells were transfected with a gE-expressing plasmid or, as control, empty vector or a GFP-expressing plasmid. At 48 h posttransfection, when gE is expressed at the cell surface (data not shown), T lymphocytes were cocultured with transfected ST cells for different time points. T lymphocytes incubated with gE-expressing ST cells showed an increased ERK1/2 activation already at 5 min postcocultivation, followed by a slow decrease to almost basal levels at 30 min postcoincubation (Fig. 4B), which was not observed in T lymphocytes incubated with control plasmid-transfected ST cells (Fig. 4B) or empty-vector-transfected ST cells (data not shown). To ensure that detected ERK1/2 phosphorylation was not derived from ST cells that might have detached from the bottom of the well during T lymphocyte collection, the same assay was performed without addition of T lymphocytes. Neither total ERK1/2 nor phosphorylated ERK1/2 could be detected (data not shown). Finally, in a third experimental assay, primary porcine T lymphocytes were brought into contact with PRV WT or PRV ΔgE (Fig. 4C). PRV WT induced rapid and substantial ERK1/2 phosphorylation, whereas PRV ΔgE led to a less prominent ERK1/2 phosphorylation than did PRV WT. Hence, also in primary porcine T lymphocytes, PRV gE triggers ERK1/2 phosphorylation.
FIG 4.

Providing gE in trans triggers ERK1/2 phosphorylation (pERK) in primary porcine T lymphocytes. (A) Primary porcine T lymphocytes were stimulated with gE recombinant (100 μg/ml) for different time points and lysed, and ERK1/2 phosphorylation was detected via Western blotting. (B) ST cells were transfected with either a control or a gE-encoding plasmid and incubated for 48 h. The first four lanes (left) correspond to primary T lymphocytes cocultured with control-transfected ST cells; the last four lanes (right) correspond to T lymphocytes cocultured with gE-transfected ST cells. After coincubation with transfected ST cells for different time points, primary T lymphocytes were collected and lysed. ERK1/2 phosphorylation was assessed via Western blotting. (C) Primary T lymphocytes were inoculated with either PRV WT or PRV ΔgE for different time points, collected, and lysed. Figures are representative of three independent biological replicates (primary T lymphocytes were isolated from 3 different porcine blood donors).
Primary T lymphocytes can transmit PRV to susceptible cells.
To be able to determine possible biological consequences of gE-mediated ERK1/2 phosphorylation in T lymphocytes, we first investigated whether PRV WT and PRVΔgE showed obvious differences in their ability to productively infect primary T lymphocytes. It has been described previously that the percentage of T lymphocytes productively infected by PRV is very low, both in vivo and in vitro (30, 42). In line with this observation, using immunofluorescence to detect different viral antigens, we did not observe obvious productive infection in T lymphocytes upon PRV inoculation, either with PRV WT or with PRV ΔgE (data not shown). Nonetheless, despite the absence of obvious productive infection, it has been demonstrated that T lymphocytes isolated from PRV-inoculated pigs carry infectious virus and can transmit infection to other cells (29). To corroborate this, primary T lymphocytes were inoculated with PRV WT or PRVΔgE for 24 h. After citrate buffer treatment to remove extracellular infectious virus, T lymphocytes were cocultivated with ST cells. At 24 h postcoculture, the formation of plaques was evident, indicative for direct spread of virus from T lymphocytes to ST cells (Fig. 5). In support of this, addition of PRV-neutralizing antibodies to citrate buffer-treated T lymphocytes did not prevent plaque formation in the cocultures (data not shown). In agreement with the established role of gE in cell-associated spread in cell cultures, plaques in ST cells were smaller using PRVΔgE than using WT PRV, but the number of plaques was similar for both viruses (data not shown). Hence, confirming earlier reports, although PRV does not lead to obvious productive infection in primary porcine T lymphocytes, PRV-inoculated T cells can transmit infection to susceptible cells. Despite the well-established role of gE in cell-cell spread, this glycoprotein appears not to play a substantial role in transmission of infection from T lymphocytes to susceptible cells.
FIG 5.

Primary T lymphocytes transmit PRV to susceptible cells. Primary T lymphocytes were inoculated with PRV WT for 24 h, treated with citrate buffer, and cocultured with ST cells for another 24 h. After coculture, ST cells either were observed directly by light microscopy (left) or were fixed, permeabilized, stained for PRV antigens, and observed by fluorescence microscopy (right). Typical PRV plaques in ST cells can be observed. Bar, 100 μm.
PRV-mediated ERK1/2 signaling in primary T cells leads to cell aggregation and migration.
Since gE appears to not affect productive infection in T cells or virus transmission from T cells to susceptible cells, we investigated whether the ability of PRV gE to trigger ERK1/2 activation would have any biological consequences on T lymphocyte behavior. One of the biological consequences of ERK1/2 signaling in T cells is homotypic cell aggregation, which is used as readout for T cell activation, migration, and adhesion (22). T cell aggregation was therefore assessed in primary porcine T lymphocytes that were exposed to either PRV WT or PRV ΔgE. Aggregation was analyzed at 24 hpi, and identified aggregates were categorized into small or large aggregates. Compared to mock-infected cells, PRV WT caused a significantly greater formation of large aggregates (from 2% to 23%, respectively) (Fig. 6), whereas PRV ΔgE caused significantly fewer cell aggregates (11%). The ERK1/2 signaling inhibitor U0126 strongly reduced PRV-induced aggregation close to background levels (3% of large aggregates). Addition of the DMSO-based U0126 diluent did not visibly affect cell aggregate formation (data not shown). These data confirm that T cell aggregation triggered by PRV is dependent on ERK1/2 signaling and to a large extent on gE.
FIG 6.

PRV-induced ERK1/2 activation leads to T cell aggregation. Primary T lymphocytes were inoculated with either PRV WT (in the absence or presence of 10 μM ERK1/2 signaling inhibitor U0126) or PRV ΔgE for 24 h. At 24 hpi, cells were analyzed by microscopy and pictures were taken. (A) Representative images. (B) Twenty random fields per condition were analyzed, and aggregates were measured and classified as large or small based on their surface area (see Materials and Methods). The graph depicts the average percentages (± standard deviations from three independent biological replicates) of large aggregates under each condition. Different letters above the bars represent statistically significant differences (P < 0.05). Bar, 100 μm.
T cell aggregation is often associated with increased cellular motility (43, 44). We therefore assessed whether PRV gE triggers T cell migration. To this end, T lymphocytes were inoculated with either PRV WT or PRV ΔgE, subsequently placed in a Transwell system with a collagen-coated coverslip in the lower chamber, and incubated further for 24 h (Fig. 7A). Afterwards, cells that migrated through the Transwell system onto the collagen-coated coverslip were counted. In line with what we observed for T cell aggregation, cell migration was significantly increased in PRV-inoculated T lymphocytes compared to mock-inoculated cells (Fig. 7B). PRV ΔgE led to a consistent trend, albeit not statistically significant (P value = 0.077), of decreased T cell migration compared to PRV WT. In line with what we observed for cell aggregation, addition of U0126 reduced migration of PRV-inoculated T lymphocytes to the level seen in mock-inoculated cells. As a control, addition of the DMSO-based U0126 diluent did not affect cell migration (data not shown). Furthermore, PRV-inoculated T lymphocytes that migrated to the bottom chamber of the Transwell system were treated with citrate buffer to remove extracellular virus. Subsequent cocultivation with ST cells resulted in obvious infection of the ST cells, indicating that migrating PRV-inoculated T lymphocytes can transmit infection to susceptible cells (Fig. 7C).
FIG 7.

PRV-induced ERK1/2 activation leads to increased motility of T lymphocytes. Primary T lymphocytes were inoculated with either PRV WT (in the absence or presence of 10 μM U0126) or PRV ΔgE for 24 h. (A) Afterwards, cells were placed onto a Transwell and incubated 24 h further to allow migration of the cells to the lower chamber, followed by microscopic quantification of migrated cells. (B) Fold increases (means ± SD from three independent biological replicates) in the number of migrated cells under the different conditions compared to mock-inoculated cells. Different letters above the bars represent statistically significant differences (P < 0.05). (C) T lymphocytes that had migrated to the lower chamber were treated with citrate buffer and incubated for 24 h with ST cells. Cells were analyzed by light microscopy and showed numerous PRV plaques (arrowheads). Bar, 100 μm.
Altogether, these results show that PRV triggers aggregation and migration of primary porcine T lymphocytes in vitro and that these effects are ERK1/2 signaling dependent and to some extent mediated by gE.
DISCUSSION
The interaction between alphaherpesviruses and ERK1/2 signaling has been studied extensively for the past years (17–21, 23). This signaling axis controls various fundamental cellular events, making it an attractive target for the virus to subvert the host, promoting viral replication and survival. The present study shows that PRV activates the ERK1/2 signaling pathway in T cells. In particular, we demonstrate that glycoprotein E of PRV has the previously uncharacterized ability to trigger ERK1/2 phosphorylation in Jurkat T cells and porcine primary T lymphocytes. Moreover, PRV-mediated ERK1/2 activation results in T cell aggregation and migration, which is in part mediated by gE.
Our results indicate that gE triggers ERK1/2 signaling via its extracellular domain. The cytoplasmic domain of gE was not involved in this process, and addition of recombinant PRV gE protein or WT PRV virus to T cells was sufficient to trigger ERK1/2 phosphorylation. One speculative explanation may be that gE binds to an unidentified cellular receptor on the surface of T lymphocytes, triggering ERK1/2 activation. Such a way of ERK1/2 activation has been reported before for HIV-1, where extracellular binding of gp120 recombinant protein to its receptor, CXCR4, transiently activates ERK1/2 in T cells (45). A cellular interaction partner for the extracellular domain of gE has been reported only for VZV (insulin-degrading enzyme) but not for any of the other alphaherpesviruses, including PRV (46).
It would be interesting to study if the novel function of gE that we report here for PRV is conserved in other alphaherpesviruses. HSV-1 has been reported to interfere with MAP kinase activity in Jurkat T cells, although it is unknown if gE is involved. In addition, HSV-1 appears to affect mainly stress-related kinases, like p38, rather than ERK1/2 in T cells (23). Future research may clarify whether these differences are virus dependent or reflect differences in experimental setup.
Primary T lymphocytes have been reported before to display limited susceptibility to productive PRV infection (30, 42). Still, cocultivation of T lymphocytes isolated from PRV-infected pigs with highly susceptible cells resulted in the formation of plaques, indicating that T lymphocytes may function as carrier cells to transmit virus to susceptible cells (29). We confirmed these earlier findings in vitro in primary T lymphocytes. In our assays, no differences in transmission efficiency of virus from T lymphocytes to ST cells were observed between PRV WT and PRC ΔgE inoculations, suggesting that gE (and gE-mediated ERK1/2 signaling) does not substantially contribute to this process. Despite this, gE and ERK1/2 signaling were found to significantly affect T lymphocyte behavior.
Homotypic T cell aggregation correlates with T cell activation. The formation of T cell aggregates upon contact with either infected cells or virus has been described before (47, 48). PRV WT caused a strong increase in T lymphocyte aggregation, whereas PRV ΔgE was significantly impaired in triggering cell aggregation. As others before, we observed that T cell aggregation depended on ERK1/2 signaling (22, 49), as the addition of an inhibitor of ERK1/2 signaling abrogated the formation of large aggregates. T cell aggregation has been described to correlate with T cell migration (43, 44). Other viruses, like human T cell lymphotropic virus type 1 (HTLV-1) and the betaherpesvirus human cytomegalovirus (HCMV), trigger migration of virus-inoculated leukocytes (50, 51). In the current study, PRV inoculation also resulted in an increased migration of T lymphocytes. In line with our results on T cell aggregation, gE contributed to some extent to PRV-induced migration of T lymphocytes, and inhibition of ERK1/2 signaling abrogated migration. Nevertheless, residual ERK1/2 signaling, T cell aggregation, and T cell migration were observed with PRV ΔgE. This may indicate that additional viral proteins may be involved in these processes. Interestingly, other viral proteins of PRV, like US2 and UL46, have been reported to modulate ERK1/2 signaling in other cell types (17–19, 52). Future research will show whether these or other viral proteins may be involved. It will also be important to consider the involvement of gI, which forms a heterodimer with gE (53, 54). The gE-gI complex acts mainly as one functional entity, although gI is able to reach the cell surface in the absence of gE and vice versa (53). Although our data using recombinant gE and gE-transfected cells indicate that gE, in the absence of gI, can trigger ERK1/2 phosphorylation, it will be interesting to determine whether complex formation with gI affects the efficiency of ERK1/2 phosphorylation and whether gI by itself may possibly affect this signaling pathway.
In our in vitro assays, migrating T lymphocytes were able to transmit PRV to ST cells. Some viruses utilize cells that are less permissive to viral replication as carrier cells, subverting signaling pathways of the host to facilitate viral spread. For instance, HIV-1 uses dendritic cells as transporters to reach one of its major target cell populations, CD4+ T cells (55). HCMV infects nonpermissive monocytes, prompting these cells to migrate to target tissues, where the virus then initiates replication upon differentiation of the monocytes to macrophages (50). Our study shows that PRV, in particular gE, activates ERK1/2 in T lymphocytes, leading to an increase in homotypic T cell aggregation and migration in vitro, and that migrating cells are able to transmit the virus to highly susceptible cells.
ACKNOWLEDGMENTS
M. S. Pontes is supported by Fundação para a Ciência e Tecnologia (FCT), scholarship SFRH/BD/70289/2010. This research was supported by grants from the F.W.O.-Vlaanderen (G.0835.09) and a Concerted Research Action of the Special Research Fund of Ghent University (01G01311).
We thank L. Enquist for virus mutants and gE-encoding plasmid, K. Bienkowska-Szewczyk for gE recombinant and polyclonal antibody against gE, and H. Nauwynck for monoclonal antibody against gE. Thank you to J. Lamote and C. Van Waesberghe for the valuable technical support.
REFERENCES
- 1.Pomeranz LE, Reynolds AE, Hengartner CJ. 2005. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev 69:462–500. doi: 10.1128/MMBR.69.3.462-500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mettenleiter TC. 2000. Aujeszky's disease (pseudorabies) virus: the virus and molecular pathogenesis—state of the art, June 1999. Vet Res 31:99–115. doi: 10.1051/vetres:2000110. [DOI] [PubMed] [Google Scholar]
- 3.Lomniczi B, Watanabe S, Ben-Porat T, Kaplan AS. 1984. Genetic basis of the neurovirulence of pseudorabies virus. J Virol 52:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mettenleiter TC, Lukàcs N, Rziha HJ. 1985. Pseudorabies virus avirulent strains fail to express a major glycoprotein. J Virol 56:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mettenleiter TC, Zsak L, Kaplan AS, Ben-Porat T, Lomniczi B. 1987. Role of a structural glycoprotein of pseudorabies in virus virulence. J Virol 61:4030–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enquist LW, Miselis RR, Card JP. 1994. Specific infection of rat neuronal circuits by pseudorabies virus. Gene Ther 1(Suppl 1):S10. [PubMed] [Google Scholar]
- 7.Babic N, Klupp B, Brack A, Mettenleiter TC, Ugolini G, Flamand A. 1996. Deletion of glycoprotein gE reduces the propagation of pseudorabies virus in the nervous system of mice after intranasal inoculation. Virology 219:279–284. doi: 10.1006/viro.1996.0247. [DOI] [PubMed] [Google Scholar]
- 8.Kritas SK, Pensaert MB, Mettenleiter TC. 1994. Role of envelope glycoproteins gI, gp63 and gIII in the invasion and spread of Aujeszky's disease virus in the olfactory nervous pathway of the pig. J Gen Virol 75:2319–2327. doi: 10.1099/0022-1317-75-9-2319. [DOI] [PubMed] [Google Scholar]
- 9.Banfield BW, Yap GS, Knapp AC, Enquist LW. 1998. A chicken embryo eye model for the analysis of alphaherpesvirus neuronal spread and virulence. J Virol 72:4580–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kratchmarov R, Kramer T, Greco TM, Taylor MP, Ch'ng TH, Cristea IM, Enquist LW. 2013. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J Virol 87:9431–9440. doi: 10.1128/JVI.01317-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Favoreel HW, Nauwynck HJ, Oostveldt PV, Mettenleiter TC, Pensaert MB. 1997. Antibody-induced and cytoskeleton-mediated redistribution and shedding of viral glycoproteins, expressed on pseudorabies virus-infected cells. J Virol 71:8254–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rizvi SM, Raghavan M. 2003. Responses of herpes simplex virus type 1-infected cells to the presence of extracellular antibodies: gE-dependent glycoprotein capping and enhancement in cell-to-cell spread. J Virol 77:701–708. doi: 10.1128/JVI.77.1.701-708.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desplanques AS, Nauwynck HJ, Tilleman K, Deforce D, Favoreel HW. 2007. Tyrosine phosphorylation and lipid raft association of pseudorabies virus glycoprotein E during antibody-mediated capping. Virology 362:60–66. doi: 10.1016/j.virol.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 14.O'Neill E, Kolch W. 2004. Conferring specificity on the ubiquitous Raf/MEK signalling pathway. Br J Cancer 90:283–288. doi: 10.1038/sj.bjc.6601488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wellbrock C, Karasarides M, Marais R. 2004. The RAF proteins take centre stage. Nat Rev Mol Cell Biol 5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 16.Chong H, Vikis HG, Guan K-L. 2003. Mechanisms of regulating the Raf kinase family. Cell Signal 15:463–469. doi: 10.1016/S0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- 17.Lyman MG, Randall JA, Calton CM, Banfield BW. 2006. Localization of ERK/MAP kinase is regulated by the alphaherpesvirus tegument protein Us2. J Virol 80:7159–7168. doi: 10.1128/JVI.00592-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang M-H, Banfield BW. 2010. Pseudorabies virus tegument protein Us2 recruits the mitogen-activated protein kinase extracellular-regulated kinase (ERK) to membranes through interaction with the ERK common docking domain. J Virol 84:8398–8408. doi: 10.1128/JVI.00794-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz KS, Liu X, Klupp BG, Granzow H, Cohen JI, Mettenleiter TC. 2014. Pseudorabies virus pUL46 induces activation of ERK1/2 and regulates herpesvirus-induced nuclear envelope breakdown. J Virol 88:6003–6011. doi: 10.1128/JVI.00501-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Li Q, Dowdell K, Fischer ER, Cohen JI. 2012. Varicella-zoster virus ORF12 protein triggers phosphorylation of ERK1/2 and inhibits apoptosis. J Virol 86:3143–3151. doi: 10.1128/JVI.06923-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chuluunbaatar U, Roller R, Mohr I. 2012. Suppression of extracellular signal-regulated kinase activity in herpes simplex virus 1-infected cells by the Us3 protein kinase. J Virol 86:7771–7776. doi: 10.1128/JVI.00622-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Layseca-Espinosa E, Pedraza-Alva G, Montiel JL, del Río R, Fierro NA, González-Amaro R, Rosenstein Y. 2003. T cell aggregation induced through CD43: intracellular signals and inhibition by the immunomodulatory drug leflunomide. J Leukoc Biol 74:1083–1093. doi: 10.1189/jlb.0303095. [DOI] [PubMed] [Google Scholar]
- 23.Sloan DD, Jerome KR. 2007. Herpes simplex virus remodels T-cell receptor signaling, resulting in p38-dependent selective synthesis of interleukin-10. J Virol 81:12504–12514. doi: 10.1128/JVI.01111-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khunkaewla P, Schiller HB, Paster W, Leksa V, Cermák L, Andera L, Horejsí V, Stockinger H. 2008. LFA-1-mediated leukocyte adhesion regulated by interaction of CD43 with LFA-1 and CD147. Mol Immunol 45:1703–1711. doi: 10.1016/j.molimm.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 25.Naci D, Aoudjit F. 2014. Alpha2beta1 integrin promotes T cell survival and migration through the concomitant activation of ERK/Mcl-1 and p38 MAPK pathways. Cell Signal 26:2008–2015. doi: 10.1016/j.cellsig.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 26.Sen N, Mukherjee G, Sen A, Bendall SC, Sung P, Nolan GP, Arvin AM. 2014. Single-cell mass cytometry analysis of human tonsil T cell remodeling by varicella zoster virus. Cell Rep 8:633–645. doi: 10.1016/j.celrep.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zerboni L, Sen N, Oliver SL, Arvin AM. 2014. Molecular mechanisms of varicella zoster virus pathogenesis. Nat Rev Microbiol 12:197–210. doi: 10.1038/nrmicro3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arvin AM, Moffat J, Sommer M, Oliver S, Che X, Vleck S, Zerboni L, Ku C-C. 2010. Varicella-zoster virus T cell tropism and the pathogenesis of skin infection. Curr Top Microbiol Immunol 342:189–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Page GR, Wang FI, Hahn EC. 1992. Interaction of pseudorabies virus with porcine peripheral blood lymphocytes. J Leukoc Biol 52:441–448. [DOI] [PubMed] [Google Scholar]
- 30.Wang FI, Pang VF, Hahn EC. 1988. Flow cytometric analysis of porcine peripheral blood leukocytes infected with pseudorabies virus. J Leukoc Biol 43:256–264. [DOI] [PubMed] [Google Scholar]
- 31.Devriendt B, Baert K, Dierendonck M, Favoreel H, De Koker S, Remon JP, De Geest BG, Cox E. 2013. One-step spray-dried polyelectrolyte microparticles enhance the antigen cross-presentation capacity of porcine dendritic cells. Eur J Pharm Biopharm 84:421–429. doi: 10.1016/j.ejpb.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 32.Tirabassi RS, Townley RA, Eldridge MG, Enquist LW. 1997. Characterization of pseudorabies virus mutants expressing carboxy-terminal truncations of gE: evidence for envelope incorporation, virulence, and neurotropism domains. J Virol 71:6455–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tirabassi RS, Enquist LW. 1999. Mutation of the YXXL endocytosis motif in the cytoplasmic tail of pseudorabies virus gE. J Virol 73:2717–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gut M, Jacobs L, Tyborowska J, Szewczyk B, Bienkowska-Szewczyk K. 1999. A highly specific and sensitive competitive enzyme-linked immunosorbent assay (ELISA) based on baculovirus expressed pseudorabies virus glycoprotein gE and gI complex. Vet Microbiol 69:239–249. doi: 10.1016/S0378-1135(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 35.Tirabassi RS, Enquist LW. 1998. Role of envelope protein gE endocytosis in the pseudorabies virus life cycle. J Virol 72:4571–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deruelle M, Geenen K, Nauwynck HJ, Favoreel HW. 2007. A point mutation in the putative ATP binding site of the pseudorabies virus US3 protein kinase prevents Bad phosphorylation and cell survival following apoptosis induction. Virus Res 128:65–70. doi: 10.1016/j.virusres.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 37.Abraham RT, Weiss A. 2004. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol 4:301–308. doi: 10.1038/nri1330. [DOI] [PubMed] [Google Scholar]
- 38.Olson JK, Bishop GA, Grose C. 1997. Varicella-zoster virus Fc receptor gE glycoprotein: serine/threonine and tyrosine phosphorylation of monomeric and dimeric forms. J Virol 71:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Favoreel HW, Nauwynck HJ, Pensaert MB. 1999. Role of the cytoplasmic tail of gE in antibody-induced redistribution of viral glycoproteins expressed on pseudorabies-virus-infected cells. Virology 259:141–147. doi: 10.1006/viro.1999.9749. [DOI] [PubMed] [Google Scholar]
- 40.Favoreel HW, Mettenleiter TC, Nauwynck HJ. 2004. Copatching and lipid raft association of different viral glycoproteins expressed on the surfaces of pseudorabies virus-infected cells. J Virol 78:5279–5287. doi: 10.1128/JVI.78.10.5279-5287.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riteau B, de Vaureix C, Lefèvre F. 2006. Trypsin increases pseudorabies virus production through activation of the ERK signalling pathway. J Gen Virol 87:1109–1112. doi: 10.1099/vir.0.81609-0. [DOI] [PubMed] [Google Scholar]
- 42.Nauwynck HJ, Pensaert MB. 1994. Virus production and viral antigen expression in porcine blood monocytes inoculated with pseudorabies virus. Arch Virol 137:69–79. doi: 10.1007/BF01311174. [DOI] [PubMed] [Google Scholar]
- 43.Jevnikar Z, Obermajer N, Bogyo M, Kos J. 2008. The role of cathepsin X in the migration and invasiveness of T lymphocytes. J Cell Sci 121:2652–2661. doi: 10.1242/jcs.023721. [DOI] [PubMed] [Google Scholar]
- 44.Pike KA, Kulkarni S, Pawson T. 2011. Immature T-cell clustering and efficient differentiation require the polarity protein Scribble. Proc Natl Acad Sci U S A 108:1116–1121. doi: 10.1073/pnas.1018224108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Popik W, Hesselgesser JE, Pitha PM. 1998. Binding of human immunodeficiency virus type 1 to CD4 and CXCR4 receptors differentially regulates expression of inflammatory genes and activates the MEK/ERK signaling pathway. J Virol 72:6406–6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Q, Ali MA, Cohen JI. 2006. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 127:305–316. doi: 10.1016/j.cell.2006.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi T, Higuchi M, Fukushi M, Oie M, Ito M, Fujii M. 2002. Homotypic cell-cell adhesion induced by human T cell leukemia virus type 1 Tax protein in T cell lines. Virology 302:132–143. doi: 10.1006/viro.2002.1629. [DOI] [PubMed] [Google Scholar]
- 48.van Velzen M, Jing L, Osterhaus ADME, Sette A, Koelle DM, Verjans GMGM. 2013. Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently infected human trigeminal ganglia. PLoS Pathog 9:e1003547. doi: 10.1371/journal.ppat.1003547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo N, Zhang K, Lv M, Miao J, Chen Z, Zhu P. 2015. CD147 and CD98 complex-mediated homotypic aggregation attenuates the CypA-induced chemotactic effect on Jurkat T cells. Mol Immunol 63:253–263. doi: 10.1016/j.molimm.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 50.Smith MS, Bentz GL, Alexander JS, Yurochko AD. 2004. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol 78:4444–4453. doi: 10.1128/JVI.78.9.4444-4453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chevalier SA, Turpin J, Cachat A, Afonso PV, Gessain A, Brady JN, Pise-Masison CA, Mahieux R. 2014. Gem-induced cytoskeleton remodeling increases cellular migration of HTLV-1-infected cells, formation of infected-to-target T-cell conjugates and viral transmission. PLoS Pathog 10:e1003917. doi: 10.1371/journal.ppat.1003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nogalski MT, Chan GCT, Stevenson EV, Collins-McMillen DK, Yurochko AD. 2013. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog 9:e1003463. doi: 10.1371/journal.ppat.1003463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Whealy ME, Card JP, Robbins AK, Dubin JR, Rziha HJ, Enquist LW. 1993. Specific pseudorabies virus infection of the rat visual system requires both gI and gp63 glycoproteins. J Virol 67:3786–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuckermann FA, Mettenleiter TC, Schreurs C, Sugg N, Ben-Porat T. 1988. Complex between glycoproteins gI and gp63 of pseudorabies virus: its effect on virus replication. J Virol 62:4622–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cavrois M, Neidleman J, Greene WC. 2008. The Achilles heel of the Trojan horse model of HIV-1 trans-infection. PLoS Pathog 4:e1000051. doi: 10.1371/journal.ppat.1000051. [DOI] [PMC free article] [PubMed] [Google Scholar]