

Abstract
Treating chronic kidney disease (CKD) has been challenging because of its pathogenic complexity. Epoxyeicosatrienoic acids (EETs) are cytochrome P-450-dependent derivatives of arachidonic acid with antihypertensive, anti-inflammatory, and profibrinolytic functions. We recently reported that genetic ablation of soluble epoxide hydrolase (sEH), an enzyme that converts EETs to less active dihydroxyeicosatrienoic acids, prevents renal tubulointerstitial fibrosis and inflammation in experimental mouse models of CKD. Here, we tested the hypothesis that pharmacological inhibition of sEH after unilateral ureteral obstruction (UUO) would attenuate tubulointerstitial fibrosis and inflammation in mouse kidneys and may provide a novel approach to manage the progression of CKD. Inhibition of sEH enhanced levels of EET regioisomers and abolished tubulointerstitial fibrosis, as demonstrated by reduced collagen deposition and myofibroblast formation after UUO. The inflammatory response was also attenuated, as demonstrated by decreased influx of neutrophils and macrophages and decreased expression of inflammatory cytokines keratinocyte chemoattractant, macrophage inflammatory protein-2, monocyte chemotactic protein-1, TNF-α, and ICAM-1 in kidneys after UUO. UUO upregulated transforming growth factor-β1/Smad3 signaling and induced NF-κB activation, oxidative stress, tubular injury, and apoptosis; in contrast, it downregulated antifibrotic factors, including peroxisome proliferator-activated receptor (PPAR) isoforms, especially PPAR-γ. sEH inhibition mitigated the aforementioned malevolent effects in UUO kidneys. These data demonstrate that pharmacological inhibition of sEH promotes anti-inflammatory and fibroprotective effects in UUO kidneys by preventing tubular injury, downregulation of NF-κB, transforming growth factor-β1/Smad3, and inflammatory signaling pathways, and activation of PPAR isoforms. Our data suggest the potential use of sEH inhibitors in treating fibrogenesis in the UUO model of CKD.
Keywords: chronic kidney disease, epoxyeicosatrienoic acid, peroxisome proliferator-activated receptor, soluble epoxide hydrolase, trans-4-{4-[3-(4-trifluoromethoxyphenyl)ureido]cyclohexyloxy}benzoic acid
chronic kidney disease (CKD) is progressive, incurable, and ultimately fatal (2). The incidence and prevalence of CKD is increasing, and the associated financial burden is overwhelming the United States healthcare system (21). Renal interstitial fibrosis and tubular atrophy are the final common pathways in CKD that lead to disease progression and, ultimately, end-stage renal disease (10). Current therapy directed at inhibiting the renin-angiotensin-aldosterone system can slow the progression but cannot prevent or cure end-stage renal disease, and, therefore, the development of novel approaches to treat CKD are required (12).
The eicosanoid metabolites of arachidonic acid are generated through the action of three classes of enzymes: cyclooxygenase, lipoxygenase, and cytochrome P-450 (CYP). Eicosanoids play important roles in physiology, including vasodilatory, anti-inflammatory, and antiapoptotic functions (9, 28, 58). CYP oxidases, mainly the CYP2C and CYP2J families, metabolize arachidonic acid into several products, including epoxyeicosatrienoic acids (EETs), by catalyzing the epoxidation of the olefinic bonds of arachidonic acid, resulting in the production of the following four regioisomeric EETs: 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET (25). Once formed, EETs are rapidly hydrated in vivo by epoxide hydrolases, primarily soluble epoxide hydrolase (sEH) in the cytosol, to their corresponding less potent diols, termed dihydroxyeicosatrienoic acids (DHETs). Therefore, sEH activity is a major determinant of EET bioavailability (68). Genetic deletion of sEH as well as its pharmacological inhibition increases EET levels in tissues and plasma, potentiates the effects of EETs, and thus elicits antihypertensive and anti-inflammatory effects (11, 27). In experimental models of diabetes, chronic administration of sEH inhibitors improves glucose homeostasis by increasing insulin release and sensitivity and also attenuates target organ damage (17, 26, 49). Inhibition of sEH has also been shown to decrease glomerular injury and renal inflammation in rodent models of angiotensin-induced and DOCA-salt-induced hypertension (45, 48, 51). Inhibition of the sEH enzyme has beneficial effects on cardiovascular disorders, including ischemia-reperfusion, heart failure, and atherosclerosis (26).
We have previously demonstrated that genetic ablation of sEH prevents inflammation and fibrogenesis in unilateral ureteral obstruction (UUO) and glomerulonephropathy models of CKD by increasing EET bioavailability (35). Given the pathophysiological role of the EET pathway, we tested the premise that pharmacological inhibition of sEH would prevent the inflammatory and fibrogenic response and thus provide a novel approach to manage the progression of the disease in the UUO model of CKD. In the present study, we characterized the efficacy of the highly potent sEH inhibitor trans-4-{4-[3-(4-trifluoromethoxyphenyl)ureido]cyclohexyloxy}benzoic acid [t-TUCB (64)] in modulating the inflammatory and fibrogenic response in the UUO kidney.
MATERIALS AND METHODS
UUO model.
Male C57BL/6J mice (age: 8–10 wk) were purchased from Jackson Laboratories (Bar Harbor, ME). All mouse experiments were performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center. UUO was conducted as previously reported (38, 39). Briefly, mice were anesthetized with an intraperitoneal injection of a cocktail containing ketamine (200 mg/kg body wt) and xylazine (16 mg/kg body wt). After exposure of the left kidney through a left flank incision, the left ureter was ligated completely near the kidney pelvis using a 5-0 silk tie. Sham-operated mice underwent the same surgical procedure without ureter ligation. For the pharmacological inhibition of sEH, t-TUCB [0.4 mg·mouse−1·day−1 (63, 65)] or vehicle (10% DMSO in 0.5% methylcellulose) was administered by oral gavage beginning 24 h before UUO. Currently, t-TUCB is an experimental tool. No side effects have been noted in >20 publications using t-TUCB and additional publications on its cis isomer. The compound is negative in Ames' assays and has been tested in scaling doses in equine medicine in vivo from 0.01 to 3 mg/kg iv with no side effects (20). It has been recently tested on hERG and CYP enzymes involved in EET synthesis and was found to have no side effects (44).
Renal blood flow measurement.
A laser-Doppler flowmeter (BLF-21D, Transonic Systems, Ithaca, NY) was used in conjunction with the fiber optic probe to detect perfusion in the renal cortex, providing a voltage output that is proportional to flow. After a 30-min stabilization period, the control value for cortical blood flow was assessed. Subsequently, UUO was induced, and, after a stabilization period, cortical renal blood flow was assessed at 10 min and 24 h after UUO in vehicle- and t-TUCB-treated mice, as previously described (31). Total renal blood flow was measured using ultrasound monitoring (Vevo 770 system, VisualSonics, Toronto, ON, Canada) at 30 min and 1 day after UUO, as previously described (4, 61).
Blood pressure measurement.
The systolic blood pressure of mice was measured by a noninvasive tail-cuff method (CODA, Kent Scientific, Torrington, CT). Mice were placed on a heated platform (30°C) in an isolated chamber, and systolic blood pressure levels were obtained.
Collagen deposition.
Collagen deposition was assessed by both Sirius red staining and hydroxyproline assay as previously described (38, 39). The area of positive Sirius red staining was measured in five randomly chosen high-power (×200 magnification) fields per kidney using ImageJ software (National Institutes of Health).
Immunohistochemistry, histology, and TUNEL assay.
Immunohistochemical staining of the kidneys was performed on paraffin-embedded sections as previously described (40). Briefly, 4% paraformaldehyde-fixed kidney sections were rehydrated and labeled with antibodies against α-smooth muscle actin (α-SMA; Sigma, St. Louis, MO), F4/80 (Abcam, Cambridge, MA), or polymorphonuclear neutrophils (PMNs; Accurate, Westbury, NY). Sections were then incubated with peroxidase-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA). The respective α-SMA-positive area and F4/80-positive area were measured in 10 randomly chosen high-power (×200 magnification) fields per kidney using ImageJ software. The number of PMN-positive cells was counted in 10 randomly chosen high-power (×200 magnification) fields per kidney. Periodic acid-Schiff-stained sections were used to determine the tubular injury score as previously described (37). Histological damage of tubular injury was scored by the percentage of tubules that displayed tubular necrosis, cast formation, and tubular dilation as follows: 0 = normal, 1 = <10%, 2 = 10–25%, 3 = 26–50%, 4 = 51–75%, and 5 = >75%. Ten randomly chosen high-power (×200 magnification) fields per kidney were used for the counting.
Western blot analysis.
We performed electrophoresis of protein extracts using Tris-glycine buffer systems and subsequent blotting as previously described (37, 39). Membranes were incubated with antibodies against α-SMA (Sigma), fibronectin (Cedarlane, Hornby, ON, Canada), phosphorylated (p-)Smad3, p-NF-κB p65 (p-p65), poly(ADP-ribose) polymerase 1 (PARP1), caspase-3 (Cell Signaling, Beverly, MA), poly(ADP-ribose) (PAR; BD Pharmingen, San Jose, CA), ICAM-1 (Santa Cruz Biotechnology, Santa Cruz, CA), TNF-α (Abcam), or sEH (Cayman). Peroxidase-conjugated secondary antibodies (Vector Laboratories) were applied, and a chemiluminescence reagent (Perkin-Elmer, Boston, MA) was used to detect proteins. Anti-β-actin antibody (Sigma) was used for loading controls on stripped membranes. Bands were quantified using Lab Works analysis software (Ultra-Violet Products, Cambridge, UK).
ELISA.
The ratios of EETs to DHETs in the kidneys were assessed using an EET/DHET ELISA kit (Detroit R&D, Detroit, MI). Levels of transforming growth factor (TGF)-β1, keratinocyte chemoattractant, macrophage inflammatory protein-2, and monocyte chemotactic protein-1 in the kidneys were measured using a multiplex immunoassay (Millipore, Bedford, MA). Activities of peroxisome proliferator-activated receptor (PPAR) isoforms in the kidneys were measured using the PPAR-α, -β/δ, and -γ transcription factor assay kit (Cayman). The level of lipid hydroperoxide in the kidneys was measured using a lipid hydroperoxide assay kit (Cayman).
Statistical analyses.
ANOVA was used to compare data among groups. Differences between two groups were assessed by a two-tailed unpaired Student's t-test. P values of <0.05 were considered statistically significant.
RESULTS
Pharmacological inhibition of sEH increases levels of EETs in UUO kidneys.
To assess the role of sEH during renal interstitial fibrogenesis in vivo, we used the UUO mouse model of obstructive nephropathy. UUO increased expression of sEH protein in a time-dependent fashion in mouse kidneys treated with vehicle or t-TUCB at 3 and 10 days after UUO (Fig. 1A). The ratio of EETs to DHETs was decreased in vehicle-treated kidneys at 10 days after UUO, but kidneys in mice treated with t-TUCB showed a significantly increased ratio of EETs to DHETs compared with that in mouse kidneys treated with vehicle (Fig. 1B). These target engagement data indicate that t-TUCB treatment is effective at inhibiting sEH catalytic activaty but not in regulating its expression level.
Fig. 1.
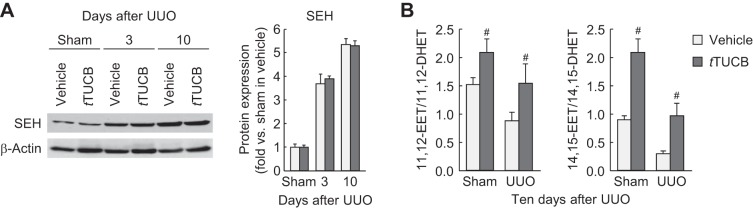
Unilateral ureteral obstruction (UUO) increases soluble epoxide hydrolase (sEH) expression and activity. A: expression of kidney sEH in vehicle- or trans-4-{4-[3-(4-trifluoromethoxyphenyl)ureido]cyclohexyloxy}benzoic acid (t-TUCB)-treated mice at 3 or 10 days after sham operation or UUO using Western blot analysis. Bands were quantified using Lab Works analysis software. B: sEH activity presented as the ratio of Epoxyeicosatrienoic acids (EETs) to dihydroxyeicosatrienoic acids (DHETs) in the kidneys using an EET/DHET ELISA kit. Values are means ± SD; n = 6. #P < 0.05 vs. vehicle.
sEH inhibition prevents renal fibrogenesis during UUO.
During UUO, vehicle-treated kidneys showed a time-dependent increase of collagen deposition, as evaluated by Sirius red-positive area and hydroxyproline level, whereas pharmacological inhibition of sEH markedly reduced collagen deposition (Fig. 2, A–C). UUO-induced tubulointerstitial expression of α-SMA, a marker of myofibroblast formation, was also diminished by sEH inhibition (Fig. 2, A and D). At 3 or 10 days after UUO, fibronectin and α-SMA expression in whole kidneys was also diminished by sEH inhibition (Fig. 2E). Since myofibroblast differentiation and activation by TGF-β signaling are critical events in renal fibrosis (7), we tested whether the TGF-β1 level was affected by sEH activation in UUO kidneys. The TGF-β1 level and its downstream signaling mediator p-Smad3 were significantly reduced in t-TUCB-treated kidneys at 3 and 10 days after UUO compared with those in vehicle-treated kidneys (Fig. 3, A and B).
Fig. 2.

sEH inhibition reduces interstitial fibrogenesis during UUO. A: collagen deposition detected by Sirius red stain and α-smooth muscle actin (α-SMA) expression stained with α-SMA monoclonal antibody on kidney sections in vehicle- or t-TUCB-treated mice at 10 days after sham operation or UUO. Scale bars = 50 μm. B: percentage of Sirius red-positive areas on kidney sections. C: collagen content represented by hydroxyproline level in kidneys. D: percentage of α-SMA-positive areas on kidney sections. E: profibrotic expression of α-SMA and fibronectin in kidneys using Western blot analysis. Bands were quantified using Lab Works analysis software. Values are means ± SD; n = 6. #P < 0.05 vs. vehicle.
Fig. 3.
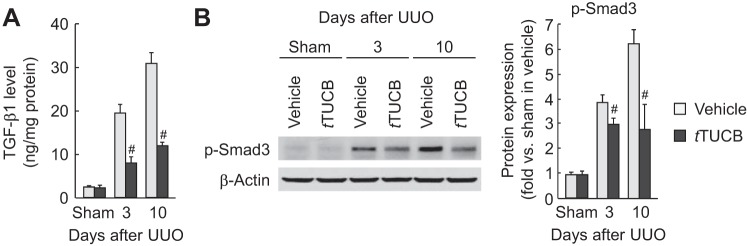
sEH inhibition reduces transforming growth factor (TGF)-β signaling during UUO. A: kidney TGF-β1 levels in vehicle- or t-TUCB-treated mice after sham operation or UUO were measured by a multiplex immunoassay. B: phosphorylated (p-)Smad3 in kidneys was analyzed by Western blot analysis. Bands were quantified using Lab Works analysis software. Values are means ± SD; n = 6. #P < 0.05 vs. vehicle.
sEH inhibition does not alter blood pressure or renal blood flow after UUO.
To determine whether the effects of t-TUCB are mediated through alterations in hemodynamics, blood pressure and renal blood flow were assessed. UUO decreased total and cortical renal blood flow, but treatment with t-TUCB did not significantly alter their levels before and after UUO (Fig. 4, A and B). Furthermore, systolic blood pressure assessed using a tail-cuff method was not different between groups over a period of 10 days (123.750 ± 6.585 mmHg in vehicle at 0 days after UUO, 120.625 ± 4.229 mmHg with t-TUCB at 0 days after UUO, 130.464 ± 8.560 mmHg with vehicle at 10 days after UUO, and 127.375 ± 12.613 mmHg with t-TUCB at 10 days after UUO). These data suggest that sEH activation contributes to tubulointerstitial fibrogenesis independent of renal hemodynamics and blood pressure.
Fig. 4.

sEH inhibition does not alter hemodynamics before and after UUO. Total (A) and cortical (B) renal blood flows were measured in vehicle- or t-TUCB-treated mice at 1 day before and after UUO using ultrasound monitoring (Vevo 770 system, VisualSonics, Toronto, NO, Canada) and laser-Doppler flowmetry, respectively. Values are means ± SD; n = 3.
sEH inhibition attenuates inflammation during UUO.
To determine whether sEH is implicated in renal inflammation in obstructive nephropathy, we next examined leukocyte influx and proinflammatory response in UUO kidneys. At 3 and 10 days after UUO, a prominent influx of both PMN-positive neutrophils and F4/80-positive macrophages occurred in vehicle-treated kidneys, but sEH inhibition significantly attenuated this influx in UUO kidneys (Fig. 5). Similarly, sEH inhibition decreased levels of keratinocyte chemoattractant, macrophage inflammatory protein-2, and monocyte chemotactic protein-1, potent leukocyte chemotactic factors upregulated by UUO (Fig. 6A). Since these chemotactic factors can be transactivated by the NF-κB transcription factor (55, 62, 66), we assessed NF-κB activation by quantifying the phosphorylation status of p-p65 in UUO kidneys. The level of p-p65 was significantly reduced in t-TUCB-treated kidneys during UUO compared with that in vehicle-treated kidneys (Fig. 6B). Among other NF-κB target gene proteins, expression levels of TNF-α and ICAM-1 were also attenuated by sEH inhibition during UUO (Fig. 6B). These data suggest that sEH upregulation exacerbates renal inflammation in an NF-κB-dependent manner during interstitial fibrogenesis.
Fig. 5.
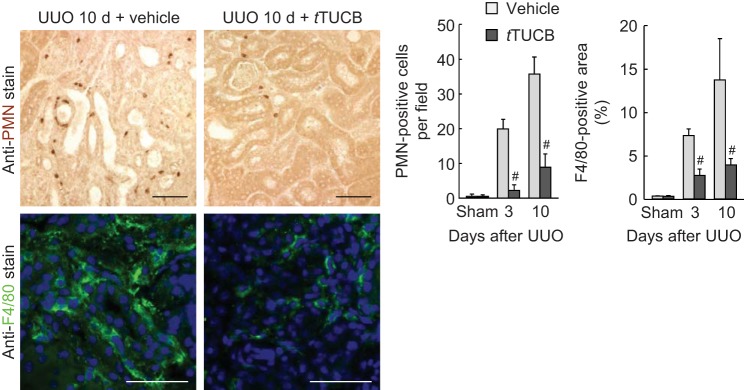
sEH inhibition attenuates the infiltration of neutrophils and macrophages during UUO. The number of polymorphonuclear neutrophil (PMN)-positive cells and percentage of F4/80-positive areas (right) on kidney sections (left) in vehicle- or t-TUCB-treated mice after sham operation or UUO using immunohistochemistry are shown. Values are means ± SD; n = 6. #P < 0.05 vs. vehicle.
Fig. 6.

sEH inhibition reduces inflammatory gene expression during UUO. A: level of keratinocyte chemoattractant (KC), macrophage inflammatory protein (MIP)-2, and monocyte chemotactic protein (MCP)-1 in kidneys in vehicle- or t-TUCB-treated mice after sham operation or UUO using a multiplex immunoassay. B: protein expression of p-p65, TNF-α, and ICAM-1 in kidneys using Western blot analysis. Bands were quantified using Lab Works analysis software. Values are means ± SD; n = 6. #P < 0.05 vs. vehicle.
sEH inhibition diminishes oxidative stress and cell death during UUO.
To explore whether sEH inhibition contributes to an antioxidative mechanism during UUO, we next evaluated lipid peroxidation in UUO kidneys with or without sEH inhibition. UUO increased lipid peroxidation, as represented by the increased level of lipid hydroperoxide in vehicle-treated kidneys at 3 and 10 days, whereas sEH inhibition ameliorated lipid peroxidation induced by UUO (Fig. 7A). Since inflammation and fibrogenesis can be initiated by cell death (54), we assessed tubular injury and cell death in UUO-subjected kidneys with or without sEH inhibition. The tubular injury score based on periodic acid-Schiff-stained kidney sections was increased at 3 and 10 days after UUO, but sEH inhibition significantly attenuated the score in UUO kidneys (Fig. 7B). sEH inhibition also reduced UUO-induced tubular cell death, including both necrosis, as demonstrated by PARP1-dependent increased PAR formation and PARP1 expression, and apoptosis, as demonstrated by cleaved PARP1 and caspase-3 expression (Fig. 7C). These data suggest that sEH activation and the resulting decline in epoxy fatty acids induces oxidative stress, leading to the progression of tubular cell death.
Fig. 7.

sEH inhibition suppresses tubular cell damage during UUO. A: lipid peroxidation indicated by the lipid hydroperoxide level in kidneys using a lipid hydroperoxide assay kit. B: periodic acid-Schiff (PAS) stain on kidney sections in vehicle- or t-TUCB-treated mice at 10 days after sham operation or UUO. Scale bars = 50 μm. The tubular injury score represented PAS stain in the kidneys. C: protein expression of poly(ADP-ribose) (PAR), poly(ADP-ribose) polymerase 1 (PARP1), cleaved PARP1, and cleaved caspase-3 in kidneys using Western blot analysis. Bands were quantified using Lab Works analysis software. Values are means ± SD; n = 6. #P < 0.05 vs. vehicle.
sEH inhibition induces PPAR activation during UUO.
PPAR isoforms play a protective role in renal interstitial fibrosis (22, 34). The activity of PPAR isoforms was time dependently decreased in vehicle-treated mouse kidneys after UUO, but sEH inhibition attenuated the decrease in activity of PPARs after UUO. PPAR-γ activity was reduced by UUO as early as 3 days and persisted at 10 days (Fig. 8), whereas PPAR-α and PPAR-β/γ were reduced only at 10 days after UUO. Inhibition of sEH significantly restored the activity of all PPAR isoforms at 10 days after UUO.
Fig. 8.
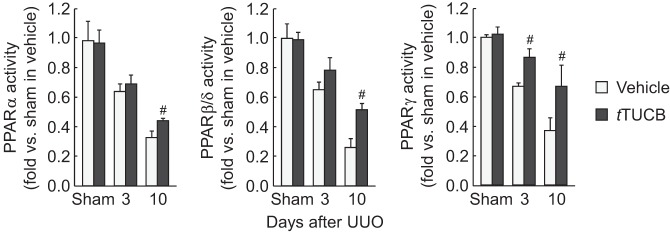
sEH inhibition restricts the reduction of peroxisome proliferator-activated receptor (PPAR) activity during UUO. Activities of PPAR isoforms in kidneys in vehicle- or t-TUCB-treated mice after sham operation or UUO using the PPAR-α, -β/δ, and -γ transcription factor assay kit are shown. Values are means ± SD; n = 6. #P < 0.05 vs. vehicle.
DISCUSSION
CKD is a major public health problem on a global scale, with enormous socioeconomic burden on families and society. Despite the desperate need for therapy, no effective treatment has been developed, mainly because of a lack of understanding of the complex pathophysiology of CKD. Regardless of the disease etiology, which includes hypertension and diabetes, tubulointerstitial fibrosis is the final common pathway in CKD that leads to disease progression and, ultimately, end-stage renal disease. The major features of tubulointerstitial fibrosis include an inflammatory cascade with infiltration of inflammatory cells and the release of inflammatory cytokines, oxidative stress, differentiation of different types of cells to myofibroblasts, deposition of extracellular matrix components, microvascular rarefaction, tubular injury, and atrophy (15, 23, 46). The major fibrogenic signaling molecule TGF-β is induced and released by the injured tubules and infiltrating cells (5, 6). Recently, TGF-β-independent signaling via other molecules, including NF-κB and ANG II, has also been implicated in renal fibrosis (30, 50, 57). However, the primary signaling pathways that activate these overlapping malevolent events and the consequent inflammatory response and fibrosis after the initial insult to the kidney remain undefined. Defining the primary signaling events may be required to design effective therapeutic strategies to prevent CKD at its onset.
We recently reported that genetic ablation of sEH could prevent the progression of renal failure in two experimental models of CKD, the UUO model and in glomerulonephropathy, by attenuating inflammation and interstitial fibrogenesis (36). In the present study, we tested the effect of pharmacological inhibition of sEH on the development of renal failure in the mouse UUO model using the highly potent sEH inhibitor t-TUCB. Our data indicate that pharmacological inhibition of sEH is equally beneficial to sEH deletion in preventing the progression of renal failure. Pharmacological inhibition of sEH increased EET levels in UUO kidneys. Furthermore, intrarenal levels of inflammatory cytokines, including TGF-β and NF-κB, chemokines, and the infiltration of both neutrophils and macrophages into the renal parenchyma, were significantly reduced. The fibrotic response was significantly attenuated, as shown by decreased collagen, fibronectin, and α-SMA deposition in UUO-induced kidneys. sEH inhibition also prevented histological damage, attenuated oxidative stress, decreased apoptosis, and significantly increased the activities of PPARs, especially the level of PPAR-γ. Collectively, these data suggest that increasing the levels of EETs inhibits a myriad of malevolent signaling events to prevent renal interstitial inflammation and fibrogenesis in the mouse UUO model.
The expression level and activity of sEH are increased in kidney proximal tubules, suggesting that EET levels and their protective effects may be modulated after UUO (18). Indeed, our data indicate that levels of EETs are decreased after UUO as a function of sEH, because its inhibition using t-TUCB significantly increased the levels of 11,12-EET and 14,15-EET regioisomers. Furthermore, the increases in the ratio of EETs to DHETs indicated the effectiveness of t-TUCB as a sEH inhibitor. EETs can elicit several physiological effects, including antioxidative functions, anti-inflammatory effects by limiting leukocyte adhesion, transmigration across the endothelium and inhibiting platelet aggregation, promoting fibrinolysis, and regulating angiogenesis (3). The precise signaling pathways transduced by EETs have not been defined because no receptors for EETs have been identified to date. However, it has been suggested that EETs may effect these functions by coupling to and activating ion channels and/or signaling proteins or transcription factors, including PPAR-γ (26).
Oxidative stress plays a major role in the pathophysiology of CKD by promoting inflammation and apoptotic signals, increasing matrix deposition, and inducing epithelial-to-myofibroblast transformation (60). Mitigation of ROS levels using a SOD mimetic or treatment with scavenging reagents attenuates renal fibrosis after UUO (13). In agreement with previous studies, our data show that UUO increases lipid peroxidation, a marker of oxidative stress. sEH inhibition attenuated oxidative stress, as shown by decreased lipid peroxidation after UUO. Oxidative stress and inflammation are tightly linked, and they can reciprocally induce each other. It is well established that ROS can induce oxidative damage to DNA and hyperactivation of PARP1 (40). PARP1 can activate the inflammatory transcription factor NF-κB by binding directly to its subunits p65 and p50. Our data indicate that pharmacological inhibition of sEH attenuates PARP1 and NF-κB activation as well as the inflammatory response in UUO-induced mouse kidneys, similar to that observed in sEH-deficient UUO-induced kidneys. These data are also in agreement with our previous report (38) showing that in the absence of PARP1, NF-κB-mediated inflammatory mediators TNF-α and ICAM-1, infiltration of inflammatory cells, and the production of cytokines are all attenuated, suggesting a PARP1-NF-κB-inflammation axis in the UUO-induced kidney. These data also agree with previous reports showing that sEH inhibition attenuated myocardial NF-κB activation in a model of cardiac hypertrophy (67) and in DOCA-salt-induced hypertension (51).
A variety of eicosanoids derived from arachidonic acid have been found to be ligands of PPAR-γ, including EETs (47). PPAR-γ is a negative regulator of profibrotic signaling and blocks matrix deposition and fibrogenesis in diabetic glomerulosclerosis (53) and CCl4-induced liver fibrosis (43). PPAR levels are downregulated in UUO kidneys, but sEH gene ablation or, as shown in the present study, its pharmacological inhibition restores levels of PPARs and attenuates UUO-induced fibrosis. It has previously been shown that the sEH-selective inhibitor adamantyl-ureido-dodecanoic acid increased PPAR-γ transcription activity in endothelial cells and 3T3-L1 preadipocytes (47). However, the molecular signaling pathways by which PPARs attenuate UUO-induced fibrosis are not well characterized. A role for PPAR-γ has been reported in regulating ROS levels in the hypothalamus in high fat diet-fed mice (14) and in LPS-induced pulpal inflammation (41). PPAR-γ has also been implicated in the modulation of TGF-β/Smad3 signaling. The PPAR-γ agonist troglitazone attenuates UUO-induced renal interstitial fibrosis and inflammation through suppression of TGF-β1 expression (33). Ligand-activated PPAR-γ prevents TGF-β-induced collagen synthesis via sequestration of the essential coactivator p300 from the TGF-β-induced p-Smad complex on the collagen gene promoter (19). It remains to be determined if EETs may attenuate ROS production, inflammatory responses, and/or TGF-β/Smad3 stimulation via PPAR-γ activation after UUO.
Hypertension is an important pathogenic factor in the progression of CKD. The role of sEH inhibition and EETs on its antihypertensive effects on renoprotection is under debate because blood pressure lowering has been demonstrated in some studies but not in others. There appear to be two major factors in this debate. In spontaneously hypertensive rats, ANG II-induced hypertension and DOCA-salt-induced and salt-sensitive hypertension models, sEH inhibition demonstrated antihypertensive effects (24, 29, 32, 48). However, the antihypertensive effect of sEH inhibitors is model dependent and was not observed in 5/6-nephrectomic mice (8), stroke-prone spontaneously hypertensive rats (16), and hypertensive Goto-Kakizaki rats (56). Can lowering blood pressure be one of the mechanisms by which sEH inhibition exerts renoprotective effects in models of hypertensive kidney injury? sEH inhibition was protective in animal models with lowered blood pressure (29, 52) as well as in those without lowered blood pressure (56, 59). The results of these studies suggest that sEH inhibitors can exert renoprotective effects independent of their blood pressure-lowering effects. A lack of effect of lowering blood pressure on renoprotection has also been observed in CKD patients. In both the African American Study of Kidney Disease and Hypertension and Modification of Diet in Renal Disease studies, blood pressure lowering to a mean arterial pressure of <92 mmHg was not effective in slowing the progression of CKD (1, 42). Our data in the present study also indicate that sEH inhibition was not effective in lowering blood pressure or altering renal blood flow but exerted renoprotective effects via its anti-inflammatory and antifibrotic functions.
Collectively, these studies demonstrate that the increased EETs present after pharmacological inhibition of sEH significantly attenuate histological damage, oxidative stress, and inflammatory responses, including reduced levels of inflammatory cytokines and chemokines, leukocyte infiltration, and adverse renal remodeling, in UUO kidneys. Our data demonstrate that EETs promote both anti-inflammatory and fibroprotective effects in UUO kidneys independent of their effects on blood pressure and renal blood flow and possibly through activation of PPAR-γ and downregulation of PARP1, NF-κB, and TGF-β1/Smad3 signaling pathways. Given the physiological and pathophysiological roles of the EET pathway, increasing EET bioavailability by inhibiting sEH or by other methods, including enhancing CYP epoxygenase expression and/or activity or administration of EET analogs, are possible strategies for preventing the progression of interstitial fibrogenesis and inflammation in CKD, including obstructive nephropathy.
GRANTS
This work was supported by American Heart Association Postdoctoral Fellowship 13POST16350005 (to J. Kim) and partially supported by National Institutes of Health (NIH) Grants R01-ES-02710 and P42-ES-04699 (to B. D. Hammock). B. J. Padanilam was supported by NIH Grant DK-083291, and J. D. Imig was supported by NIH Grant DK-38226.
DISCLOSURES
B. D. Hammock is a member of the EicOsis Animal Health, working to take t-TUCB to the clinic for a variety of indications. B. D. Hammock and S.-H. Hwang hold patents of sEH inhibitor synthesis and use through the University of California.
AUTHOR CONTRIBUTIONS
Author contributions: J.K. and B.J.P. conception and design of research; J.K. and S.P.Y. performed experiments; J.K., J.D.I., S.H.H., B.D.H., and B.J.P. analyzed data; J.K., M.L.T., J.D.I., S.H.H., B.D.H., and B.J.P. interpreted results of experiments; J.K. prepared figures; J.K. and B.J.P. drafted manuscript; J.K., M.L.T., J.D.I., B.D.H., and B.J.P. edited and revised manuscript; J.K., S.P.Y., M.L.T., J.D.I., S.H.H., B.D.H., and B.J.P. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Youngsu Cho for technical assistance with the Western blot analysis and immunohistochemistry and Kelly E. Long and Sherry N. Westphal for mouse care. The authors also thank to Dr. Erika I. Boesen and Dr. Irving H. Zucker for helping with ultrasound imaging experiments and Dr. Hanjun Wang for helping with laser-Doppler experiments.
REFERENCES
- 1.Appel LJ, Wright JT Jr, Greene T, Agodoa LY, Astor BC, Bakris GL, Cleveland WH, Charleston J, Contreras G, Faulkner ML, Gabbai FB, Gassman JJ, Hebert LA, Jamerson KA, Kopple JD, Kusek JW, Lash JP, Lea JP, Lewis JB, Lipkowitz MS, Massry SG, Miller ER, Norris K, Phillips RA, Pogue VA, Randall OS, Rostand SG, Smogorzewski MJ, Toto RD, Wang X; AASK Collaborative Research Group. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med 363: 918–929, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakris G, Vassalotti J, Ritz E, Wanner C, Stergiou G, Molitch M, Nesto R, Kaysen GA, Sowers JR. National Kidney Foundation consensus conference on cardiovascular and kidney diseases and diabetes risk: an integrated therapeutic approach to reduce events. Kidney Int 78: 726–736, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Bellien J, Joannides R. Epoxyeicosatrienoic acid pathway in human health and diseases. J Cardiovasc Pharmacol 61: 188–196, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Boesen EI, Crislip GR, Sullivan JC. Use of ultrasound to assess renal reperfusion and P-selectin expression following unilateral renal ischemia. Am J Physiol Renal Physiol 303: F1333–F1340, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor β1. Nature 346: 371–374, 1990. [DOI] [PubMed] [Google Scholar]
- 6.Borges FT, Melo SA, Ozdemir BC, Kato N, Revuelta I, Miller CA, Gattone VH 2nd LeBleu VS, Kalluri R. TGF-β1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J Am Soc Nephrol 24: 385–392, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bottinger EP. TGF-β in renal injury and disease. Semin Nephrol 27: 309–320, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Chamberlain RM, Shirley DG. Time course of the renal functional response to partial nephrectomy: measurements in conscious rats. Exp Physiol 92: 251–262, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Chen JK, Capdevila J, Harris RC. Cytochrome P450 epoxygenase metabolism of arachidonic acid inhibits apoptosis. Mol Cell Biol 21: 6322–6331, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int 75: 1145–1152, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Chiamvimonvat N, Ho CM, Tsai HJ, Hammock BD. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol 50: 225–237, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Decleves AE, Sharma K. Novel targets of antifibrotic and anti-inflammatory treatment in CKD. Nat Rev Nephrol 10: 257–267, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dendooven A, Ishola DA Jr, Nguyen TQ, Van der Giezen DM, Kok RJ, Goldschmeding R, Joles JA. Oxidative stress in obstructive nephropathy. Int J Exp Pathol 92: 202–210, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S, Kelly K, Gyengesi E, Arbiser JL, Belsham DD, Sarruf DA, Schwartz MW, Bennett AM, Shanabrough M, Mobbs CV, Yang X, Gao XB, Horvath TL. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med 17: 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Djamali A. Oxidative stress as a common pathway to chronic tubulointerstitial injury in kidney allografts. Am J Physiol Renal Physiol 293: F445–F455, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Dorrance AM, Rupp N, Pollock DM, Newman JW, Hammock BD, Imig JD. An epoxide hydrolase inhibitor, 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA), reduces ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol 46: 842–848, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elmarakby AA, Faulkner J, Al-Shabrawey M, Wang MH, Maddipati KR, Imig JD. Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. Am J Physiol Regul Integr Comp Physiol 301: R1307–R1317, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem 52: 447–454, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh AK, Bhattacharyya S, Wei J, Kim S, Barak Y, Mori Y, Varga J. Peroxisome proliferator-activated receptor-γ abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J 23: 2968–2977, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guedes AG, Morisseau C, Sole A, Soares JH, Ulu A, Dong H, Hammock BD. Use of a soluble epoxide hydrolase inhibitor as an adjunctive analgesic in a horse with laminitis. Vet Anaesth Analg 40: 440–448, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamer RA, El Nahas AM. The burden of chronic kidney disease. BMJ 332: 563–564, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han JY, Kim YJ, Kim L, Choi SJ, Park IS, Kim JM, Chu YC, Cha DR. PPARγ agonist and angiotensin II receptor antagonist ameliorate renal tubulointerstitial fibrosis. J Korean Med Sci 25: 35–41, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogaboam CM, Steinhauser ML, Chensue SW, Kunkel SL. Novel roles for chemokines and fibroblasts in interstitial fibrosis. Kidney Int 54: 2152–2159, 1998. [DOI] [PubMed] [Google Scholar]
- 24.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol 289: F496–F503, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev 92: 101–130, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 8: 794–805, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol 7: 2364–2370, 1996. [DOI] [PubMed] [Google Scholar]
- 28.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 39: 690–694, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension 46: 975–981, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inoue T, Takenaka T, Hayashi M, Monkawa T, Yoshino J, Shimoda K, Neilson EG, Suzuki H, Okada H. Fibroblast expression of an IκB dominant-negative transgene attenuates renal fibrosis. J Am Soc Nephrol 21: 2047–2052, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishii N, Fujiwara K, Lane PH, Patel KP, Carmines PK. Renal cortical nitric oxide synthase activity during maturational growth in the rat. Pediatr Nephrol 17: 591–596, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension 45: 759–765, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, Kohno N, Yorioka N. PPAR-γ agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-β. Lab Invest 89: 47–58, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Kim J, Imig JD, Yang J, Hammock BD, Padanilam BJ. Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am J Physiol Renal Physiol 307: F971–F980, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Long KE, Tang K, Padanilam BJ. Poly(ADP-ribose) polymerase 1 activation is required for cisplatin nephrotoxicity. Kidney Int 82: 193–203, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Kim J, Padanilam BJ. Loss of poly(ADP-ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am J Physiol Renal Physiol 301: F450–F459, 2011. [DOI] [PubMed] [Google Scholar]
- 39.Kim J, Padanilam BJ. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J Am Soc Nephrol 24: 229–242, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Seok YM, Jung KJ, Park KM. Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am J Physiol Renal Physiol 297: F461–F470, 2009. [DOI] [PubMed] [Google Scholar]
- 41.Kim JC, Lee YH, Yu MK, Lee NH, Park JD, Bhattarai G, Yi HK. Anti-inflammatory mechanism of PPARγ on LPS-induced pulp cells: role of the ROS removal activity. Arch Oral Biol 57: 392–400, 2012. [DOI] [PubMed] [Google Scholar]
- 42.Klahr S, Levey AS, Beck GJ, Caggiula AW, Hunsicker L, Kusek JW, Striker G. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of Diet in Renal Disease Study Group. N Engl J Med 330: 877–884, 1994. [DOI] [PubMed] [Google Scholar]
- 43.Kon K, Ikejima K, Hirose M, Yoshikawa M, Enomoto N, Kitamura T, Takei Y, Sato N. Pioglitazone prevents early-phase hepatic fibrogenesis caused by carbon tetrachloride. Biochem Biophys Res Commun 291: 55–61, 2002. [DOI] [PubMed] [Google Scholar]
- 44.Lee KSS, Liu JY, Wagner KM, Pakhomova S, Dong H, Morisseau C, Fu SH, Yang J, Wang P, Ulu A, Mate CA, Nguyen LV, Hwang SH, Edin ML, Mara AA, Wulff H, Newcomer ME, Zeldin DC, Hammock BD. Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. J Med Chem 57: 7016–7030, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Carroll MA, Chander PN, Falck JR, Sangras B, Stier CT. Soluble epoxide hydrolase inhibitor, AUDA, prevents early salt-sensitive hypertension. Front Biosci 13: 3480–3487, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Lindenmeyer MT, Kretzler M, Boucherot A, Berra S, Yasuda Y, Henger A, Eichinger F, Gaiser S, Schmid H, Rastaldi MP, Schrier RW, Schlondorff D, Cohen CD. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol 18: 1765–1776, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: the role of PPARγ, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci USA 102: 16747–16752, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loch D, Hoey A, Morisseau C, Hammock BO, Brown L. Prevention of hypertension in DOCA-salt rats by an inhibitor of soluble epoxide hydrolase. Cell Biochem Biophys 47: 87–98, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luria A, Bettaieb A, Xi Y, Shieh GJ, Liu HC, Inoue H, Tsai HJ, Imig JD, Haj FG, Hammock BD. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc Natl Acad Sci USA 108: 9038–9043, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma LJ, Yang H, Gaspert A, Carlesso G, Barty MM, Davidson JM, Sheppard D, Fogo AB. Transforming growth factor-β-dependent and -independent pathways of induction of tubulointerstitial fibrosis in β6−/− mice. Am J Pathol 163: 1261–1273, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, Hammock BD, Imig JD. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol 297: F740–F748, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCarthy KJ, Routh RE, Shaw W, Walsh K, Welbourne TC, Johnson JH. Troglitazone halts diabetic glomerulosclerosis by blockade of mesangial expansion. Kidney Int 58: 2341–2350, 2000. [DOI] [PubMed] [Google Scholar]
- 54.Mehal W, Imaeda A. Cell death and fibrogenesis. Semin Liver Dis 30: 226–231, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohmori Y, Fukumoto S, Hamilton TA. Two structurally distinct κB sequence motifs cooperatively control LPS-induced KC gene transcription in mouse macrophages. J Immunol 155: 3593–3600, 1995. [PubMed] [Google Scholar]
- 56.Olearczyk JJ, Quigley JE, Mitchell BC, Yamamoto T, Kim IH, Newman JW, Luria A, Hammock BD, Imig JD. Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin Sci (Lond) 116: 61–70, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Egido J, Ruiz-Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-β-independent mechanism. Circulation 111: 2509–2517, 2005. [DOI] [PubMed] [Google Scholar]
- 58.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA 102: 9772–9777, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem 275: 40504–40510, 2000. [DOI] [PubMed] [Google Scholar]
- 60.Small DM, Coombes JS, Bennett N, Johnson DW, Gobe GC. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 17: 311–321, 2012. [DOI] [PubMed] [Google Scholar]
- 61.Sullivan JC, Wang B, Boesen EI, D'Angelo G, Pollock JS, Pollock DM. Novel use of ultrasound to examine regional blood flow in the mouse kidney. Am J Physiol Renal Physiol 297: F228–F235, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y, Okubo T. NF-κB and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol 153: 2052–2063, 1994. [PubMed] [Google Scholar]
- 63.Ulu A, Appt S, Morisseau C, Hwang SH, Jones PD, Rose TE, Dong H, Lango J, Yang J, Tsai HJ, Miyabe C, Fortenbach C, Adams MR, Hammock BD. Pharmacokinetics and in vivo potency of soluble epoxide hydrolase inhibitors in cynomolgus monkeys. Br J Pharmacol 165: 1401–1412, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang L, Yang J, Guo L, Uyeminami D, Dong H, Hammock BD, Pinkerton KE. Use of a soluble epoxide hydrolase inhibitor in smoke-induced chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 46: 614–622, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang YX, Ulu A, Zhang LN, Hammock B. Soluble epoxide hydrolase in atherosclerosis. Curr Atheroscler Rep 12: 174–183, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Widmer U, Manogue KR, Cerami A, Sherry B. Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1α, and MIP-1β, members of the chemokine superfamily of proinflammatory cytokines. J Immunol 150: 4996–5012, 1993. [PubMed] [Google Scholar]
- 67.Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RK, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci USA 103: 18733–18738, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res 87: 992–998, 2000. [DOI] [PubMed] [Google Scholar]