

Abstract
Thick ascending limbs reabsorb 30% of the filtered NaCl load. Nitric oxide (NO) produced by NO synthase 3 (NOS3) inhibits NaCl transport by this segment. In contrast, chronic angiotensin II (ANG II) infusion increases net thick ascending limb transport. NOS3 activity is regulated by changes in expression and phosphorylation at threonine 495 (T495) and serine 1177 (S1177), inhibitory and stimulatory sites, respectively. We hypothesized that NO production by thick ascending limbs is impaired by chronic ANG II infusion, due to reduced NOS3 expression, increased phosphorylation of T495, and decreased phosphorylation of S1177. Rats were infused with 200 ng·kg−1·min−1 ANG II or vehicle for 1 and 5 days. ANG II infusion for 5 days decreased NOS3 expression by 40 ± 12% (P < 0.007; n = 6) and increased T495 phosphorylation by 147 ± 26% (P < 0.008; n = 6). One-day ANG II infusion had no significant effect. NO production in response to endothelin-1 was blunted in thick ascending limbs from ANG II-infused animals [ANG II −0.01 ± 0.06 arbitrary fluorescence units (AFU)/min vs. 0.17 ± 0.02 AFU/min in controls; P < 0.01]. This was not due to reduced endothelin-1 receptor expression. Phosphatidylinositol 3,4,5-triphosphate (PIP3)-induced NO production was also reduced in ANG II-infused rats (ANG II −0.07 ± 0.06 vs. 0.13 ± 0.04 AFU/min in controls; P < 0.03), and this correlated with an impaired ability of PIP3 to increase S1177 phosphorylation. We conclude that in ANG II-induced hypertension NO production by thick ascending limbs is impaired due to decreased NOS3 expression and altered phosphorylation.
Keywords: kidney, renal medulla, blood pressure, salt sensitivity
angiotensin ii (ANG II) affects many systems, including the cardiovascular, renal, and central nervous systems. In experimental models, doses of ANG II that have no measurable effects on blood pressure, urinary volume, and urinary Na excretion acutely have profound effects on these parameters when infused chronically (14, 27, 49, 64). Such models of ANG II-induced hypertension have been widely used to study both the causes (1, 2, 12, 17, 27, 44) and consequences (9, 14, 28, 30) of hypertension.
A large range of doses has been used to induce ANG II-dependent hypertension in rats. Generally, lower doses of ANG II (5–200 ng·kg−1·min−1) produce a salt-sensitive form of hypertension (75), a disease in which dietary salt increases blood pressure, whereas higher doses of ANG II (>400 ng·mg−1·min−1) produce immediate increases in blood pressure that no longer depend on salt intake (75). In salt-sensitive forms of hypertension induced by ANG II, animals are in positive Na balance from 3 to 10 days (25, 75). The Na retention may be at least in part due to reductions in the effects on nitric oxide (NO) in the kidney, since the prohypertensive actions of ANG II are specifically mitigated by NO (5, 12, 54, 76). In addition, decreased NO production and signaling in the renal medulla are associated with salt-sensitive hypertension (18, 26, 38, 50, 51, 65, 80).
The thick ascending limb of the loop of Henle reabsorbs ∼25% of the filtered NaCl load, and NO acts as an autacoid regulating this process (23, 57, 58, 61, 68–70, 83). All isoforms of NO synthases (NOS1, 2, and 3) are expressed in thick ascending limbs. Stimulation of NO production with the general NOS substrate l-arginine decreases transport in wild-type mice (69). This effect is preserved in NOS1 or NOS2 knockout mice, but is blunted in NOS3 knockout mice (62, 69). These data indicate that NO from NOS3 regulates transport in this nephron segment (13, 62, 66, 69).
NOS3 activity can be regulated by changes in expression and phosphorylation in the thick ascending limb (35, 73). We showed that ANG II reduces (72) while a high-salt diet initially increases (33, 35), and then diminishes NOS3 expression (35). Several factors acutely alter phosphorylation including endothelin-1 (34, 71), ATP (6, 78), and luminal flow (6, 59, 60). These factors stimulate thick ascending limb NO production via phosphatidylinositol 3,4,5-tri-phosphate (PIP3) and phosphorylation of NOS3 at serine 1177 (S1177). In contrast, phosphorylation at threonine 495 (T495) by protein kinase C (PKC) reduces NO production (21). Although defects in thick ascending limb NO production and/or signaling cause salt-sensitive hypertension (22, 35, 38, 50, 51), and thick ascending limb transport is enhanced acutely (19) and chronically (24, 79) by ANG II, it is not known whether NO production is impaired in thick ascending limbs of ANG II-induced hypertensive animals and the mechanisms involved.
We hypothesized that NO production in the thick ascending limb is impaired by chronic ANG II infusion, due to reduced NOS3 expression, increased phosphorylation of T495, and decreased phosphorylation at S1177.
METHODS
Drugs and buffers.
Unless specified, all drugs and reagents were obtained from Sigma (St. Louis, MO). The NO-sensitive dye 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM diacetate) was obtained from Invitrogen (Grand Island, NY). Coomasie Plus Protein Assay Reagent was obtained from Thermo-Scientific (Rockford, IL). ANG II and endothelin-1 were obtained from Bachem (Torrance, CA). Phosphatidylinositol 3,4,5-triphosphate (PIP3) and neomycin sulfate (carrier) were obtained from Echelon Biosciences (Salt Lake City, UT).
HEPES-buffered physiological saline (physiological saline) used for kidney perfusion and to generate thick ascending limb suspensions contained (in mmol/l) 10 HEPES (pH 7.5), 130 NaCl, 4 KCl, 2.5 NaH2PO4, 1.2 MgSO4, 5.5 glucose, 6.0 DL-alanine, 2.0 Ca(lactate)2, and 1.0 Na3citrate. Osmolality was adjusted to 290 ± 5 mosmol/kgH2O.
Lysis buffer contained 20 mmol/l HEPES (pH 7.5), 2 mmol/l EDTA, 300 mmol/l sucrose, 1.0% Igepal, 0.1% Na dodecyl sulfate, 5 mg/l antipain, 10 mg/l aprotinin, 5 mg/l leupeptin, 5 mg/l chymostatin, 5 mg/l pepstatin A, 4 mmol/l benzamidine, and 116 mmol/l pf-block (Sigma). Milk blocking buffer contained 50 g/l nonfat milk dissolved in 20 mmol/l Tris (pH 7.6), 137 mmol/l NaCl, and 0.1% Tween-20. The blocking buffer Blok-PO, the phosphatase inhibitor Cocktail Set II, and the Luminata luminol-based chemiluminescent substrate for horseradish peroxidase were obtained from EDM Millipore (San Diego, CA).
Animal model.
This study was approved by the Institutional Animal Care and Use Committee of the Henry Ford Hospital, Wayne State University, and Case Western Reserve University. All studies were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male Sprague-Dawley rats (Charles River Breeding Laboratories, Wilmington, MA) weighing 90–120 g were infused with either vehicle (0.01 mol/l acetic acid) or ANG II (200 ng·kg−1·min−1, Bachem, dissolved in 0.01 mol/l acetic acid) via osmotic minipumps (ALZET 1007D, Cupertino, CA) for 1 or 5 days. Rats of this size were used so that thick ascending limbs could be dissected after ANG II infusion. Minipump implantation was performed under isofluorane anesthesia (1.5% in O2, flow rate 0.5 l/min). At day 5, animals were anesthetized with ketamine (100 mg/kg body wt ip) and xylazine (20 mg/kg body wt ip). Animals used to prepare thick ascending limb suspensions were also given 2 IU heparin ip. Mean arterial blood pressure (MAP) was measured under anesthesia as we reported before (24).
Thick ascending limb suspension preparation.
Thick ascending limb suspensions were generated as we described (24, 73). Briefly, the abdominal cavity was opened and the kidneys were flushed with cold physiological saline containing 2.5 U/ml of heparin and 0.1% type I collagenase via retrograde perfusion of the aorta. Perfused kidneys were excised and cut in coronal slices. The inner stripe of the outer medulla was dissected, minced, and digested in 0.1% type I collagenase for 30 min at 37°C. During each 5-min period, the preparation was agitated and gassed with 100% O2. Then, the sample was centrifuged (100 g, 2 min, 4°C) and the pellet was resuspended in fresh physiological saline and stirred on ice for 30 min. After being stirred, the tubules were filtered through a 250-μm nylon mesh, recovered from the filtrate by centrifugation (100 g, 2 min, 4°C) and rinsed two times with cold physiological saline. The resulting product yields ∼95% pure suspension of thick ascending limbs (10, 32).
PIP3 treatment in thick ascending limb suspensions.
Thick ascending limbs were resuspended in physiological saline and 2 aliquots of 300 μl incubated at 37°C for 10 min, oxygenating every 5 min. They were treated with vehicle (2.5 μmol/l carrier) or PIP3 (2.5 μmol/l + carrier) for 8 min, oxygenating every 4 min. Suspensions were rapidly cooled by adding 2 volumes (600 μl) of cold physiological saline containing 1:35 phosphatase inhibitor cocktail, and put on ice. Tubules were recovered by centrifugation (100 g, 2 min, 4°C) and dissolved in lysis buffer containing 1:100 phosphatase inhibitor cocktail.
Western blot.
Western blots were conducted using similar techniques as before (20, 24, 72). Unless otherwise noted, samples were processed as follows: thick ascending limb suspensions were dissolved in lysis buffer and the debris was removed by centrifugation (5,600 g, 5 min, 4°C). Protein was determined by a colorimetric assay. Polyacrylamide gels (6%) were used. Equal amounts of protein from freshly prepared lysates were loaded per lane. Samples from vehicle and ANG II-infused rats were processed in pairs and loaded in the same gel, so each gel had its own control. For ETB receptor samples, a 4% stacking/9% running gel combination was used. For T495 phosphorylation, paired samples were loaded in nonadjacent lanes of the same gel. Optic density of each band was expressed as a fraction of the total density. Values were corrected for loading using the same procedure as depicted below. Western blots were conducted as specified in Table 1. For loading control, the membranes were stripped with 200 mmol/l glycine (pH 2.8) for 30 min and reblotted for β-tubulin in the case of NOS3 and ETB receptor, and NOS3 in the case of phospho-NOS3 (S1177) and (T495).
Table 1.
Antibodies and blotting conditions
| Conditions |
||||||||
|---|---|---|---|---|---|---|---|---|
| Antibody | Provider | Catalog Number | Source | Blocking Buffer | Dilution | Buffer | Time | Temp. |
| NOS3 | BD Bioscience | 610296 | Mouse | 5% Milk† | 1:1,000 | 5% Milk | 1 h | Room T. |
| pNOS3 (S1177) | BD Bioscience | 612392 | Mouse | 5% Milk | 1:2,000 | 5% Milk | 1 h | Room T. |
| pNOS3 (T495) | CellSignaling | 9574 | Rabbit | BlokTM-PO* | 1:500 | BlokTM-PO | 15 h | 4°C |
| ETB receptor | Alomone | AER-002 | Rabbit | 5% Milk | 1:1,250 | 5% Milk | 1 h | Room T. |
| β-Tubulin | Abcam | ab6046 | Rabbit | 5% Milk | 1:10,000 | 5% Milk | 1 h | Room T. |
| 2ry anti-rabbit-HRP | GE Healthcare | NA9340V | Donkey | 1:2,500 | 5% Milk | 1 h | Room T. | |
| 2ry anti-mice-HRP | GE Healthcare | NA931V | Sheep | 1:1,000 | 5% Milk | 1 h | Room T. | |
Blok TM-PO (Millipore, cat. no.: WBAVDP001).
5% Milk: 50 g/l nonfat milk dissolved in 20 mM Tris (pH 7.6), 137 mM NaCl, and 0.1% Tween 20.
Protein detection and densitometry.
The signal was detected by exposing a Fuji Super RX film to the membranes. Films were scanned using an EPSON Expression 1680 scanner with EPSON-Scan software (positive film, 16-bit grayscale, 600 dpi). Densitometry of the bands was measured using a custom-made software. The exposure times for the films were standardized so that the mean optical density of the bands was between 0.40 and 1.00.
Measurement of NO production.
NO production was measured by fluorescence microscopy as we have done before (7, 8, 31, 60, 62). In brief, thick ascending limbs were manually dissected, transferred to a temperature-controlled chamber (37 ± 1°C), and held by two glass pipettes without luminal flow. The bath contained 100 μmol/l l-arginine. Under these conditions, basal NO production by the tubules is negligible (59, 60). Tubules were loaded with DAF-FM (4 μmol/l) for 20 min and then rinsed for 20 min. For imaging, a ×40 oil immersion objective was used. The dye was excited by a xenon arc lamp with a 488-nm band pass filter. The fluorescence emitted by the NO-bound dye (>500 nm) was measured using Metafluor software (Molecular Devices). Measurements were recorded once every 30 s for a 10-min control period. Then treatments were applied, and fluorescence was measured under the same conditions during a 10-min experimental period. Given that dye binding to NO is irreversible, the change in fluorescence with time represents NO production. A linear regression of the 5 min of maximal stimulation during the experimental period was performed and compared with the last 5 min of the control period. The difference was expressed as arbitrary fluorescence units (AFU)/min and represents the NO response to the treatment. Treatments were as follows: 1) 1 nmol/l endothelin-1 (dissolved in 0.005% acetic acid) vs. vehicle (0.005% acetic acid); 2) 2.5 μmol/l PIP3 vs. vehicle (2.5 μmol/l carrier).
Data analysis.
Results are expressed as means ± SE. Paired t-test and 2-sample Wilcoxon tests were used as appropriate. When multiple pair-wise comparisons were made, Hochsberg's method for multiple test of significance was used. P < 0.05 was considered significant.
RESULTS
Rats infused with 200 ng·kg−1·min−1 of ANG II for 5 days had increased MAP compared with vehicle-infused rats (Vehicle: 96 ± 4 mmHg, n = 5 vs. ANG II: 120 ± 8 mmHg in ANG II, n = 4; P < 0.04). One-day infusion of ANG II did not produce any measurable change in MAP (Table 2).
Table 2.
Blood pressure values at the end of each experimental period
| Control, mmHg | ANG II, mmHg | n | P | |
|---|---|---|---|---|
| 1 Day | 96 ± 4 | 100 ± 5 | 5 | < 0.57 |
| 5 Day | 96 ± 4 | 120 ± 8 | 4 | < 0.03 |
Values are means ± SE.
We previously showed that in vitro ANG II treatment decreases NOS3 expression in primary cultures of thick ascending limbs (72); therefore, we next measured NOS3 expression in thick ascending limbs from ANG II-infused and control animals. We found that infusion of ANG II for 5 days decreased NOS3 expression by 40 ± 12% (P < 0.007; n = 6; Fig. 1) compared with controls infused with vehicle. ANG II had no significant effect when infused for 1 day (Δ = −7 ± 18%; n = 5).
Fig. 1.

Nitric oxide synthase 3 (NOS3) expression in thick ascending limb suspensions obtained from ANG II- and vehicle-infused rats after 5 days of treatment. Paired vehicle and ANG II samples were loaded in the same gel. NOS3 expression levels were corrected by β-tubulin and values were expressed as percentage of vehicle. ANG II infusion for 5 days decreased NOS3 expression by 40 ± 12% (P < 0.007; n = 6).
Several studies have shown that PKC mediates at least some of the effects of ANG II in thick ascending limbs (3, 36, 79, 85). Since PKC targets NOS3 at the inhibitory site T495 (37, 48), we next measured NOS3 phosphorylation at this residue. We found that 5-day infusion of ANG II increased NOS3 phosphorylation at T495 by 147 ± 26% (P < 0.008; n = 6) compared with the controls infused with vehicle (Fig. 2). In contrast, T495 phosphorylation did not change in thick ascending limbs from rats infused with ANG II for 1 day (Δ = 13 ± 20%; n = 5).
Fig. 2.

Phospho (P)-NOS3 (T495) levels in thick ascending limb suspensions obtained from ANG II- and vehicle-infused rats after 5 days of treatment. Paired samples were loaded in nonadjacent lanes in the same gel. Values were expressed as P-NOS3 (T495)/NOS3 ratio. ANG II infusion for 5 days increased NOS3 phosphorylation at T495 by 174 ± 26% (P < 0.008; n = 6) compared with vehicle.
Given that ANG II infusion altered NOS3 expression and phosphorylation only after 5 days of infusion, we studied whether these effects reduce NO production in response to physiological stimuli at this time point. We tested whether the ability of 1 nmol/l endothelin-1 to stimulate NO production was impaired in thick ascending limbs from ANG II hypertensive rats. Endothelin-1 increased NO production in thick ascending limbs from vehicle-infused rats by 0.17 ± 0.02 AFU/min (n = 8). In contrast, endothelin-1 did not significantly affect NO production in tubules from ANG II-infused animals (−0.01 ± 0.06 AFU/min; n = 7; vehicle vs. ANG II: P < 0.01; Fig. 3).
Fig. 3.
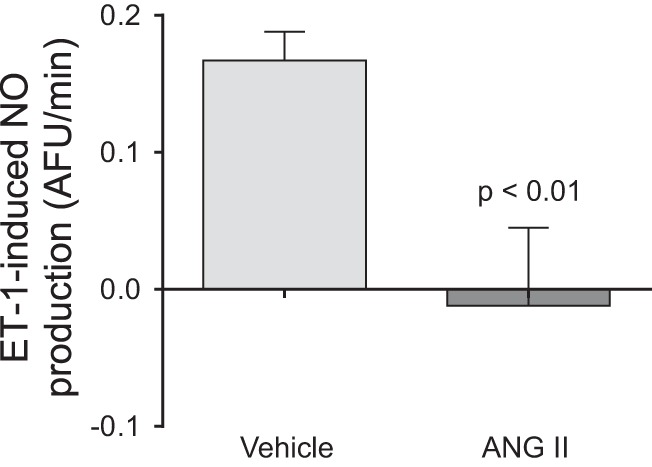
Endothelin-1-induced NO production as measured by DAF-FM fluorescence in isolated thick ascending limbs obtained from ANG II- and vehicle-infused rats after 5 days of treatment. The NO response to endothelin-1 stimulation was blunted in thick ascending limbs from rat infused ANG II (−0.01 ± 0.06; n = 7) compared with controls (0.17 ± 0.06; n = 8, P < 0.01).
Endothelin-1 increases NO by binding ETB receptors. Therefore, the diminished response to endothelin-1 could be a result of a reduction in ETB receptors. We found that chronic ANG II infusion did not change thick ascending limb ETB receptor expression compared with controls (Δ = 10 ± 10%; n = 6; Fig. 4).
Fig. 4.

ETB receptor expression in thick ascending limb suspensions obtained from ANG II- and vehicle-infused rats after 5 days of treatment. Paired vehicle and ANG II samples were loaded in the same gel. ETB receptor levels were corrected by β-tubulin and values were expressed as percentage of vehicle. ANG II had no effect on ETB receptor expression (Δ = 10 ± 10; ns, n = 6).
PIP3 mediates the stimulatory action of many factors that activate NOS3 in thick ascending limbs and use of PIP3 as a NOS3 activator bypasses many of the signaling steps that could be altered by ANG II-induced hypertension. Thus, we next tested whether PIP3-induced NO production is reduced by ANG II infusion. In thick ascending limbs from vehicle-treated animals PIP3 increased NO production by 0.13 ± 0.04 AFU/min (n = 4); however, it had no significant effect on NO production in tubules from ANG II-infused animals (Δ = −0.07 ± 0.06 AFU/min, n = 4; Control vs. ANG II: P < 0.03; Fig. 5).
Fig. 5.
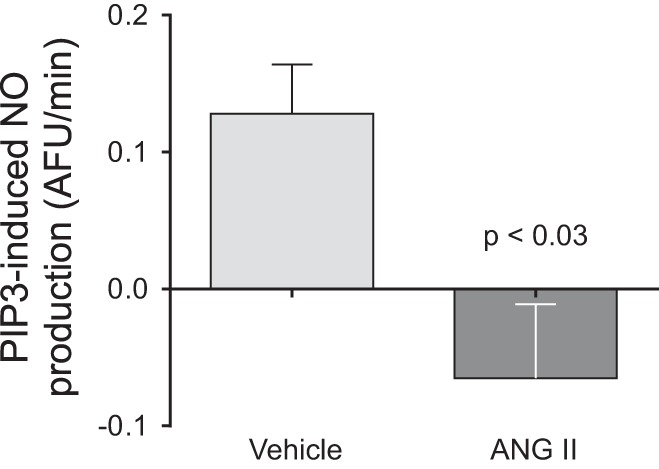
Phosphatidylinositol 3,4,5-triphosphate (PIP3)-induced NO production as measured by DAF-FM fluorescence in isolated thick ascending limbs obtained from ANG II- and vehicle-infused rats after 5 days of treatment. The NO response to PIP3 stimulation was blunted in thick ascending limbs from ANG II-infused animals (−0.07 ± 0.06; n = 4) compared with controls (0.13 ± 0.04; n = 4, P < 0.03).
The final step in NOS3 activation by PIP3 is phosphorylation at S1177. Thus, we tested whether PIP3-induced phosphorylation of NOS3 at S1177 was reduced in ANG II-induced hypertension. In vehicle-treated rats, PIP3 augmented phosphorylation by 18 ± 4% (P < 0.02; n = 5; Fig. 6A). In contrast, PIP3 had no significant effect on thick ascending limbs from ANG II-treated animals (Δ = −1 ± 18%; Fig. 6B).
Fig. 6.

Effect of PIP3 stimulation on NOS3 phosphorylation at S1177. Thick ascending limb suspensions obtained from ANG II- and vehicle-infused rats after 5 days of treatment were split, incubated at 37°C for 10 min, and then treated with either PIP3 plus carrier or carrier alone for 8 min. Phospho (P)-NOS3 (S1177) levels were corrected by NOS3 expression and results were expressed as percentage of P-NOS3 (S1177) in thick ascending limbs treated with carrier. A: PIP3 stimulation increased P-NOS3 (S1177) in thick ascending limbs from vehicle-infused animals by 18 ± 4% (P < 0.02; n = 5). B: PIP3 stimulation had no effect on P-NOS3 (S1177) in thick ascending limbs from ANG II-infused animals compared with carrier (ns; n = 5).
DISCUSSION
NO production by NOS3 in thick ascending limbs is a critical factor in blood pressure and salt handling by the kidney. Since NO antagonizes the actions of ANG II in the kidney, we hypothesized that NO production by thick ascending limbs is impaired by chronic ANG II infusion due to downregulation in NOS3 expression and altered phosphorylation.
First, we measured NOS3 expression in medullary thick ascending limb suspensions by Western blot. We found that chronic ANG II infusion decreased NOS3 expression by 40% compared with vehicle at day 5, but had no effect at day 1. This raises the question of whether the decrease in NO is a result of ANG II accumulation or the associated increase in blood pressure. Some insights to answer this question come from the 2 kidney-1 clip hypertension model, where each kidney is exposed to similar ANG II levels (84) but different perfusion pressures. In this model, NOS3 decreased to the same extent in the medulla of the clipped and contralateral kidneys (86). These data indicate that some factor other than blood pressure reduces NOS3 expression, likely the elevated ANG II. In addition, ANG II decreased NOS3 expression in primary cultures of thick ascending limbs (72) where blood pressure, shear stress, and hypoxia are absent. Taken together, these data suggest that chronic elevations in ANG II rather than the associated increase in blood pressure are responsible for decreasing NOS3 expression in the thick ascending limb.
Our data showing that ANG II decreases NOS3 expression at 5 days contrast with those of others showing that ANG II infusion did not change (11, 54, 86) or increased (53) NOS3 in the renal medulla. They are also different from data showing that chronic ANG II infusions increase NOS3 expression in the renal cortex (11, 53, 54). Some insights into the variability of the results come from analyzing the effects on ANG II on other tissues. On one hand, ANG II increased NOS3 expression in the rat aorta (52) and primary cultures of bovine pulmonary artery endothelial cells (56), but not in bovine coronary artery endothelial cells (56). On the other hand, there are reports showing the opposite, ANG II decreases NOS3 expression in human coronary artery endothelial cells (45) and ANG II receptor blockade increases NOS3 expression in rat aortic endothelial cells (87). Disparate results have been also reported in the heart (42, 53, 81). Taken together, these results seem to indicate that the effects of ANG II are dose- and time-dependent.
Directly to this point, McDonough and colleagues (55) recently showed that even though ANG II acutely stimulates Na transport all along the nephron, 2-wk infusion of pressor doses of ANG II results in segment-specific effects. These investigators postulated that initially ANG II infusion produces a direct increase in salt reabsorption in all nephron segments. However, with time the increase in blood pressure reduces proximal nephron reabsorption via a pressure-natriuretic mechanism while distal nephron Na reabsorption remains elevated. In support of this, 2-wk infusion of pressor doses of ANG II results in elevations in cortical NO (11, 12) concomitantly with the pressure natriuretic response. Thus, these data suggest that increased NO is a compensatory response to the elevation of blood pressure, not a direct effect of ANG II.
Given that NOS3 is regulated by both expression and phosphorylation, we next studied whether phosphorylation of NOS3 at the inhibitory site T495 was affected by ANG II infusion. We found that phosphorylation of T495 increased by 147% in thick ascending limbs from animals infused with ANG II for 5 days but did not significantly change after 1 day. These data suggest that ANG II accumulation is necessary for the increase in T495 phosphorylation. In this regard, our lab showed that acute and chronic activation of angiotensin receptor 1 (AT1) increases superoxide production via PKC-α (36, 79). Since PKC-α phosphorylates NOS3 at T495 (39, 77), it is possible that the increase in T495 phosphorylation observed in our study is a result of ANG II-induced PKC activation rather than increased blood pressure.
Both lines of evidence, reduced NOS3 expression and increased phosphorylation at the inhibitory site T495, indicate that NO production by the thick ascending limbs is impaired in ANG II-induced hypertension. To test this possibility, we measured the capacity of a physiological stimulus to increase NO production by isolated thick ascending limbs. Endothelin-1 increased NO production in control tubules but not in thick ascending limbs from ANG II-treated rats.
Since endothelin-1 stimulates NO production by binding ETB receptors in the thick ascending limb (68), it is possible that the reduced response to endothelin-1 in ANG II-infused rats was due to downregulation of the receptor. Therefore, we tested whether ETB receptor expression was affected by ANG II infusion. We were unable to find any differences in ETB receptor expression among groups. Thus, reduced receptor expression cannot explain the lack of response to endothelin-1.
ATP (6, 78), α2 adrenergic agonists (67), endothelin (34, 71), and luminal flow (6, 59, 60) stimulate NO production by NOS3 in thick ascending limbs via activation of phosphatidyl inositol 3 kinase and PIP3. Since many factors that activate NOS3 in thick ascending limbs do so by elevating PIP3 (21), and use of PIP3 would avoid further confounding issues of receptor number, etc., we next examined the ability of PIP3 to stimulate NO production. We found that the increase in NO production present in control animals was absent in animals infused with ANG II. These data indicate that ANG II infusion impairs the capacity of NOS3 to respond to any physiological stimulus that acts via PIP3.
The final step in NOS3 activation by PIP3 is phosphorylation of the stimulatory site S1177. There is growing evidence of regulation of NOS3 activity by reciprocal phosphorylation of T495 and S1177 upon application of several stimuli (29, 47, 77, 82), including blockade of the AT1 receptor (4, 74). In addition, changes in NOS3 phosphorylation at S1177 have been shown in several diseases including hypertension (16, 40, 41, 43, 46). Thus, we next tested whether phosphorylation at S1177 was also affected by ANG II infusion.
Our results indicate that PIP3-induced phosphorylation of NOS3 at S1177 was impaired in thick ascending limbs from ANG II-induced hypertensive animals. Consistent with these findings in the thick ascending limb, phosphorylation of NOS3 at S1177 was found to be decreased in the aorta (87) and cerebral cortex (63) of spontaneously hypertensive rats, whereas this was restored by AT1 blockade (63, 87), indicating that ANG II signaling is involved in this process.
The data presented here indicate that changes in phosphorylation of NOS3, and thus activity, caused by ANG II are time-dependent. This hypothesis is consistent with previously published reports. We and others have shown that acute in vitro ANG II treatment stimulates NO in the thick ascending limb (15, 31). Intravenous infusion of 200 ng·kg−1·min−1 for 3 h to rats also produced an increase on medullary NO; however, NO production returned to values not different from controls at day 3 of infusion (53). Building on these results, our current study shows that NO production and phosphorylation of T495 are affected by day 5 of infusion of slow-pressor doses of ANG II.
In the thick ascending limb, NOS3/NO/cGMP pathway regulates salt reabsorption by retrieving NKCC2 from the plasma membrane, thereby reducing its activity. In addition, we previously reported that net transport is increased by the thick ascending limbs in ANG II-induced hypertension (79) and that the increase in thick ascending limb's Na-K-ATPase in this model depends on a chronically increased NKCC2-dependent Na reabsorption (24). We now report that NO production is impaired in the thick ascending limbs during ANG II-induced hypertension. We speculate that defects in NO signaling are responsible for the chronic increase in transport by the thick ascending limbs in this model.
In summary, our data indicate that in ANG II-induced hypertension NO production is impaired in the thick ascending limbs, due to decreased NOS3 expression and altered phosphorylation. Lack of NO in the renal medulla contributes to the development and maintenance of salt-sensitive hypertension (22, 26, 35, 38, 50, 65), therefore, reduced NO production by this segment might be a contributing factor to increased blood pressure in this model of hypertension.
GRANTS
This work was supported in part by National Institutes of Health Grant HL28982P4 to J. L. Garvin and from American Heart Association (11PRE7510005) to V. D. Ramseyer.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: V.D.R., A.G.-V., O.A.C., and J.L.G. conception and design of research; V.D.R. and A.G.-V. performed experiments; V.D.R., A.G.-V., O.A.C., and J.L.G. analyzed data; V.D.R., A.G.-V., O.A.C., and J.L.G. interpreted results of experiments; V.D.R., A.G.-V., O.A.C., and J.L.G. prepared figures; V.D.R., A.G.-V., O.A.C., and J.L.G. drafted manuscript; V.D.R., A.G.-V., O.A.C., and J.L.G. edited and revised manuscript; V.D.R., A.G.-V., O.A.C., and J.L.G. approved final version of manuscript.
REFERENCES
- 1.Agarwal R, Campbell RC, Warnock DG. Oxidative stress in hypertension and chronic kidney disease: role of angiotensin II. Semin Nephrol 24: 101–114, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Alexander BT, Cockrell KL, Rinewalt AN, Herrington JN, Granger JP. Enhanced renal expression of preproendothelin mRNA during chronic angiotensin II hypertension. Am J Physiol Regul Integr Comp Physiol 280: R1388–R1392, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Amlal H, LeGoff C, Vernimmen C, Soleimani M, Paillard M, Bichara M. ANG II controls Na+-K+(NH4+)-2Cl− cotransport via 20-HETE and PKC in medullary thick ascending limb. Am J Physiol Cell Physiol 274: C1047–C1056, 1998. [DOI] [PubMed] [Google Scholar]
- 4.Barauna VG, Mantuan PR, Magalhaes FC, Campos LC, Krieger JE. AT1 receptor blocker potentiates shear-stress induced nitric oxide production via modulation of eNOS phosphorylation of residues Thr(495) and Ser(1177). Biochem Biophys Res Commun 441: 713–719, 2013. [DOI] [PubMed] [Google Scholar]
- 5.Beierwaltes WH, Potter DL, Carretero OA, Sigmon DH. Nitric oxide synthesis inhibition blocks reversal of two-kidney, one clip renovascular hypertension after unclipping. Hypertension 25: 174–179, 1995. [DOI] [PubMed] [Google Scholar]
- 6.Cabral PD, Garvin JL. Luminal flow regulates NO and O2− along the nephron. Am J Physiol Renal Physiol 300: F1047–F1053, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabral PD, Hong NJ, Garvin JL. ATP mediates flow-induced NO production in thick ascending limbs. Am J Physiol Renal Physiol 303: F194–F200, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cabral PD, Hong NJ, Garvin JL. Shear stress increases nitric oxide production in thick ascending limbs. Am J Physiol Renal Physiol 299: F1185–F1192, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cervenka L, Kramer HJ, Maly J, Heller J. Role of nNOS in regulation of renal function in angiotensin II-induced hypertension. Hypertension 38: 280–285, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Chamberlin ME, LeFurgey A, Mandel LJ. Suspension of medullary thick ascending limb tubules from the rabbit kidney. Am J Physiol Renal Fluid Electrolyte Physiol 247: F955–F964, 1984. [DOI] [PubMed] [Google Scholar]
- 11.Chin SY, Pandey KN, Shi SJ, Kobori H, Moreno C, Navar LG. Increased activity and expression of Ca2+-dependent NOS in renal cortex of ANG II-infused hypertensive rats. Am J Physiol Renal Physiol 277: F797–F804, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin SY, Wang CT, Majid DS, Navar LG. Renoprotective effects of nitric oxide in angiotensin II-induced hypertension in the rat. Am J Physiol Renal Physiol 274: F876–F882, 1998. [DOI] [PubMed] [Google Scholar]
- 13.De Miguel C, Foster JM, Carmines PK, Pollock JS. Interaction between NO synthase and NADPH oxidase in control of sodium transport by the renal thick ascending limb during diabetes. Acta Physiol (Oxf) 209: 148–155, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeClue JW, Guyton AC, Cowley AW Jr, Coleman TG, Norman RA Jr, McCaa RE. Subpressor angiotensin infusion, renal sodium handling, and salt-induced hypertension in the dog. Circ Res 43: 503–512, 1978. [DOI] [PubMed] [Google Scholar]
- 15.Dickhout JG, Mori T, Cowley AW Jr. Tubulovascular nitric oxide crosstalk: buffering of angiotensin II-induced medullary vasoconstriction. Circ Res 91: 487–493, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Dolinsky VW, Chakrabarti S, Pereira TJ, Oka T, Levasseur J, Beker D, Zordoky BN, Morton JS, Nagendran J, Lopaschuk GD, Davidge ST, Dyck JR. Resveratrol prevents hypertension and cardiac hypertrophy in hypertensive rats and mice. Biochim Biophys Acta 1832: 1723–1733, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Edwards BS, Schwab TR, Zimmerman RS, Heublein DM, Jiang NS, Burnett JC Jr. Cardiovascular, renal, and endocrine response to atrial natriuretic peptide in angiotensin II mediated hypertension. Circ Res 59: 663–667, 1986. [DOI] [PubMed] [Google Scholar]
- 18.Feng D, Yang C, Geurts AM, Kurth T, Liang M, Lazar J, Mattson DL, O'Connor PM, Cowley AW Jr. Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab 15: 201–208, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC. Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Physiol Renal Physiol 274: F148–F155, 1998. [DOI] [PubMed] [Google Scholar]
- 20.Fink C, Morgan F, Loew LM. Intracellular fluorescent probe concentrations by confocal microscopy. Biophys J 75: 1648–1658, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fleming I. Molecular mechanisms underlying the activation of eNOS. Pflügers Arch 459: 793–806, 2010. [DOI] [PubMed] [Google Scholar]
- 22.Garcia NH, Plato CF, Stoos BA, Garvin JL. Nitric oxide-induced inhibition of transport by thick ascending limbs from Dahl salt-sensitive rats. Hypertension 34: 508–513, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Garvin JL, Hong NJ. Nitric oxide inhibits sodium/hydrogen exchange activity in the thick ascending limb. Am J Physiol Renal Physiol 277: F377–F382, 1999. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Vicente A, Garvin JL. Angiotensin II-induced hypertension increases plasma membrane Na pump activity by enhancing Na entry in rat thick ascending limbs. Am J Physiol Renal Physiol 305: F1306–F1314, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, Seth DM, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ACE protects against hypertension. J Clin Invest 123: 2011–2023, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Granger JP, Alexander BT. Abnormal pressure-natriuresis in hypertension: role of nitric oxide. Acta Physiol Scand 168: 161–168, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Granger JP, Schnackenberg CG. Renal mechanisms of angiotensin II-induced hypertension. Semin Nephrol 20: 417–425, 2000. [PubMed] [Google Scholar]
- 28.Hamaguchi A, Kim S, Yano M, Yamanaka S, Iwao H. Activation of glomerular mitogen-activated protein kinases in angiotensin II-mediated hypertension. J Am Soc Nephrol 9: 372–380, 1998. [DOI] [PubMed] [Google Scholar]
- 29.Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric oxide synthase in response to bradykinin stimulation. J Biol Chem 276: 16587–16591, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Harrison-Bernard LM, El-Dahr SS, O'Leary DF, Navar LG. Regulation of angiotensin II type 1 receptor mRNA and protein in angiotensin II-induced hypertension. Hypertension 33: 340–346, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Herrera M, Garvin JL. Angiotensin II stimulates thick ascending limb NO production via AT(2) receptors and Akt1-dependent nitric oxide synthase 3 (NOS3) activation. J Biol Chem 285: 14932–14940, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herrera M, Garvin JL. Endothelin stimulates endothelial nitric oxide synthase expression in the thick ascending limb. Am J Physiol Renal Physiol 287: F231–F235, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Herrera M, Garvin JL. A high-salt diet stimulates thick ascending limb eNOS expression by raising medullary osmolality and increasing release of endothelin-1. Am J Physiol Renal Physiol 288: F58–F64, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Herrera M, Hong NJ, Ortiz PA, Garvin JL. Endothelin-1 inhibits thick ascending limb transport via Akt-stimulated nitric oxide production. J Biol Chem 284: 1454–1460, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrera M, Silva G, Garvin JL. A high-salt diet dissociates NO synthase-3 expression and NO production by the thick ascending limb. Hypertension 47: 95–101, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Herrera M, Silva GB, Garvin JL. Angiotensin II stimulates thick ascending limb superoxide production via protein kinase Cα-dependent NADPH oxidase activation. J Biol Chem 285: 21323–21328, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirata K, Kuroda R, Sakoda T, Katayama M, Inoue N, Suematsu M, Kawashima S, Yokoyama M. Inhibition of endothelial nitric oxide synthase activity by protein kinase C. Hypertension 25: 180–185, 1995. [DOI] [PubMed] [Google Scholar]
- 38.Hong NJ, Garvin JL. Angiotensin II type 2 receptor-mediated inhibition of NaCl absorption is blunted in thick ascending limbs from Dahl salt-sensitive rats. Hypertension 60: 765–769, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang X, Yang F, Tan H, Liao D, Bryan RM Jr, Randhawa JK, Rumbaut RE, Durante W, Schafer AI, Yang X, Wang H. Hyperhomocystinemia impairs endothelial function and eNOS activity via PKC activation. Arterioscler Thromb Vasc Biol 25: 2515–2521, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kashiwagi S, Atochin DN, Li Q, Schleicher M, Pong T, Sessa WC, Huang PL. eNOS phosphorylation on serine 1176 affects insulin sensitivity and adiposity. Biochem Biophys Res Commun 431: 284–290, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim YH, Hwang JH, Noh JR, Gang GT, Kim do H, Son HY, Kwak TH, Shong M, Lee IK, Lee CH. Activation of NAD(P)H:quinone oxidoreductase ameliorates spontaneous hypertension in an animal model via modulation of eNOS activity. Cardiovasc Res 91: 519–527, 2011. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi N, Mori Y, Nakano S, Tsubokou Y, Kobayashi T, Shirataki H, Matsuoka H. TCV-116 stimulates eNOS and caveolin-1 expression and improves coronary microvascular remodeling in normotensive and angiotensin II-induced hypertensive rats. Atherosclerosis 158: 359–368, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Kolluru GK, Siamwala JH, Chatterjee S. eNOS phosphorylation in health and disease. Biochimie 92: 1186–1198, 2010. [DOI] [PubMed] [Google Scholar]
- 44.Kopkan L, Castillo A, Navar LG, Majid DS. Enhanced superoxide generation modulates renal function in ANG II-induced hypertensive rats. Am J Physiol Renal Physiol 290: F80–F86, 2006. [DOI] [PubMed] [Google Scholar]
- 45.Li D, Tomson K, Yang B, Mehta P, Croker BP, Mehta JL. Modulation of constitutive nitric oxide synthase, bcl-2 and Fas expression in cultured human coronary endothelial cells exposed to anoxia-reoxygenation and angiotensin II: role of AT1 receptor activation. Cardiovasc Res 41: 109–115, 1999. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Xie ZZ, Tang YB. Genistein prevents myocardial hypertrophy in 2-kidney 1-clip renal hypertensive rats by restoring eNOS pathway. Pharmacology 86: 240–248, 2010. [DOI] [PubMed] [Google Scholar]
- 47.Liu J, Gao Y, Negash S, Longo LD, Raj JU. Long-term effects of prenatal hypoxia on endothelium-dependent relaxation responses in pulmonary arteries of adult sheep. Am J Physiol Lung Cell Mol Physiol 296: L547–L554, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsubara M, Hayashi N, Jing T, Titani K. Regulation of endothelial nitric oxide synthase by protein kinase C. J Biochem 133: 773–781, 2003. [DOI] [PubMed] [Google Scholar]
- 49.McCubbin JW, DeMoura RS, Page IH, Olmsted F. Arterial hypertension elicited by subpressor amounts of angiotensin. Science 149: 1394–1395, 1965. [DOI] [PubMed] [Google Scholar]
- 50.Miyata N, Cowley AW Jr. Renal intramedullary infusion of l-arginine prevents reduction of medullary blood flow and hypertension in Dahl salt-sensitive rats. Hypertension 33: 446–450, 1999. [DOI] [PubMed] [Google Scholar]
- 51.Miyata N, Zou AP, Mattson DL, Cowley AW Jr. Renal medullary interstitial infusion of l-arginine prevents hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 275: R1667–R1673, 1998. [DOI] [PubMed] [Google Scholar]
- 52.Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res 90: E58–E65, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Moreno C, Lopez A, Llinas MT, Rodriguez F, Lopez-Farre A, Nava E, Salazar FJ. Changes in NOS activity and protein expression during acute and prolonged ANG II administration. Am J Physiol Regul Integr Comp Physiol 282: R31–R37, 2002. [DOI] [PubMed] [Google Scholar]
- 54.Navar LG, Ichihara A, Chin SY, Imig JD. Nitric oxide-angiotensin II interactions in angiotensin II-dependent hypertension. Acta Physiol Scand 168: 139–147, 2000. [DOI] [PubMed] [Google Scholar]
- 55.Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol 305: F510–F519, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olson SC, Dowds TA, Pino PA, Barry MT, Burke-Wolin T. ANG II stimulates endothelial nitric oxide synthase expression in bovine pulmonary artery endothelium. Am J Physiol Lung Cell Mol Physiol 273: L315–L321, 1997. [DOI] [PubMed] [Google Scholar]
- 57.Ortiz PA, Garvin JL. Autocrine effects of nitric oxide on HCO3− transport by rat thick ascending limb. Kidney Int 58: 2069–2074, 2000. [DOI] [PubMed] [Google Scholar]
- 58.Ortiz PA, Garvin JL. NO inhibits NaCl absorption by rat thick ascending limb through activation of cGMP-stimulated phosphodiesterase. Hypertension 37: 467–471, 2001. [DOI] [PubMed] [Google Scholar]
- 59.Ortiz PA, Hong NJ, Garvin JL. Luminal flow induces eNOS activation and translocation in the rat thick ascending limb. Am J Physiol Renal Physiol 287: F274–F280, 2004. [DOI] [PubMed] [Google Scholar]
- 60.Ortiz PA, Hong NJ, Garvin JL. Luminal flow induces eNOS activation and translocation in the rat thick ascending limb. II. Role of PI3-kinase and Hsp90. Am J Physiol Renal Physiol 287: F281–F288, 2004. [DOI] [PubMed] [Google Scholar]
- 61.Ortiz PA, Hong NJ, Garvin JL. NO decreases thick ascending limb chloride absorption by reducing Na+-K+-2Cl− cotransporter activity. Am J Physiol Renal Physiol 281: F819–F825, 2001. [DOI] [PubMed] [Google Scholar]
- 62.Ortiz PA, Hong NJ, Wang D, Garvin JL. Gene transfer of eNOS to the thick ascending limb of eNOS-KO mice restores the effects of l-arginine on NaCl absorption. Hypertension 42: 674–679, 2003. [DOI] [PubMed] [Google Scholar]
- 63.Oyama N, Yagita Y, Sasaki T, Omura-Matsuoka E, Terasaki Y, Sugiyama Y, Sakoda S, Kitagawa K. An angiotensin II type 1 receptor blocker can preserve endothelial function and attenuate brain ischemic damage in spontaneously hypertensive rats. J Neurosci Res 88: 2889–2898, 2010. [DOI] [PubMed] [Google Scholar]
- 64.Passmore JC, Jimenez AE. Effect of chloride on renal blood flow in angiotensin II induced hypertension. Can J Physiol Pharmacol 69: 507–511, 1991. [DOI] [PubMed] [Google Scholar]
- 65.Patel AR, Granger JP, Kirchner KA. l-Arginine improves transmission of perfusion pressure to the renal interstitium in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 266: R1730–R1735, 1994. [DOI] [PubMed] [Google Scholar]
- 66.Perez-Rojas JM, Kassem KM, Beierwaltes WH, Garvin JL, Herrera M. Nitric oxide produced by endothelial nitric oxide synthase promotes diuresis. Am J Physiol Regul Integr Comp Physiol 298: R1050–R1055, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Plato CF, Garvin JL. α2-Adrenergic-mediated tubular NO production inhibits thick ascending limb chloride absorption. Am J Physiol Renal Physiol 281: F679–F686, 2001. [DOI] [PubMed] [Google Scholar]
- 68.Plato CF, Pollock DM, Garvin JL. Endothelin inhibits thick ascending limb chloride flux via ETB receptor-mediated NO release. Am J Physiol Renal Physiol 279: F326–F333, 2000. [DOI] [PubMed] [Google Scholar]
- 69.Plato CF, Shesely EG, Garvin JL. eNOS mediates l-arginine-induced inhibition of thick ascending limb chloride flux. Hypertension 35: 319–323, 2000. [DOI] [PubMed] [Google Scholar]
- 70.Plato CF, Stoos BA, Wang D, Garvin JL. Endogenous nitric oxide inhibits chloride transport in the thick ascending limb. Am J Physiol Renal Physiol 276: F159–F163, 1999. [DOI] [PubMed] [Google Scholar]
- 71.Ramseyer VD, Cabral PD, Garvin JL. Role of endothelin in thick ascending limb sodium chloride transport. Contrib Nephrol 172: 76–83, 2011. [DOI] [PubMed] [Google Scholar]
- 72.Ramseyer VD, Garvin JL. Angiotensin II decreases nitric oxide synthase 3 expression via nitric oxide and superoxide in the thick ascending limb. Hypertension 53: 313–318, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ramseyer VD, Hong NJ, Garvin JL. Tumor necrosis factor alpha decreases nitric oxide synthase type 3 expression primarily via Rho/Rho kinase in the thick ascending limb. Hypertension 59: 1145–1150, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rios NB, Esparragon FR, Rodriguez Perez JC. Telmisartan-induced eNOS gene expression is partially independent of its PPAR-gamma agonist property. Clin Invest Med 35: E55–E64, 2012. [DOI] [PubMed] [Google Scholar]
- 75.Rugale C, Delbosc S, Cristol JP, Mimran A, Jover B. Sodium restriction prevents cardiac hypertrophy and oxidative stress in angiotensin II hypertension. Am J Physiol Heart Circ Physiol 284: H1744–H1750, 2003. [DOI] [PubMed] [Google Scholar]
- 76.Sigmon DH, Beierwaltes WH. Influence of nitric oxide in the chronic phase of two-kidney, one clip renovascular hypertension. Hypertension 31: 649–656, 1998. [DOI] [PubMed] [Google Scholar]
- 77.Signorello MG, Segantin A, Passalacqua M, Leoncini G. Homocysteine decreases platelet NO level via protein kinase C activation. Nitric Oxide 20: 104–113, 2009. [DOI] [PubMed] [Google Scholar]
- 78.Silva GB, Garvin JL. Akt1 mediates purinergic-dependent NOS3 activation in thick ascending limbs. Am J Physiol Renal Physiol 297: F646–F652, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Silva GB, Garvin JL. Angiotensin II-dependent hypertension increases Na transport-related oxygen consumption by the thick ascending limb. Hypertension 52: 1091–1098, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Szentivanyi M Jr, Zou AP, Mattson DL, Soares P, Moreno C, Roman RJ, Cowley AW Jr. Renal medullary nitric oxide deficit of Dahl S rats enhances hypertensive actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol 283: R266–R272, 2002. [DOI] [PubMed] [Google Scholar]
- 81.Tambascia RC, Fonseca PM, Corat PD, Moreno H Jr, Saad MJ, Franchini KG. Expression and distribution of NOS1 and NOS3 in the myocardium of angiotensin II-infused rats. Hypertension 37: 1423–1428, 2001. [DOI] [PubMed] [Google Scholar]
- 82.Thomas SR, Chen K, Keaney JF Jr. Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem 277: 6017–6024, 2002. [DOI] [PubMed] [Google Scholar]
- 83.Varela M, Herrera M, Garvin JL. Inhibition of Na-K-ATPase in thick ascending limbs by NO depends on O2− and is diminished by a high-salt diet. Am J Physiol Renal Physiol 287: F224–F230, 2004. [DOI] [PubMed] [Google Scholar]
- 84.Von Thun AM, Vari RC, el-Dahr SS, Navar LG. Augmentation of intrarenal angiotensin II levels by chronic angiotensin II infusion. Am J Physiol Renal Fluid Electrolyte Physiol 266: F120–F128, 1994. [DOI] [PubMed] [Google Scholar]
- 85.Wang M, Luan H, Wu P, Fan L, Wang L, Duan X, Zhang D, Wang WH, Gu R. Angiotensin II stimulates basolateral 50-pS K channels in the thick ascending limb. Am J Physiol Renal Physiol 306: F509–F516, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wickman A, Andersson IJ, Jia J, Hedin L, Bergstrom G. Endothelial nitric oxide synthase protein is reduced in the renal medulla of two-kidney, one-clip hypertensive rats. J Hypertens 19: 1665–1673, 2001. [DOI] [PubMed] [Google Scholar]
- 87.Xu L, Liu Y. Administration of telmisartan reduced systolic blood pressure and oxidative stress probably through the activation of PI3K/Akt/eNOS pathway and NO release in spontaneously hypertensive rats. Physiol Res 62: 351–359, 2013. [DOI] [PubMed] [Google Scholar]