

ABSTRACT
The influenza virus hemagglutinin (HA) envelope protein mediates virus entry by first binding to cell surface receptors and then fusing viral and endosomal membranes during endocytosis. Cleavage of the HA precursor (HA0) into a surface receptor-binding subunit (HA1) and a fusion-inducing transmembrane subunit (HA2) by host cell enzymes primes HA for fusion competence by repositioning the fusion peptide to the newly created N terminus of HA2. We previously reported that the influenza virus M2 protein enhances pandemic 2009 influenza A virus [(H1N1)pdm09] HA-pseudovirus infectivity, but the mechanism was unclear. In this study, using cell-cell fusion and HA-pseudovirus infectivity assays, we found that the ion channel function of M2 was required for enhancement of HA fusion and HA-pseudovirus infectivity. The M2 activity was needed only during HA biosynthesis, and proteolysis experiments indicated that M2 proton channel activity helped to protect (H1N1)pdm09 HA from premature conformational changes as it traversed low-pH compartments during transport to the cell surface. While M2 has previously been shown to protect avian influenza virus HA proteins of the H5 and H7 subtypes that have polybasic cleavage motifs, this study demonstrates that M2 can protect HA proteins from human H1N1 strains that lack a polybasic cleavage motif. This finding suggests that M2 proton channel activity may play a wider role in preserving HA fusion competence among a variety of HA subtypes, including HA proteins from emerging strains that may have reduced HA stability.
IMPORTANCE Influenza virus infects cells when the hemagglutinin (HA) surface protein undergoes irreversible pH-induced conformational changes after the virus is taken into the cell by endocytosis. HA fusion competence is primed when host cell enzymes cleave the HA precursor. The proton channel function of influenza virus M2 protein has previously been shown to protect avian influenza virus HA proteins that contain a polybasic cleavage site from pH-induced conformational changes during biosynthesis, but this effect is less well understood for human influenza virus HA proteins that lack polybasic cleavage sites. Using assays that focus on HA entry and fusion, we found that the M2 protein also protects (H1N1)pdm09 influenza A virus HA from premature conformational changes as it transits low-pH compartments during biosynthesis. This work suggests that M2 may play a wider role in preserving HA function in a variety of influenza virus subtypes that infect humans and may be especially important for HA proteins that are less stable.
INTRODUCTION
The influenza virus hemagglutinin (HA) envelope protein mediates virus entry by binding to cell surface receptors, followed by endocytosis and fusion of the viral and endosomal membranes. Cellular proteases cleave the HA precursor (HA0) to generate the HA1 surface subunit, which mediates binding to cell surface sialic acid receptors, and the HA2 transmembrane subunit, which mediates membrane fusion between viral and endosomal membranes during endocytosis (reviewed in references 1 to 3).
The M2 protein plays an important role in several steps of influenza virus infection. During viral entry, the proton-selective ion channel function of M2 protein promotes uncoating of the influenza virus ribonucleoprotein core after membrane fusion. The low pH in the endosomes activates the ion channel activity of M2 protein. This protein channel pumps protons into the interior of the virion, making the M1-viral RNP (vRNP) interaction weak and allowing the release of the vRNP into the host cell cytoplasm (4). In the biosynthetic pathway, the ion channel activity of M2 protein may also regulate the pH balance between the acidic lumen of the trans-Golgi network (TGN) and the pH of the cytoplasm. For HA proteins with a polybasic cleavage site, such as influenza Rostock virus (H7N1) and H5 virus HA proteins, in which HA cleavage can occur in the exocytic pathway, the M2 activity can protect HA from premature pH-induced conformational changes in the TGN (5–13).
The M2 protein is also important for viral assembly and release. The cytosolic tail of M2 protein participates in genome packaging and facilitates virus production. During viral release, M2 is located at the neck of the budding virion. The C-terminal amphipathic helix of M2 protein alters the membrane curvature in a cholesterol-dependent manner and assists the membrane scission process independently of the host ESCRT (endosomal sorting complex required for transport) machinery (14–16). More recently, M2 was implicated in facilitating the formation of filamentous forms of influenza virus (17).
We previously showed that the M2 protein enhanced (H1N1)pdm09 HA-pseudovirus infectivity when M2 was coexpressed with HA during pseudovirus production (18). However, the mechanism responsible for this effect remained unknown. Unlike influenza Rostock virus H7N1 HA, (H1N1)pdm09 HA does not have a polybasic cleavage site, and to our knowledge, M2 protection of HA from low-pH-induced conformational changes in the TGN has not been reported for HA proteins that lack polybasic cleavage motifs. In this report, we analyzed possible mechanisms that might account for the M2-mediated enhancement of HA fusion. We found that M2-mediated enhancement of HA fusion depended on the ion channel function of M2 during HA biosynthesis. This finding indicates that M2 protects (H1N1)pdm09 HA from premature conformational changes during HA transport to the cell surface. Because (H1N1)pdm09 HA is less stable than many other H1N1 HA proteins (19–21), our data suggest that M2 proton channel activity may be especially important for HA proteins that are less stable, regardless of subtype or cleavage site motif.
MATERIALS AND METHODS
Plasmids and cell lines.
As previously described (22), the full-length Q223R HA open reading frame (ORF) from A/Mexico/4108/2009 (MX HA) (GenBank accession no. GQ223112), which was previously reported to enhance pseudovirus infectivity (18), and the wild-type NA ORF from A/California/04/2009 (GenBank accession no. FJ966084) were amplified from the respective viruses by reverse transcription-PCR (RT-PCR) and placed into the CMV/R 8κB expression plasmid, encoding HA from A/PR/8/34 (PR HA) (GenBank accession no. CY009444), which was obtained from Gary J. Nabel (National Institutes of Health [NIH], Bethesda, MD). Full-length wild-type M2 genes from A/Mexico/4115/2009 (MX M2) (GenBank accession no. ACQ99588.1) and A/PR/8/34 (PR M2) were synthesized by Integrated DNA Technologies (Coralville, IA) and placed into the pcDNA 3.1(+) plasmid (Invitrogen, Carlsbad, CA). A30P, T27V, and N31S mutations in the M2 genes were introduced using a QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) and confirmed by sequencing. HIV Gag/Pol (pCMV ΔR8.2) and luciferase reporter (pHR'CMV-Luc) plasmids, which were described previously (23, 24), were obtained from Gary J. Nabel (NIH, Bethesda, MD). The codon-optimized human airway trypsin-like protease (HAT) gene expression construct pCAGGS-HATcop (HATcop) was described previously (18). The HIV-1 JRCSF Env gene expression construct pCMV/R-Env was described previously (25). pCI4070A, encoding the amphotropic murine leukemia virus (A-MLV) envelope protein, was described previously (26). A vesicular stomatitis virus glycoprotein (VSV G) gene expression construct (27) was provided by Theodore Friedmann (University of California at San Diego, La Jolla, CA). A β-galactosidase (β-Gal) α subunit expression plasmid and 293T cells stably expressing the β-Gal ω subunit (28) were provided by Nathaniel Landau (New York University, New York, NY).
293T cells and 293T cells expressing the β-Gal ω subunit were cultured in Dulbecco's modified Eagle medium (DMEM) with high glucose, l-glutamine, minimal essential medium (MEM) nonessential amino acids, penicillin-streptomycin, and 10% fetal calf serum. Human primary glioblastoma cells (U87.CD4.CXCR4) expressing CD4 and CXCR4 were obtained from Dan Littman (New York University, New York, NY) and grown in the same medium as that for 293T cells, with the addition of 1 μg/ml puromycin (Sigma, St. Louis, MO) and 300 μg/ml G418 sulfate (Mediatech, Inc., Manassas, VA).
Antibodies.
Mouse anti-(H1N1)pdm09 HA monoclonal antibody 4F8 was described previously (29). Rabbit polyclonal antibodies against PR M2 and actin were purchased from Thermo Scientific (Rockford, IL). Rabbit antisera against H1N1 HA1 were produced via immunization with A/New Caledonia/20/1999 HA1 peptides (eENZYME, Gaithersburg, MD) as described previously (30). Rabbit antisera against the H1N1 HA2 C helix were produced by immunization with an A/New Caledonia/20/1999 HA2 C helix peptide (RMENLNKKVDDGFLDIWTYNAELLVLLENERTLDFHDSNVKNLYEKVKSQLKNNA, with CSGSGSG residues added to the N terminus) conjugated to keyhole limpet hemocyanin (KLH) (Pierce) and mixed with Freund's adjuvant.
Pseudovirus production and infectivity assay.
Pseudoviruses were produced in 293T cells as described previously (18). Briefly, to make HA-pseudovirus, 3 μg of MX HA plasmid or 0.5 μg of PR HA plasmid, 2 μg of MX M2 or PR M2 plasmid, 2 μg of HATcop plasmid, 4 μg of CA NA plasmid, 5 μg of pCMV ΔR8.2, and 5 μg of pHR'CMV-Luc were cotransfected by use of FuGENE 6 (Promega, Madison, WI). For some experiments, the amounts of plasmids transfected varied as indicated in the figures. To make A-MLV Env-, VSV G-, or HIV-1 Env-pseudovirus, 2 μg of A-MLV Env, VSV G, or HIV-1 Env plasmid, 2 μg of MX M2 or PR M2 plasmid, 5 μg of pCMV ΔR8.2, and 5 μg of pHR'CMV-Luc were cotransfected by use of FuGENE 6. Pseudoviruses were harvested at 48 h posttransfection and then used immediately for electron microscopy studies, filtered through a 0.45-μm low-protein-binding filter and used immediately, or stored at −80°C. HA-, A-MLV Env-, and VSV G-pseudovirus titers were determined by infecting 293T cells with these pseudoviruses for 48 h prior to measuring luciferase activity by using a luciferase assay system (Promega), and the titers were normalized by use of the p24 enzyme-linked immunosorbent assay (ELISA) level as described previously (18). HIV-1 Env-pseudovirus titers were determined by infecting U87.CD4.CXCR4 cells. Pseudovirus titers were normalized to fold changes with respect to pseudoviruses not containing M2.
Pseudoviruses were quantified by HIV-1 p24 Gag ELISA (AIDS Vaccine Program, NCI-Frederick Cancer Research and Development Center, Frederick, MD) as described previously (30). HA and M2 expression and incorporation into pseudoviruses were quantified by immunoblot analysis using rabbit HA1 antisera and PR M2 polyclonal antibodies.
Electron microscopy.
HA-pseudoviruses were concentrated from unfiltered, clarified culture supernatant by centrifugation at 30,590 × g and 4°C for 3 h through a 20% sucrose cushion, followed by resuspension in 1× Dulbecco's phosphate-buffered saline (DPBS) (Mediatech, Inc.). Aliquots were placed on 400-mesh carbon-coated grids (Electron Microscopy Sciences, Hatfield, PA), fixed with 2% formaldehyde and 2% glutaraldehyde in DPBS, and washed gently with distilled water. The samples were stained with 2% methylamine tungstate and examined with a Zeiss EM 912 transmission electron microscope.
Cell-cell fusion assay.
Cell-cell fusion was quantified using a reporter system based on β-Gal complementation (31). Briefly, 293T cells were transfected with the HA expression plasmid (pCMV/R-HA) along with M2, HAT, and β-Gal α subunit expression plasmids by use of FuGENE 6 reagent. Forty-eight hours after transfection, the transfected 293T cells were detached using a nonenzymatic cell dissociation solution (Sigma) and washed with DMEM. A total of 6 × 104 cells per well were then added to β-Gal ω subunit-expressing 293T target cells that had been seeded the night before at 3 × 104 cells per well in a 96-well plate. Cells were cocultivated for 3 h at 37°C. The culture supernatants were then removed and replaced with DPBS previously adjusted to the desired pH with 0.1 M citric acid. The cells were treated for 4 min in the DPBS-citrate buffer and then cultured with DMEM. Eighteen hours later, cell-cell fusion was scored by measuring β-Gal activity in coculture cell lysates by use of a Galacto-Star kit (Applied Biosystems, Carlsbad, CA) according to the manufacturer's instructions.
For HA-induced cell-cell fusion with trypsin-activated HA, NIH/3T3 cells were transfected with MX HA (pCMV/R-HA), β-Gal α subunit, and wild-type MX M2 expression plasmids by use of FuGENE 6. Forty-eight hours after transfection, NIH/3T3 cells were washed twice with DPBS and treated with 3 μg/ml of trypsin treated with l-1-tosylamide-2-phenylmethyl chloromethyl ketone (TPCK-trypsin) (Thermo Scientific) for 5 min at 37°C. TPCK-trypsin was removed, and 10% fetal calf serum-containing medium was added. 293T cells expressing the β-Gal ω subunit and NIH/3T3 cells were cocultivated for 3 h at 37°C. Cells in coculture were treated with acidic DPBS (pH 5.0) for 4 min at 37°C. Acidic DPBS was replaced with medium, and 18 h later, cell-cell fusion was scored by measuring β-Gal activity in coculture cell lysates by use of a Galacto-Star kit (Applied Biosystems) according to the manufacturer's instructions.
β-Gal activity was expressed in relative luminescence units (RLU). The fusion level was normalized to that of the controls, as indicated in the figure legends. Amantadine inhibition of cell-cell fusion was evaluated by adding amantadine to cells, to a final concentration of 10 μM, at different time points during the fusion process.
FACS analysis.
Transfected 293T cells were detached by use of cell dissociation solution (Sigma), washed, and resuspended in fluorescence-activated cell sorter (FACS) staining buffer (DPBS with 2% fetal bovine serum [FBS]). The cells were then incubated with appropriate primary monoclonal antibodies on ice for 30 min, followed by washing with FACS staining buffer. The cells were then resuspended in FACS staining buffer and incubated with a phycoerythrin (PE)-conjugated anti-mouse IgG secondary antibody in the dark on ice for 30 min. After washing 3 times with DPBS, the cells were fixed with DPBS containing 2% paraformaldehyde. The number of cells expressing HA was determined by flow cytometric analysis on an LSR II flow cytometer (BD Biosciences). The postacquisition data analysis was performed using FlowJo software v9 (Tree Star).
Protease sensitivity.
To detect pH-induced conformational changes in HA, HA-pseudoviruses made in serum-free medium in the absence of HAT were subjected to limited proteolysis by trypsin as described previously (32–37). In brief, HA-pseudovirus supernatant samples were mixed with 10% n-dodecyl β-d-maltoside (DDM) to a final concentration of 1% DDM and incubated at 37°C for 1 h. The samples were then digested with TPCK-treated trypsin (Pierce) at a final concentration of 100 μg/ml at room temperature for 20 h. The trypsin-digested samples were then resolved by nonreducing SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA) for Western blot analysis. The blots were probed with rabbit antisera against the HA2 C helix and with horseradish peroxidase-linked goat anti-rabbit antibodies and detected by use of Lumiglo Reserve (KPL, Gaithersburg, MD).
Data analysis.
Data reported were from at least three independent experiments. t tests for paired-data comparisons, one-way and two-way analyses of variance (ANOVA) for group data, and corresponding P values were analyzed using GraphPad Prism software. P values of <0.05 were considered statistically significant.
RESULTS
M2 enhances infectivity of HA-pseudovirus.
Our earlier report showed that M2 enhances the infectivity of (H1N1)pdm09 HA-pseudovirus (18). To confirm and extend this observation, pseudoviruses bearing HA from A/Mexico/4108/2009 (MX) or A/PR/8/34 (PR) were produced in 293T cells, with or without MX or PR M2. As shown in Fig. 1A, M2 significantly enhanced the infectivity of pseudoviruses bearing either MX or PR HA (t test; P < 0.001), with greater enhancement for the MX HA-pseudovirus. The HA levels (Fig. 1A, bottom panels) and total p24 levels (data not shown) in HA-pseudoviruses were similar whether or not M2 was included in producing the HA-pseudoviruses, indicating that addition of M2 did not affect the production or incorporation of HA into pseudoviruses. As a control, addition of M2 did not enhance the infectivity of other pseudoviruses, bearing the envelope protein from HIV-1, A-MLV, or VSV (Fig. 1B), indicating that M2 enhancement of pseudovirus infectivity is specific to HA function and does not play a role in enhancing infectivity by enhancing capsid uncoating. Additionally, M2 was incorporated into pseudoviruses, as shown by M2 Western blotting of pseudoviruses (Fig. 1C).
FIG 1.
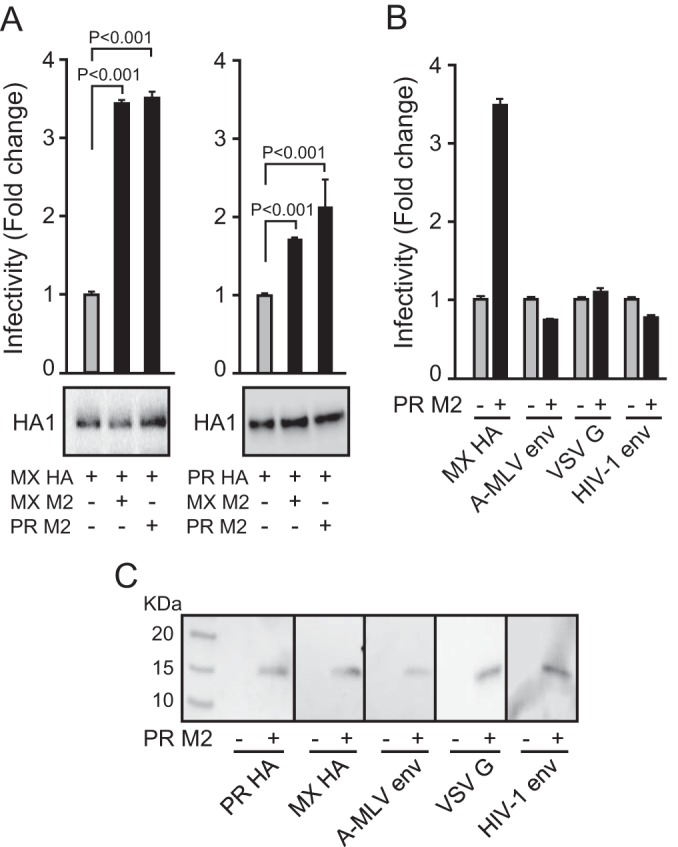
Infectivities of HA-pseudoviruses with and without M2. (A) Infectivities of pseudoviruses bearing A/Mexico/4108/2009 (MX) HA (left) or A/PR/8/34 (PR) HA (right), with and without MX or PR M2. Data are shown as means and standard deviations for three independent experiments. (B) Infectivities of pseudoviruses bearing A-MLV Env, HIV-1 Env, or VSV G, with and without PR M2. (C) Western blots of M2 incorporation into pseudoviruses bearing MX HA, PR HA, A-MLV Env, VSV G, or HIV-1 Env. PR M2 was detected with an M2-specific rabbit antiserum.
M2 does not affect HA-pseudovirus morphology and HA incorporation.
It has been reported that M2 influences the morphogenesis of influenza virus (17, 38, 39), so we investigated whether M2 could also affect HA-pseudovirus morphology. We therefore examined our HA-pseudoviruses by transmission electron microscopy and found no apparent differences, with or without M2. All HA-pseudoviruses were sphere-like particles with diameters of approximately 100 nm, with no obvious differences in the number of HA spikes (Fig. 2). Western blot analysis of HA-pseudovirus also suggested that M2 did not alter HA incorporation into viral particles (Fig. 1A). Thus, M2 enhancement of HA-pseudovirus infectivity is not due to alterations in HA incorporation or changes in HA-pseudovirus morphology.
FIG 2.
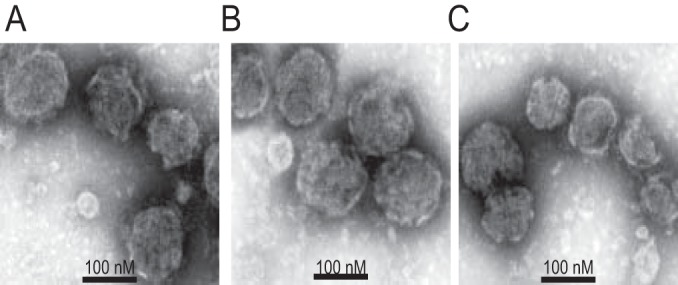
Transmission electron microscopy of HA-pseudoviruses with negative staining. (A) MX HA-pseudovirus without M2; (B) MX HA-pseudovirus with MX M2; (C) MX HA-pseudovirus with PR M2.
M2 enhances HA-mediated cell-cell fusion.
To further understand how M2 enhances HA-pseudovirus infectivity, we assessed HA-mediated cell-cell fusion to rule out M2 effects during the postfusion steps of viral entry, including uncoating and endocytosis. In the cell-cell fusion assay, we coexpressed HA and HAT to induce HA-mediated cell-cell fusion, similar to the case in the pseudovirus assay. As expected, both PR (data not shown) and MX HA-mediated cell-cell fusion depended on low-pH treatment, and higher levels of HA resulted in more fusion (one-way ANOVA; P < 0.001) (Fig. 3A). M2 enhanced HA-mediated cell-cell fusion in a dose-dependent manner (one-way ANOVA; P < 0.01) (Fig. 3B, right panel), with more pronounced enhancement when M2 levels were high and HA levels were low (Fig. 3B, left panel). M2 expression did not change total levels of HA (Fig. 3D) or the level of HA presented on the cell surface (Fig. 3C).
FIG 3.

M2 enhancement of HA-mediated cell-cell fusion. (A) Various levels of MX HA expressed in effector cells (0.5 to 4.5 μg of plasmid transfected) induced cell-cell fusion at pHs 7.4 and 5.0. The bottom left panel shows a Western blot of HA expression in effector cells. The right panels show a flow cytometry analysis of cell surface levels of HA in effector cells (0.5 to 4.5 μg of plasmid transfected). MFI, mean fluorescence intensity. (B) M2 enhanced MX HA-mediated cell-cell fusion at pH 5.0. (Left) A constant level of MX and PR M2 expression (6 μg of plasmid transfected) was tested with various levels of MX HA expression (1 to 4.5 μg of plasmid transfected). The fusion levels were normalized to the individual fusion levels induced by various HA levels without M2 expression. (Right) Various levels of MX and PR M2 expression (0 to 6 μg of plasmid transfected) were tested with a constant level of MX HA expression (1 μg of plasmid transfected). The fusion levels were normalized to the fusion level induced by HA alone without M2 expression. (C) Flow cytometry analysis of cell surface levels of HA in effector cells from the right part of panel B. The MX HA levels on the cell surface with various levels of MX M2 expression are shown in the top panels. The bottom panels show MX HA levels on the cell surface with various levels of PR M2 expression. (D) Western blots of total HA and M2 expression in effector cells from the right part of panel B. The top panels show MX M2 expression with various amounts of transfected DNA. The bottom panels show PR M2 expression with various amounts of transfected DNA. HA0 and HA1 were detected with rabbit HA1 antiserum. MX and PR M2 proteins were detected by use of M2-specific rabbit antiserum. Cell-cell fusion data are shown as means and standard deviations for three independent experiments.
PR and MX M2 enhancement of HA-mediated cell-cell fusion occurred across a range of pH values (Fig. 4A). As observed before (Fig. 3B), we found that 3 μg M2 enhanced 1-μg-HA-mediated cell-cell fusion about 3-fold (Fig. 4A). Relative fusion, in which the absolute fusion was normalized to the maximum fusion obtained with MX M2 (Fig. 4A, top panel) or PR M2 (Fig. 4A, bottom panel), was used to compare the fusion levels with and without M2. To more easily compare the pH values of half-maximal fusion for the groups, a fusion index was calculated by normalizing fusion to the maximum fusion for each group (Fig. 4B). For all groups, the presence of M2 did not alter the fusion index curves (two-way ANOVA; P > 0.1). Half-maximal cell-cell fusion was achieved at approximately pH 5.4, regardless of the presence of M2 (Fig. 4B). These results indicate that M2 does not alter the pH curve for HA-mediated cell-cell fusion or the HA response to pH. Thus, M2 enhancement of HA-mediated cell-cell fusion does not appear to be due to an interaction with HA that alters the threshold or sensitivity of HA to pH-induced conformational changes. Finally, because the total amount of HA on the cell surface was unchanged in the presence or absence of M2 (Fig. 4C), our data indicate that M2 enhanced fusion activity by increasing the amount of fusion-competent HA on the cell surface.
FIG 4.

M2 enhancement of MX HA-mediated cell-cell fusion across a range of pH values. (A) Relative cell-cell fusion levels mediated by MX HA at various pHs in the presence or absence of MX M2 (top) or PR M2 (bottom). For determinations of relative fusion, the fusion levels were normalized to the maximum fusion obtained with MX M2 (top) or PR M2 (bottom). (B) Fusion index comparisons. Fusion levels in each curve from panel A were normalized to the maximum fusion level for that curve. For example, the individual fusion levels at different pHs in the curve for MX HA were normalized to the maximum fusion level in the curve for MX HA. (C) Flow cytometry analysis of cell surface expression of HA in the effector cells used for panels A and B. Fusion data are shown as means and standard deviations for three independent experiments. Plasmids used for transfection included 1 μg of MX HA plasmid and 3 μg of M2 plasmid.
M2 proton channel function is needed to enhance HA-mediated cell-cell fusion.
We next tested whether M2 proton channel function is needed to enhance HA-mediated cell-cell fusion. Previously, it was reported that an alanine-to-proline substitution at position 30 (A30P) in M2 impairs the proton channel function of influenza A/Udorn/72 (H3N2) virus M2 (40). Like A/Udorn/72 M2, both MX and PR M2 proteins bear the A30 residue (Fig. 5A). Therefore, we introduced an A30P mutation into both MX and PR M2. Both M2 proteins with the A30P mutation were expressed at the same levels as that for wild-type M2, and the M2 mutation did not change the HA levels on the cell surface (data not shown). Importantly, M2 with the A30P mutation was no longer able to enhance cell-cell fusion (two-way ANOVA for comparison to HA without M2; P > 0.1) (Fig. 5B and C, left panels), demonstrating that the proton channel function of M2 is necessary for HA protection and enhancement of HA-mediated cell-cell fusion.
FIG 5.

M2 proton channel function requirement for enhancement of MX HA-mediated cell-cell fusion. (A) Alignment of amino acids 21 to 35 of A/Udorn/72 (Udorn), PR, and MX M2 proteins. (B) MX HA-mediated cell-cell fusion at various pHs, with and without MX M2 proton channel function. (C) MX HA-mediated cell-cell fusion at various pHs, with and without PR M2 proton channel function. (Left) M2 proteins and M2 A30P mutants with proton channel dysfunction. (Middle) Controls (HA or HA plus M2), with and without amantadine. (Right) M2 mutants sensitive to amantadine, with and without amantadine. AM, 10 μM amantadine. Fusion data are shown as means and standard deviations for three independent experiments. Plasmids used for transfection included 1 μg of MX HA plasmid and 3 μg of M2 plasmid.
Since amantadine blocks the proton channel activity of amantadine-sensitive M2 (41), we further studied the effect of M2 proton channel function on HA-mediated cell-cell fusion by treatment with amantadine. It was previously reported that the serine 31 (S31) cluster is the high-affinity binding site for amantadine (42–44) and that residues S31 and valine 27 (V27) in M2 confer M2 sensitivity to amantadine (45–53). Wild-type MX M2 contains residues N31 and V27, while PR M2 contains N31 and threonine 27 (T27) (Fig. 5A). Both wild-type MX and PR M2 proteins were resistant to amantadine, and the amantadine treatment did not affect M2 enhancement of HA-mediated cell-cell fusion (two-way ANOVA for comparison to HA with M2; P > 0.1) (Fig. 5B and C, middle panels).
To make M2 gain sensitivity to amantadine, an asparagine-to-serine mutation at position 31 (N31S) was introduced into MX M2, while N31S and a threonine-to-valine substitution at position 27 (T27V) were introduced into PR M2, such that both mutated MX and PR M2 proteins had residues S31 and V27. MX M2 containing the N31S mutation (MX M2-N31S) and PR M2 containing the N31S and T27V mutations (PR M2-T27V-N31S) still enhanced HA-mediated cell-cell fusion (two-way ANOVA for comparison to HA without M2; P < 0.001), at levels similar to those for wild-type MX and PR M2 (Fig. 5B and C, right panels). At high concentrations (>0.1 mM), amantadine neutralizes the acidic intracellular environment, thereby preventing low-pH-induced conformational changes in HA. At low concentrations (<10 μM), amantadine is believed to block the M2 channels (11). Thus, a low concentration (10 μM) of amantadine was used to block the proton channel activity of M2 in the cell-cell fusion assay by maintaining amantadine with HA- and M2-transfected cells from 0.5 h posttransfection (after adding plasmids to the cells) until the end of the fusion assay, when cells were lysed. As expected, the presence of 10 μM amantadine during transfections with M2 nearly completely abolished the MX M2-N31S enhancement of HA-mediated cell-cell fusion (two-way ANOVA for comparison to HA with M2-N31S; P < 0.001) (Fig. 5B, right panel), while the amantadine treatment itself had no effect on HA-mediated cell-cell fusion in the control without M2 (two-way ANOVA for comparison to HA without M2; P > 0.1) (Fig. 5B and C, middle panels). The same treatments also led to a 70% reduction of the PR M2-T27V-N31S enhancement of HA-mediated cell-cell fusion (two-way ANOVA for comparison to HA with M2-T27V-N31S; P < 0.001) (Fig. 5C, right panel). The more modest reduction of PR M2-T27V-N31S enhancement of HA-mediated cell-cell fusion by amantadine was probably due to differences in residues between the MX and PR M2 proteins that may also modulate M2's sensitivity to amantadine. M2 and its mutants were expressed at the same levels, and their expression did not change the level of HA presented on the cell surface (data not shown). Altogether, these observations demonstrate that M2 proton channel function is needed for the enhancement of HA-mediated cell-cell fusion.
M2 proton channel function during HA biosynthesis accounts for the enhancement of HA-mediated cell-cell fusion.
Since our HA-mediated cell-cell fusion assay involves several steps, including HA biosynthesis, pH treatment to trigger HA conformational changes, and cell-cell fusion scored by β-Gal activity, we next tried to determine at which step the M2 proton channel function is needed to enhance overall fusion. Thus, 10 μM amantadine, an inhibitor of M2 activity for sensitive strains, was maintained at the following time points during the cell-cell fusion assay (Fig. 6A): (i) from 0.5 h post-HA transfection (after adding plasmids to the cells) of effector cells and thereafter, until cells were lysed and scored for β-Gal activity; (ii) from the time of low-pH treatment (amantadine was added to low-pH buffer) of effector-target cocultures and thereafter, until cells were lysed and scored for β-Gal activity; and (iii) from the point right after the pH treatment (amantadine was added to culture medium) of effector-target cocultures and thereafter, until cells were lysed and scored for β-Gal activity. Previous experiments showed that amantadine is stable at acidic pH (41, 54–56), so amantadine function was not affected at the various time points when it was used. We found that amantadine abrogated M2 enhancement of HA-mediated cell-cell fusion only when amantadine was present during the period from HA transfection of effector cells and thereafter, until cell lysis (t test; P < 0.001), but not in the period from low-pH treatment to cell lysis (t test; P > 0.1) (Fig. 6B). Therefore, M2 proton channel function is required during HA expression in effector cells in order to see the M2 enhancement of HA-mediated cell-cell fusion. This finding strongly suggests that M2 proton channel function preserves HA fusion competence during HA biosynthesis and transport to the cell surface.
FIG 6.
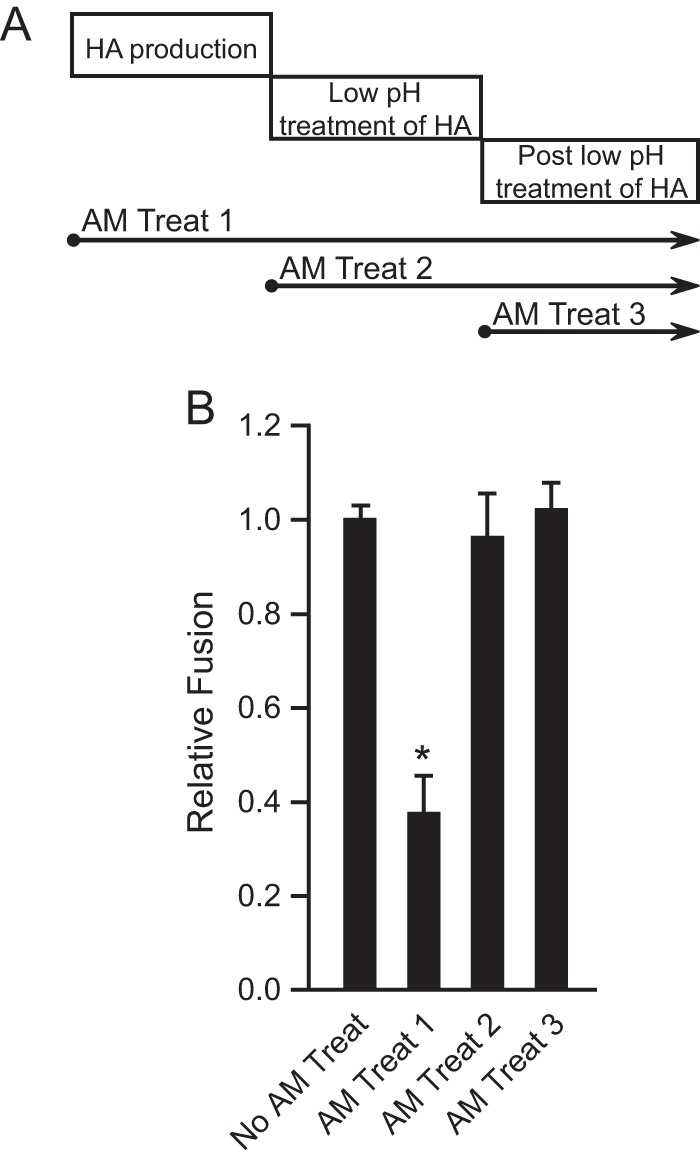
M2 proton channel function during HA biosynthesis accounts for the enhancement of HA-mediated cell-cell fusion. (A) Amantadine (AM) treatment windows. Treat 1, amantadine was maintained from 0.5 h post-HA and -M2 transfection of effector cells and thereafter, until the fusion assay ended, at the point of lysing cells; treat 2, amantadine was maintained from the time of low-pH treatment of effector-target cocultures and thereafter, until the fusion assay ended, at the point of lysing cells; treat 3, amantadine was maintained from the point right after the pH treatment of effector-target cocultures and thereafter, until the fusion assay ended, at the point of lysing cells. (B) Effects of amantadine treatment on MX M2-N31S enhancement of MX HA-mediated cell-cell fusion at pH 5.0. Amantadine (10 μM final concentration) was maintained according to treatments 1, 2, and 3. Fusion data are shown as means and standard deviations for three independent experiments. Plasmids used for transfection included 1 μg of MX HA plasmid and 3 μg of MX M2-N31S plasmid. *, P < 0.001 (compared to no AM treatment; t test).
M2 protects (H1N1)pdm09 HA from premature conformational changes during transport to the cell surface.
Since HAT activity may not be restricted to the cell surface when HAT is overexpressed and may also cleave HA during transit to the cell surface, we next assessed whether M2 enhancement of HA-mediated cell-cell fusion depends on HAT expression. We thus expressed HA0 without HAT and instead cleaved HA0 at the cell surface by adding TPCK-trypsin. Because TPCK-trypsin treatment was mildly toxic to 293T cells and gave less robust results (30; data not shown), we expressed HA in NIH/3T3 cells and subjected the undetached cells to TPCK-trypsin treatment. Again, as seen with HAT expression, M2 enhanced HA-mediated cell-cell fusion (t test; P < 0.0001) when MX HA was cleaved on the cell surface by TPCK-trypsin (Fig. 7A). Thus, M2-enhanced HA-mediated cell-cell fusion is not the result of HAT expression and does not depend on intracellular processing of HA0. Because HA0 may undergo some degree of pH-induced conformational changes that may lead to HA inactivation (34), our data suggest that M2 protects HA0 during biosynthesis.
FIG 7.
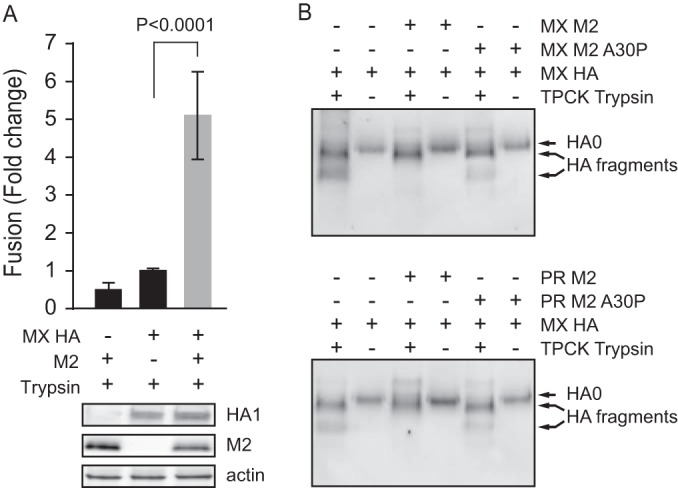
M2 enhancement of TPCK-trypsin-activated HA-mediated fusion and M2 protection of HA from premature conformational changes. (A) TPCK-trypsin-digested HA induced cell-cell fusion at pH 5.0 in the presence or absence of MX M2. The fusion levels were normalized to the MX HA-mediated fusion level with TPCK-trypsin digestion. The bottom panels show Western blots of expression of HA, M2, and actin. Plasmids used for transfection included 1 μg of MX HA plasmid and 3 μg of MX M2 plasmid. (B) Nonreducing Western blots of TPCK-trypsin-digested pseudovirus HA in the presence or absence of MX or PR M2 or M2 with the A30P mutation. HA0 and HA fragments were detected with antisera against the HA2 C helix. Cell-cell fusion data are shown as means and standard deviations for three independent experiments. HA fragments were disulfide-bonded HA1 and HA2 proteolytic fragments.
To further elucidate how M2 protects HA fusion competence, we directly evaluated the effect of M2 on premature HA conformational changes during biosynthesis. HA conformations can be assessed by differential sensitivity to extensive proteolysis. Virus particles and purified HA proteins have previously been used to demonstrate HA sensitivity to proteolysis after acidic pH-induced HA conformational changes (32–37). Following acidic pH-induced conformational changes in HA, TPCK-treated trypsin partially digests the HA1 subunit but not the HA2 subunit (32). We therefore produced HA-pseudoviruses in serum-free medium without HAT coexpression and subsequently digested HA with TPCK-trypsin. In this case, we found that without acidic pH treatment, TPCK-trypsin partially digested MX HA0 from pseudovirions into two major fragments, as detected by nonreducing SDS-PAGE followed by Western blotting with antisera against the HA2 C helix (Fig. 7B). However, when either MX M2 (Fig. 7B, top panel) or PR M2 (Fig. 7B, bottom panel) was coexpressed with HA during pseudovirus production, there was less extensive digestion of HA by TPCK-trypsin. The relative protection of HA from digestion by the M2 proteins further depended on the channel function activity, because the M2 A30P mutation did not confer protection from digestion. These results indicate that MX HA0 undergoes some degree of conformational change during transport to the cell surface. We concluded that M2 channel activity protects MX HA from premature conformational changes during biosynthesis.
(H1N1)pdm09 HA undergoes pH-induced conformational changes at a higher pH than that for PR HA.
Because our data indicated that M2 proton channel activity was required during HA biosynthesis in order to enhance HA-mediated fusion, we next assessed whether (H1N1)pdm09 HA was more sensitive than PR HA to pH-induced conformational changes. Indeed, in directly comparing the fusion levels of the two strains at various pHs (Fig. 8), we found that significant fusion started at about pH 5.7 for (H1N1)pdm09 HA, with half-maximal fusion occurring at about pH 5.4. In contrast, PR HA initiated fusion at about pH 5.4, with half-maximal fusion occurring at about pH 5.2. Thus, (H1N1)pdm09 HA is more sensitive than PR HA to pH-induced conformational changes, consistent with our finding that M2 has a greater effect on enhancing fusion of (H1N1)pdm09 HA than PR HA.
FIG 8.
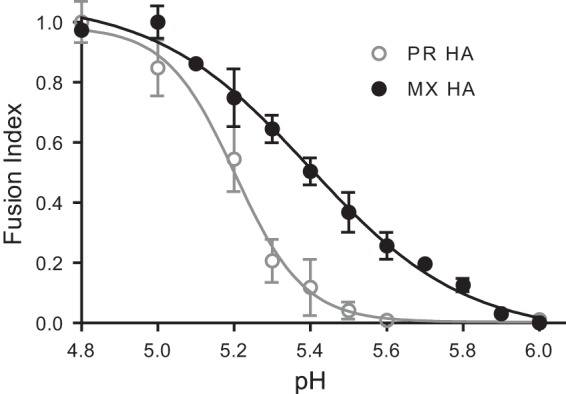
Comparison of pH dependences of MX and PR HA-mediated cell-cell fusion. Fusion responses to pH were compared for MX and PR HA proteins. The fusion response to pH was normalized to the maximum fusion for that curve. Fusion data are shown as means and standard deviations for three independent experiments.
DISCUSSION
The M2 protein can promote influenza virus infection during several steps in the viral life cycle, including viral assembly and release (14–17), uncoating of the ribonucleoprotein core (4), and biosynthesis of certain H5 and H7 subtype HA proteins that contain a polybasic cleavage motif (5–13). Previously, we found that M2 enhanced the infectivity of (H1N1)pdm09 HA-pseudovirus (18). However, the mechanism was unclear because this HA does not contain a polybasic cleavage motif, and it was not known whether M2 could play a role during (H1N1)pdm09 HA biosynthesis.
Our current studies show that M2 enhancement of HA-mediated fusion and HA-pseudovirus infectivity specifically acts on HA and does not affect HA-pseudovirus assembly and release (Fig. 2). M2 improved the infectivity of two different H1N1 HA-pseudoviruses, albeit to different extents, but there was no enhancement of pseudoviruses bearing the envelope from HIV-1, A-MLV, or VSV (Fig. 1), indicating that M2 specifically enhances HA function rather than nonspecifically promoting virus binding to target cells, altering the viral membrane's propensity to fuse with target cell membranes, or promoting endocytosis and uncoating. Furthermore, M2 enhanced HA-mediated cell-cell fusion and protected HA from premature conformational changes, demonstrating that M2 has a direct effect on HA function (Fig. 3 to 7). We additionally showed that M2 enhancement depends on the proton-selective ion channel function of M2. M2 enhancement of HA-mediated fusion was abrogated by either introducing a channel-impairing A30P mutation into M2 or treating cells expressing an amantadine-sensitive M2 protein with amantadine. Similarly, M2 containing the loss-of-function A30P mutation did not improve the infectivity of HA-pseudovirus (data not shown).
Significantly, amantadine treatment at various time points during the cell-cell fusion assay further showed that M2 proton channel function is needed during HA biosynthesis but not during the fusion process itself or afterwards. This finding suggested that M2 most likely protects HA during its transport through the exocytic pathway. However, the protective effect of M2 ion channel activity on HA has been reported only for HA proteins from subtypes H5 and H7 with a polybasic cleavage site, which are cleaved in the exocytic pathway (9–11, 13). In these cases, it is believed that the M2 ion channel activity regulates the pH balance between the acidic lumen of the trans-Golgi network (TGN) and the pH of the cytoplasm to protect HA from premature low-pH-induced conformational changes in the TGN (5–13).
In our HA-pseudovirus, human airway trypsin-like protease (HAT) was coexpressed with HA to promote cleavage because TPCK-trypsin was mildly toxic to the 293T cells used for pseudovirus production (30; data not shown). The location of HAT cleavage of HA is not fully understood, though it has been reported that HAT can cleave HA at the cell surface (57). HAT activity may be dependent on the cell type, influenza virus strain (58), and other unknown factors, but it is nonetheless a relevant in vivo protease for cleaving HA. If HAT cleaves HA at the cell surface, then HA might be expected to be less sensitive to low-pH-induced conformational changes, because uncleaved HA0 may not undergo low-pH-induced irreversible conformational changes to the same extent as mature HA, which is cleaved into HA1 and HA2 subunits (34). However, we found that M2 enhanced HA-mediated cell-cell fusion when HA was cleaved by either HAT or TPCK-trypsin, indicating that M2-mediated enhancement of fusion is not specifically due to HAT expression. Importantly, (H1N1)pdm09 HA is known to be less stable than many other human influenza virus H1 HA proteins (19–21). Our data therefore suggest that M2 protects (H1N1)pdm09 HA from undergoing some degree of pH-induced conformational change that impairs its function even prior to cleavage into mature HA.
However, MX HA still mediated cell-cell fusion without M2 (Fig. 3), indicating that only a fraction of the total HA was protected by M2 proton channel activity. The reduced stability of (H1N1)pdm09 HA likely makes it more susceptible to pH or other environmental stresses. Several groups have shown that the stability of HA varies greatly among different virus strains that have adapted to different environmental conditions and hosts (21, 58–66). HA proteins from viruses of the same or different subtypes can exhibit different sensitivities to proteases and pH activation of membrane fusion. For example, pH 5.0 triggers fusion for A/Victoria/3/1975 (H3N2) HA, while pH 5.5 is needed for A/duck/Ukraine/1/1963 (H3N8) HA (58). H1N1 subtypes also vary in their pH of fusion activation. A relatively high pH of 5.7 is needed to trigger fusion of A/Pennsylvania/08/2008 (H1N1) HA, while pH 5.1 triggers fusion for A/PR/8/1934 (H1N1) HA and pH 5.6 triggers fusion for A/duck/Alberta/35/1976 (H1N1) HA (58). Pathogenic H7 HA proteins can vary up to 0.6 pH unit in their pH of fusion activation (9).
Viruses adapted to different hosts can also develop mutations that affect HA stability and the pH of conformational changes. For example, H7N3 viruses from turkeys can mediate fusion below pH 6, but H7N3 viruses from ducks can mediate fusion at pH 6 (67). H3, H5, and H7 HA proteins with a lysine-to-isoleucine substitution at position 58 (K58I; H3 numbering) in HA2 have been shown to decrease the pH for membrane fusion by 0.6 to 0.7 pH unit (12, 64–66). In addition, HA mutants with less stability in the presence of acid were rapidly selected when viruses passaged in mammalian cells were subsequently passaged in embryonated chicken eggs (62, 63). An H5 HA mutation with increased acid stability rendered an H5N1 virus more efficient in airborne transmission in ferrets (68, 69). The more recent (H1N1)pdm09 HA acquired some mutations that conferred greater stability in acid (fusion at lower pH) than that of isolates from 2009 (21, 60), suggesting that early isolates were adapted to the porcine host but additional adaptation was needed for the human host. Indeed, we found that MX HA initiated cell-cell fusion at a higher pH than that for PR HA (Fig. 8), irrespective of M2 function. This is consistent with previous reports (58). The absolute level of MX HA-mediated cell-cell fusion is lower than that mediated by PR HA (data not shown), possibly reflecting the instability of MX HA and the resulting lower level of fusion-competent HA on the cell surface. In this case, M2 would be expected to have a greater impact on fusion when fusion-competent HA is limiting than when fusion-competent HA is abundant, consistent with our findings (Fig. 3). Perhaps a higher level of fusion-competent PR HA on the cell surface explains the smaller impact of M2 on PR HA-mediated fusion, which occurs at a high level with or without M2.
M2 protection of HA during exocytosis, previously shown in the context of influenza virus HA subtypes H5 and H7 (5–13), has not been reported for influenza virus strains with HA proteins with a monobasic cleavage site. The data presented here provide direct evidence that M2 can also protect HA from conformational changes in human H1N1 strains that lack the polybasic cleavage motif. Recently, O'Donnell et al. reported that M2 function affects the acid and heat stability of HA proteins of some pandemic strains, including (H1N1)pdm09 (70), but the mechanism was not directly demonstrated. Although M2 enhancement of MX HA function is less pronounced than that of an acid-sensitive H7 HA (9–11), our data with two different H1N1 HA proteins indicate that M2 may promote infection by viruses with non-H7 subtype HA in a strain-dependent manner. We suggest that M2 proton channel activity may be especially significant for HA proteins that are less stable, which may be the case for emerging strains that are less well adapted to their new hosts.
ACKNOWLEDGMENTS
We thank Xianghong Jing and Vladimir Lugovstev (U.S. FDA, Center for Biologics Evaluation and Research, Silver Spring, MD) for critical readings of the manuscript.
This work was supported by institutional funds from the U.S. FDA.
Research described in this report contributed in part to the fulfillment of requirements for a Ph.D. for Esmeralda Alvarado-Facundo.
We do not have commercial or other associations that might pose a conflict of interest.
REFERENCES
- 1.Rogers GN, Paulson JC, Daniels RS, Skehel JJ, Wilson IA, Wiley DC. 1983. Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature 304:76–78. doi: 10.1038/304076a0. [DOI] [PubMed] [Google Scholar]
- 2.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 3.Wharton SA, Calder LJ, Ruigrok RW, Skehel JJ, Steinhauer DA, Wiley DC. 1995. Electron microscopy of antibody complexes of influenza virus haemagglutinin in the fusion pH conformation. EMBO J 14:240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palese P, Shaw ML. 2007. Orthomyxoviridae: the viruses and their replication, p 1648–1689 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Grambas S, Bennett MS, Hay AJ. 1992. Influence of amantadine resistance mutations on the pH regulatory function of the M2 protein of influenza A viruses. Virology 191:541–549. doi: 10.1016/0042-6822(92)90229-I. [DOI] [PubMed] [Google Scholar]
- 6.Ciampor F, Bayley PM, Nermut MV, Hirst EM, Sugrue RJ, Hay AJ. 1992. Evidence that the amantadine-induced, M2-mediated conversion of influenza A virus hemagglutinin to the low pH conformation occurs in an acidic trans Golgi compartment. Virology 188:14–24. doi: 10.1016/0042-6822(92)90730-D. [DOI] [PubMed] [Google Scholar]
- 7.Sakaguchi T, Leser GP, Lamb RA. 1996. The ion channel activity of the influenza virus M2 protein affects transport through the Golgi apparatus. J Cell Biol 133:733–747. doi: 10.1083/jcb.133.4.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeuchi K, Lamb RA. 1994. Influenza virus M2 protein ion channel activity stabilizes the native form of fowl plague virus hemagglutinin during intracellular transport. J Virol 68:911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grambas S, Hay AJ. 1992. Maturation of influenza A virus hemagglutinin—estimates of the pH encountered during transport and its regulation by the M2 protein. Virology 190:11–18. doi: 10.1016/0042-6822(92)91187-Y. [DOI] [PubMed] [Google Scholar]
- 10.McKay TM, Patel M, Pickles RJ, Johnson LG, Olsen JC. 2006. Influenza M2 envelope protein augments avian influenza hemagglutinin pseudotyping of lentiviral vector. Gene Ther 13:715–724. doi: 10.1038/sj.gt.3302715. [DOI] [PubMed] [Google Scholar]
- 11.Ohuchi M, Cramer A, Vey M, Ohuchi R, Garten W, Klenk HD. 1994. Rescue of vector-expressed fowl plague virus hemagglutinin in biologically active form by acidotropic agents and coexpressed M2 protein. J Virol 68:920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinhauer DA, Wharton SA, Skehel JJ, Wiley DC, Hay AJ. 1991. Amantadine selection of a mutant influenza virus containing an acid-stable hemagglutinin glycoprotein: evidence for virus-specific regulation of the pH of glycoprotein transport vesicles. Proc Natl Acad Sci U S A 88:11525–11529. doi: 10.1073/pnas.88.24.11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey R, Martin AC, Zambon M, Barclay WS. 2004. Restrictions to the adaptation of influenza A virus H5 hemagglutinin to the human host. J Virol 78:502–507. doi: 10.1128/JVI.78.1.502-507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossman JS, Jing X, Leser GP, Lamb RA. 2010. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossman JS, Lamb RA. 2011. Influenza virus assembly and budding. Virology 41:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen BJ, Leser GP, Jackson D, Lamb RA. 2008. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol 82:10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossman JS, Jing X, Leser GP, Balannik V, Pinto LH, Lamb RA. 2010. Influenza virus M2 ion channel protein is necessary for filamentous virion formation. J Virol 84:5078–5088. doi: 10.1128/JVI.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W, Castelán-Vega JA, Jiménez-Alberto A, Vassell R, Ye Z, Weiss CD. 2010. A mutation in the receptor binding site enhances infectivity of 2009 H1N1 influenza hemagglutinin pseudotypes without changing antigenicity. Virology 407:374–380. doi: 10.1016/j.virol.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 19.Farnsworth A, Cyr TD, Li C, Wang J, Li X. 2011. Antigenic stability of H1N1 pandemic vaccines correlates with vaccine strain. Vaccine 29:1529–1533. doi: 10.1016/j.vaccine.2010.12.120. [DOI] [PubMed] [Google Scholar]
- 20.Feshchenko E, Rhodes DG, Felberbaum R, McPherson C, Rininger JA, Post P, Cox MM. 2012. Pandemic influenza vaccine: characterization of A/California/07/2009 (H1N1) recombinant hemagglutinin protein and insights into H1N1 antigen stability. BMC Biotechnol 12:77. doi: 10.1186/1472-6750-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang H, Chang JC, Guo Z, Carney PJ, Shore DA, Donis RO, Cox NJ, Villanueva JM, Klimov AI, Stevens J. 2014. Structural stability of influenza A(H1N1)pdm09 virus hemagglutinins. J Virol 88:4828–4838. doi: 10.1128/JVI.02278-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang W, Anderson CM, De Feo CJ, Zhuang M, Yang H, Vassell R, Xie H, Ye Z, Scott D, Weiss CD. 2011. Cross-neutralizing antibodies to pandemic 2009 H1N1 and recent seasonal H1N1 influenza A strains influenced by a mutation in hemagglutinin subunit 2. PLoS Pathog 7:e1002081. doi: 10.1371/journal.ppat.1002081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 24.Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 25.Wang W, De Feo CJ, Zhuang M, Vassell R, Weiss CD. 2011. Selection with a peptide fusion inhibitor corresponding to the first heptad repeat of HIV-1 gp41 identifies two genetic pathways conferring cross-resistance to peptide fusion inhibitors corresponding to the first and second heptad repeats (HR1 and HR2) of gp41. J Virol 85:12929–12938. doi: 10.1128/JVI.05391-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, Jobbagy Z, Bird TH, Eiden MV, Anderson WB. 2005. Cell signaling through the protein kinases cAMP-dependent protein kinase, protein kinase Cepsilon, and RAF-1 regulates amphotropic murine leukemia virus envelope protein-induced syncytium formation. J Biol Chem 280:16772–16783. doi: 10.1074/jbc.M411537200. [DOI] [PubMed] [Google Scholar]
- 27.Guibinga GH, Hall FL, Gordon EM, Ruoslahti E, Friedmann T. 2004. Ligand-modified vesicular stomatitis virus glycoprotein displays a temperature-sensitive intracellular trafficking and virus assembly phenotype. Mol Ther 9:76–84. doi: 10.1016/j.ymthe.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 28.Holland AU, Munk C, Lucero GR, Nguyen LD, Landau NR. 2004. Alpha-complementation assay for HIV envelope glycoprotein-mediated fusion. Virology 319:343–352. doi: 10.1016/j.virol.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 29.Schmeisser F, Friedman R, Besho J, Lugovtsev V, Soto J, Wang W, Weiss C, Williams O, Xie H, Ye Z, Weir JP. 2013. Neutralizing and protective epitopes of the 2009 pandemic influenza H1N1 hemagglutinin. Influenza Other Respir Viruses 7:480–490. doi: 10.1111/irv.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W, Butler EN, Veguilla V, Vassell R, Thomas JT, Moos M Jr, Ye Z, Hancock K, Weiss CD. 2008. Establishment of retroviral pseudotypes with influenza hemagglutinins from H1, H3, and H5 subtypes for sensitive and specific detection of neutralizing antibodies. J Virol Methods 153:111–119. doi: 10.1016/j.jviromet.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 31.Rossi F, Charlton CA, Blau HM. 1997. Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc Natl Acad Sci U S A 94:8405–8410. doi: 10.1073/pnas.94.16.8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skehel JJ, Bayley PM, Brown EB, Martin SR, Waterfield MD, White JM, Wilson IA, Wiley DC. 1982. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc Natl Acad Sci U S A 79:968–972. doi: 10.1073/pnas.79.4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruigrok RW, Cremers AF, Beyer WE, de Ronde-Verloop FM. 1984. Changes in the morphology of influenza particles induced at low pH. Arch Virol 82:181–194. doi: 10.1007/BF01311162. [DOI] [PubMed] [Google Scholar]
- 34.Boulay F, Doms RW, Wilson I, Helenius A. 1987. The influenza hemagglutinin precursor as an acid-sensitive probe of the biosynthetic pathway. EMBO J 6:2643–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carr CM, Chaudhry C, Kim PS. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc Natl Acad Sci U S A 94:14306–14313. doi: 10.1073/pnas.94.26.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vanderlinden E, Goktas F, Cesur Z, Froeyen M, Reed ML, Russell CJ, Cesur N, Naesens L. 2010. Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J Virol 84:4277–4288. doi: 10.1128/JVI.02325-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu R, Wilson IA. 2011. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J Virol 85:5172–5182. doi: 10.1128/JVI.02430-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nayak DP, Balogun RA, Yamada H, Zhou ZH, Barman S. 2009. Influenza virus morphogenesis and budding. Virus Res 143:147–161. doi: 10.1016/j.virusres.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwatsuki-Horimoto K, Horimoto T, Noda T, Kiso M, Maeda J, Watanabe S, Muramoto Y, Fujii K, Kawaoka Y. 2006. The cytoplasmic tail of the influenza A virus M2 protein plays a role in viral assembly. J Virol 80:5233–5240. doi: 10.1128/JVI.00049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holsinger LJ, Nichani D, Pinto L, Lamb RA. 1994. Influenza A virus M2 ion channel protein: a structure-function analysis. J Virol 68:1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C, Takeuchi K, Pinto LH, Lamb RA. 1993. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J Virol 67:5585–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schnell JR, Chou JJ. 2008. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML. 2010. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc Natl Acad Sci U S A 107:15075–15080. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. 2008. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Astrahan P, Arkin IT. 2011. Resistance characteristics of influenza to amino-adamantyls. Biochim Biophys Acta 1808:547–553. doi: 10.1016/j.bbamem.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 46.Balannik V, Carnevale V, Fiorin G, Levine BG, Lamb RA, Klein ML, Degrado WF, Pinto LH. 2010. Functional studies and modeling of pore-lining residue mutants of the influenza A virus M2 ion channel. Biochemistry 49:696–708. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kolocouris A, Tzitzoglaki C, Johnson FB, Zell R, Wright AK, Cross TA, Tietjen I, Fedida D, Busath DD. 2014. Aminoadamantanes with persistent in vitro efficacy against H1N1 (2009) influenza A. J Med Chem 57:4629–4639. doi: 10.1021/jm500598u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abed Y, Goyette N, Boivin G. 2005. Generation and characterization of recombinant influenza A (H1N1) viruses harboring amantadine resistance mutations. Antimicrob Agents Chemother 49:556–559. doi: 10.1128/AAC.49.2.556-559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barr IG, Deng YM, Iannello P, Hurt AC, Komadina N. 2008. Adamantane resistance in influenza A(H1) viruses increased in 2007 in South East Asia but decreased in Australia and some other countries. Antiviral Res 80:200–205. doi: 10.1016/j.antiviral.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 50.Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, Zhang Y, Shu Y, Gubareva LV, Cox NJ, Klimov AI. 2007. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis 196:249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 51.Saito R, Suzuki Y, Li D, Zaraket H, Sato I, Masaki H, Kawashima T, Hibi S, Sano Y, Shobugawa Y, Oguma T, Suzuki H. 2008. Increased incidence of adamantane-resistant influenza A(H1N1) and A(H3N2) viruses during the 2006–2007 influenza season in Japan. J Infect Dis 197:630–632. doi: 10.1086/525055. [DOI] [PubMed] [Google Scholar]
- 52.Saito R, Sakai T, Sato I, Sano Y, Oshitani H, Sato M, Suzuki H. 2003. Frequency of amantadine-resistant influenza A viruses during two seasons featuring cocirculation of H1N1 and H3N2. J Clin Microbiol 41:2164–2165. doi: 10.1128/JCM.41.5.2164-2165.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng PK, Leung TW, Ho EC, Leung PC, Ng AY, Lai MY, Lim WW. 2009. Oseltamivir- and amantadine-resistant influenza viruses A (H1N1). Emerg Infect Dis 15:966–968. doi: 10.3201/eid1506.081357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Askal HF, Khedr AS, Darwish IA, Mahmoud RM. 2008. Quantitative thin-layer chromatographic method for determination of amantadine hydrochloride. Int J Biomed Sci 4:155–160. [PMC free article] [PubMed] [Google Scholar]
- 55.Jing X, Ma C, Ohigashi Y, Oliveira FA, Jardetzky TS, Pinto LH, Lamb RA. 2008. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc Natl Acad Sci U S A 105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stouffer AL, Ma C, Cristian L, Ohigashi Y, Lamb RA, Lear JD, Pinto LH, DeGrado WF. 2008. The interplay of functional tuning, drug resistance, and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure 16:1067–1076. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Böttcher-Friebertshäuser E, Freuer C, Sielaff F, Schmidt S, Eickmann M, Uhlendorff J, Steinmetzer T, Klenk HD, Garten W. 2010. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J Virol 84:5605–5614. doi: 10.1128/JVI.00140-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galloway SE, Reed ML, Russell CJ, Steinhauer DA. 2013. Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: implications for host range and adaptation. PLoS Pathog 9:e1003151. doi: 10.1371/journal.ppat.1003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murakami S, Horimoto T, Ito M, Takano R, Katsura H, Shimojima M, Kawaoka Y. 2012. Enhanced growth of influenza vaccine seed viruses in Vero cells mediated by broadening the optimal pH range for virus membrane fusion. J Virol 86:1405–1410. doi: 10.1128/JVI.06009-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cotter CR, Jin H, Chen Z. 2014. A single amino acid in the stalk region of the H1N1pdm influenza virus HA protein affects viral fusion, stability and infectivity. PLoS Pathog 10:e1003831. doi: 10.1371/journal.ppat.1003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakowitsch S, Wolschek M, Morokutti A, Ruthsatz T, Krenn BM, Ferko B, Ferstl N, Triendl A, Muster T, Egorov A, Romanova J. 2011. Mutations affecting the stability of the haemagglutinin molecule impair the immunogenicity of live attenuated H3N2 intranasal influenza vaccine candidates lacking NS1. Vaccine 29:3517–3524. doi: 10.1016/j.vaccine.2011.02.100. [DOI] [PubMed] [Google Scholar]
- 62.Nakowitsch S, Waltenberger AM, Wressnigg N, Ferstl N, Triendl A, Kiefmann B, Montomoli E, Lapini G, Sergeeva M, Muster T, Romanova JR. 2014. Egg- or cell culture-derived hemagglutinin mutations impair virus stability and antigen content of inactivated influenza vaccines. Biotechnol J 9:405–414. doi: 10.1002/biot.201300225. [DOI] [PubMed] [Google Scholar]
- 63.Lin YP, Wharton SA, Martin J, Skehel JJ, Wiley DC, Steinhauer DA. 1997. Adaptation of egg-grown and transfectant influenza viruses for growth in mammalian cells: selection of hemagglutinin mutants with elevated pH of membrane fusion. Virology 233:402–410. doi: 10.1006/viro.1997.8626. [DOI] [PubMed] [Google Scholar]
- 64.Krenn BM, Egorov A, Romanovskaya-Romanko E, Wolschek M, Nakowitsch S, Ruthsatz T, Kiefmann B, Morokutti A, Humer J, Geiler J, Cinatl J, Michaelis M, Wressnigg N, Sturlan S, Ferko B, Batishchev OV, Indenbom AV, Zhu R, Kastner M, Hinterdorfer P, Kiselev O, Muster T, Romanova J. 2011. Single HA2 mutation increases the infectivity and immunogenicity of a live attenuated H5N1 intranasal influenza vaccine candidate lacking NS1. PLoS One 6:e18577. doi: 10.1371/journal.pone.0018577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reed ML, Yen HL, DuBois RM, Bridges OA, Salomon R, Webster RG, Russell CJ. 2009. Amino acid residues in the fusion peptide pocket regulate the pH of activation of the H5N1 influenza virus hemagglutinin protein. J Virol 83:3568–3580. doi: 10.1128/JVI.02238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reed ML, Bridges OA, Seiler P, Kim JK, Yen HL, Salomon R, Govorkova EA, Webster RG, Russell CJ. 2010. The pH of activation of the hemagglutinin protein regulates H5N1 influenza virus pathogenicity and transmissibility in ducks. J Virol 84:1527–1535. doi: 10.1128/JVI.02069-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giannecchini S, Campitelli L, Calzoletti L, De Marco MA, Azzi A, Donatelli I. 2006. Comparison of in vitro replication features of H7N3 influenza viruses from wild ducks and turkeys: potential implications for interspecies transmission. J Gen Virol 87:171–175. doi: 10.1099/vir.0.81187-0. [DOI] [PubMed] [Google Scholar]
- 68.Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S, Li C, Kawakami E, Yamada S, Kiso M, Suzuki Y, Maher EA, Neumann G, Kawaoka Y. 2012. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486:420–428. doi: 10.1038/nature10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2012. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336:1534–1541. doi: 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O'Donnell CD, Vogel L, Matsuoka Y, Jin H, Subbarao K. 2014. The matrix gene segment destabilizes the acid and thermal stability of the hemagglutinin of pandemic live attenuated influenza virus vaccines. J Virol 88:12374–12384. doi: 10.1128/JVI.01107-14. [DOI] [PMC free article] [PubMed] [Google Scholar]