

ABSTRACT
Interferon-induced Mx proteins show strong antiviral activity against influenza A viruses (IAVs). We recently demonstrated that the viral nucleoprotein (NP) determines resistance of seasonal and pandemic human influenza viruses to Mx, while avian isolates retain Mx sensitivity. We identified a surface-exposed cluster of amino acids in NP of pandemic A/BM/1/1918 (H1N1), comprising isoleucine-100, proline-283, and tyrosine-313, that is essential for reduced Mx sensitivity in cell culture and in vivo. This cluster has been maintained in all descendant seasonal strains, including A/PR/8/34 (PR/8). Accordingly, two substitutions in the NP of PR/8 [PR/8(mut)] to the Mx-sensitive amino acids (P283L and Y313F) led to attenuation in Mx1-positive mice. Serial lung passages of PR/8(mut) in Mx1 mice resulted in a single exchange of tyrosine to asparagine at position 52 in NP (in close proximity to the amino acid cluster at positions 100, 283, and 313), which partially compensates loss of Mx resistance in PR/8(mut). Intriguingly, the NP of the newly emerged avian-origin H7N9 virus also contains an asparagine at position 52 and shows reduced Mx sensitivity. N52Y substitution in NP results in increased sensitivity of the H7N9 virus to human Mx, indicating that this residue is a determinant of Mx resistance in mammals. Our data strengthen the hypothesis that the human Mx protein represents a potent barrier against zoonotic transmission of avian influenza viruses. However, the H7N9 viruses overcome this restriction by harboring an NP that is less sensitive to Mx-mediated host defense. This might contribute to zoonotic transmission of H7N9 and to the severe to fatal outcome of H7N9 infections in humans.
IMPORTANCE The natural host of influenza A viruses (IAVs) are aquatic birds. Occasionally, these viruses cross the species barrier, as in early 2013 when an avian H7N9 virus infected humans in China. Since then, multiple transmissions of H7N9 viruses to humans have occurred, leaving experts puzzled about molecular causes for such efficient crossing of the species barrier compared to other avian influenza viruses. Mx proteins are known restriction factors preventing influenza virus replication. Unfortunately, some viruses (e.g., human IAV) have developed some resistance, which is associated with specific amino acids in their nucleoproteins, the target of Mx function. Here, we demonstrate that the novel H7N9 bird IAV already carries a nucleoprotein that overcomes the inhibition of viral replication by human MxA. This is the first example of an avian IAV that is naturally less sensitive to Mx-mediated inhibition and might explain why H7N9 viruses transmitted efficiently to humans.
INTRODUCTION
In 2013 a new H7N9 variant of avian influenza A virus (IAV) was transmitted from birds to humans, causing more than 300 cases of severe disease with often fatal outcomes (1, 2). The unusual, relatively high number of human infections caused by this virus correlated with several adaptations to the mammalian host, including changes in the hemagglutinin (HA), neuraminidase (NA), and components of the polymerase complex (summarized in reference 3) that overcome restrictions in receptor usage and cellular cofactors needed for efficient viral entry and replication (4, 5). Here, we investigate if changes in sensitivity to innate restriction factors could also contribute to adaptation of avian influenza A virus in mammalian hosts.
Interferons (IFNs) are key cytokines in the innate immune response and induce a broad antiviral state in the mammalian host (6). Shortly upon infection, IFN is secreted from the infected cells, and hundreds of IFN-stimulated genes (ISGs) are upregulated in the surrounding tissues (7). This innate immune reaction allows for an early block of viral replication, with the virus transiently halted by the IFN system until the adaptive immunity steps in to efficiently eliminate the pathogen. Specific knockout studies and ISG overexpression screens have identified IFN-induced effector molecules that most efficiently block virus replication (8). Among them are the myxovirus resistance genes coding for the Mx proteins (9).
Mx proteins are large GTPases that are expressed exclusively under the control of type I and type III IFNs (10, 11). They are synthesized in all tissues upon exposure to IFN and have a strong antiviral effect against a number of RNA and DNA viruses, including influenza A viruses (12, 13). This effect is best demonstrated in Mx1-positive mice that are well protected from otherwise lethal infections with highly pathogenic influenza viruses (14, 15), whereas mouse strains that lack Mx1 expression due to inactivating mutations in the Mx1 gene succumb to the infection (16). Accordingly, the human homologue encoded by the MX1 gene, here called MxA, is likewise active against influenza A virus in cell culture (17).
Influenza A virus is a prominent target of Mx action. The virus replicates its segmented RNA genome in the nucleus. Upon endocytosis, the viral nucleocapsids consisting of the genomic RNA encapsidated by the viral nucleoprotein (NP) and associated to the viral polymerase complex are released into the cytoplasm. These viral ribonucleoprotein complexes (vRNPs) are targets of Mx action. The cytoplasmic human MxA blocks nuclear entry of the vRNPs (18, 19) or a later cytoplasmic replication step after primary transcription (17). In contrast, the nuclear murine Mx1 inhibits the vRNP-associated viral polymerase activity at the level of primary transcription (17).
Analysis of the Mx sensitivity of various influenza A virus strains revealed that the polymerase complexes of avian isolates, such as highly pathogenic H5N1 viruses, were efficiently blocked by human MxA and mouse Mx1, whereas polymerase complexes of human viruses showed an almost 10-fold-reduced sensitivity to Mx inhibition (20). By exchanging the components of the viral minireplicon systems, viral NP, the main structural component of vRNPs, was identified to determine Mx sensitivity of influenza viruses (21). The subsequent structural analysis identified in the body domain of NP a cluster of surface-exposed amino acids that, when exchanged between the strains, made a human isolate sensitive whereas an avian virus gained resistance to Mx action (22).
To evaluate the capability of human influenza A viruses to escape Mx restriction, we here modified the NP of a mouse-adapted, human strain of influenza A virus, A/PR/8/34 (PR/8), to an Mx-sensitive phenotype. During passaging of this virus in Mx1-positive mice, a tyrosine-to-asparagine exchange at position 52 in NP emerged that overcomes the virus's Mx sensitivity. Intriguingly, the same asparagine-52 is found in the NP of human isolates of the 2013 H7N9 virus and mediates Mx resistance. This indicates that, in addition to adaptive mutations in the HA and polymerase genes, the newly emerged avian H7N9 virus harbors substitutions in the NP gene that allow the virus to overcome restriction by mammalian Mx and to efficiently replicate in the human host.
(This work was conducted by David Riegger and Dominik Dornfeld in partial fulfillment of the requirements for a Ph.D. degree from the Faculty of Biology of the University of Freiburg, Germany, 2015.)
MATERIALS AND METHODS
Cells.
Madin-Darby canine kidney, MDCKII, cells were purchased from the ATCC. Human alveolar epithelial A549 control and A549 MxA-expressing cells were described previously (19). Human lung adenocarcinoma Calu-3 and human HEK 293T cells were purchased from the ATCC. All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (HiClone).
Generation of recombinant viruses.
Recombinant hvPR/8 (H1N1) (where hv is highly virulent) viruses were rescued and handled under biosafety level 2 (BSL2) conditions. All work involving SH/1 (H7N9) viruses was performed in the animal biosafety level 3 (ABSL3) laboratory of the Icahn School of Medicine at Mount Sinai in strict accordance with CDC guidelines for BSL3 agents.
hvPR/8 plasmids were cloned into the ambisense expression vector pDZ (23) as described previously (14). The pDZ-NP plasmid encoding the entire segment 5 was used for site-directed mutagenesis. Two independent isolates were generated for each mutant, and the presence of the mutations was confirmed by sequencing segment 5 of the individual virus stocks.
All eight viral segments of the influenza A/Shanghai/1/2013 (H7N9) strain (SH/1; Global Initiative on Sharing Avian Influenza Data [GISAID] accession numbers EPI 439486 to EPI439494) were in vitro synthesized by GeneArt Gene Synthesis (Invitrogen) and subcloned into pDZ (23, 24). For biosafety reasons, the neuraminidase inhibitor-sensitizing mutation [NA(K292R)] (24) was introduced in segment 6. The N52Y substitution in NP was introduced in segment 5 via site-directed mutagenesis.
For virus rescue, a mixture of the eight pDZ plasmids was transfected into a coculture of 293T and MDCKII cells. All recombinant viruses were plaque purified on MDCKII cells, and virus stocks were prepared in embryonated chicken eggs. Successful introduction of the mutations in segment 5 was confirmed by whole-genome sequencing of the viruses.
Ethics statement.
All research studies involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC, protocol LA08-00594) of the Icahn School of Medicine at Mount Sinai and were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals or according to the guidelines of the Animal Care Committee at the Regierungspraesidium, Freiburg, Germany (35-9185.81/G-12/48).
Animal experiments.
Six- to 10-week-old C57BL/6 mice from Jackson Laboratories or Janvier (Strasbourg) and congenic B6.A2G-Mx1 mice carrying the functional Mx1 allele (25) were used. Mice were anesthetized by intraperitoneal injection of a mixture of ketamine (100 μg per gram of body weight) and xylazine (5 μg per gram) and inoculated via the intranasal route (i.n.) with the indicated doses of viruses in 40 μl of phosphate-buffered saline (PBS) containing 0.3% bovine serum albumin (BSA). When indicated, mice were pretreated with 20,000 U of universal type I interferon alpha (IFN-α) (PBL, Piscataway, NJ) by the intranasal route 24 h before infection (see Fig. 5E and F). Mice were monitored daily for weight loss and clinical signs. Animals with severe symptoms or more than 25% weight loss were euthanized. The 50% minimum lethal dose (MLD50) values were calculated as described previously (26) by using five animals for each dilution.
FIG 5.

Amino acid 52N in H7N9 NP contributes to reduced sensitivity to murine Mx1. (A) Direct comparison of the Mx1 sensitivity of SH/1-NP(wt), SH/1-NP(N52Y), and VN/04 in the VN/04 polymerase reconstitution system and increasing amounts of Mx1 as described in the legend of Fig. 4C. Expression of Mx1 and viral PB2 was monitored by Western blotting. (B) Introduction of SH/1-NP(N52Y) or VN/04-NP does not alter virus growth of SH/1 in MDCKII cells. Cells were infected at an MOI of 0.01 with the indicated viruses in the presence of 0.5 μg/ml trypsin. At the indicated time points postinfection (p.i.), virus titers were determined by plaque assay. Error bars indicate the standard deviations of one experiment performed in duplicates. (C and D) Lung titers of H7N9 SH/1 viruses encoding an N52Y exchange in NP or the NP of VN/04. C57BL/6 (C) and B6.A2G-Mx1 (D) mice (n = 3) were infected intranasally with 100 PFU. At 3 and 6 days postinfection, virus titers were determined in lung homogenates by plaque assay. (E and F) Lung titers of H7N9 in IFN-pretreated animals. C57BL/6 (E) and B6.A2G-Mx1 (F) mice (n = 3) were intranasally pretreated with 20,000 units of IFN-α or PBS (ctrl). After 24 h the animals were infected intranasally with 200 PFU of the indicated viruses for 2 days, and virus titers were determined in lung homogenates by plaque assay. Student's t test was performed to determine the P values. *, P < 0.05; **P < 0.01; ***, P < 0.001; ns, not significant. D, day.
Lung homogenates were prepared using a FastPrep24 system (MP Biomedicals). After addition of 800 ml of PBS containing 0.3% BSA, lungs were subjected to two rounds of mechanical treatment for 10 s each at 6.5 m/s. Tissue debris was removed by low-speed centrifugation, and virus titers in supernatants were determined by performing 10-fold serial dilutions in PBS with 0.3% BSA followed by plaque assay on MDCKII cells. RNA was isolated from the lung homogenates using peqGoldTriFast, and the sequences were determined from segment-specific reverse transcription-PCR (RT-PCR) products. Sequence alignments were conducted using Geneious, version 6, created by Biomatters. The extracted RNA was further used for quantitative detection of Mx1 induction in the infected lungs by Mx1-specific quantitative RT-PCR (qRT-PCR).
qRT-PCR.
RNA was isolated from lung homogenates using peqGOLD TriFast (peqlab). Samples were then processed through NucleoSpin columns (Macherey-Nagel). RT was performed using QuantiScript reverse transcriptase (Qiagen) with 1 μg of DNase-treated RNA in a final volume of 20 μl. One microliter of cDNA was used with the QuantiTect SYBR green system (Qiagen). The QuantiTect Primers for detection of murine Mx1 (QT01743308) and murine GAPDH (QT01658692) were purchased from Qiagen. Samples were measured in a LightCycler apparatus (Roche).
Virus growth curves.
Calu-3 cells seeded in six-well plates were incubated with virus at a multiplicity of infection (MOI) of 0.001 in PBS containing 0.3% BSA for 1 h at 37°C. The inoculum was removed, and 3 ml of infection medium (DMEM supplemented with 0.3% BSA and 10 mM HEPES) containing 0.5 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin was added.
For analysis of single-round replication (see Fig. 6C), A549 control and A549 MxA-expressing cells were infected for 1 h at 37°C at an MOI of 2 with the indicated viruses in PBS containing 0.3% BSA. After 1 h of inoculation cells were washed thoroughly and incubated in DMEM supplemented with 0.3% BSA without addition of TPCK-trypsin for an additional 16 h. Virus titers in cell culture supernatants were determined at the indicated time points by plaque assay on MDCKII cells and are expressed as PFU (PFU per ml).
FIG 6.

Amino acid 52N in H7N9 NP contributes to resistance against human MxA. (A) MxA sensitivity of SH/1-NP(wt), SH1-NP(N52Y), and VN/04 was determined in the VN/04 polymerase reconstitution system with increasing amounts of MxA expression plasmid as described in the legend of Fig. 4D. Relative polymerase activity was calculated as the ratio of luciferase activity of wild-type MxA and MxA(T103A). The activity in the absence of MxA was set to 100%. Error bars indicate standard deviations of three independent experiments. Student's t test was performed to determine the P values. **, P < 0.01; ***, P < 0.001. Western blot analysis shows the expression levels of MxA and PB2 in the cell lysates. (B) Expression of MxA limits replication of H7N9 NP(N52Y). Control A549 cells and A549 cells constitutively expressing MxA were infected at an MOI of 2 with the indicated viruses. At 16 h p.i. cells were lysed and analyzed for viral NS1, MxA, and β-actin by Western blotting. The second panel shows a longer exposure of the anti-NS1 blot. The Western blot signals of NS1 were quantified using ImageJ program and normalized to the signals for actin in the cell lysates (graphs). (C) Viral titers in A549 cells stably expressing MxA and control cells. Cells were infected for 16 h at an MOI of 2 with indicated viruses in the absence of TPCK-trypsin. Viral titers are depicted as PFU/ml from three independent samples; error bars represent standard deviations.
Western blot analysis of infected cells was performed as described previously (27).
Minireplicon assay.
To determine hvPR/8 polymerase activity, HEK 293T cells seeded in 12-well plates were transfected using Nanofectin (PAA Laboratories). Ten nanograms of pDZ plasmids encoding PB2, PB1, and PA and 50 ng of NP-encoding plasmid were cotransfected with 50 ng of the firefly luciferase-encoding viral minigenome construct pPolI-FFLuc-RT, which is flanked by the noncoding regions of segment 8 of influenza A virus (28). Some cells were also cotransfected with 60 ng of wild-type (wt) Mx1 or with a pcDNA3.1 expression vector expressing a K49A substitution in Mx1 [Mx1(K49A)] (21). The transfection mixture also contained 10 ng of pRL-SV40, a plasmid constitutively expressing Renilla luciferase under the control of the simian virus 40 (SV40) promoter to normalize variations in transfection efficiency. To achieve equal amounts of transfected DNA, an empty vector plasmid was added. Cells were lysed at 24 h posttransfection. The experiments were performed twice with triplicates.
For the A/Vietnam/1203/2004 (VN/04) polymerase reconstitution assays (see Fig. 4C and D, 5A, and 6A), HEK 293T cells were seeded in 24-well plates at 24 h prior to transfection with 50 ng of pCAGGS-PB1, -PB2, and -PA (all VN/04), 100 ng of the indicated pCAGGS-NP expression plasmids, 50 ng of pCAGGS-Renilla luciferase, 100 ng of pPolI-FFLuc-RT, and various amounts of pCAGGS-MxA or pCAGGS-Mx1. Expression constructs for the antiviral inactive MxA(T103A) (29) and Mx1(K49A) (30) were used as controls. Transfection had no obvious effects on cell viability as controlled by bright-field microscopy at 24 h posttransfection. Medium was changed at 24 h posttransfection, and reporter assays were performed at 48 h posttransfection.
FIG 4.

Sensitivity of A/SH/01/13 to murine and human Mx. (A) Amino acid differences in NP of A/BM/1/18 (BM/18), A/PR/8/34 (PR/8), A/WSN/33, A/AH/1/13 (AH/1), A/SH/01/13 (SH/1), and A/Vietnam/1203/04 (VN/04). Blue boxes highlight positions increasing Mx resistance; red boxes indicate differences between AH/1 and SH/1 relative to VN/04. (B) Structural models of NP with the positions critical for Mx resistance highlighted in blue for BM/18 and PR/8 and the positions of SH/1 that differ from VN/04 highlighted in red. (C and D) Polymerase reconstitution assay with the polymerase subunits of VN/04 combined with expression plasmids encoding NP (100 ng) of SH/1, VN/04, or WSN/33. The assays were performed as described in the legend of Fig. 1B with increasing amounts of cotransfected expression plasmids for mouse Mx1 (C) or human MxA (D). Relative polymerase activity was calculated as the ratio of wild-type Mx1 or MxA to the inactive Mx1(K49A) or MxA(T103A), respectively. The activity with wild-type NP in the absence of Mx was set to 100%. Error bars indicate standard deviations of three independent experiments. Student's t test was performed to determine the P values. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Western blot analysis shows the expression levels of Mx proteins, viral PB2 and beta-actin in the cell lysates.
Firefly and Renilla luciferase activities were measured using a dual-luciferase reporter assay (Promega) according to the manufacturer's protocol. Relative polymerase activity was calculated as the ratio of firefly luciferase to Renilla luciferase activity. Mx-dependent inhibition was normalized to polymerase activity in the presence of equal amounts of inactive mutants of either Mx1 or MxA. The control in which empty vector was transfected was set to 100%. Statistical analysis was done using GraphPad Prism, version 4, software. The expression of proteins was monitored by Western blotting using monoclonal mouse antibody M143 (31) directed against Mx proteins, rabbit polyclonal antibody (32) directed against PR/8 NP, and antibodies directed against viral PB2 (33) and NS1 (34) and against β-actin (Sigma).
Molecular modeling.
The program PyMOL (www.pymol.org) was used to assign the indicated positions in the structural model of the NP of A/HK/483/97 (H5N1) (Protein Data Bank [PDB] code 2Q06).
Alignments.
Alignments were performed using the NCBI Influenza Virus Resource (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html). Additional sequence data were obtained from the Influenza Research Database (IRD).
RESULTS
Mx sensitivity-enhancing mutations in NP of A/PR/8/34.
We recently described three positions (100I, 283P, and 313Y) in NP of A/Brevig Mission/1/1918 (BM/18) that are highly conserved in human strains and determine the resistance of the viral polymerase to the antiviral effect of murine Mx1 and human MxA (22). Accordingly, the PR/8 virus, whose NP shows a high identity of 96% to that of BM/18, including the three Mx resistance-enhancing amino acids at positions 100, 283, and 313, revealed weak sensitivity to the expression of Mx1 compared to avian viruses. To analyze this in a viral polymerase reconstitution system, we cotransfected expression plasmids for the components of the viral polymerase complex and NP of a mouse-adapted laboratory strain, called hvPR/8 (14), together with an artificial viral minigenome encoding firefly luciferase flanked by the noncoding regions of segment 8. Firefly luciferase activity measured in the cell lysates reflects the activity of the reconstituted viral polymerase complex (35). When an expression plasmid for murine Mx1 was cotransfected, the viral polymerase activity was reduced to about 60% of that of the control (Fig. 1A). This reduction reflects the phenotype of a human virus-derived polymerase complex with reduced sensitivity to Mx. In contrast, the polymerase activity in the presence of an avian virus NP was reduced by Mx expression to about 25% (22). Coexpression of the antivirally inactive mutant, Mx1(K49A) (30), showed no polymerase-reducing effect (Fig. 1A) and was used as a control to normalize the data in the following experiments.
FIG 1.
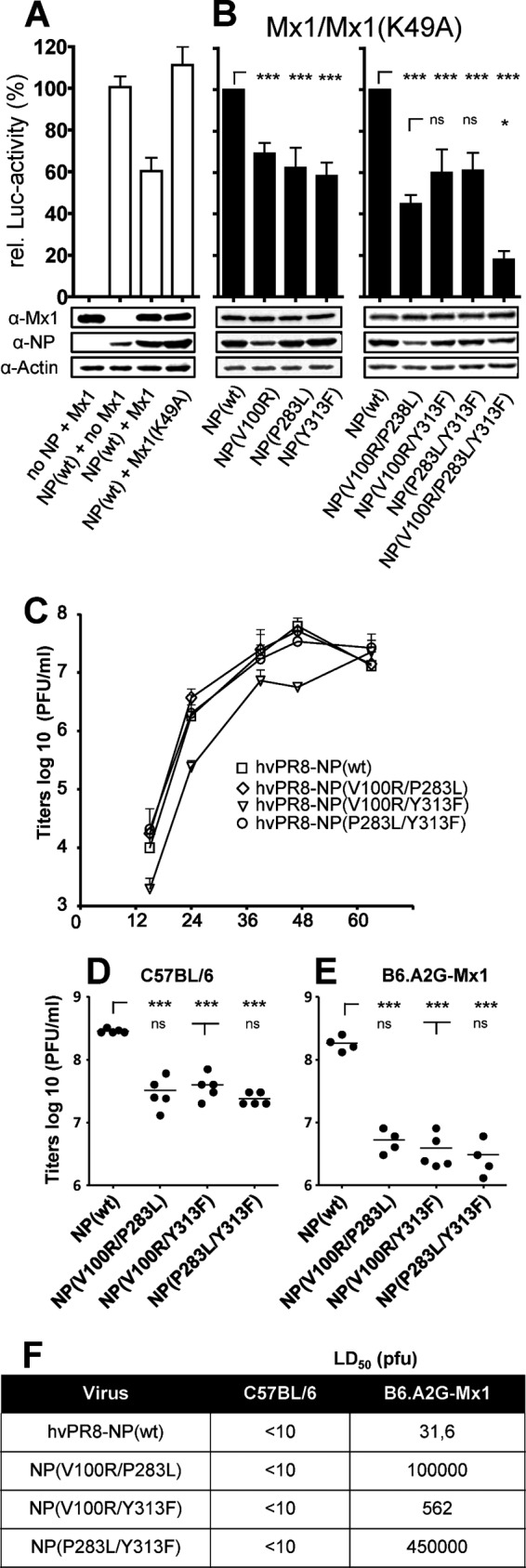
Mx sensitivity of PR/8 NP mutants at positions 100, 283, and 313. (A and B) Viral polymerase reconstitution assay. HEK 293T cells were transfected with expression plasmids coding for the polymerase subunits PB2, PB1, PA, and the indicated NP protein of hvPR/8, a firefly luciferase minigenome under the control of the RNA polymerase I promoter and the Mx1-encoding plasmid. In addition, a plasmid encoding Renilla luciferase under the control of the SV40 promoter was cotransfected to normalize variations in transfection efficiency. Mx1(K49A) is an inactive mutant that was used as a control to normalize the data obtained by coexpression of wild-type Mx1. Relative polymerase activity was calculated as the ratio of luciferase activity in the presence of wild-type Mx1 compared to the luciferase activity in the presence of antiviral inactive Mx1(K49A). The polymerase activity with wild-type NP was set to 100%. Bars represent mean values with standard deviations of two independent experiments performed in triplicates. Two-way analysis of variance was performed to calculate P values. ns, not significant; *, P < 0,01; ***, P < 0,001. A representative Western blot analysis shows the expression levels of wild-type Mx1 (72 kDa), NP (56 kDa), and β-actin (42 kDa) in the cell lysates. (A) Inhibition of the viral polymerase activity by Mx1. Relative luciferase activity was determined in the presence of Mx1 or Mx1(K49A). (B) Point mutations in NP influence Mx1 sensitivity of the viral polymerase. Wild-type NP or NP encoding single or combinations of mutations were used in the replicon assay. Luciferase activity obtained in the presence of wild-type Mx1 was normalized to the activity in the presence of Mx1(K49A). (C) Growth kinetics of hvPR/8 encoding either wild-type NP or NP with the indicated mutations. Calu-3 cells were infected with an MOI of 0.001 in the presence of 0.5 μg/ml trypsin and were incubated at 37°C. At the indicated time points, virus titers in the cell supernatants were determined by plaque assay. Error bars indicate the standard deviation of one experiment performed in duplicates. (D and E) Lung titers of hvPR/8 encoding wild-type NP compared to the viruses encoding the indicated NP mutants in C57BL/6 (D) or B6.A2G-Mx1 (E) mice. Mice were infected intranasally with 200 PFU for 48 h. Viral titers of the lung homogenates were determined by plaque assay. (F) MLD50 values of hvPR/8 encoding wild-type NP or the indicated NP mutants determined in C57BL/6 and B6.A2G-Mx1 mice (n = 5/group). α, anti.
To further characterize Mx sensitivity of hvPR/8, we introduced single or multiple mutations into NP of hvPR/8 at positions 100, 283, and 313 that were recently identified to increase Mx sensitivity (22). We compared the polymerase activity in the presence of wild-type NP under the Mx1-restricted conditions (set to 100%) with the activity in the presence of mutant NP. The single amino acid substitutions caused a reduction in activity of about 40% in the presence of Mx1 (Fig. 1B). Two or all three substitutions resulted in a reduction of polymerase activity of up to 80% (Fig. 1B, right panel), indicating heightened Mx1 sensitivity of the hvPR8 polymerase complex. For further analysis, recombinant hvPR/8 viruses were generated encoding NPs with three different combinations of double exchanges. Despite several attempts, we were not able to rescue an hvPR/8 virus encoding NP with all three exchanges. However, viruses with double mutations in NP could be rescued and grew to titers of about 1 × 108 PFU/ml in 10-day-old embryonated chicken eggs (data not shown), which was comparable to the titer of hvPR/8-NP(wt), encoding wild-type NP. These mutant viruses showed comparable growth in human lung epithelial Calu-3 cells, with a slight attenuation of hvPR/8-NP(V100R/Y313F) (Fig. 1C).
When C57BL/6 mice were infected with a low dose (200 PFU) of recombinant viruses, the relatively Mx insensitive hvPR/8-NP(wt) replicated to high titers in lungs within 48 h (Fig. 1D), and these titers were not remarkably reduced in congenic Mx1-positive B6.A2G-Mx1 animals (Fig. 1E). However, the lung titers observed after infection with the mutant viruses were slightly reduced (7- to 12-fold) in C57BL/6 mice (Fig. 1D) but to a stronger extent in Mx1-positive animals (Fig. 1E) than with wild-type virus infection (34- to 60-fold).
Next, we evaluated the infectious dose that leads to death of 50% of the animals (MLD50) as a measure of virulence for all four viruses in the absence and presence of Mx1. As shown in Fig. 1F, all four viruses were highly virulent for Mx1-negative C57BL/6 mice. Intriguingly, the NP mutant viruses showed increased MLD50 values in Mx1-positive animals. The reduced mortality did not necessarily correlate with the in vivo replication efficiency of the different viral variants and is a discrepancy that has been observed by other investigators (21, 36, 37). Since hvPR/8-NP(P283L/Y313F) showed a 14,000-fold higher MLD50 in Mx1-positive mice than hvPR/8-NP(wt), we used this recombinant virus for further analysis of the Mx1-mediated resistance in vivo.
Escape of an Mx-sensitive PR/8 virus from Mx restriction in vivo.
hvPR/8 wild type is highly adapted to replicate in Mx1-positive mice (14). To test whether an Mx1-sensitive mutant version of this virus [hvPR/8-NP(P283L/Y313F)] can regain Mx1 resistance, we performed serial lung passages in Mx1-positive mice. We performed two independent series of 10 consecutive lung passages (series A and B) by repetitive intranasal infection with 100 PFU. The lung homogenates of each passage were analyzed at 48 h p.i. for viral titers and genome sequence. The viral titers in the lung homogenates increased constantly during the passages from about 8 × 105 PFU/ml to about 5 × 107 PFU/ml (Fig. 2A). To analyze the passaged viruses for adaptive mutations, we focused on the components of the viral nucleocapsid, the confirmed target of Mx restriction, and did not consider possible changes in other segments that might also contribute to enhanced viral replication. No nucleotide changes in the polymerase genes could be detected in the genomes of the passaged viruses by direct sequencing of RT-PCR amplification products. However, in the NP gene of series A, one nucleotide mutation in codon 52 (TAT to AAT) emerged early during the passaging, resulting in the amino acid change of tyrosine to asparagine (Fig. 2B). In series B a transient nucleotide exchange in codon 309 of the NP open reading frame (ORF) appeared (AAC to AAA) after the sixth passage, resulting in an asparagine-to-lysine exchange, but did not become dominant up to passage 10 (Fig. 2B).
FIG 2.
Passaging Mx-sensitive hvPR/8-NP(P283L/Y313F) in B6.A2G-Mx1 mice. Two independent lines of Mx1-positive mice were infected intranasally with 100 PFU of hvPR/8-NP(P283L/Y313F) for 48 h. Then lungs were harvested, and virus titers were determined. The lung homogenates were diluted appropriately with PBS and directly used for the next passage. (A) Lung virus titers of each step in the passage lines A and B. The first appearance of NP mutations in codon 52 in line A and codon 309 in line B is indicated. (B) RNA was isolated from the lung homogenates and used for segment 5-specific RT-PCR and sequencing.
To analyze the effect of these mutations on viral Mx1 sensitivity, we introduced the escape mutations into NP(wt) or NP(P283L/Y313F) expression constructs and performed viral polymerase reconstitution assays as described in the legend of Fig. 1B. Introduction of Y52N into NP(wt) had a weak but nonsignificant effect on the polymerase activity in the presence of Mx1 compared to NP(wt) (Fig. 3A). However, when introduced into the Mx1-sensitive NP(P283L/Y313F), the Y52N exchange reduced the inhibitory effect of Mx1 to levels measured with NP(wt) (Fig. 3A). The N309K exchange did not alter the activity of NP and showed no effect on the Mx1 sensitivity of the double mutant (Fig. 3A). Even when the N309K mutant was combined with Y52N, the sensitivity of the reconstituted polymerase complex was not altered (Fig. 3A), indicating that the N309K exchange has, at best, a minor influence on the Mx-mediated antiviral effect. Therefore, only the Y52N exchange in NP was further analyzed in vivo.
FIG 3.

Mx sensitivity of NP mutants at positions 52 and 309. (A) A polymerase reconstitution assay was performed as described in the legend of Fig. 1B, including the compensatory NP mutants. Relative polymerase activity was calculated as the ratio of luciferase activity in the presence of wild-type Mx1 compared to the luciferase activity in the presence of inactive Mx1(K49A). The activity with wild-type NP was set to 100%. Bars represent mean values with standard deviations of two independent experiments performed in triplicates. Two-way analysis of variance was performed to calculate P values. ns, not significant; ***, P < 0.001. Western blot analysis shows the expression levels of Mx1, NP, and β-actin in the cell lysates. (B) Growth kinetics of the recombinant hvPR/8 viruses on Calu-3 cells infected at an MOI of 0.001 in the presence of 0.5 μg/ml trypsin. Error bars indicate the standard deviations of one experiment performed in duplicates. (C and D) Lung titers of hvPR/8 viruses in C57BL/6 (C) and B6.A2G-Mx1 (D) mice upon intranasal infection with 200 PFU. At 48 h postinfection virus titers were determined in lung homogenates. Student's t test was performed to calculate P values. (E) MLD50 values of hvPR/8 encoding wild-type NP or the indicated NP mutants determined in B6.A2G-Mx1 mice (n = 5/group). (F) Detection of Mx1 expression by quantitative RT-PCR. Lung homogenates of B6.A2G-Mx1 mice (n = 5) infected with 200 PFU for 48 h were used to extract RNA and determine the expression of Mx1 and GAPDH by qRT-PCR. The Mx1 signals were normalized to the GAPDH levels. qRT-PCR results were calculated as Mx1-to-GAPDH ratio by the 2ΔCT method (59). The relative induction of Mx1 in mock-infected animals was set to 1. Bars represent mean values with standard deviations using data from five animals each (three animals in the mock control). Two-way analysis of variance was performed to calculate P values. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. α, anti.
Recombinant hvPR/8 viruses harboring the additional Y52N mutation were generated in the background of NP(wt) or NP(P283L/Y313F), resulting in hvPR/8-NP(Y52N) and hvPR/8-NP(Y52N/P283L/Y313F). Both viruses showed growth kinetics in Calu-3 cell cultures comparable to those of the control viruses (Fig. 3B). Infection of C57BL/6 and B6.A2G-Mx1 mice revealed slightly lower viral lung titers with hvPR/8-NP(Y52N) than with wt virus at day 2 postinfection, indicating an Mx-independent effect of this substitution on viral replication (Fig. 3C and D). However, introduction of Y52N into the Mx1-sensitive hvPR/8-NP(P283L/Y313F) virus partially reverted its attenuation, as judged by viral lung titers in C57BL/6 mice, and was more pronounced in B6.A2G-Mx1 mice (Fig. 3C and D). More strikingly, determination of the MLD50 revealed that the strong attenuation and reduced lethality of hvPR/8-NP(P283L/Y313F) in Mx1-positive animals were severely compensated by introduction of Y52N. The MLD50 of hvPR/8-NP(Y52N/P283L/Y313F) was reduced to levels comparable to those of hvPR/8-NP(wt) and hvPR/8-NP(Y52N) (Fig. 3E).
To exclude the possibility that introduction of Y52N into hvPR/8-NP(Y52N/P283L/Y313F) might influence the IFN-inducing capacity of the virus and, as a consequence, Mx1 expression, we determined Mx1 transcripts by quantitative RT-PCR in lungs of infected animals. At 48 h p.i., hvPR/8 infection enhanced Mx1 transcript levels about 300-fold. No significant difference in Mx1 induction by the four different virus strains could be detected (Fig. 3F), indicating their robust capacity to induce type I IFN. These results indicate that introduction of asparagine at position 52 in the NP of hvPR/8-NP(P283L/Y313F) helps to reduce the sensitivity of the P283L/Y313F virus to the antiviral effect of Mx1 in vivo.
Newly emerged avian H7N9 viruses with asparagine-52 in NP.
To determine whether viruses encoding NP containing asparagine-52 circulate in nature, we screened influenza A virus sequence databases (Influenza Research Database [IRD], an NCBI influenza virus resource). Strikingly, a tyrosine residue is mostly found at this position in human and avian isolates (Fig. 4A). However, consistent with other reports (3), we found N-52 to be predominant in the recently emerged H7N9 isolates that have been repeatedly transmitted to humans in southern China (1, 2). As shown in Fig. 4A, the NPs of the H7N9 isolates A/Shanghai/1/13 (SH/1) and A/Anhui/01/13 (AH/1) do not possess any of the previously identified amino acids that confer the Mx resistance (blue) of the human pandemic isolate A/BM/01/1918 (BM/18) and its descendants like PR/8 although they differ in several amino acids (Fig. 4A, red) from the Mx-sensitive H5N1 isolate A/Vietnam/1203/2004 (VN/04). In total, recent H7N9 NPs harbor three common changes in amino acid positions from VN/04 (Fig. 4A) (3, 38). Analysis of the predicted crystal structure of SH/1-NP revealed that position 52 is surface exposed and is found in the center of the amino acid cluster determining Mx resistance (Mx patch) of BM/18 and descendant human strains, including PR/8 (22, 38) (Fig. 4B).
Because of the effect of N-52 on PR/8 Mx sensitivity, we wondered whether NP of SH/1 containing N-52 would exhibit increased Mx resistance compared to the NP of VN/04. To test this, we compared the Mx sensitivity of NP of SH/1 with that of VN/04 and the human WSN/33 isolate in a VN/04-based polymerase reconstitution assay. Increasing amounts of murine Mx1 or human MxA suppressed viral polymerase activity when the polymerase complex is reconstituted in the presence of VN/04-NP, whereas the NP of WSN/33 rendered the system rather insensitive (Fig. 4C and D). Interestingly, NP of SH/1 showed an intermediate phenotype with only larger amounts of Mx proteins clearly reducing polymerase activity (Fig. 4C and D), indicating that SH/1-NP indeed harbors partial Mx resistance. Because of the antigenic variability between the different NP proteins, PB2 was analyzed by Western blotting to show constant transfection efficiency of the minireplicon components.
Asparagine-52 in NP contributes to the reduced Mx sensitivity of H7N9.
The NP of the H7N9 isolate SH/1 exhibits increased Mx resistance compared to VN/04 NP. Thus, we assumed that it encodes specific polymorphisms to escape Mx restriction. While SH/1-NP harbors a total of seven amino acid differences with respect to VN/04 (Fig. 4A), only three positions, including N-52, are common among recent H7N9 isolates (3). We decided to focus on the substitution at position 52 since a mutation at this position reverted the Mx sensitivity of hvPR/8-NP(P283L/Y313F). The N52Y mutation in SH/1-NP resulted in loss of Mx1 resistance in viral polymerase assays. We quantitatively analyzed the effect of N52Y by transfecting increasing amounts of an Mx1-expressing plasmid in the VN/04-based polymerase reconstitution system where the N52Y exchange in SH/1-NP increased Mx1 sensitivity to a level observed with VN/04-NP (Fig. 5A).
In order to test the effect of NP(N52Y) in the context of the SH/1 virus, we used a previously established SH/1 rescue system (24) to generate SH/1-NP(N52Y). Additionally, we generated SH/1-NP(VN/04), an SH/1-based virus with the NP of VN/04, to obtain a highly Mx-sensitive control. All three recombinant viruses showed comparable replication kinetics in multicycle growth curves on MDCKII cells (Fig. 5B), indicating no significant impact of NP(N52Y) or VN/04-NP on viral growth in these cells. For in vivo evaluation of the viruses, C57BL/6 and B6.A2G-Mx1 mice were infected intranasally with 100 PFU, and viral lung titers were determined 3 and 6 days p.i. (Fig. 5C and D). In accordance with the growth curve obtained in MDCK cells, SH/1-NP(N52Y) and SH/1-NP(VN/04) replicated comparably to SH/1-NP(wt) in Mx1-negative mice (Fig. 5C). Intriguingly, we observed 10- to 100-fold-reduced viral lung titers for SH/1-NP(VN/04) in Mx1-positive mice, again implying that high Mx sensitivity was conferred by the H5N1 NP (Fig. 5D). In contrast, replication of SH/1-NP(wt) as well as SH/1-NP(N52Y) was only marginally reduced by about 10-fold, suggesting low Mx sensitivity of SH/1 that seems not to be considerably affected by the N52Y exchange in NP under these conditions (Fig. 5D). In order to create more stringent Mx-inhibitory conditions for SH/1 virus replication in vivo, groups of three Mx1-positive mice were pretreated with IFN-α for 24 h to increase endogenous Mx1 levels. As a control for Mx1-independent, IFN-induced antiviral effects, C57BL/6 mice were similarly pretreated 24 h before infection. IFN treatment showed only a weak suppression of SH/1-NP(wt) replication even in Mx1-positive animals at 48 h p.i., confirming its relative low sensitivity to the effect of Mx1 (Fig. 5E and F). However, replication of SH/1-NP(N52Y) was reduced more than 1 log in IFN-prestimulated Mx1-positive mice, confirming the effectiveness of the IFN treatment and indicating that the N52Y exchange enhances Mx1 sensitivity of SH/1 in vivo. Accordingly, we observed a 27-fold increase in the MLD50 of SH/1-NP(N52Y) in Mx1-positive mice compared to SH/1-NP(wt)-infected animals (Table 1). Taken together, these data implicate that asparagine 52 of H7N9 NP is required to maintain low Mx1 sensitivity in vivo although additional positions in SH/1-NP might contribute to the reduced Mx sensitivity of the H7N9 virus.
TABLE 1.
Comparison of MLD50s of H7N9 wt and indicated NP mutants in C57BL/6 and B6.A2G-Mx1 mice
| Virus | MLD50 (PFU) |
|
|---|---|---|
| C57BL/6 | B6.A2G-Mx1 | |
| SH/1-NP(wt) | 3.16 × 101 | 1.78 × 104 |
| SH/1-NP(N52Y) | 1.47 × 102 | 4.81 × 105 |
| SH/1-NP(VN/04) | 3.16 × 102 | 4.81 × 105 |
Finally, we tested if asparagine at position 52 of NP also confers reduced sensitivity of SH/1 to human MxA. Polymerase reconstitution assays revealed an increased susceptibility of the viral polymerase complex of the SH/1-NP(N52Y) to MxA action. The inhibitory effect of MxA on NP(N52Y) was comparable to that of the NP(VN/04)-containing replicon system (Fig. 6A). For analysis of the recombinant SH/1 viruses, we used human lung epithelial A549 cells that constitutively overexpress MxA (19). These cells were infected at an MOI of 2 with the recombinant viruses, and their intracellular replication was monitored 16 h later by Western blotting of the expression of the viral NS1 protein. As indicated in Fig. 6B, infection of A549 control cells with the three viruses resulted in comparable accumulation levels of the viral NS1 protein. Viral infection did not lead to induction of significant amounts of endogenous MxA. However, ectopic expression of MxA strongly suppressed NS1 expression in SH/1-NP(N52Y)- and SH/1-NP(VN/04)-infected cells, whereas SH/1-NP(wt) virus infection resulted in only a slightly reduced accumulation of viral NS1 (Fig. 6B). Accordingly, titers of SH/1-NP(N52Y) and SH/1-NP(VN/04) were reduced in the supernatant of MxA-expressing cells (Fig. 6C). These data are consistent with the effects of Mx1 in the mouse model and support our assumption that asparagine at position 52 in H7N9 NP confers enhanced resistance to human MxA.
DISCUSSION
Zoonotic transmissions of H7N9 viruses from birds to humans were first reported in March 2013, with >400 laboratory-confirmed infections (39). Taking into account the number of H5N1 cases in a comparable time frame (globally, ∼50 cases from 1 January 2013 until 27 July 272014 [40]), the efficacy of transmission of H7N9 viruses appears to be dramatically higher. Successful replication of avian viruses in human hosts requires adaptations in multiple viral gene products. Hemagglutinin of human-derived H7N9 has been shown to bind to human sialic acid receptors in vitro (41, 42) and in organ explants (43). The majority of H7N9 isolates from human patients carried the well-described PB2(E627K) adaptation that allows efficient replication in mammalian cells (44). This PB2 adaptation was also previously described to occur in H5N1-infected human patients (45) and to confer enhanced replication in mammalian hosts. Adaptations to overcome species-specific factors of the innate immune system are less well characterized in zoonotic transmission of IAV from avian to mammalian hosts. Mx proteins are key restriction factors of influenza A viruses in mice and humans. Albeit the exact mechanism is a matter of ongoing research, recent work has identified IAV NP as the main determinant for sensitivity to Mx antiviral action (21, 22). We found several surface-exposed residues in NP, including glutamic acid or aspartic acid at position 53, that discriminate between Mx-sensitive H5N1 and Mx-resistant H1N1 NPs. Here, we demonstrate that the NP of the H7N9 isolate makes the viral polymerase complex less sensitive to inhibition by Mx than the H5N1 NP due to an asparagine residue at position 52 found in the H7N9 NP. Replacement of this residue with tyrosine, which is prevalent in the NP of highly Mx sensitive H5N1 isolates, results in higher MLD50s in Mx-positive mice. Viral replication of SH/1-NP(N52Y) was affected only in Mx-positive mice upon IFN type I pretreatment. Influenza A viruses were previously described to successfully suppress early innate host responses in vivo, allowing the virus to outrun the type I interferon response (46). It is thus likely that the amounts of Mx1 expressed early after infection are not sufficient to affect SH/1-NP(N52Y) replication. Pretreatment of mice with IFN-α would increase the amounts of Mx1 and the antiviral effect it mediates. In contrast, accumulation of Mx1 over time could have a stronger effect as supported by the observed changes in MLD50 values. Overall, our in vivo data strengthen the hypothesis of a surface patch on NP that governs sensitivity to the antiviral function of Mx. The location of this patch in NP is so far not described to be important for binding of viral RNA, NP oligomerization, or interaction with polymerase subunits (47, 48). Modeling of the N52Y substitution onto the structure of H5N1 VN/04-NP showed that this residue is located in the body domain of NP close to the interface of two NPs from antiparallel strands in the viral RNP (49). Currently, there is no defined function in viral replication for this interface.
However, our data indicate that mutations in NP affecting Mx sensitivity can alter viral replication in vivo, even in the absence of Mx1. This indicates that the NP surface patch plays a role in viral polymerase function during a yet undefined step of viral replication and that the balance of amino acids within this patch is critical.
Mx was recently shown to interfere with the interaction of PB2 and NP (50, 51). Data from our lab did not confirm direct interaction of MxA with either NP or PB2, and we found no evidence for consistent colocalization of Mx with NP in H7N9-infected cells, as determined by confocal laser scanning microscopy (our unpublished observations). We cannot exclude the possibility that Mx requires other cellular factors to act antivirally at this stage. Thus, the molecular mechanism of Mx-dependent inhibition of viral replication and transcription currently remains elusive.
Novel H7N9 originated by multiple reassortment events of avian isolates in East Asia (1). The NP segment originated from avian H9N2 isolates. H9N2 internal gene cassettes (PB2, PB1, PA, NP, M, and NS) gave rise to several zoonotic viruses in East Asia in recent years (2, 52, 53). H5N1 isolates from 1997 and H7N9 and, more recently, H10N8 isolates harbor the six internal segments from H9N2. Sequence comparison of avian H9N2 isolates revealed that recent H9N2 isolates predominantly carry asparagine (conferring reduced Mx sensitivity) at position 52, while isolates before 2009 predominantly carry tyrosine. It is currently unclear what promoted this switch. So far, none of the avian Mx proteins tested under laboratory conditions inhibits IAV polymerase activity (54, 55).
However, we cannot exclude that the H9N2 NP carrying 52N evolved in swine or yet undetermined birds with potential antiviral-acting Mx proteins. Moreover, we do not exclude the possibility that Y52N substitution harbors an Mx-independent advantage for the virus. Intriguingly, NP(Y52N) mutations were described in mouse-adapted H9N2 viruses isolated from BALB/c mice, which do not carry a functional Mx1 gene (56–58). The authors demonstrate that this adaptation contributes to viral replication and transmission of H9N2 viruses in Kunming mice. It is currently unclear if this mouse strain encodes a functional, antiviral Mx1 protein, which could explain the enhanced replication and transmission rates. This would be in accordance with the results we obtained from passaging Mx-sensitive hvPR8 mutants in Mx1-positive mice. The appearance of an N309K exchange in NP during our second independent passage of hvPR8-NP(P283L/Y313F) suggests alternative strategies of influenza A viruses to increase replication efficiency under Mx1 restriction. As N309K did not change Mx sensitivity in the subsequent analysis, we did not further characterize this mutant in the present study.
In summary, we provide evidence that the early acquisition of an NP harboring asparagine-52 predisposed the avian H7N9 viruses for crossing the species barrier and to productively infect humans. The reduced Mx sensitivity might contribute to efficient replication in the human host without being adequately controlled by the innate immune system and thereby provokes the severe pathogenic effects in infected individuals. Therefore, our data highlight a novel facet in the intriguing arms race of viral virulence and host restriction factors and reveal the need of constant surveillance efforts including residues in the Mx patch of NP as novel biomarkers pointing to potentially zoonotic influenza A viruses.
ACKNOWLEDGMENTS
We thank David Jackson and Rick Randall, University of St. Andrews, for providing A549 cells expressing human MxA. We thank Valentina Wagner, Freiburg, Germany, as well as Richard Cadagan and Osman Lizardo at the Icahn School of Medicine for excellent technical support.
Parts of this work were supported by a grant from the Deutsche Forschungsgemeinschaft (Ko 1579/8-2) to G.K. and by NIAID grants (U19AI083025 and P01AI058113) and by CRIP, an NIAID-funded Center for Research in Influenza Pathogenesis (grant HHSN266200700010C to A.G.-S.).
REFERENCES
- 1.Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, Li Y, Yao X, Zhang Y, Wu H, Zheng S, Diao H, Xia S, Zhang Y, Chan KH, Tsoi HW, Teng JL, Song W, Wang P, Lau SY, Zheng M, Chan JF, To KK, Chen H, Li L, Yuen KY. 2013. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet 381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, Xu X, Lu H, Zhu W, Gao Z, Xiang N, Shen Y, He Z, Gu Y, Zhang Z, Yang Y, Zhao X, Zhou L, Li X, Zou S, Zhang Y, Li X, Yang L, Guo J, Dong J, Li Q, Dong L, Zhu Y, Bai T, Wang S, Hao P, Yang W, Zhang Y, Han J, Yu H, Li D, Gao GF, Wu G, Wang Y, Yuan Z, Shu Y. 2013. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 3.Neumann G, Macken CA, Kawaoka Y. 2014. Identification of amino acid changes that may have been critical for the genesis of A(H7N9) influenza viruses. J Virol 88:4877–4896. doi: 10.1128/JVI.00107-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kageyama T, Fujisaki S, Takashita E, Xu H, Yamada S, Uchida Y, Neumann G, Saito T, Kawaoka Y, Tashiro M. 2013. Genetic analysis of novel avian A(H7N9) influenza viruses isolated from patients in China, February to April 2013. Euro Surveill 18:20453 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20453. [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q, Shi J, Deng G, Guo J, Zeng X, He X, Kong H, Gu C, Li X, Liu J, Wang G, Chen Y, Liu L, Liang L, Li Y, Fan J, Wang J, Li W, Guan L, Li Q, Yang H, Chen P, Jiang L, Guan Y, Xin X, Jiang Y, Tian G, Wang X, Qiao C, Li C, Bu Z, Chen H. 2013. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science 341:410–414. doi: 10.1126/science.1240532. [DOI] [PubMed] [Google Scholar]
- 6.Samuel CE. 2001. Antiviral actions of interferons. Clin Microbiol Rev 14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borden EC, Williams BR. 2011. Interferon-stimulated genes and their protein products: what and how? J Interferon Cytokine Res 31:1–4. doi: 10.1089/jir.2010.0129. [DOI] [PubMed] [Google Scholar]
- 8.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. 1986. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell 44:147–158. doi: 10.1016/0092-8674(86)90493-9. [DOI] [PubMed] [Google Scholar]
- 10.Holzinger D, Jorns C, Stertz S, Boisson-Dupuis S, Thimme R, Weidmann M, Casanova JL, Haller O, Kochs G. 2007. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J Virol 81:7776–7785. doi: 10.1128/JVI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, Klucher K, Staeheli P. 2008. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog 4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haller O, Kochs G. 2011. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J Interferon Cytokine Res 31:79–87. doi: 10.1089/jir.2010.0076. [DOI] [PubMed] [Google Scholar]
- 13.Verhelst J, Hulpiau P, Saelens X. 2013. Mx proteins: antiviral gatekeepers that restrain the uninvited. Microbiol Mol Biol Rev 77:551–566. doi: 10.1128/MMBR.00024-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grimm D, Staeheli P, Hufbauer M, Koerner I, Martinez-Sobrido L, Solorzano A, Garcia-Sastre A, Haller O, Kochs G. 2007. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc Natl Acad Sci U S A 104:6806–6811. doi: 10.1073/pnas.0701849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tumpey TM, Szretter KJ, Van Hoeven N, Katz JM, Kochs G, Haller O, Garcia-Sastre A, Staeheli P. 2007. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J Virol 81:10818–10821. doi: 10.1128/JVI.01116-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staeheli P, Grob R, Meier E, Sutcliffe JG, Haller O. 1988. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol Cell Biol 8:4518–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pavlovic J, Haller O, Staeheli P. 1992. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J Virol 66:2564–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matzinger SR, Carroll TD, Dutra JC, Ma ZM, Miller CJ. 2013. Myxovirus resistance gene A (MxA) expression suppresses influenza A virus replication in alpha interferon-treated primate cells. J Virol 87:1150–1158. doi: 10.1128/JVI.02271-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao H, Killip MJ, Staeheli P, Randall RE, Jackson D. 2013. The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J Virol 87:13053–13058. doi: 10.1128/JVI.02220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dittmann J, Stertz S, Grimm D, Steel J, Garcia-Sastre A, Haller O, Kochs G. 2008. Influenza A virus strains differ in sensitivity to the antiviral action of Mx-GTPase. J Virol 82:3624–3631. doi: 10.1128/JVI.01753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimmermann P, Manz B, Haller O, Schwemmle M, Kochs G. 2011. The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J Virol 85:8133–8140. doi: 10.1128/JVI.00712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manz B, Dornfeld D, Gotz V, Zell R, Zimmermann P, Haller O, Kochs G, Schwemmle M. 2013. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog 9:e1003279. doi: 10.1371/journal.ppat.1003279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinlivan M, Zamarin D, Garcia-Sastre A, Cullinane A, Chambers T, Palese P. 2005. Attenuation of equine influenza viruses through truncations of the NS1 protein. J Virol 79:8431–8439. doi: 10.1128/JVI.79.13.8431-8439.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hai R, Schmolke M, Leyva-Grado VH, Thangavel RR, Margine I, Jaffe EL, Krammer F, Solorzano A, Garcia-Sastre A, Palese P, Bouvier NM. 2013. Influenza A(H7N9) virus gains neuraminidase inhibitor resistance without loss of in vivo virulence or transmissibility. Nat Commun 4:2854. doi: 10.1038/ncomms3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Staeheli P, Dreiding P, Haller O, Lindenmann J. 1985. Polyclonal and monoclonal antibodies to the interferon-inducible protein Mx of influenza virus-resistant mice. J Biol Chem 260:1821–1825. [PubMed] [Google Scholar]
- 26.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg (Lond) 27:493–497. [Google Scholar]
- 27.Schmolke M, Manicassamy B, Pena L, Sutton T, Hai R, Varga ZT, Hale BG, Steel J, Perez DR, Garcia-Sastre A. 2011. Differential contribution of PB1-F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog 7:e1002186. doi: 10.1371/journal.ppat.1002186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rolling T, Koerner I, Zimmermann P, Holz K, Haller O, Staeheli P, Kochs G. 2009. Adaptive mutations resulting in enhanced polymerase activity contribute to high virulence of influenza A virus in mice. J Virol 83:6673–6680. doi: 10.1128/JVI.00212-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ponten A, Sick C, Weeber M, Haller O, Kochs G. 1997. Dominant-negative mutants of human MxA protein: domains in the carboxy-terminal moiety are important for oligomerization and antiviral activity. J Virol 71:2591–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pitossi F, Blank A, Schroder A, Schwarz A, Hussi P, Schwemmle M, Pavlovic J, Staeheli P. 1993. A functional GTP-binding motif is necessary for antiviral activity of Mx proteins. J Virol 67:6726–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flohr F, Schneider-Schaulies S, Haller O, Kochs G. 1999. The central interactive region of human MxA GTPase is involved in GTPase activation and interaction with viral target structures. FEBS Lett 463:24–28. doi: 10.1016/S0014-5793(99)01598-7. [DOI] [PubMed] [Google Scholar]
- 32.Koerner I, Matrosovich MN, Haller O, Staeheli P, Kochs G. 2012. Altered receptor specificity and fusion activity of the haemagglutinin contribute to high virulence of a mouse-adapted influenza A virus. J Gen Virol 93:970–979. doi: 10.1099/vir.0.035782-0. [DOI] [PubMed] [Google Scholar]
- 33.Heaton NS, Leyva-Grado VH, Tan GS, Eggink D, Hai R, Palese P. 2013. In vivo bioluminescent imaging of influenza a virus infection and characterization of novel cross-protective monoclonal antibodies. J Virol 87:8272–8281. doi: 10.1128/JVI.00969-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solorzano A, Webby RJ, Lager KM, Janke BH, Garcia-Sastre A, Richt JA. 2005. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J Virol 79:7535–7543. doi: 10.1128/JVI.79.12.7535-7543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lutz A, Dyall J, Olivo PD, Pekosz A. 2005. Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. J Virol Methods 126:13–20. doi: 10.1016/j.jviromet.2005.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belser JA, Wadford DA, Pappas C, Gustin KM, Maines TR, Pearce MB, Zeng H, Swayne DE, Pantin-Jackwood M, Katz JM, Tumpey TM. 2010. Pathogenesis of pandemic influenza A (H1N1) and triple-reassortant swine influenza A (H1) viruses in mice. J Virol 84:4194–4203. doi: 10.1128/JVI.02742-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu X, Renshaw M, Tumpey TM, Kelly GD, Hu-Primmer J, Katz JM. 2001. Immunity to influenza A H9N2 viruses induced by infection and vaccination. J Virol 75:4896–4901. doi: 10.1128/JVI.75.10.4896-4901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng AK, Zhang H, Tan K, Li Z, Liu JH, Chan PK, Li SM, Chan WY, Au SW, Joachimiak A, Walz T, Wang JH, Shaw PC. 2008. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J 22:3638–3647. doi: 10.1096/fj.08-112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.World Health Organization. 2014. Confirmed human cases of avian influenza A(H7N9) reported to WHO. Report 18: data in WHO/HQ as of 14 July 2014. World Health Organization, Geneva, Switzerland: http://www.who.int/influenza/human_animal_interface/influenza_h7n9/18_reportwebh7n9number_20140714.pdf?ua=1. [Google Scholar]
- 40.World Health Organization. 27July2014. Cumulative number of confirmed human cases for avian influenza A(H5N1) reported to WHO, 2003–2014. World Health Organization, Geneva, Switzerland: http://www.who.int/influenza/human_animal_interface/EN_GIP_20140727CumulativeNumberH5N1cases.pdf?ua=1. [Google Scholar]
- 41.Ramos I, Krammer F, Hai R, Aguilera D, Bernal-Rubio D, Steel J, Garcia-Sastre A, Fernandez-Sesma A. 2013. H7N9 influenza viruses interact preferentially with α2,3-linked sialic acids and bind weakly to α2,6-linked sialic acids. J Gen Virol 94:2417–2423. doi: 10.1099/vir.0.056184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong X, Martin SR, Haire LF, Wharton SA, Daniels RS, Bennett MS, McCauley JW, Collins PJ, Walker PA, Skehel JJ, Gamblin SJ. 2013. Receptor binding by an H7N9 influenza virus from humans. Nature 499:496–499. doi: 10.1038/nature12372. [DOI] [PubMed] [Google Scholar]
- 43.van Riel D, Leijten LM, de Graaf M, Siegers JY, Short KR, Spronken MI, Schrauwen EJ, Fouchier RA, Osterhaus AD, Kuiken T. 2013. Novel avian-origin influenza A (H7N9) virus attaches to epithelium in both upper and lower respiratory tract of humans. Am J Pathol 183:1137–1143. doi: 10.1016/j.ajpath.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hudjetz B, Gabriel G. 2012. Human-like PB2 627K influenza virus polymerase activity is regulated by importin-α1 and -α7. PLoS Pathog 8:e1002488. doi: 10.1371/journal.ppat.1002488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subbarao EK, London W, Murphy BR. 1993. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J Virol 67:1761–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moltedo B, Lopez CB, Pazos M, Becker MI, Hermesh T, Moran TM. 2009. Cutting edge: stealth influenza virus replication precedes the initiation of adaptive immunity. J Immunol 183:3569–3573. doi: 10.4049/jimmunol.0900091. [DOI] [PubMed] [Google Scholar]
- 47.Torreira E, Schoehn G, Fernandez Y, Jorba N, Ruigrok RW, Cusack S, Ortin J, Llorca O. 2007. Three-dimensional model for the isolated recombinant influenza virus polymerase heterotrimer. Nucleic Acids Res 35:3774–3783. doi: 10.1093/nar/gkm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye Q, Krug RM, Tao YJ. 2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444:1078–1082. doi: 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 49.Arranz R, Coloma R, Chichon FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortin J, Martin-Benito J. 2012. The structure of native influenza virion ribonucleoproteins. Science 338:1634–1637. doi: 10.1126/science.1228172. [DOI] [PubMed] [Google Scholar]
- 50.Turan K, Mibayashi M, Sugiyama K, Saito S, Numajiri A, Nagata K. 2004. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res 32:643–652. doi: 10.1093/nar/gkh192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verhelst J, Parthoens E, Schepens B, Fiers W, Saelens X. 2012. Interferon-inducible protein Mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J Virol 86:13445–13455. doi: 10.1128/JVI.01682-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen H, Yuan H, Gao R, Zhang J, Wang D, Xiong Y, Fan G, Yang F, Li X, Zhou J, Zou S, Yang L, Chen T, Dong L, Bo H, Zhao X, Zhang Y, Lan Y, Bai T, Dong J, Li Q, Wang S, Zhang Y, Li H, Gong T, Shi Y, Ni X, Li J, Zhou J, Fan J, Wu J, Zhou X, Hu M, Wan J, Yang W, Li D, Wu G, Feng Z, Gao GF, Wang Y, Jin Q, Liu M, Shu Y. 2014. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. Lancet 383:714–721. doi: 10.1016/S0140-6736(14)60111-2. [DOI] [PubMed] [Google Scholar]
- 53.Guan Y, Shortridge KF, Krauss S, Webster RG. 1999. Molecular characterization of H9N2 influenza viruses: were they the donors of the “internal” genes of H5N1 viruses in Hong Kong? Proc Natl Acad Sci U S A 96:9363–9367. doi: 10.1073/pnas.96.16.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bazzigher L, Schwarz A, Staeheli P. 1993. No enhanced influenza virus resistance of murine and avian cells expressing cloned duck Mx protein. Virology 195:100–112. doi: 10.1006/viro.1993.1350. [DOI] [PubMed] [Google Scholar]
- 55.Bernasconi D, Schultz U, Staeheli P. 1995. The interferon-induced Mx protein of chickens lacks antiviral activity. J Interferon Cytokine Res 15:47–53. doi: 10.1089/jir.1995.15.47. [DOI] [PubMed] [Google Scholar]
- 56.Wu R, Sui Z, Liu Z, Liang W, Yang K, Xiong Z, Xu D. 2010. Transmission of avian H9N2 influenza viruses in a murine model. Vet Microbiol 142:211–216. doi: 10.1016/j.vetmic.2009.09.068. [DOI] [PubMed] [Google Scholar]
- 57.Wu R, Zhang H, Yang K, Liang W, Xiong Z, Liu Z, Yang X, Shao H, Zheng X, Chen M, Xu D. 2009. Multiple amino acid substitutions are involved in the adaptation of H9N2 avian influenza virus to mice. Vet Microbiol 138:85–91. doi: 10.1016/j.vetmic.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 58.Qiu M, Fang F, Chen Y, Wang H, Chen Q, Chang H, Wang F, Wang H, Zhang R, Chen Z. 2006. Protection against avian influenza H9N2 virus challenge by immunization with hemagglutinin- or neuraminidase-expressing DNA in BALB/c mice. Biochem Biophys Res Commun 343:1124–1131. doi: 10.1016/j.bbrc.2006.03.088. [DOI] [PubMed] [Google Scholar]
- 59.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]