

Abstract
Sepsis is a life-threatening clinical condition that is particularly serious among the elderly who experience considerably higher mortality rates compared with younger patients. Using a sterile endotoxemia model, we previously reported age-dependent mortality in conjunction with enhanced coagulation and insufficient levels of anti-coagulant factor activated protein C (aPC). The purpose of the present study was to further investigate the mechanisms for age-dependent coagulation and aPC insufficiency during experimental sepsis. Intra-abdominal sepsis was induced by cecal ligation and puncture (CLP) using 21 or 16 gauge (G) needles (double-puncture) on young (4 to 6 mo old) and aged (20 to 25 mo old) male C57BL/6 mice. When compared with young mice, aged mice showed significantly increased mortality (92% vs. 28%), systemic inflammation, and coagulation in the lung and kidney after 21G CLP. Young mice with more severe CLP (16G) showed a mortality rate and inflammation equivalent to aged mice with 21G CLP; however, enhanced coagulation and kidney dysfunction were significant only in the aged. In young mice, increased levels of aPC after CLP were coupled with reduced levels of protein C (PC), suggesting the conversion of PC to aPC; however, PC and aPC levels remained unchanged in aged mice, indicating a lack of PC to aPC conversion. Activation of fibrinolysis, determined by plasma d-dimer levels, was similar regardless of age or CLP severity, and plasminogen activator inhibitor-1, an inhibitor of fibrinolysis, showed severity-dependent induction independent of age. These results suggest that enhanced coagulation in aged mice during sepsis is due to dysfunction of the PC activation mechanism.
Keywords: aging, cecal ligation and puncture, coagulation, protein C, renal dysfunction, sepsis
sepsis is an infection-initiated systemic inflammatory syndrome that can be catastrophic for the body, causing multi-organ failure and leading to death in severe cases. There are ∼900,000 cases of sepsis annually in the United States, among which 60% include a diagnosis of severe sepsis with organ failure (8, 15). An estimated 58–65% of sepsis cases in the United States occur in the over-65-age group (2, 8, 21) with mortality rates of 30–40% (2, 21). Although incidence rates and deaths due to sepsis are well documented to increase with age (2, 8, 15, 21), the underlying mechanisms for this age-associated vulnerability are not well understood (32). Sepsis can be caused by either Gram-positive or Gram-negative bacteria (20). Despite the growing incidence and prevalence of Gram-positive bacterial infection in sepsis cases, Gram-negative bacterial infections are more common in the elderly (21). The epidemiological data support the idea that mechanisms of infection and injury may be different in aging patients.
The clinical manifestations of sepsis are highly variable and include fever or hypothermia, tachycardia, tachypnea, hypotension, leukocytosis or leucopenia, elevated inflammatory mediators, and an array of organ dysfunction variables including coagulation abnormalities (7, 12). Pathologically, sepsis is characterized by a systemic inflammatory response resulting in tissue damage and cell death, along with a procoagulant state leading to disseminated intravascular coagulation (DIC) (3, 16). Abundant clinical data suggest that there is an age-related increase in both inflammation and thrombosis, putting elderly patients at a higher risk of developing severe sepsis and succumbing to the disease (6, 11, 13, 24, 32). Studies using laboratory animals have reproduced these findings, including our previous study, which suggests that the microvasculature from aged mice is more sensitive to inflammatory mediators present in the circulation of patients with sepsis (34).
The protein C (PC) pathway is a negative feedback mechanism that regulates coagulation (10, 17, 18, 37, 39). The main player of this pathway is activated protein C (aPC), which proteolytically cleaves coagulation factors Va and VIIIa to arrest thrombin generation and subsequent fibrin formation. In addition to its role as an anticoagulant, aPC also has anti-inflammatory and cytoprotective functions. Inactive PC is converted to aPC by the binding of thrombin; this activation process is augmented more than 1,000-fold when thrombin binds to its receptor, thrombomodulin (TM), and a further 10- to 20-fold when PC binds to endothelial protein C receptor (EPCR) (39).
Using a sterile endotoxemia model induced by intraperitoneal injection with lipopolysaccharide (LPS) to young and aged mice, we previously demonstrated age-dependent mortality, in conjunction with enhanced fibrin deposition and insufficient activation of the anti-coagulant PC pathway (33). The age-dependent enhanced coagulation and loss of PC pathway function held true even when young mice were given a far more lethal dose of LPS, which induced a mortality rate equivalent to that of the aged mice. Specifically, we observed profound downregulation of TM by vascular cells and significantly low plasma concentrations of aPC in aged mice, compared with young, with endotoxemia (33).
Although our previous study showed distinct differences in the level of fibrin deposition and PC pathway activation in endotoxic mice by aging, two important questions remain unanswered. First, it is not known whether enhanced coagulation in the aged is a result of suppressed PC pathway function (reduced anti-coagulant activity resulting in excess fibrin deposition) (33) or reduced fibrinolysis (impaired fibrin clearance, known to occur in the aged) (40). It is also unclear whether low aPC levels in the aged are a result of reduced PC activation or increased aPC consumption relative to demand. Additionally, confirmation that our previous observation where we used a sterile endotoxemia model is reproducible in bacterial sepsis has not yet been reported. In the present study, we attempted to resolve these questions using a more clinically relevant animal model of intra-abdominal polymicrobial sepsis induced by cecal-ligation and puncture (CLP).
In this study, we compared several aspects of sepsis pathophysiology (including inflammation, hypothermia, tissue injury, and coagulation) between young and aged mice with abdominal sepsis induced by CLP with a 21-gauge (G) needle. We previously demonstrated that aged mice, compared with young mice that received an equally severe injury by CLP, exhibit higher mortality rates accompanied with more severe systemic inflammation and hypothermia (30). Therefore, in the current study, we included an additional group of young mice that received a more severe injury by CLP with a 16G needle. Young mice with 16G-CLP showed a similar mortality rate as aged mice with 21G-CLP; this enabled us to determine whether age-associated differences in sepsis pathophysiology are caused by advanced age or secondary to age-associated mortality/severity of sepsis.
MATERIALS AND METHODS
Animals.
Young (4 to 6 mo old) and aged (20 to 25 mo old) male C57BL/6 mice were obtained from the National Institute on Aging. All mice were maintained in pressurized intraventilated (PIV) cages (maximum 5 per cage) in an environment under controlled temperature (21–23°C), humidity (30–70%), and lighting (14 h light/10 h dark) with free access to drinking water and chow (Rodent Diet No. 2500; LabDiet, St. Louis, MO). Mice were acclimated for at least 7 days before experimentation. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Kentucky and performed in accord with the National Institutes of Health guidelines for ethical animal treatment.
Animal model of sepsis.
A commonly used mouse model of acute peritonitis induced by CLP was used in this study. For the CLP procedure, mice were deeply anesthetized by isoflurane inhalation, the abdominal cavity was opened, and the distal 1 cm of the cecum ligated with suture and punctured twice with a needle [21-gauge (21G) or 16-gauge (16G)]. After surgery, all mice received a 1-ml subcutaneous injection of warmed physiologic saline. Sham-operated mice, as controls, received the same procedures except for ligation and puncture. Body temperature, a parameter for the severity of sepsis (30), was assessed by rectal temperature probe with a digital thermometer (Precision Thermometer 4600; YSI). For survival studies, mortality of each mouse was monitored daily for 10 days after CLP or sham surgery. Major organs from dead mice were examined, and animals with any evident signs of tumor were excluded from the study.
Sample collection.
For tissue protein analysis, mice were deeply anesthetized by isoflurane inhalation and intraperitoneally injected with heparin (50 units per mouse; APP Pharmaceuticals, Schaumburg, IL) 5 min before laparotomy. Heparin was administered to prevent coagulation during blood collection and tissue harvesting. Immediately after laparotomy, blood was collected from the inferior vena cava with a heparin-coated syringe. Subsequently, the inferior vena cava was cut, and the entire vasculature was perfused with physiological saline through the cardiac ventricles, for the purpose of eliminating circulating cells, and each tissue was harvested, flash frozen in liquid nitrogen, and stored at −80°C. For analysis of coagulation-related factors such as aPC, heparin was not injected and blood was collected with 10% volume of 0.1 M sodium citrate. The collected blood was immediately centrifuged at 4°C to obtain plasma, which was stored at −80°C.
White blood cell count.
Whole blood from each mouse was mixed with Türk's solution, and red blood cells were lysed. The number of white blood cells (WBC) in each sample were determined by using a hemocytometer under a microscope (Nikon Eclipse E200; Nikon Instruments, Melville, NY).
Plasma analyses.
Plasma creatinine and alanine aminotransferase (ALT) concentrations were measured by colorimetric method using QuantiChrom Creatinine assay kit (Bioassay Systems, Hayward, CA) and ALT (SGPT) Reagent (BQ Kits, San Diego, CA), respectively. Plasma concentrations of IL-6, d-dimer, and plasminogen activator inhibitor-1 (PAI-1) were measured by enzyme-linked immunosorbent assay (ELISA) using kits purchased from Thermo Scientific (Rockford, IL), Stago (Asnie′res, France), and Molecular Innovations (Novi, MI), respectively.
Western blot analysis.
Protein was extracted from individual frozen tissues and Western blot analysis performed as previously described (36). Plasma was diluted 25× in lysis buffer with protease inhibitors before running on the gel. The membranes were incubated with anti-fibrin monoclonal antibody 59D8 (29), anti-TM antibody (sc-7079; Santa Cruz), anti-EPCR antibody (sc-18062), anti-PC antibody (sc-73466), or anti-SP-D antibody (sc-13980). Chemiluminescent detection was performed using ECL detection reagents (GE Healthcare). All membranes were reprobed with anti-GAPDH (sc-20357) or anti-β-actin antibody (A1978; Sigma) for normalization. Intensity of each protein band was determined by densitometric analysis (Carestream Molecular Imaging Software, Carestream, New Haven, CT). Although Western blot images shown in this article represent pooled samples run on mini gels (15 wells) using the XCell SureLock Mini-Cell Apparatus (Invitrogen); individual samples were run together on larger gels (25 wells) using the XCell SureLock Midi-Cell Apparatus for densitometric and statistical analysis.
aPC immunocapture assay.
As previously described, blood samples were taken from the inferior vena cava and mixed with 0.1 M sodium citrate and 0.3 M benzamidine-HCl (Sigma-Aldrich) at a ratio of 1/10 and centrifuged at 2,000 g for 10 min at 4°C (33). The concentration of plasma aPC was determined by using an immunocapture assay (19) except for the substitution of chromogenic substrate S2366 (0.46 mM in TBS, pH 7.5) (DiaPharma, West Chester, OH) for Spectrozyme PCA as we previously described (33).
Statistical analysis.
All data are expressed as means and SD. Survival curves were analyzed by Kaplan Meier LogRank test using SigmaPlot Statistical Software version 11.0 (Systat Software). ANOVA models were fit using JMP (SAS Institute, Cary, NC) without assuming equal variance, thus Welch's test was used to determine statistical significance. A P value <0.05 was considered statistically significant.
RESULTS
Severity of sepsis in young and aged mice after CLP.
Young (4–6 mo, n = 18) and aged (20–25 mo, n = 12) male C57BL/6 mice were subjected to double puncture CLP with a 21G needle. Another group of young mice (n = 13) received a more severe injury by double-puncture CLP with a larger needle (16G). Survival of mice in these three groups was monitored for at least 10 days (Fig. 1A). No mortality was observed in young mice for the first 3 days after 21G CLP; by the end of the 10-day observation period, mortality in young mice with 21G CLP rose to 28%. The mortality rate of young mice with 16G CLP was 23% within 24 h, 85% by day 2, and 92% by day 10. The mortality rate of aged mice with 21G CLP was 0% within 24 h, 42% by day 2, and 92% by day 10. The survival patterns of young mice with 16G CLP and aged mice with 21G CLP were statistically similar (P = 0.189), and both patterns were statistically distinct from the survival pattern of young mice with 21G CLP (P < 0.001). No mortality was observed in age-matched young and aged sham-operated mice used as controls (data not shown). To compare the severity of sepsis among the groups, body temperature (Fig. 1B), white blood cell (WBC) count (Fig. 1C), and plasma level of IL-6 (Fig. 1D) were measured 24 h after CLP in a separate set of mice. All mice that received CLP surgery exhibited signs of hypothermia (P < 0.001 for all CLP groups at both 6 and 24 h). Young mice with 16G CLP and aged mice with 21G CLP exhibited significantly more profound hypothermia than young mice with 21G CLP, with average body temperatures of 29°C and 31°C, respectively. There was no significant difference in the body temperatures of young mice with 16G CLP and aged mice with 21G CLP (P = 0.102). WBC count in both young and aged mice were markedly decreased 24 h after CLP regardless of age or mortality (P < 0.001, compared with sham operation). There was no significant difference in WBC count among the three CLP groups.
Fig. 1.

Severity of sepsis in young and aged mice after cecal ligation and puncture (CLP). A: survival study demonstrating age-dependent mortality during CLP-induced sepsis. Young and aged male C57BL/6 mice received CLP surgery with a 16-gauge (16G) or 21-gauge (21G) needle, and survival was monitored for 10 days (N = 12–18/group). There was no statistical difference between the survival curve patterns of aged mice with 21G CLP and young mice with 16G CLP (P = 0.189). No mortality was observed in age-matched sham-operated mice (data not shown). B: hypothermia of CLP-operated mice 0, 6, and 24 h after surgery. There was no statistical difference between body temperatures of aged mice with 21G CLP and young mice with 16G CLP (P = 0.102 and P = 0.380 for 6 and 24 h, respectively). C: white blood cell (WBC) count of sham- and CLP-operated mice 24 h after surgery. There was no statistical difference among the CLP groups (P = 0.68). D: circulating levels of IL-6 in sham- and CLP-operated mice 24 h after surgery. There was no statistical difference between plasma IL-6 level of aged mice with 21G CLP and young mice with 16G CLP (P = 0.27). Values are means ± SD; n = 5–8 for each group. *Statistical significance compared with sham-operated mice of the same age; †statistical significance compared with young mice with 21G CLP. One, two, or three symbols signify P < 0.05, 0.01, or 0.001, respectively.
The plasma level of inflammatory cytokine IL-6 increased 24 h after 21G CLP in young mice (P = 0.050, compared with young sham), 21G CLP in aged mice (P = 0.059, compared with aged sham), and 16G CLP in young mice (P = 0.045, compared with young sham). Although the average value was higher in young mice with 16G CLP, there was no statistical difference in IL-6 level between young mice with 16G CLP and aged mice with 21G CLP (P = 0.140; Fig. 1D).
Age-dependent organ dysfunction during CLP-induced sepsis.
To assess the extent of organ injury in mice after CLP, we measured plasma levels of creatinine (a kidney injury marker), ALT (a liver injury marker), and surfactant protein-D (SP-D, a lung injury marker) (25, 38). Plasma creatinine concentration was significantly elevated only in aged mice 24 h after 21G CLP (P = 0.041; Fig. 2A). A few (2 of 5) young mice with 16G CLP showed elevated creatinine levels, whereas no young mice with 21G CLP showed increased creatinine levels compared with sham-operated mice. Plasma ALT level was slightly elevated in all CLP groups at 24 h; however, none reached significance (Fig. 2B). Plasma SP-D, measured by Western blot analysis, was significantly increased by CLP in all groups; there was no significant difference among the three CLP groups (i.e., young with 21G, aged with 21G, and young with 16G) (Fig. 2C).
Fig. 2.

Age-dependent organ dysfunction during CLP-induced sepsis. Young and aged male C57BL/6 mice received CLP surgery with a 16G or 21G needle. Age-matched sham-operated mice were used as controls. Concentrations of creatinine (A), alanine aminotransferase (ALT; B), and relative levels of surfactant protein-D (SP-D; C) were measured in the plasma 24 h after CLP or sham surgery. Values are means ± SD; n = 4–8 for each group. *Statistical significance compared with sham-operated mice of the same age; †statistical significance compared with young mice with 21G CLP. One or two symbols signify P < 0.05 or 0.01, respectively.
Coagulation is uniquely increased in the lung and kidney of aged animals after CLP.
Fibrin formation in the lung and kidney during CLP-induced sepsis was assessed to examine age-associated changes in microvascular coagulation. Western blot analysis was performed to detect fibrin formation using a fibrin-specific monoclonal antibody 59D8 (Fig. 3A). Fibrin formation in the lung (Fig. 3B) and kidney (Fig. 3C) clearly increased in aged mice 24 h after 21G CLP; fibrin formation was not observed in young mice with 21G or 16G CLP. The age-associated increase in fibrin deposition was statistically significant in the lung (P < 0.05) and kidney (P < 0.01) compared with young mice with 21G or 16G CLP. These results suggest that aged mice are significantly more prone to sepsis-induced coagulation than young mice, regardless of the mortality rate.
Fig. 3.

Age-dependent coagulation in the lung and kidney during sepsis. Young and aged male C57BL/6 mice received CLP surgery with a 16G or 21G needle. Age-matched sham-operated mice were used as controls. A–C: fibrin deposition in lung (B) and kidney (C) 24 h after CLP or sham surgery was assessed by Western blot analysis and quantified by densitometric analysis of individually run protein samples. Blot images shown represent pooled protein samples derived equally from 4 to 5 mice; however, individual samples were run and blotted separately for statistical analyses. Membranes were reprobed for GAPDH to assure equal protein loading, and quantified fibrin levels were adjusted for GAPDH. This analysis was performed twice in different sets of mice with the same result. Plasma levels of fibrinolytic markers d-dimer (D) and plasminogen activator inhibitor (PAI)-1 (E) at 6 and 24 h after CLP or sham surgery are shown. Values are means ± SD; n = 4–8. *Statistical significance compared with sham-operated mice of the same age; †statistical significance compared with young mice with 21G CLP; #statistical significance compared with aged mice with 21G CLP. One, two, or three symbols signify P < 0.05, 0.01, or 0.001, respectively.
To determine whether enhanced coagulation in the aged during sepsis was a result of inhibition of fibrinolysis, plasma levels of fibrinolytic markers d-dimer (a fibrin degradation product) and PAI-1 (an inhibitor of fibrinolysis) were measured. d-dimer levels were elevated in all groups 24 h after CLP, although only aged mice showed a statistically significant elevation (6.5 ± 5.1 ng/ml to 94.5 ± 30.3 ng/ml, P = 0.0002; Fig. 3D). d-dimer levels of young mice rose from 6.2 ± 2.5 ng/ml in sham-operated mice to 64.1 ± 57.7 ng/ml with 21G CLP and 42.3 ± 29.2 ng/ml with 16G CLP (P = 0.074). d-dimer levels at 6 h showed no significant induction compared with sham for either age-group. Plasma levels of PAI-1 were elevated in all animals after CLP-surgery with a trend toward higher levels in more severe sepsis (aged with 21G CLP and young with 16G CLP). At 24 h after CLP, young mice with 21G CLP and aged mice with 21G CLP showed significantly elevated PAI-1 levels (P = 0.003 and P = 0.015, respectively; Fig. 3E). PAI-1 induction in young mice with 16G CLP was not significant (P = 0.077). There was no significant difference in PAI-1 levels among the three CLP groups.
Reduced activation of protein C (PC) in the aged during CLP-induced sepsis.
To assess whether activated protein C (aPC) levels are altered by age during sepsis, we measured the level of aPC in plasma samples obtained from young and aged mice after CLP (Fig. 4). Plasma aPC levels of young and aged mice after sham operation were low (2.4 ± 1.5 ng/ml and 3.0 ± 2.3 ng/ml, respectively), and there was no significant difference between these groups (P = 0.84). Plasma aPC levels increased significantly in young mice after 21G CLP (8.2 ± 1.7 ng/ml; P = 0.001 compared with young mice after sham operation). This CLP-induced aPC elevation was further augmented when young mice were subjected to 16G CLP (20.2 ± 10.3 ng/ml; P = 0.018 compared with young mice after sham operation; P = 0.059 compared with young mice after 21G CLP). However, elevation of aPC was not seen in aged mice after 21G CLP (2.1 ± 1.3 ng/ml; P = 0.474 compared with aged sham). Thus, after CLP, plasma aPC levels of aged mice were significantly lower than those of young mice with CLP (P = 0.001 compared with young 21G CLP; P = 0.017 compared with young 16G CLP).
Fig. 4.
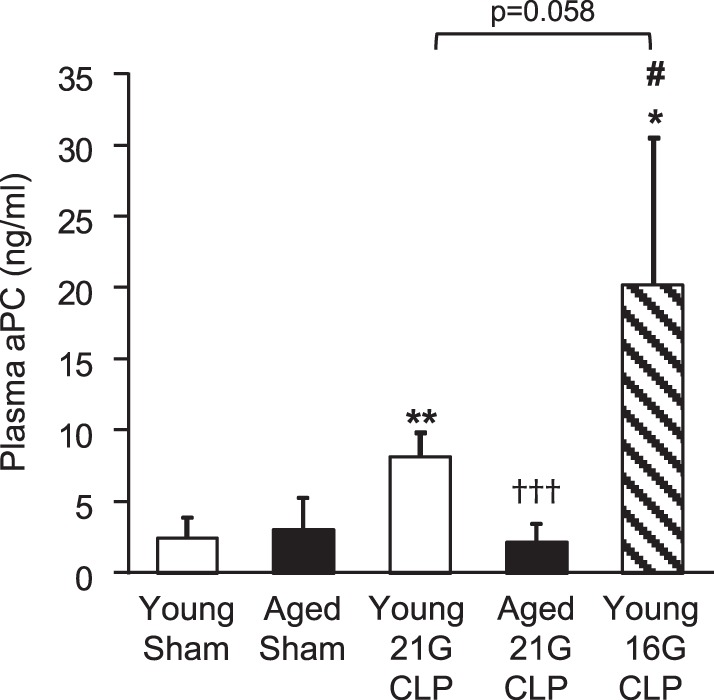
Plasma levels of activated protein C (aPC) in young and aged mice during sepsis. Young and aged male C57BL/6 mice received CLP surgery with a 16G or 21G needle. Age-matched sham-operated mice were used as controls. Plasma aPC level 24 h after surgery was measured by immunocapture assay. This analysis was performed twice in different sets of mice with the similar results. Values are means ± SD; n = 4 to 5. *Statistical significance compared with sham-operated mice of the same age; †statistical significance compared with young mice with 21G CLP; #statistical significance compared with aged mice with 21G CLP. One, two, or three symbols signify P < 0.05, 0.01, or 0.001, respectively.
Analysis of PC pathway components during CLP-induced sepsis.
To evaluate whether reduced PC activation in the aged during sepsis was due to altered expression of PC pathway components, we compared the level of TM in the lungs of young and aged mice 24 h after CLP by Western blotting (Fig. 5A). We analyzed TM in the lungs because this organ is the major site for TM production and expression. In young mice, the level of TM did not change after 21G CLP but showed a modest 10% decrease after 16G CLP; this decrease was not statistically significant (P = 0.302). In aged mice, the level of TM in the lungs after 21G CLP decreased 10% compared with the sham level (P = 0.017). We also examined the levels of endothelial protein C receptor (EPCR) in these tissue samples. Although EPCR level did not change in young mice regardless of severity of CLP, there was a nearly twofold increase in EPCR levels in aged mice after CLP (P = 0.031). We then assessed the level of plasma PC in young and aged mice 24 h after CLP by Western blotting (Fig. 5D). Plasma PC levels were decreased 67% after young 21G CLP (P = 0.059) and 98% after young 16G CLP (P = 0.042). Aged mice showed no change in PC level after 21G CLP (P = 0.861).
Fig. 5.

Change in levels of protein C (PC) pathway components during sepsis. Young and aged male C57BL/6 mice received CLP surgery with a 16G or 21G needle. Age-matched sham-operated mice were used as controls. A–C: thrombomodulin (TM) and endothelial protein C receptor (EPCR) levels in the lung 24 h after surgery were assessed by Western blot analysis (pooled blot shown) and quantified by densitometric analysis of individually run protein samples. Each lane shown represents a pooled protein sample derived equally from 4 to 5 mice. Membranes were reprobed for β-actin to assure equal protein loading, and quantified levels were adjusted for β-actin. This analysis was performed twice in different sets of mice with the same result. D and E: PC level in the plasma 24 h after surgery was assessed by Western blot analysis (pooled blot shown) and quantified by densitometric analysis of individually run protein samples. This analysis was performed twice in different sets of mice with the same result. Values are means ± SD; n = 4 to 5. *Statistical significance compared with sham-operated mice of the same age; †statistical significance compared with young mice with 21G CLP; #statistical significance compared with aged mice with 21G CLP. One symbol signifies P < 0.05.
DISCUSSION
The present study describes, for the first time, age-dependent coagulation resulting from inefficient activation of PC in a clinically relevant polymicrobial mouse model of sepsis. Although age-dependent fibrin deposition and reduced PC activation were previously shown by our group using a sterile endotoxemia model with LPS (derived from Gram-negative bacteria), the mechanisms of enhanced coagulation and PC pathway dysfunction in the aged have not been investigated until now. The major purpose of this study was to answer two important questions: 1) Is enhanced coagulation in the aged a result of suppressed PC pathway function (reduced anti-coagulant activity resulting in excess fibrin deposition) or reduced fibrinolysis (impaired fibrin clearance); and 2) Are low aPC levels in the aged a result of inefficient PC activation or excessive aPC consumption. Additionally, as the mortality rate of aged mice is significantly higher than that of young mice given the same injury, an additional experimental group (Y 16G, young mice with a more severe injury yielding a mortality rate equivalent to aged mice) was added to distinguish whether the observed age-dependent responses are indeed a function of age rather than a secondary effect of increased mortality in the aged. This experimental design clearly demonstrated which aspects of CLP-induced sepsis physiology are truly age dependent (Table 1).
Table 1.
Comparison of sepsis physiology in young and aged mice with CLP-induced sepsis, indicating unique age-dependent conditions
| Survival | Temperature | White Blood Cell | IL-6 | Creatinine | Surfactant Protein-D | Alanine Aminotransferase | Fibrin | d-Dimer | Plasminogen Activator Inhibitor-1 | Activated Protein C | Protein C | Endothelial Protein C Receptor | Thrombomodulin | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Young mice | ||||||||||||||
| With 21G CLP | ↓ | ↓ | ↓ | ↑ | ↔ | ↑ | (↑) | ↔ | (↑) | ↑ | ↑ | (↓) | ↔ | ↔ |
| With 16G CLP | ↓↓ | ↓↓ | ↓ | ↑↑ | (↑) | ↑ | (↑) | ↔ | (↑) | (↑) | ↑↑ | ↓↓ | ↔ | ↔ |
| Aged mice with 21G CLP | ↓↓ | ↓↓ | ↓ | (↑↑) | ↑* | ↑ | (↑) | ↑↑* | ↑ | ↑ | ↔* | ↔* | ↑* | ↓* |
CLP, cecal-ligation and puncture; 21G, 21-gauge needle; 16G, 16-gauge needle. (↑) or (↓) A modest change without statistical significance;
a condition unique to aged mice.
Circulating d-dimer levels, a fibrin degradation product, were similarly elevated in both young and aged mice with CLP regardless of mortality rate, indicating active fibrinolysis in both age-groups. Circulating PAI-1 levels, a major pro-coagulant factor that inhibits fibrin degradation, were also increased by CLP without significant differences due to age or mortality. Despite similar d-dimer levels and similar levels of the fibrinolysis inhibitor PAI-1 in young and aged mice, only the aged showed enhanced fibrin deposition, suggesting that altered fibrinolysis is not a major factor contributing to exaggerated coagulation in the aged with CLP-induced sepsis. A recent study performing 20G CLP on young and aged mice also showed no difference in plasma PAI-1 levels 12 h after CLP (4). Sterile endotoxemia models appear to have a different effect on fibrinolysis. Yamamoto et al. (40) previously reported that circulating levels, and kidney and liver mRNA expression, of PAI-1 were increased in aged mice with endotoxemia, suggesting that enhanced coagulation in the aged is mediated by inhibited fibrinolysis. We also recently reported age-associated augmentation of PAI-1 mRNA expression in adipose tissue of endotoxic mice (31) and an age-dependent increased in PAI-1 protein concentration in the circulation of mice with acute pancreatitis (23). Taken together, these data indicate that inhibition of fibrinolysis may not play a major role in the age-dependent thrombosis observed during intra-abdominal sepsis.
The PC anti-coagulant pathway includes a number of factors that aid in the conversion of PC to aPC by proteolytic cleavage. Our previous study (33) using a sterile endotoxemia model indicated that profound downregulation (>80%) of TM by pulmonary vascular cells of aged mice was a major factor for low aPC levels and enhanced age-associated coagulation; however, the same mechanism does not appear to be in place with intra-abdominal sepsis. Although the levels of TM (and EPCR) appear sufficient in aged mice with CLP, aPC levels remained low, suggesting that another factor may be responsible for the low aPC levels observed in aged mice with CLP. These variations could be attributable to a number of differences due to the choice of animal model; for example, endotoxemia is induced by a Gram-negative bacterial component without an actual infection, whereas CLP causes a polymicrobial infection.
Low aPC levels could be the result of low aPC generation (i.e., reduced conversion of PC to aPC) or rapid aPC consumption (i.e., aPC produced but rapidly consumed). To verify these possibilities, we determined the levels of circulating inactivated PC in young and aged mice with sham or CLP operation. Because the half-life of inactive PC is relatively long (6–8 h) and the half-life of aPC very short (15–20 min) (14), reduced levels of PC due to consumption after CLP should be readily apparent. Although slightly lower in aged sham-operated mice, plasma PC levels were not significantly different by aging, indicating that baseline PC levels in aged mice are sufficient. After CLP, PC levels were significantly reduced in young mice in a severity-dependent fashion; however, PC levels remained constant in the aged. This result suggests that in young mice, the conversion of PC to aPC (aPC generation) results in reduced circulating PC levels, indicating consumption. However, in aged mice PC levels were not reduced, indicating that aging affects the conversion of PC to aPC, limiting aPC generation. We therefore conclude that PC is not sufficiently activated in aged mice during sepsis. This finding may explain why some elderly patients, despite controlled inflammation and significant fluid resuscitation, die from unmanageable thrombosis.
Several parameters for organ dysfunction in young and aged mice after CLP were evaluated in this study; among lung, liver, and kidney, only the kidney showed significant age-associated damage. Although we did not measure these organ dysfunction variables after LPS in our previous study (33), it seems reasonable that LPS, which causes high age-associated mortality much earlier than CLP, induced significant age-associated injury to both the lung and kidney, whereas age-related organ damage after CLP was predominant only in the kidney. Another recent study using a CLP model of severe sepsis in very young mice (2 mo old) showed that mortality was strongly associated with kidney injury (5). The data from these animal studies bear resemblance to clinical studies in which patients with an intra-abdominal source of infection are more likely to develop acute kidney injury (AKI) (26); AKI is also more common in the elderly with severe sepsis (27, 41). Predominant kidney injury as a result of CLP-induced sepsis may explain our finding that pulmonary TM levels were not so profoundly reduced after CLP as they were after LPS injection (a model known to produce lung injury) (33).
Although final mortality rates were similar between young mice with 16G CLP and aged mice with 21G CLP, young mice with a more severe injury would likely have a more severe acute infection at an earlier time-point than the aged mice with a milder injury. This is supported by the survival curve of these two groups, which shows higher mortality rates on day 1 and day 2 following CLP surgery, and higher plasma IL-6 and PAI-1 levels 24 h after CLP in the young mice with 16G CLP. We did not examine these parameters in aged mice with 16G CLP because they all die within 12 h of surgery (data not shown). We measured plasma IL-6 levels at 24 h, rather than the more frequently examined 6-h timepoint, which was suggested to predict later mortality (28). This choice of timepoint was based on our intention to assess the severity of systemic inflammation at the same timepoint, which shows significant age-associated differences in coagulation (Fig. 3). Although 24 h may not be the optimal timepoint for IL-6 analysis, our previous studies showed that IL-6 levels were similar at 6 and 24 h after CLP; thus earlier timepoints are not necessarily better (30). A previous study using the CLP-model on young and aged mice reported that high plasma cytokine levels correlated with high mortality rate, independent of age (35). Our data supports this finding, since we also found plasma IL-6 levels to be statistically similar between young mice with 16G CLP and aged mice with 21G CLP.
Despite similar levels of inflammatory markers, enhanced coagulation and renal dysfunction were unique to aged mice, indicating that causes of death between young and aged mice may be different even though mortality rates are similar. Although there is no direct evidence that increased thrombosis in aged mice is the main cause of mortality, Ely et al. (9) reported that recombinant aPC administration to patients with severe sepsis was more effective in terms of short- and long-term survival for elderly patients than for young patients, consistent with the premise that thrombosis during systemic inflammation is a major contributing factor for mortality in the aged. Furthermore, in human endotoxemia experiments, which induces mild systemic inflammation without affecting levels of anti-coagulant proteins (1), administration of recombinant aPC did not show significant effects on thrombin generation or fibrinolysis (22), supporting the notion that aPC treatment may have benefits as an anticoagulant therapy only in patients with coagulation abnormalities. Thus, specifically targeting aged individuals exhibiting coagulation abnormalities with similar agents may prove beneficial for their survival.
An obvious limitation of this study is that mice are not men; thus data obtained in this study may not be directly applied to clinical sepsis at this point. Mouse models using the CLP procedure have failed to show the same occurrence or severity of acute lung injury as humans with severe abdominal sepsis. Furthermore, among different mouse strains, differences in the inflammatory response have been reported which could suggest that data reported in this study are specific to the C57BL/6 strain. Although differences in strain and species are not to be taken for granted, we and others have shown an age-dependent increase in mortality and similarly increased expression of inflammatory cytokines in both sexes of multiple mouse strains and rats after LPS and CLP (32). That being said, certain aspects of this research are consistent with what is observed clinically and further research in this area may be able to bring our results more in line with a clinical resolution.
In conclusion, CLP-induced sepsis caused a significant increase in age-associated mortality, enhanced coagulation, and renal dysfunction, which were prominent in aged mice. Enhanced coagulation in aged mice was associated with loss of PC pathway function, resulting in little to no generation of aPC. Although the mechanisms for PC pathway dysfunction are still unclear, the data presented here provide important information with clinical implications for the reasons behind age-associated susceptibility to sepsis.
GRANTS
This work was supported by National Institutes of Health Grants R01-AG025908 and R01-AG039732 (to H. Saito), UM1 HL120877 (to C. T. Esmon), and UL1TR000117 (to A. J. Stromberg).
DISCLOSURES
C. T. Esmon is a consultant for Portola and Bayer. The remaining authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Author contributions: M.E.S., J.U., and H.S. conception and design of research; M.E.S., H.T., D.O., B.A.Z., A.A.M., J.U., and H.S. performed experiments; M.E.S., H.T., D.O., J.U., A.J.S., and H.S. analyzed data; M.E.S., D.O., B.A.Z., J.U., B.M.E., C.T.E., and H.S. interpreted results of experiments; M.E.S., H.T., D.O., and H.S. prepared figures; M.E.S. and H.S. drafted manuscript; M.E.S., A.A.M., A.J.S., B.M.E., C.T.E., and H.S. edited and revised manuscript; M.E.S., H.T., D.O., B.A.Z., A.A.M., J.U., A.J.S., B.M.E., C.T.E., and H.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Donna Gilbreath of the Markey Cancer Center Research Communications Office for illustrative assistance.
REFERENCES
- 1.Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem 15: 1697–1705, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29: 1303–1310, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med 369: 2063, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Coletta C, Modis K, Olah G, Brunyanszki A, Herzig DS, Sherwood ER, Ungvari Z, Szabo C. Endothelial dysfunction is a potential contributor to multiple organ failure and mortality in aged mice subjected to septic shock: preclinical studies in a murine model of cecal ligation and puncture. Crit Care 18: 511, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craciun FL, Iskander KN, Chiswick EL, Stepien DM, Henderson JM, Remick DG. Early murine polymicrobial sepsis predominantly causes renal injury. Shock 41: 97–103, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Gaudio AR, Rinaldi S, Chelazzi C, Borracci T. Pathophysiology of sepsis in the elderly: clinical impact and therapeutic considerations. Curr Drug Targets 10: 60–70, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke R, Osborn TM, Nunnally ME, Townsend SR, Reinhart K, Kleinpell RM, Angus DC, Deutschman CS, Machado FR, Rubenfeld GD, Webb SA, Beale RJ, Vincent JL, Moreno R. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 41: 580–637, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med 35: 1244–1250, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Ely EW, Angus DC, Williams MD, Bates B, Qualy R, Bernard GR. Drotrecogin alfa (activated) treatment of older patients with severe sepsis. Clin Infect Dis 37: 187–195, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Esmon CT. Protein C anticoagulant pathway and its role in controlling microvascular thrombosis and inflammation. Crit Care Med 29: S48–S51, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Girard TD, Opal SM, Ely EW. Insights into severe sepsis in older patients: from epidemiology to evidence-based management. Clin Infect Dis 40: 719–727, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C, Remick DG. Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol Rev 93: 1247–1288, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kale SS, Yende S. Effects of aging on inflammation and hemostasis through the continuum of critical illness. Aging Dis 2: 501–511, 2011. [PMC free article] [PubMed] [Google Scholar]
- 14.Knoebl PN. Severe congenital protein C deficiency: the use of protein C concentrates (human) as replacement therapy for life-threatening blood-clotting complications. Biologics 2: 285–296, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med 40: 754–761, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Levi M. The coagulant response in sepsis and inflammation. Hamostaseologie 30: 10–16, 2010. [PubMed] [Google Scholar]
- 17.Levi M, de Jonge E, van der Poll T, ten Cate H. Disseminated intravascular coagulation. Thromb Haemost 82: 695–705, 1999. [PubMed] [Google Scholar]
- 18.Levi M, van der Poll T. The role of natural anticoagulants in the pathogenesis and management of systemic activation of coagulation and inflammation in critically ill patients. Semin Thromb Hemost 34: 459–468, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Zheng X, Gu J, Hunter J, Ferrell GL, Lupu F, Esmon NL, Esmon CT. Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost 3: 1351–1359, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348: 1546–1554, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Martin GS, Mannino DM, Moss M. The effect of age on the development and outcome of adult sepsis. Crit Care Med 34: 15–21, 2006. [DOI] [PubMed] [Google Scholar]
- 22.O′Brien JM Jr, Abraham E. Human models of endotoxemia and recombinant human activated protein C. Crit Care Med 32: S202–S208, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Okamura D, Starr ME, Lee EY, Stromberg AJ, Evers BM, Saito H. Age-dependent vulnerability to experimental acute pancreatitis is associated with increased systemic inflammation and thrombosis. Aging Cell 11: 760–769, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Opal SM, Girard TD, Ely EW. The immunopathogenesis of sepsis in elderly patients. Clin Infect Dis 41, Suppl 7: S504–S512, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Pan T, Nielsen LD, Allen MJ, Shannon KM, Shannon JM, Selman M, Mason RJ. Serum SP-D is a marker of lung injury in rats. Am J Physiol Lung Cell Mol Physiol 282: L824–L832, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Plataki M, Kashani K, Cabello-Garza J, Maldonado F, Kashyap R, Kor DJ, Gajic O, Cartin-Ceba R. Predictors of acute kidney injury in septic shock patients: an observational cohort study. Clin J Am Soc Nephrol 6: 1744–1751, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Poukkanen M, Vaara ST, Pettila V, Kaukonen KM, Korhonen AM, Hovilehto S, Inkinen O, Laru-Sompa R, Kaminski T, Reinikainen M, Lund V, Karlsson S. Acute kidney injury in patients with severe sepsis in Finnish Intensive Care Units. Acta Anaesthesiol Scand 57: 863–872, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 17: 463–467, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Runge MS, Bode C, Matsueda GR, Haber E. Antibody-enhanced thrombolysis: targeting of tissue plasminogen activator in vivo. Proc Natl Acad Sci USA 84: 7659–7662, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saito H, Sherwood ER, Varma TK, Evers BM. Effects of aging on mortality, hypothermia, and cytokine induction in mice with endotoxemia or sepsis. Mech Ageing Dev 124: 1047–1058, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Starr ME, Hu Y, Stromberg AJ, Carmical JR, Wood TG, Evers BM, Saito H. Gene expression profile of mouse white adipose tissue during inflammatory stress: age-dependent upregulation of major procoagulant factors. Aging Cell 12: 194–206, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Starr ME, Saito H. Sepsis in old age: review of human and animal studies. Aging Dis 5: 126–136, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Starr ME, Ueda J, Takahashi H, Weiler H, Esmon CT, Evers BM, Saito H. Age-dependent vulnerability to endotoxemia is associated with reduction of anticoagulant factors activated protein C and thrombomodulin. Blood 115: 4886–4893, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tucsek Z, Gautam T, Sonntag WE, Toth P, Saito H, Salomao R, Szabo C, Csiszar A, Ungvari Z. Aging exacerbates microvascular endothelial damage induced by circulating factors present in the serum of septic patients. J Gerontol A Biol Sci Med Sci 68: 652–660, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turnbull IR, Clark AT, Stromberg PE, Dixon DJ, Woolsey CA, Davis CG, Hotchkiss RS, Buchman TG, Coopersmith CM. Effects of aging on the immunopathologic response to sepsis. Crit Care Med 37: 1018–1023, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ueda J, Starr ME, Takahashi H, Du J, Chang LY, Crapo JD, Evers BM, Saito H. Decreased pulmonary extracellular superoxide dismutase during systemic inflammation. Free Radic Biol Med 45: 897–904, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev 93: 327–358, 2013. [DOI] [PubMed] [Google Scholar]
- 38.Ware LB, Koyama T, Zhao Z, Janz DR, Wickersham N, Bernard GR, May AK, Calfee CS, Matthay MA. Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit Care 17: R253, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiler H. Multiple receptor-mediated functions of activated protein C. Hamostaseologie 31: 185–195, 2011. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto K, Shimokawa T, Yi H, Isobe K, Kojima T, Loskutoff DJ, Saito H. Aging accelerates endotoxin-induced thrombosis: increased responses of plasminogen activator inhibitor-1 and lipopolysaccharide signaling with aging. Am J Pathol 161: 1805–1814, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yegenaga I, Hoste E, Van Biesen W, Vanholder R, Benoit D, Kantarci G, Dhondt A, Colardyn F, Lameire N. Clinical characteristics of patients developing ARF due to sepsis/systemic inflammatory response syndrome: results of a prospective study. Am J Kidney Dis 43: 817–824, 2004. [DOI] [PubMed] [Google Scholar]