

Abstract
Electrospray ionization (ESI) in the negative ion mode was used to create anionic, gas-phase oxo-molybdenum complexes with dithiolene ligands. By varying ESI and ion transfer conditions, both doubly and singly charged forms of the complex, with identical formulas, could be observed. Collision-induced dissociation (CID) of the dianion generated exclusively the monoanion, while fragmentation of the monoanion involved decomposition of the dithiolene ligands. The intrinsic structure of the monoanion and the dianion were determined by using wavelength-selective infrared multiple-photon dissociation (IRMPD) spectroscopy and density functional theory calculations. The IRMPD spectrum for the dianion exhibits absorptions that can be assigned to (ligand) C=C, C–S, C—C≡N, and Mo=O stretches. Comparison of the IRMPD spectrum to spectra predicted for various possible conformations allows assignment of a pseudo square pyramidal structure with C2v symmetry, equatorial coordination of MoO2+ by the S atoms of the dithiolene ligands, and a singlet spin state. A single absorption was observed for the oxidized complex. When the same scaling factor employed for the dianion is used for the oxidized version, theoretical spectra suggest that the absorption is the Mo=O stretch for a distorted square pyramidal structure and doublet spin state. A predicted change in conformation upon oxidation of the dianion is consistent with a proposed bonding scheme for the bent-metallocene dithiolene compounds [Lauher, J. W.; Hoffmann, R. J. Am. Chem. Soc. 1976, 98, 1729−1742], where a large folding of the dithiolene moiety along the S···S vector is dependent on the occupancy of the in-plane metal d-orbital.
Introduction
Among the mononuclear molybdenum enzymes, the DMSO reductase family is the most diverse in terms of their structure and function. Members of this family are involved in global C, S, N, and As cycling; for example, formate dehydrogenase catalyzes the transformation of formate to CO2, DMSO reductase catalyzes the reduction of dimethyl sulfoxide (DMSO) to dimethyl sulfide (DMS), nitrate reductase catalyzes the reduction of nitrate to nitrite, and arsenite oxidase catalyzes the oxidation of arsenite to arsenate.1−4 These substrate transformations require transfer of two electrons, and during catalysis the Mo center shuttles between the +4 and +6 oxidation states. The catalytically competent Mo center is regenerated by two one-electron steps, thereby transiently passing though the +5 state. The different states of the metal center have been spectroscopically (e.g., EXAFS, EPR, and resonance Raman) characterized.5−9 In all cases, in the fully oxidized state, the Mo center is coordinated by two ene-dithiolate moieties from pyranopterin cofactors (Figure 1). In most cases, a terminal oxo group occupies the fifth coordination site, and depending on the enzyme, the Mo center is coordinated by additional ligands such as serine (in DMSO reductase), cysteine (in nitrate reductase), selenocysteine (in formate dehydrogenase), or hydroxide (in arsenite oxidase). These coordination modes have been confirmed by protein crystallography.10,11
Figure 1.
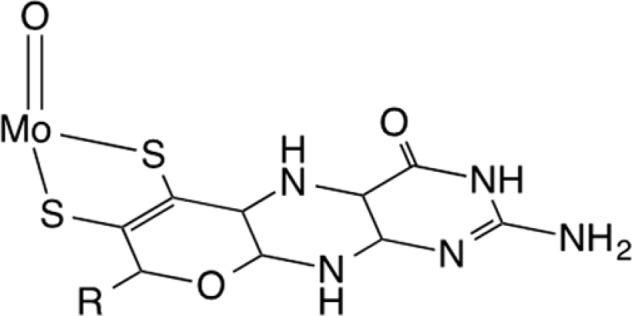
Mo center with one of two coordinating ene-dithiolate pyranopterin cofactors present in mononuclear Mo enzymes.
The spectroscopic and structural studies to date have provided the motivation to develop synthetic molecules with the {Mo(O)S4} core as a vehicle to understand the intricate details of the electronic structure of the enzymatic Mo center.12−15 The terminal oxo coordination provides a strong ligand field that orients the redox active dx2–y2 orbital in the equatorial plane, which implies that the ene-dithiolene moieties are involved in the electron transfer pathway to and from the Mo center. Mo compounds possessing a paramagnetic {MoVOS4} core have been used to probe S → Mo charge transfer transitions by a variety of spectroscopic approaches such as EPR, absorption, MCD, and resonance Raman spectroscopy.
Early work by Lauher and Hoffmann provided a bonding scheme for bent-metallocene dithiolene compounds,16 where a large folding of the dithiolene moiety along the S···S vector has been described. They proposed that the magnitude of the folding is dependent on the occupancy of the in-plane metal d-orbital; i.e., the more occupied the orbital is, the less folding is observed (Figure 2). Enemark and co-workers described the folding of the dithiolene ligand in oxo-Mo centers as a contributing factor to modulation of electron transfer in mononuclear molybdenum enzymes during the regeneration of the catalytically competent state.17−19 In this case, the redox orbital interacts with the symmetric combination of out-of-plane orbitals of the dithiolene sulfur.
Figure 2.
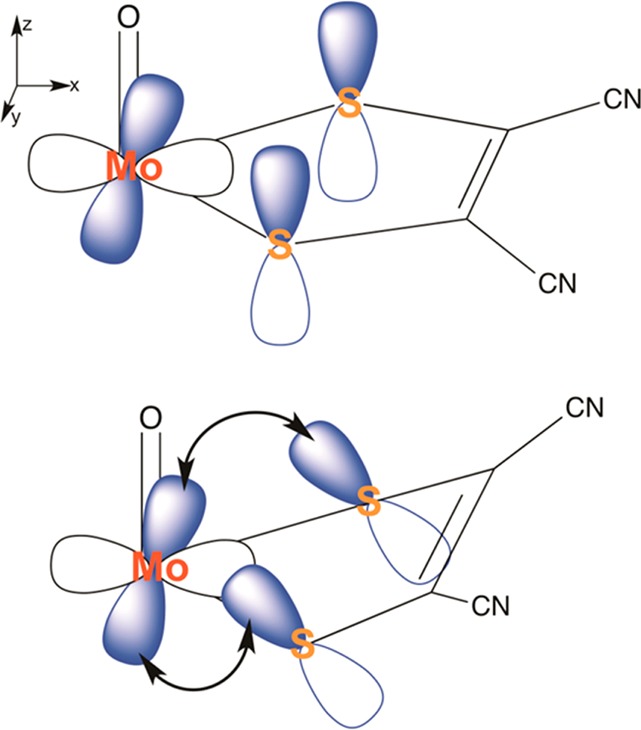
Depiction of ligand “folding” proposed to modulate electron transfer in mononuclear molybdenum enzymes.
The intrinsic propensity of folding of a dithiolene ligand as a function of the oxidation state of the metal can be measured best in discrete model oxo–metal–ligand complexes, without any influences from counterion, solvent, or other condensed-phase effects. Vibrational spectroscopy in the gas phase provides a means to study the intrinsic structure and behavior of metal–ligand complexes, and it is now well-known that infrared spectra of ionic species confined to the gas-phase environment of a mass spectrometer can be collected using the combination of tandem mass spectrometry and (wavelength-selective) infrared multiple-photon dissociation (IRMPD) spectroscopy [for reviews, see refs (20−26) and the literature cited therein]. Much of the work involving IRMPD spectroscopy and metal complexes has focused on probing the interaction(s) between cations and biologically relevant molecules.27−50 In many cases, the attention has been on the intrinsic structure and bonding interactions of inorganic and organometallic species.51−64
In this study we used electrospray ionization (ESI) to generate discrete, gas-phase anions from a model oxo-molybdenum complex with dithiolene ligands with formula [MoO(mnt)2]n−, where n = 1 or 2 (mnt2– = 1,2-dicyanoethylenedithiolate; Figure 2). The ions were then investigated in the gas phase using collision-induced dissociation (CID) and IRMPD. In the IRMPD spectroscopy experiment used in this work, gas-phase ions are irradiated at mid-IR wavelengths using a free electron laser (FEL). When the FEL wavelength matches the energy of a vibrational transition, absorption of multiple photons raises the vibrational energy of the trapped ion to the dissociation threshold. An IR spectrum is generated by measuring the fragmentation induced by this process as a function of photon wavelength. Vibrational mode assignment and structural determination is made with the assistance of frequencies predicted by density functional theory (DFT) or related computational methods.
Our primary goals in this study were (a) to use wavelength-selective infrared multiple-photon photodissociation spectroscopy to determine the structure of the dianionic complex [MoIVO(mnt)2]2– and (b) measure the intrinsic frequency of the Mo=O stretch within the complex. The results obtained will pave the way for more detailed investigations of the nature of metal–ligand interactions using a library of similar complexes designed to mimic the important oxo-transfer chemistry of the pyranopterin enzymes.
Experimental Methods
Collision-Induced-Dissociation (CID) Experiments
The tetraethylammonium salt of the molybdenum(IV) complex [Et4N]2[MoIVO(mnt)2] was synthesized using an established procedure described in detail elsewhere.65 Preliminary ESI and CID experiments were performed on a ThermoScientific (San Jose, CA) LTQ-XL linear ion trap mass spectrometer (MS) equipped with an Ion Max ESI source. For the ESI experiments, a stock solution (approximately 0.001 M) of the [MoIVO(mnt)2]2– complex as the tetraethylammonium salt was prepared in acetonitrile. The solution was infused into the ESI-MS instrument using the incorporated syringe pump at a flow rate of 10–15 μL/min. The atmospheric pressure ionization stack settings for the LTQ (lens voltages, quadrupole and octopole voltage offsets, etc.) were optimized for maximum transmission of singly or doubly charged anions to the ion trap mass analyzer by using the autotune routine within the LTQ Tune program. In general, harsher ESI and ion transmission conditions (i.e., higher tube lens and skimmer voltages) result in higher yields of the singly charged ion, which is presumably created by electron detachment from the dianion. Helium was used as the bath/buffer gas to improve trapping efficiency and as the collision gas for CID experiments.
Because of the complex isotopic pattern of Mo, precursor ions were isolated for the initial dissociation stage (MS/MS) using an isolation width of 0.9–1.2 mass to charge (m/z) units centered on the 98Mo isotopic peak. Product ions selected for subsequent CID (MS3 experiments) were isolated using slightly greater widths (1.2–1.5 m/z units) to improve trapping and fragmentation efficiency. For each stage, the exact width was chosen empirically to produce the best compromise between high precursor ion intensity and the ability to isolate a single isotopic peak. The (mass) normalized collision energy (NCE, as defined by ThermoScientific) was set between 25 and 35%, which corresponds to the application of roughly 0.55–0.68 V tickle voltage to the end-cap electrodes with the current instrument calibration. The activation Q, which defines the frequency of the applied rf potential, was set at 0.30. In all cases, the activation time employed was 30 ms. Spectra displayed represent the accumulation and averaging of at least 30 isolation, dissociation, and ejection/detection steps.
ESI FT-ICR Mass Spectrometry
As in the preliminary ESI studies, a stock solution (approximately 1.0 × 10–4 M) of the [MoO(mnt)2]2– complex was prepared in acetonitrile for the IRMPD experiments. Previously established methods used by our group for generation of ions and the subsequent collection of IRMPD spectra58−64 were used here. Briefly, ESI was performed using a Micromass (now a component of Waters Corp., Milford, MA) Z-Spray source in the negative ion mode. Dry nitrogen (∼80 °C) was used to assist in the desolvation process. Ions were injected into a home-built Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer described in detail elsewhere.66 Ions were accumulated for the duration of the previous FT-ICR cycle (6 s) in an external hexapole and injected into the ICR cell via a quadrupole deflector and an octapole rf ion guide. Instrument operating parameters, such as desolvation temperature, cone voltage, and ion accumulation and transfer optics voltages, were optimized to maximize the formation of either singly or doubly charged anions and transfer of the species to the ICR cell.
Infrared Multiple Photon Dissociation (IRMPD)
Infrared spectra were recorded by measuring the photodissociation yield as a function of photon wavelength. Precursor anions were irradiated using 23 FELIX macropulses (35 mJ/macropulse, 5 μs pulse duration, full width at half-maximum (fwhm) bandwidth ∼0.5% of central λ). In the IRMPD process, a photon is absorbed when the laser frequency matches a vibrational mode of the gas-phase ion and its energy is subsequently distributed over all vibrational modes by intramolecular vibrational redistribution (IVR). The IVR process allows the energy of each photon to be dissipated before the ion absorbs another, which leads to promotion of ion internal energy toward the dissociation threshold via multiple photon absorption.67 It is important to note that infrared spectra obtained using IRMPD are comparable to those collected using linear absorption techniques.68,69 For these experiments, the FEL wavelength was tuned between 5.7 and 14 μm in 0.03–0.1 μm increments. The intensities of product and undissociated precursor ions were obtained from an averaged mass spectrum measured using the excite/detect sequence of the FT-ICR-MS after each IRMPD step. The IRMPD yield was normalized to the total ion current.
DFT Geometry and Frequency Calculations
All DFT calculations were performed using the Gaussian 09 group of programs.70 Initial optimization of [MoIVO(mnt)2]2– and [MoVO(mnt)2]− was performed at the B3LYP/3-21G* level of theory using geometries in which dithiolene ligands were randomly arranged around a {MoOn+} core and coordination of the metal was by either the thiolate or cyano groups. Minima identified after the initial calculations were then subjected to full optimization using the same functional, effective core potential, and associated basis on Mo (MWB28) and the 6-311+G(d) basis set on C, N, O, and S.
To test the general consistency of relative rankings of energy for various minima, calculations were also performed with the 6-311+G(3df) basis set on C, N, O, and S. An exhaustive survey of models, functionals, and basis sets is beyond the scope of this investigation. However, to check general agreement between experiment and theory, bond lengths and vibrational frequencies were also calculated using the M06-L functional. The hybrid B3LYP functional, a standard in IRMPD investigations of gas-phase ions, is an approximation to the exchange–correlation energy functional which includes some portion of exact exchange from Hartree–Fock theory with exchange and correlation from other sources.71−74 M06-L is one of a group of meta-GGA functionals.75,76 We have found in prior studies of gas-phase metal complex thermochemistry that the M06 functional accurately reproduced trends in ligand exchange and addition to uranyl species.77 The M06-L is fully local, with no Hartree–Fock exchange, and is also reported to be effective for metal ions and inorganic and organometallic species.75
The DFT calculations performed here were primarily to assist with assignment of vibrational modes and determinations of intrinsic structures. Our intent was not to model fragmentation energetics or rigorously determine the differences in energy of possible spin states of the [MoO(mnt)2]n− complexes. Therefore, no corrections were made for possible errors in the energies for the respective species due to differences in spin–orbit coupling for [MoIVO(mnt)2]2– and [MoVO(mnt)2]−. In any case, any error that may be due to differences in spin–orbit coupling is expected to be minor78 (on the order of 200–600 cm–1), particularly when compared to the overall electronic energy differences between [MoIVO(mnt)2]2– and [MoVO(mnt)2]− or difference spin states for the latter species, and should not hinder the qualitative determination of the preference for a given geometry or spin state.
Scaling factors were chosen empirically by bringing the predicted stretching frequencies in the C=C stretch region (ca. 1460 cm–1) into agreement with the IRMPD spectrum. For a given functional/basis set combination, the same scaling factor, 0.96 at the B3LYP/6-311+G(d) level, was used for the spectra predicted for [MoIVO(mnt)2]2– and [MoVO(mnt)2]−.
Results and Discussion
ESI and CID of [MoIVO(mnt)2]2– and [MoVO(mnt)2]−
ESI mass spectrometry has been employed to investigate oxo-molybdenum complexes with or without dithiolene ligands. For example, Dessapt and co-workers studied the formation of novel Mo(V) dithiolene compounds created by adding alkynes to solutions of MoO2S22– in a mixture of MeOH and NH3.79 Llusar and co-workers reported a combined ESI mass spectrometry and DFT study of sulfur-based reactions in Mo2S74+ and Mo3S44+ clusters that included dithiolene ligands.80 The structure of a phosphine oxide bound intermediate molecule originating from a dioxo-molybdenum(VI) complex was investigated by ESI mass spectrometry and surface induced dissociation (SID).81 In addition, ESI mass spectrometry has been used in probing dynamics in oxygen atom transfer reactions.82,83 Important to our study is the fact that coupling of an ESI source to a photoelectron spectrometer84 has allowed a number of negatively charged inorganic species including a series of gas-phase oxo-molybdenum(V)-tetrathiolato and -bis(dithiolene) anions,85 and [MIVO(mnt)2]2– (M = Mo and W),86 to be investigated by photodetachment photoelectron spectroscopy.
ESI mass spectra generated in our study from [MoIVO(mnt)2]2– dissolved in acetonitrile are shown in Figure 3. In Figure 3a, the ESI and ion transfer conditions/voltages were tuned with the LTQTune program to maximize the intensity of the doubly charged anion (m/z 194–199), which was observed as the most abundant ion. Also observed was a species that corresponds to a tetraethylammonium adduct to the dianion at m/z 518–528. The inset in Figure 3a shows an expanded view of the high-resolution scan in the region of m/z 194–199, which reveals the isotopic distribution expected for Mo, and a peak spacing of 0.5 mass unit to confirm the assignment of the −2 charge state to the anion.
Figure 3.

ESI mass spectra generated from acetonitrile solution of [MoO(mnt)2]2– as the tetraethylammonium salt: (a) conditions optimized for production of doubly charged anion and (b) conditions retuned for optimized yield of the oxidized species [MoO(mnt)2]−. The insets are high-resolution scans to show the expected Mo isotope patterns for the two ions.
The spectrum in Figure 3b was generated by instead tuning the ESI and ion transfer voltages to optimize the yield of the singly charged anion (ca. m/z 394). Under these conditions, the singly charged anion was the dominant species in the ESI spectrum. Minor peaks (less than 10% relative intensity) were observed in the region of m/z 125–200 which may correspond to Mo with a mixture of oxygen and sulfur atoms. The resolving power and mass measurement accuracy is not sufficient to distinguish oxygen from sulfur coordination in these ions. In Figure 3b, other minor peaks in the region m/z 200–350 correspond to fragments of the monoanion. The tetraethylammonium adduct of the dianion (m/z 518–528) and a lower mass species at m/z 417–427 were also observed. CID (MS/MS stage, Figure S1a in the Supporting Information) of the species at m/z 524 generates the ion at m/z 423 through a neutral loss of 101 mass units (u). The product ion at m/z 423 in turn is 29 u higher in mass than the species at m/z 394. The loss of 101 u from the precursor at m/z 524 is consistent with elimination of triethyl amine, and transfer of an ethyl group to the MoO-dithiolene anion. Subsequent CID of the product ion at m/z 423 (MS3 stage, Figure S1b in the Supporting Information) furnishes the ion at m/z 394. It is not clear from the MS/MS experiments whether the ethyl group is transferred to the Mo metal center, or instead to a dithiolene ligand.
Our primary interest in this study were the doubly and singly charged forms of the MoO-dithiolene complex. CID (MS/MS stage, Figure S2a in the Supporting Information) of the peak at m/z 197 produces exclusively the singly charged anion at m/z 394, presumably through electron detachment from one of the dithiolene ligands. Similar behavior of similar doubly charged anions containing Mo and dithiolene ligands was reported by Llusar and co-workers.80 Subsequent CID (MS3 stage, Figure S2b in the Supporting Information) of the product ion at m/z 394 generated fragment ions at m/z 308, 312, 318, 330, and 366. The peak at m/z 366 is derived from a neutral loss of 28 u, which is attributed to elimination of CO by a mechanism that, while not clear, presumably involves decomposition of a dithiolene ligand and reaction with the oxygen atom of the MoO center. The loss of 64 u to generate the product ion at m/z 330 likely involves elimination of two S atoms. Likewise the fragment ion at m/z 318 may be generated by elimination of S2 and a C atom from the dithiolene ligand. The neutral losses associated with generation of the fragment ions at m/z 308 and 312 are consistent with elimination of S, CN, and CO (86 u) and S, C2, and CN (82 u), respectively. The apparent decomposition of the dithiolene ligands during CID warrants further study, and determination of the fragmentation pathways and their mechanisms would require isotope labeling.
DFT Calculations of Ion Structure
Potential structures for [MoIVO(mnt)2]2– and [MoVO(mnt)2]− are shown in Figure 4. Relative energies for the respective species are provided in Table 1. Initial optimization of [MoIVO(mnt)2]2– was done using a singlet spin state. Three minima were identified, structures I, III, and IV, which differed in coordination of MoIVO2+ by the dithiolene ligands. Structures in which the ligands coordinate the metal center through the cyano groups (structures III and IV), were investigated for the sake of completeness and were found to be ∼49–105 kcal/mol higher in energy at the B3LYP/6-311+G(d) level of theory than structure I (S coordination). Because the complexes with coordination by the cyano groups are unlikely for the mnt ligands and energetically noncompetitive, and the predicted IR spectra are in poor agreement with the IRMPD results described below, further investigation using other functional and basis set combinations was not pursued.
Figure 4.

Predicted structures for [MoO(mnt)2]2– and [MoO(mnt)2]−. Spin states and energies for the species are provided in Table 1. Energies shown in the figure are relative to structure I and are the result of calculations at the B3LYP/6-311+G(d) level of theory.
Table 1. Electronic Energies for [Mo(mnt)2]2– and [Mo(mnt)2]−a.
| [MoO(mnt)2]2– struct | spin state | functional/basis set | E | ZPE | E + ZPE | ΔE (kcal/mol) |
|---|---|---|---|---|---|---|
| I | singlet | B3LYP/6-31G(d) | –2260.251 154 | 0.068 537 | –2260.182 617 | 0.0 |
| II | triplet | B3LYP/6-31G(d) | –2260.193 877 | 0.066 698 | –2260.127 179 | 34.8 |
| I | singlet | B3LYP/6-311+G(d) | –2260.557 88 | 0.0679 84 | –2260.489 896 | 0.0 |
| II | triplet | B3LYP/6-311+G(d) | –2260.505 557 | 0.066 573 | –2260.438 984 | 31.9 |
| III | singlet | B3LYP/6-311+G(d) | –2260.479 474 | 0.067 545 | –2260.411 93 | 48.9 |
| IV | singlet | B3LYP/6-311+G(d) | –2260.390 217 | 0.066 893 | –2260.323 324 | 104.5 |
| I | singlet | B3LYP/6-311+G(3df) | –2260.627 36 | 0.068 287 | –2260.559 073 | 0.0 |
| II | triplet | B3LYP/6-311+G(3df) | –2260.573 471 | 0.066 631 | –2260.506 84 | 32.8 |
| I | singlet | M06-L/6-311+G(d) | –2260.428 426 | 0.068 133 | –2260.360 292 | 0.0 |
| II | triplet | M06-L/6-311+G(d) | –2260.380 189 | 0.066 237 | –2260.313 952 | 29.1 |
| [MoO(mnt)2]− struct | spin state | functional/basis set | E | ZPE | E + ZPE | ΔE (kcal/mol) |
|---|---|---|---|---|---|---|
| V | doublet | B3LYP/6-31G(d) | –2260.230 045 | 0.068 777 | –2260.161 268 | 13.4 |
| VI | quartet | B3LYP/6-31G(d) | –2260.159 497 | 0.067 211 | –2260.092 286 | 56.7 |
| V | doublet | B3LYP/6-311+G(d) | –2260.525 821 | 0.068 414 | –2260.457 407 | 20.4 |
| VI | quartet | B3LYP/6-311+G(d) | –2260.456 007 | 0.066 897 | –2260.389 11 | 63.2 |
| V | doublet | B3LYP/6-311+G(3df) | –2260.597 675 | 0.068 554 | –2260.529 121 | 18.8 |
| VI | quartet | B3LYP/6-311+G(3df) | –2260.526 069 | 0.067 013 | –2260.459 057 | 62.8 |
| V | doublet | M06-L/6-311+G(d) | –2260.405 617 | 0.068 364 | –2260.337 253 | 14.5 |
| VI | quartet | M06-L/6-311+G(d) | –2260.338 395 | 0.065 037 | –2260.273 358 | 54.6 |
The ΔE values are relative to the lowest energy structure for [Mo(mnt)2]2–.
Regardless of the functional or basis set used, the calculations performed here predict that the lowest energy structure for [MoIVO(mnt)2]2– is a pseudo square pyramidal conformation in which the oxo ligand occupies an axial position. The Mo atom lies slightly above the plane defined by the four (equatorial) S atoms of the dithiolene ligands. The experimental structural parameters for [MoIVO(mnt)2]2– from the Cambridge Structural Database, and the structural parameters computed in this work for both [MoIVO(mnt)2]2– and [MoVO(mnt)2]−, are given in Table 2. Dihedral angles for the square pyramidal structure, measured using Oaxial–Mo–Sligand–Cligand, are provided in Table S1 of the Supporting Information. The experimental Mo=O distances for [MoIVO(mnt)2]2– vary from 1.669 to1.714 Å, a range of 0.045 Å. The calculated distance of 1.701 Å matches well with the experimental data, with the maximum deviation from the experimental values being 0.032 Å. As expected, in the Mo(V) complex the calculated Mo=O distance shortens by 0.007 Å. Because of the symmetry all four Mo–S distances in [MoIVO(mnt)2]2– are equal. In the crystallographically determined structure of [MoIVO(mnt)2]2– the Mo–S distances vary from 2.370 to2.417 Å, a range of 0.047 Å. The calculated Mo–S distance in [MoIVO(mnt)2]2– is 2.443 Å, which is slightly longer than any of the experimental values with a maximum deviation from the experimental value of 0.07 Å. In the case of calculated C–S and C=C lengths for [MoIVO(mnt)2]2–, the maximum deviations from the experimental values are 0.086 and 0.060 Å, respectively, while the maximum deviation among the experimental values is 0.090 Å in both cases. For the fold angles in [MoIVO(mnt)2]2–, the experimental values range from 10.36 to 17.34°, a difference of 6.98°, while the maximum difference between the computational and experimental values is 4.49°. Optimized structures are dependent on many factors, such as the basis set and methodology used, e.g., ab initio, semiempirical, DFT, or molecular mechanics, while experimental structures may exhibit condensed-phase effects such as packing forces that result in deviations from gas-phase structures. While there are no well-defined criteria for comparing the “quality” of computed geometries, the differences in bond lengths and angles between the calculated and experimentally determined structures are similar to the variations observed among the experimental structures. We have discussed this issue in detail elsewhere.87,88
Table 2. Bond Distances and Fold Angles of [MoO(mnt)2]2– Complexes Determined by Crystallography,a and Calculated Distances and Fold Angles (Bottom Two Rows).
| distance (Å) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CSD ref codea | Mo=O | Mo–S1 | Mo–S2 | Mo–S3 | Mo–S4 | S1–C1 | S2–C3 | S3–C5 | S4–C7 | C1=C3 | C5=C7 | fold angle (deg) |
| HOGLIY | 1.67 | 2.393 | 2.386 | 2.378 | 2.389 | 1.78 | 1.72 | 1.73 | 1.75 | 1.343 | 1.345 | 11.53, 12.74 |
| HOGLIYO1 | 1.674 | 2.370 | 2.392 | 2.417 | 2.379 | 1.77 | 1.69 | 1.76 | 1.71 | 1.374 | 1.435 | 12.00, 11.91 |
| BAYNOFb | 1.714 | 2.373 | 2.376 | 2.373 | 2.377 | 1.753 | 1.742 | 1.760 | 1.750 | 1.343 | 1.354 | 17.34, 12.12 |
| ZOMJOA | 1.669 | 2.380 | 2.381 | 2.384 | 2.375 | 1.77 | 1.72 | 1.76 | 1.75 | 1.352 | 1.350 | 13.24, 10.84 |
| ZOMJUG | 1.71 | 2.38 | 2.38 | 2.38 | 2.38 | 1.758 | 1.744 | 1.76 | 1.74 | 1.366 | 1.366 | 12.97, 12.97 |
| ZOMKANa | 1.697 | 2.379 | 2.381 | 2.380 | 2.383 | 1.754 | 1.747 | 1.746 | 1.746 | 1.348 | 1.349 | 11.45, 12.59 |
| ZOMKANb | 1.692 | 2.389 | 2.383 | 2.382 | 2.381 | 1.763 | 1.757 | 1.756 | 1.732 | 1.343 | 1.349 | 12.39, 10.36 |
| [MoO(mnt)2]− | 1.694 | 2.425 | 2.425 | 2.436 | 2.436 | 1.767 | 1.767 | 1.758 | 1.758 | 1.374 | 1.372 | 29.88, 6.6 |
| [MoO(mnt)2]2– | 1.701 | 2.443 | 2.443 | 2.443 | 2.443 | 1.776 | 1.776 | 1.776 | 1.776 | 1.374 | 1.374 | 12.84, 12.86 |
From the Cambridge Structural Database on May 5, 2014.
Terminal oxo group is hydrogen bonded to a pyridinium group.
A search was also conducted for a minimum corresponding to [MoIVO(mnt)2]2– in a triplet spin state. Structure II was found using both the B3LYP and M06-L functionals. A change in dihedral angles to 108.6 and 99.24° reflects rotation of the dithiolene ligands such that a structure more reminiscent of a distorted trigonal bipyramid is created. DFT predicts the triplet state to be ∼32–34 kcal/mol higher in energy relative to the singlet state regardless of the functional or basis set used. However, we found that the triplet structure has one imaginary frequency, regardless of the level of theory or convergence criterion used, which indicates that it represents a saddle point rather than a true minimum. On the basis of this observation, we conclude that the most probable gas-phase conformation for [MoIVO(mnt)2]2– is pseudo square pyramidal, which is consistent with the local coordination environment in the Mo enzymes such as sulfite oxidase and members of the DMSO reductase family in catalytically competent Mo(IV) and Mo(V) states as revealed by X-ray absorption and EPR spectroscopies and X-ray crystal structures.5,6,11,89,90
Our results are in accord with more comprehensive calculations that were part of a photodetachment photoelectron spectroscopy study of the [MoIVO(mnt)2]2– by Wang, Wedd, and co-workers,86 in which the geometry of [MIVO(mnt)2]2– was optimized under constrained C2v point symmetry. Using their calculations, the highest occupied molecular orbitals (HOMOs) of [MIVO(mnt)2]2– were predicted to be of a1 symmetry and based upon the metal dx2–y2 orbital. The next six molecular orbitals are different linear combinations of ligand sulfur 3p atomic orbitals that are symmetry-adapted for interaction with M and O atomic orbitals, some also with contributions from C 2p atomic orbitals involved in the C=C bond of the dithiolene unit (π or π*).86
Using conventional dithiolene coordination in our calculations, optimized structures for doublet and quartet spin states were identified for the singly charged anion (structures V and VI, respectively, in Figure 4). The lowest energy structure corresponds to the doublet, which features a pseudo square pyramidal conformation similar to the doubly charged anion except for a change in the overall orientation of one dithiolene ligand, which results in conversion from C2v to Cs symmetry. This is in accord with the previous photoelectron spectroscopy study,86 in which the oxidized complex was modeled in a doublet state (considered the likely configuration based on spin selection rule, ΔS = ±1/2). The calculations suggested that the single unpaired electron in [MoVO(mnt)2] occupies the a1 dx2–y2-based molecular orbital that corresponds to the doubly occupied HOMO of the parent dianions.
In our study, we also considered the quartet configuration for [MoVO(mnt)2]−. As for the doubly charged anion, increasing the spin state of the singly charged anion resulted in a shift to a distorted trigonal-bipyramidal structure. Our calculations suggest that the quartet spin state is 43–45 kcal/mol less favorable than the doublet state when compared to the lowest energy structure for the doubly charged anion. As noted earlier, we assume that the differences in energy between the mono- and dianionic species are significantly greater than any error associated with differences in spin–orbit coupling.
Unlike [MoIVO(mnt)2]2–, no crystal structure data exists for [MoVO(mnt)2]−, thus preventing an assessment of the accuracy of the calculated structures in terms of bond lengths and angles and folding angle. The DFT calculations predict a change in fold angle with oxidation. As noted earlier, Lauher and Hoffmann presented a bonding scheme for bent-metallocene dithiolene compounds,16 and Enemark and co-workers later suggested that large folding of the dithiolene moiety along the S···S vector may be dependent on the occupancy of the in-plane metal d-orbital. The change in conformation predicted by our calculations likely represents the same type of ligand folding and is in accord with a change in conformation predicted in the photoelectron spectroscopy study,86 in which the Cs geometries with differentially folded dithiolene ligands were found to be favored by approximately 2 kcal/mol over the C2v structure. The reduction from C2v to Cs point symmetry apparently allows significant covalent interaction between the vacant metal-based dx2–y2 orbital and the highest energy S(π) orbital. The same orbitals cannot mix under C2v point symmetry.
IRMPD Spectroscopy
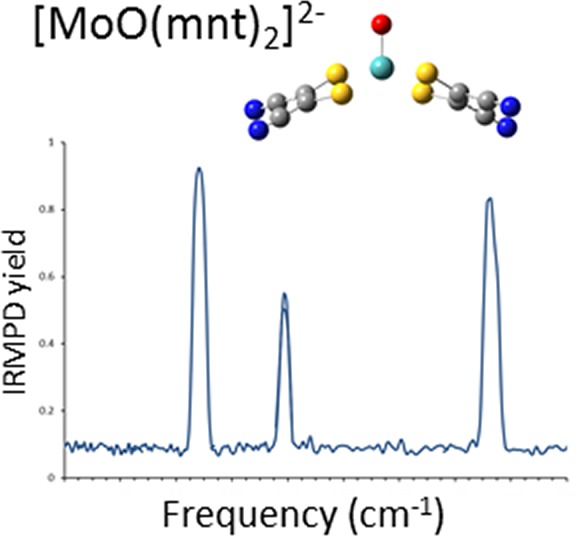
The IRMPD spectrum for [MoIVO(mnt)2]2– was generated by monitoring formation of [MoVO(mnt)2]− from electron detachment of [MoIVO(mnt)2]2– as a function of IR photon frequency and is shown in Figure 5a. The vibrational spectrum predicted for structure I, [MoIVO(mnt)2]2– with S coordination in the singlet state, is indicated by the dark trace in Figure 5b with the IRMPD spectrum included in gray to facilitate comparison. The spectrum obtained using the B3LYP functional and the 6-311+G(d) basis set on S, C, O, and N is used in Figure 5b because of the good qualitative and quantitative agreement with the IRMPD spectrum. Results obtained using the M06-L functional, and calculations at the B3LYP/6-311+G(3df) level of theory were in reasonable agreement with the experiment and are included in the Supporting Information.
Figure 5.

(a) Experimental IRMPD and (b) predicted spectra for [MoO(mnt)2]2–. Theoretical frequencies were generated at B3LYP/6-311+G(d) level of theory using a singlet spin state and are scaled by a factor of 0.96.
As noted earlier, the structure identified for the triplet state appears to be a saddle point rather than a true minimum, so consideration of its vibrational features here is not appropriate. Comparisons of the IRMPD spectrum of [MoIVO(mnt)2]2– to spectra predicted for structures III and IV, those with coordination by one or both dithiolene ligands through the cyano groups, are provided in Figure S3 of the Supporting Information. As noted earlier, structures III and IV are inconsistent with spectroscopic and crystallographic experiments in the condensed phase and are predicted to be significantly higher in energy by DFT.
Because of experimental limitations, the laser frequency could not be scanned into the IR region of the −C≡N stretch. However, three strong absorptions are observed in the IRMPD spectrum for [MoIVO(mnt)2]2– at 941, 1093, and 1464 cm–1. Based on inspection of the vibrational modes predicted by DFT, these absorptions can be assigned to the Mo=O stretch, antisymmetric C–C≡N stretch, and antisymmetric (between the two ligands) C=C stretch, respectively.
Two additional vibrations for [MoIVO(mnt)2]2– were predicted by DFT to appear at 838.5 and 966.9 cm–1, and these modes correspond to antisymmetric and symmetric C–S stretches, respectively. It is clear from the predicted spectrum shown in Figure 5b that both C–S stretches are of low intensity. The reason for the absence of these peaks in the (experimental) IRMPD spectrum is not clear. We note that, while DFT in general may predict accurately the positions of absorptions in the IRMPD experiment, the predictions of intensity are less accurate.91 This observation may be attributed in part to the approximate nature of the harmonic frequency calculations. In addition, an IRMPD action spectrum is not identical to a linear absorption spectrum. While it is assumed that a linear proportionality between IRMPD yield and IR absorption intensity is a useful approximation,69 caution should be exercised in interpreting IRMPD spectra by comparison with calculated linear absorption spectra. These difficulties may include general red shifts and broadening of bands and changes in the relative intensities of bands due to the presence of nearby bands.69 Both general effects may be the result of anharmonicities of the vibrational modes in combination with the large number of photons (i.e., typically tens to hundreds) that are absorbed in the IRMPD process.
The IRMPD spectrum of [MoVO(mnt)2]−, shown in Figure 6a, was collected by monitoring the loss of 28 and 76 u from [MoVO(mnt)2]− as a function of IR frequency (these fragmentation channels are consistent with those observed using CID). The spectrum for [MoVO(mnt)2]− contains only a single peak at 960.6 cm–1. Using the same scaling factor employed for the spectra predicted for [MoIVO(mnt)2]2–, the predicted frequencies for [MoVO(mnt)2]− are plotted in parts b and c of Figure 6 for the structures in the doublet and quartet spin states, respectively. With the scaling factor employed, there is good agreement between the position of the single absorption in the IRMPD spectrum and the position predicted for the Mo=O stretch in the oxidized complex with doublet spin state.
Figure 6.

(a) Experimental IRMPD and (b and c) predicted spectra for [MoO(mnt)2]−. Theoretical frequencies were generated at the B3LYP/6-311+G(d) level of theory and are scaled by a factor of 0.96. The predicted spectrum in (b) is for a doublet spin state, while the one in (c) is for a quartet spin state.
One explanation for the single absorption apparent in the spectrum of [MoVO(mnt)2]− is that the barriers for photodissociation may be sufficiently high that they are only accessible via the intense Mo–O stretch. A similar explanation was used in an earlier study of the photodissociation of strongly bound uranyl anion complexes.63 In particular, the spectrum generated by loss of OCH3 from uranyl methoxide contained only the antisymmetric uranyl peak, presumably because the energetics for the neutral loss were high and only accessed via the high-intensity ν3 uranyl absorption. We note that, in the present study, significantly higher relative collision energies were required to fragment the monoanion compared to the dianion, even when factoring in the difference in charge state. This would suggest that the barrier height to dissociation may play a role in determining the features of the IRMPD spectrum for [MoVO(mnt)2]−.
With a single peak in the IRMPD experiment, is not possible to unambiguously assign the structure for [MoVO(mnt)2]−. However, based on the general intensities of absorptions predicted for the quartet spin state (with the caveat about predicted intensities noted above), one would expect more features to have appeared in the IRMPD spectrum if the quartet had been generated. More importantly, a shift of the Mo=O stretching frequency of 20 cm–1 is predicted if one assumes oxidation to result in formation of the doublet state, in excellent agreement with the observed shift in the frequency in the experimental spectra. The shift to the blue by 20 cm–1 upon oxidation of the complex can then be rationalized by the decrease in donation by a dithiolene group to the metal center, thus strengthening the Mo=O bond.
Conclusions
To summarize, ESI in the negative ion mode was used to generate gas-phase doubly and singly charged anions of an oxo-molybdenym(IV) complex with dithiolene ligands. CID of the dianion generated exclusively the monoanion by electron detachment. Dissociation of the monoanion occurred through multiple pathways that include ligand elimination and ligand decomposition.
Attempts were made to determine the structures of both the dianion and monoanion using IRMPD spectroscopy and DFT calculations. The photodissociation experiments, with comparison to predicted vibrational patterns, strongly suggest that the gas-phase structure of the dianion is a distorted square planar configuration, with the oxo ligand occupying an axial position and equatorial coordination of Mo by the dithiolene ligands through sulfur atoms. The particular conformation is in excellent agreement with the structures of other model compounds for DMSO reductase (and related enzymes) active sites and molybdenum enzymes as revealed by spectroscopic and crystallography studies. One peak was observed in the IRMPD spectrum for the oxidized, monoanionic species, thus preventing an unequivocal assignment of structure to either the doublet or quartet state structures predicted for the ion. However, the single absorption may reflect the fact that the majority of vibrational modes predicted for the lower-energy doublet state are low intensity, and the barrier for photodissociation reactions for the ion may be high. In addition, based on the general intensities of absorptions predicted for the quartet spin state, one might expect more features to have appeared in the IRMPD spectrum if the quartet had been generated. We note that a shift of the Mo=O stretching frequency of 20 cm–1 is predicted by DFT if one assumes oxidation to result in formation of the doublet state, in excellent agreement with the observed shift in the frequency in the experimental spectrum. The quartet state is predicted to lie significantly higher in energy than the doublet, which also argues in favor of assignment of the latter as the spin state. Finally, there is strong evidence to support the doublet state with “folded” geometry that comes from analysis of molecular orbital energies revealed by measurements of vertical and adiabatic electron binding energies by photoelectron spectroscopy.86 We may therefore conclude that the peak in the IRMPD spectrum of [MoVO(mnt)2]− can be assigned to the Mo=O stretch of the complex in a doublet spin state. The present investigation suggests that the folding of the dithiolene ligand is present in the gas phase, and thus that it is intrinsic to metal–dithiolene moieties that modulate the reactivity of pyranopterin enzymes. In this way, our experimental results complement those of an earlier photodetachment photoelectron spectroscopy study of similar systems.86
Acknowledgments
M.J.V. acknowledges support for this work in the form of startup funding from Duquesne University and the Bayer School of Natural and Environmental Sciences, and the National Science Foundation (CHE-0963450). P.B. acknowledges the National Institutes of Health (GM 061555) for partial support of this research. The work of J.K.G. was fully supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Heavy Element Chemistry, at LBNL under Contract No. DE-AC02-05CH11231. J.O. acknowledges The Netherlands Organisation for Scientific Research (NWO) for Vici-Grant 724.011.002 and the Stichting Physica. Construction and shipping of the FT-ICR-MS was made possible through funding from the National High Field FT-ICR Facility (Grant CHE-9909502) at the National High Magnetic Field Laboratory, Tallahassee, FL. The excellent support by Dr. B. Redlich and others of the FELIX staff is gratefully acknowledged.
Supporting Information Available
Additional CID spectra generated from [MoO(mnt)2]2–, comparison of IRMPD and predicted spectra for structures III and IV, and comparisons of IRMPD and theoretical spectra for alternative functionals and basis sets. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hille R. The Mononuclear Molybdenum Enzymes. Chem. Rev. 1996, 96, 2757–2816. [DOI] [PubMed] [Google Scholar]
- Schwarz G.; Mendel R. R.; Ribbe M. W. Molybdenum Cofactors, Enzymes and Pathways. Nature (London) 2009, 460, 839–847. [DOI] [PubMed] [Google Scholar]
- Sparacino-Watkins C.; Stolz J. F.; Basu P. Nitrate and Periplasmic Nitrate Reductases. Chem. Soc. Rev. 2014, 43, 676–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enemark J. H.; Astashkin A. V.; Raitsimring A. M. Investigation of the Coordination Structures of the Molybdenum(V) Sites of Sulfite Oxidizing Enzymes by Pulsed EPR Spectroscopy. Dalton Trans. 2006, 3501–3514. [DOI] [PubMed] [Google Scholar]
- Hille R.; Hall J.; Basu P. The Mononuclear Molybdenum Enzymes. Chem. Rev. 2014, 114, 3963–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushie M. J.; George G. N. Spectroscopic Studies of Molybdenum and Tungsten Enzymes. Coord. Chem. Rev. 2011, 255, 1055–1084. [Google Scholar]
- Pushie M. J.; Cotelesage J. J.; George G. N. Molybdenum and Tungsten Oxygen Transferases—Structural and Functional Diversity Within a Common Active Site Motif. Metallomics 2014, 6, 15–24. [DOI] [PubMed] [Google Scholar]
- Johnson M. K.; Garton S. D.; Oku H. Resonance Raman as a Direct Probe for the Catalytic Mechanism of Molybdenum Oxotransferases. JBIC, J. Biol. Inorg. Chem. 1997, 2, 797–803. [Google Scholar]
- Kirk M. L. Magnetic Circular Dichroism Spectroscopy of Pyranopterin Molybdenum Enzymes. ACS Symp. Ser. 2003, 858, 340–357. [Google Scholar]
- Dobbek H. Structural Aspects of Mononuclear Mo/W-Enzymes. Coord. Chem. Rev. 2011, 255, 1104–1116. [Google Scholar]
- Romao M. J. Molybdenum and Tungsten Enzymes: A Crystallographic and Mechanistic Overview. Dalton Trans. 2009, 4053–4068. [DOI] [PubMed] [Google Scholar]
- Basu P.; Burgmayer S. J. N. Pterin Chemistry and its Relationship to the Molybdenum Cofactor. Coord. Chem. Rev. 2011, 255, 1016–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enemark J. H.; Cooney J. J. A.; Wang J.-J.; Holm R. H. Synthetic Analogues and Reaction Systems Relevant to the Molybdenum and Tungsten Oxotransferases. Chem. Rev. 2004, 104, 1175–1200. [DOI] [PubMed] [Google Scholar]
- Majumdar A.; Sarkar S. Bioinorganic Chemistry of Molybdenum and Tungsten Enzymes: A Structural-Functional Modeling Approach. Coord. Chem. Rev. 2011, 255, 1039–1054. [Google Scholar]
- Sugimoto H.; Tsukube H. Chemical Analogues Relevant to Molybdenum and Tungsten Enzyme Reaction Centres Toward Structural Dynamics and Reaction Diversity. Chem. Soc. Rev. 2008, 37, 2609–2619. [DOI] [PubMed] [Google Scholar]
- Lauher J. W.; Hoffmann R. Structure and Chemistry of Bis(cyclopentadienyl)-MLn Complexes. J. Am. Chem. Soc. 1976, 98, 1729–1742. [Google Scholar]
- Joshi H. K.; Cooney J. J. A.; Inscore F. E.; Gruhn N. E.; Lichtenberger D. L.; Enemark J. H. Investigation of Metal-Dithiolate Fold Angle Effects: Implications for Molybdenum and Tungsten Enzymes. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3719–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi H. K.; Enemark J. H. Geometrical Control of the Active Site Electronic Structure of Pyranopterin Enzymes by Metal-Dithiolate Folding: Aldehyde Oxidase. J. Am. Chem. Soc. 2004, 126, 11784–11785. [DOI] [PubMed] [Google Scholar]
- Wiebelhaus N. J.; Cranswick M. A.; Klein E. L.; Lockett L. T.; Lichtenberger D. L.; Enemark J. H. Metal-Sulfur Valence Orbital Interaction Energies in Metal-Dithiolene Complexes: Determination of Charge and Overlap Interaction Energies by Comparison of Core and Valence Ionization Energy Shifts. Inorg. Chem. 2011, 50, 11021–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlesse L.; Maitre P. Infrared Spectroscopy of Organometallic Ions in the Gas Phase: From Model to Real World Complexes. Mass Spectrom. Rev. 2007, 26, 583–605. [DOI] [PubMed] [Google Scholar]
- Dunbar R. C. Photodissociation of Trapped Ions. Int. J. Mass Spectrom. 2000, 200, 571–589. [Google Scholar]
- Duncan M. A. Frontiers in the Spectroscopy of Mass-Selected Molecular Ions. Int. J. Mass Spectrom. 2000, 200, 545–569. [Google Scholar]
- Polfer N. C.; Oomens J. Reaction Products in Mass Spectrometry Elucidated with Infrared Spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 3804–3817. [DOI] [PubMed] [Google Scholar]
- Polfer N. Infrared Multiple Photon Dissociation Spectroscopy of Trapped Ions. Chem. Soc. Rev. 2011, 40, 2211–2221. [DOI] [PubMed] [Google Scholar]
- Polfer N. C.; Oomens J. Vibrational Spectroscopy of Bare and Solvated Ionic Complexes of Biological Relevance. Mass Spectrom. Rev. 2009, 28, 468–494. [DOI] [PubMed] [Google Scholar]
- Eyler J. R. Infrared Multiple Photon Dissociation Spectroscopy of Ions in Penning Traps. Mass Spectrom. Rev. 2009, 28, 448–467. [DOI] [PubMed] [Google Scholar]
- Austin C. A.; Chen T.; Kaczan C. M.; Berden G.; Oomens J.; Rodgers M. T. Infrared Multiple Photon Dissociation Spectroscopy of Alkali Metal Cation-Cyclen Complexes: Effects of Alkali Metal Cation Size on Gas-Phase Conformation. Int. J. Mass Spectrom. 2013, 354–355, 346–355. [Google Scholar]
- Yang B.; Wu R. R.; Polfer N. C.; Berden G.; Oomens J.; Rodgers M. T. IRMPD Action Spectroscopy of Alkali Metal Cation-Cytosine Complexes: Effect of Alkali Metal Cation Size on Gas Phase Conformation. J. Am. Soc. Mass Spectrom. 2013, 24, 1523–1533. [DOI] [PubMed] [Google Scholar]
- Armentrout P. B.; Rodgers M. T.; Oomens J.; Steill J. D. Infrared Multiphoton Dissociation Spectroscopy of Cationized Serine: Effects of Alkali-Metal Cation Size on Gas-Phase Conformation. J. Phys. Chem. A 2008, 112, 2248–2257. [DOI] [PubMed] [Google Scholar]
- Rodgers M. T.; Armentrout P. B.; Oomens J.; Steill J. D. Infrared Multiple Photon Dissociation Spectroscopy of Cationized Threonine: Effects of Alkali-Metal Cation Size on Gas-Phase Conformation. J. Phys. Chem. A 2008, 112, 2258–2267. [DOI] [PubMed] [Google Scholar]
- Citir M.; Hinton C. S.; Oomens J.; Steill J. D.; Armentrout P. B. Infrared Multiple Photon Dissociation Spectroscopy of Cationized Histidine: Effects of Metal Cation Size on Gas-Phase Conformation. J. Phys. Chem. A 2012, 116, 1532–1541. [DOI] [PubMed] [Google Scholar]
- Hofstetter T. E.; Howder C.; Berden G.; Oomens J.; Armentrout P. B. Structural Elucidation of Biological and Toxicological Complexes: Investigation of Monomeric and Dimeric Comlexes of Histidine with Multiple Charged Transition Metal (Zn and Cd) Cations using IR Action Spectroscopy. J. Phys. Chem. B 2011, 115, 12648–12661. [DOI] [PubMed] [Google Scholar]
- Cooper T. E.; Carl D. R.; Oomens J.; Steill J. D.; Armentrout P. B. Infrared Spectroscopy of Divalent Zinc and Cadmium Crown Ether Systems. J. Phys. Chem. A 2011, 115, 5408–5422. [DOI] [PubMed] [Google Scholar]
- Dunbar R. C.; Berden G.; Oomens J. How Does a Small Peptide Choose How to Bind a Metal Ion? IRMPD and Computational Survey of CS versus Iminol Binding Preferences. Int. J. Mass Spectrom. 2013, 354–355, 356–364. [Google Scholar]
- Dunbar R. C.; Oomens J.; Berden G.; Lau J. K.-C.; Verkert U. H.; Hopkinson A. C.; Siu K. W. M. Metal Ion Complexes with HisGly: Comparison with PhePhe and PheGly. J. Phys. Chem. A 2013, 117, 5335–5343. [DOI] [PubMed] [Google Scholar]
- Dunbar R. C.; Polfer N. C.; Berden G.; Oomens J. Metal Ion Binding to Peptides: Oxygen or Nitrogen Sites?. Int. J. Mass Spectrom. 2012, 330–332, 71–77. [Google Scholar]
- Dunbar R. C.; Steill J. D.; Polfer N. C.; Oomens J. Metal Cation Binding to Gas-Phase Pentaalanine: Divalent Ions Restructure the Complex. J. Phys. Chem. A 2013, 117, 1094–1101. [DOI] [PubMed] [Google Scholar]
- Dunbar R. C.; Steill J. D.; Oomens J. Encapsulation of Metal Cations by the PhePhe Ligand: A Cation-π Ion Cage. J. Am. Chem. Soc. 2011, 133, 9376–9386. [DOI] [PubMed] [Google Scholar]
- Piccirillo S.; Ciavardini A.; Bodo E.; Rondino F.; Scuderi D.; Steinmetz V.; Paladini A. Probing the Competition among Different Coordination Motifs in Metal-Ciprofloxacin Complexes through IRMPD Spectroscopy and DFT Calculations. Inorg. Chem. 2013, 52, 103–112. [DOI] [PubMed] [Google Scholar]
- Chiavarino B.; Crestoni M. E.; Fornarini S.; Taioli S.; Mancini I.; Tosi P. Infrared Spectroscopy of Copper-Resveratrol Complexes: A Joint Experimental and Theoretical Study. J. Chem. Phys. 2012, 137, 024307. [DOI] [PubMed] [Google Scholar]
- Bellina B.; Compagnon I.; MacAlesse L.; Chirot F.; Lemoine J.; Maitre P.; Broyer M.; Antoine R.; Lukesza A.; Mitric R.; et al. Binding Motifs of Silver in Prion Octarepeat Model Peptides: A Joint Ion Mobility, IR and UV Spectroscopies, and Theoretical Approach. Phys. Chem. Chem. Phys. 2012, 14, 11433–11440. [DOI] [PubMed] [Google Scholar]
- Drayss M. K.; Blunk D.; Oomens J.; Gao B.; Wyttenbach T.; Bowers M. T.; Schaefer M. Systematic Study of the Structures of Potassiated Tertiary Amino Acids: Salt Bridge Structures Dominate. J. Phys. Chem. A 2009, 113, 9543–9550. [DOI] [PubMed] [Google Scholar]
- Lagutschenkov A.; Lorenz U. J.; Dopfer O. IR Spectroscopy of Isolated Metal-organic Complexes of Biocatalytic Interest: Evidence for Coordination Number Four for Zn2+(imidazole)4. Int. J. Mass Spectrom. 2011, 308, 316–329. [Google Scholar]
- Chakraborty S.; Dopfer O. Infrared Spectrum of the Ag+-(Pyridine)2 Ionic Complex: Probing Interactions in Artificial Metal-mediated Base Pairing. ChemPhysChem 2011, 12, 1999–2008. [DOI] [PubMed] [Google Scholar]
- Rajabi K.; Gillis E. A. L.; Fridgen T. D. Structure of Alkali Metal Ion-Adenine Complexes and Hydrated Complexes by IRMPD Spectroscopy and Electronic Structure Calculations. J. Phys. Chem. A 2010, 114, 3449–3456. [DOI] [PubMed] [Google Scholar]
- Bush M. F.; Oomens J.; Saykally R. J.; Williams E. R. Alkali Metal Ion Binding to Glutamine and Glutamine Derivatives Investigated by Infrared Action Spectroscopy and Theory. J. Phys. Chem. A 2008, 112, 8578–8584. [DOI] [PubMed] [Google Scholar]
- Prell J. S.; Flick T. G.; Oomens J.; Berden G.; Williams E. R. Coordination of Trivalent Metal Cations to Peptides: Results from IRMPD Spectroscopy and Theory. J. Phys. Chem. A 2010, 114, 854–860. [DOI] [PubMed] [Google Scholar]
- Bush M. F.; Oomens J.; Saykally R. J.; Williams E. R. Effects of Alkaline Earth Metal Ion Complexation on Amino Acid Zwitterion Stability: Results from Infrared Action Spectroscopy. J. Am. Chem. Soc. 2008, 130, 6463–6471. [DOI] [PubMed] [Google Scholar]
- Bush M. F.; Forbes M. W.; Jockusch R. A.; Oomens J.; Polfer N. C.; Saykally R. J.; Williams E. R. Infrared Spectroscopy of Cationized Lysine and ε-N-Methyllysine in the Gas-Phase: Effects of Alkali-Metal Ion Size and Proton Affinity on Zwitterion Stability. J. Phys. Chem. A 2007, 111, 7753–7760. [DOI] [PubMed] [Google Scholar]
- Lanucara F.; Scuderi D.; Chiavarino B.; Fornarini S.; Maitre P.; Crestoni M. E. IR Signature of NO Binding to a Ferrous Heme Center. J. Phys. Chem. Lett. 2013, 4, 2414–2417. [Google Scholar]
- Skriba A.; Jasikova L.; Rothova J. Silver(I) and Gold(I) Complexes of Diethyl Malonate. Int. J. Mass Spectrom. 2012, 330–332, 226–232. [Google Scholar]
- Brueckmann L.; Tyrra W.; Mathus S.; Berden G.; Oomens J.; Meiher A. J. H. M.; Schaefer M. Examination of the Coordination Sphere of Al(III) in Trifluoromethyl-Heteroarylalkenato Complex Ions by Gas-Phase IRMPD Spectroscopy and Computational Modelling. ChemPhysChem 2012, 13, 2037–2045. [DOI] [PubMed] [Google Scholar]
- Lagutschenkov A.; Sinha R. K.; Maitre P.; Dopfer O. Structure and Infrared Spectrum of the Ag+-Phenol Ionic Complex. J. Phys. Chem. A 2010, 114, 11053–11059. [DOI] [PubMed] [Google Scholar]
- Lagutschenkov A.; Springer A.; Lorenz U. J.; Maitre P.; Dopfer O. Structure of Zirconocene Complexes Relevant for Olefin Catalysis: Infrared Fingerprint of the Zn(C5H5)2(OH)(CH3CN)+ Cation in the Gas Phase. J. Phys. Chem. A 2010, 114, 2073–2079. [DOI] [PubMed] [Google Scholar]
- Bakker J. M.; Besson T.; Lemaire J.; Scuderi D.; Maitre P. Gas-phase Structure of a π-Allyl-Palladium Complex: Efficient Infrared Spectroscopy in a 7 T Fourier Transform Mass Spectrometer. J. Phys. Chem. A 2007, 111, 13415–13424. [DOI] [PubMed] [Google Scholar]
- Dunbar R. C.; Moore D. T.; Oomens J. IR-Spectroscopic Characterization of Acetophenone Complexes with Fe+, Co+ and Ni+ Using Free-Electron-Laser IRMPD. J. Phys. Chem. A 2006, 110, 8316–8326. [DOI] [PubMed] [Google Scholar]
- Moore D. T.; Oomens J.; Eyler J. R.; von Helden G.; Meijer G.; Dunbar R. C. Infrared Spectroscopy of Gas-Phase Cr+ Coordination Complexes: Determination of Binding Sites and Electronic States. J. Am. Chem. Soc. 2005, 127, 7243–7254. [DOI] [PubMed] [Google Scholar]
- Dain R. P.; Gresham G.; Groenewold G. S.; Steill J. D.; Oomens J.; Van Stipdonk M. J. Infrared Multiple Photon Dissociation Spectroscopy of Group I and Group II Metal Complexes with Boc-Hydroxylamine. Rapid Commun. Mass Spectrom. 2013, 27, 1867–1872. [DOI] [PubMed] [Google Scholar]
- Groenewold G. S.; van Stipdonk M. J.; Oomens J.; de Jong W. A.; McIlwain M. E. The Gas-Phase Bis-Uranyl Nitrate Complex [(UO2)2(NO3)5]−: Infrared Spectrum and Structure. Int. J. Mass Spectrom. 2011, 308, 175–180. [Google Scholar]
- Dain R. P.; Gresham G.; Groenewold G. S.; Steill J. D.; Oomens J.; van Stipdonk M. J. Infrared Multiple-Photon Dissociation Spectroscopy of Group II Metal Complexes With Salicylate. Rapid Commun. Mass Spectrom. 2011, 25, 1837–1846. [DOI] [PubMed] [Google Scholar]
- Groenewold G. S.; de Jong W. A.; Oomens J.; Van Stipdonk M. J. Variable Denticity in Carboxylate Binding to the Uranyl Coordination Complexes. J. Am. Soc. Mass Spectrom. 2010, 21, 719–727. [DOI] [PubMed] [Google Scholar]
- Groenewold G. S.; Oomens J.; de Jong W. A.; Gresham G. L.; McIlwain M. E.; Van Stipdonk M. J. Vibrational Spectroscopy of Anionic Nitrate Complexes of UO22+ and Eu3+ Isolated In The Gas Phase. Phys. Chem. Chem. Phys. 2008, 10, 1192–1202. [DOI] [PubMed] [Google Scholar]
- Groenewold G. S.; Gianotto A. K.; McIlwain M. E.; Van Stipdonk M. J.; Kullman M.; Moore D. T.; Polfer N.; Oomens J.; Infante I.; Visscher L.; et al. Infrared Spectroscopy of Discrete Uranyl Anion Complexes. J. Phys. Chem. A 2008, 112, 508–521. [DOI] [PubMed] [Google Scholar]
- Groenewold G. S.; Gianotto A. K.; Cossel K. C.; Van Stipdonk M. J.; Moore D. T.; Polfer N.; Oomens J.; de Jong W. A.; Visscher L. Vibrational Spectroscopy of Mass-Selected [UO2(ligand)n]2+ Complexes in the Gas Phase: Comparison with Theory. J. Am. Chem. Soc. 2006, 128, 4802–4813. [DOI] [PubMed] [Google Scholar]
- Das S. K.; Chaudhury P. K.; Biswas D.; Sarkar S. Modeling for the Active Site of Sulfite Oxidase: Synthesis, Characterization, and Reactivity of [MoVIO2(mnt)2]2– (mnt2– = 1,2-Dicyanoethylenedithiolate). J. Am. Chem. Soc. 1994, 116, 9061–9067. [Google Scholar]
- Valle J. J.; Eyler J. R.; Oomens J.; Moore D. T.; van der Meer A. F. G.; von Helden G.; Meijer G.; Hendrickson C. L.; Marshall A. G.; Blakney G. T. Free Electron Laser-Fourier Transform Ion Cyclotron Resonance Mass Spectrometry Facility for Obtaining Infrared Multiphoton Dissociation Spectra of Gaseous Ions. Rev. Sci. Instrum. 2005, 76, 023103. [Google Scholar]
- Bagratashvili V. N.; Letokov V. A.; Makarov A. A.; Ryabov E. A.. Multiple Photon Infrared Laser Photophysics and Photochemistry; Harwood: Chur, Switzerland, 1985. [Google Scholar]
- Moore D. T.; Oomens J.; Eyler J. R.; von Helden G.; Meijer G.; Dunbar R. C. Infrared Spectroscopy of Gas-Phase Cr+ Coordination Complexes: Determination of Binding Sites and Electronic States. J. Am. Chem. Soc. 2005, 127, 7243–7254. [DOI] [PubMed] [Google Scholar]
- Oomens J.; Tielens A. G. G. M.; Sartakov B. G.; von Helden G.; Meijer G. Laboratory Infrared Spectroscopy of Cationic Polycyclic Aromatic Hydrocarbon Molecules. Astrophys. J. 2003, 591, 968–985. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; et al. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula Into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- Vosko S. H.; Wilk L.; Nusair M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited states, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Zhao Y.; Truhlar D. G. Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States. J. Phys. Chem. A 2006, 110, 13126–13130. [DOI] [PubMed] [Google Scholar]
- Leavitt C. M.; Bryantsev V. S.; de Jong W. A.; Diallo M. S.; Goddard W. A. III; Groenewold G. S.; Van Stipdonk M. J. Addition of H2O and O2 to Acetone and Dimethylsulfoxide Ligated Uranyl(V) Dioxocations. J. Phys. Chem. A 2009, 113, 2350–2358. [DOI] [PubMed] [Google Scholar]
- Gunion R. F.; Dixon-Warren S. J.; Lineberger W. C.; Morse M. D. Ultraviolet Photoelectron Spectroscopy of Molybdenum and Molybdenum Monoxide Anions. J. Chem. Phys. 1996, 104, 1765–1773. [Google Scholar]
- Dessapt T.; Simonnet-Jegat C.; Mallard A.; Lavanant H.; Marrot J.; Secheresse F. Novel Mo(V)-dithiolene Compounds: Characterization of Nonsymmetric Dithiolene Complexes by Electrospray Ionization Mass Spectrometry. Inorg. Chem. 2003, 42, 6425–6431. [DOI] [PubMed] [Google Scholar]
- Llusa R.; Polo V.; Velez E.; Vicent C. Sulfur-based Redox Reactions in Mo3S74+ and Mo3S44+ Clusters Bearing Halide and 1,2-dithiolene Ligands: A Mass Spectrometric and Density Functional Theory Study. Inorg. Chem. 2010, 49, 8045–8055. [DOI] [PubMed] [Google Scholar]
- Nemykin V. N.; Laskin J.; Basu P. Isolation, Characterization of an Intermediate in an Oxygen Atom-Transfer Reaction, and the Determination of the Bond Dissociation Energy. J. Am. Chem. Soc. 2004, 126, 8604–8605. [DOI] [PubMed] [Google Scholar]
- Nemykin V. N.; Davie S. R.; Mondal S.; Rubie N.; Kirk M. L.; Somogyi A.; Basu P. An Analogue System Displaying All the Important Processes of the Catalytic Cycles Involving Monooxomolybdenum(VI) and Desoxomolybdenum(IV) Centers. J. Am. Chem. Soc. 2002, 124, 756–757. [DOI] [PubMed] [Google Scholar]
- Nemykin V. N.; Basu P. A Bifurcated Pathway of Oxygen Atom Transfer Reactions from a Monooxo Molybdenum(VI) Complex Under Electrospray Ionisation Mass Spectrometric Conditions. Dalton Trans. 2004, 1928–1933. [DOI] [PubMed] [Google Scholar]
- Wang L. S.; Ding C. F.; Wang X. B.; Barlow S. E. Photodetachment Photoelectron Spectroscopy of Multiply Charged Anions Using Electrospray Ionization. Rev. Sci. Instrum. 1999, 70, 1957–1966. [Google Scholar]
- Wang X.-B.; Inscore F. E.; Yang X.; Cooney J. J. A.; Enemark J. H.; Wang L.-S. Probing the Electronic Structure of [MoOS4]− Centers Using Anionic Photoelectron Spectroscopy. J. Am. Chem. Soc. 2002, 124, 10182–10191. [DOI] [PubMed] [Google Scholar]
- Waters T.; Wang X.-B.; Yang X.; Zhang L.; O’Hair R. A. J.; Wang L.-S.; Wedd A. G. Photoelectron Spectroscopy of the Doubly-Charged Anions [MIVO(mnt)2]2– (M = Mo, W; mnt = S2C2(CN)22–): Access to the Ground and Excited States of the [MVO(mnt)2]− Anion. J. Am. Chem. Soc. 2004, 126, 5119–5129. [DOI] [PubMed] [Google Scholar]
- Nemykin V. N.; Basu P. Comparative Theoretical Investigation of the Vertical Excitation Energies and the Electronic Structure of [MoVOCl4]−: Influence of Basis Set and Geometry. Inorg. Chem. 2003, 42, 4046–4056. [DOI] [PubMed] [Google Scholar]
- Hadt R. G.; Nemykin V. N.; Olsen J. G.; Basu P. Comparative Calculation of EPR Spectral Parameters in [MoVOX4]−, [MoVOX5]2–, and [MoVOX4(H2O)]− Complexes. Phys. Chem. Chem. Phys. 2009, 11, 10377–10384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romao M. J. Molybdenum and Tungsten Enzymes: A Crystallographic and Mechanistic Overview. Dalton Trans. 2009, 4053–4068. [DOI] [PubMed] [Google Scholar]
- Cramer S. P.; Gray H. B.; Dori Z.; Bino A. The Molybdenum Site of Sulfite Oxidase. Structural Information From X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 1979, 101, 2772–2774. [Google Scholar]
- Polfer N. C.; Oomens J.; Dunbar R. C. IRMPD Spectroscopy of Metal-ion/Tryptophan Complexes. Phys. Chem. Chem. Phys. 2006, 8, 2744–2751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.