

Abstract
Pulmonary vascular remodeling, mainly attributable to enhanced pulmonary arterial smooth muscle cell proliferation and migration, is a major cause for elevated pulmonary vascular resistance and pulmonary arterial pressure in patients with pulmonary hypertension. The signaling cascade through Akt, comprised of three isoforms (Akt1–3) with distinct but overlapping functions, is involved in regulating cell proliferation and migration. This study aims to investigate whether the Akt/mammalian target of rapamycin (mTOR) pathway, and particularly which Akt isoform, contributes to the development and progression of pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension (HPH). Compared with the wild-type littermates, Akt1−/− mice were protected against the development and progression of chronic HPH, whereas Akt2−/− mice did not demonstrate any significant protection against the development of HPH. Furthermore, pulmonary vascular remodeling was significantly attenuated in the Akt1−/− mice, with no significant effect noted in the Akt2−/− mice after chronic exposure to normobaric hypoxia (10% O2). Overexpression of the upstream repressor of Akt signaling, phosphatase and tensin homolog deleted on chromosome 10 (PTEN), and conditional and inducible knockout of mTOR in smooth muscle cells were also shown to attenuate the rise in right ventricular systolic pressure and the development of right ventricular hypertrophy. In conclusion, Akt isoforms appear to have a unique function within the pulmonary vasculature, with the Akt1 isoform having a dominant role in pulmonary vascular remodeling associated with HPH. The PTEN/Akt1/mTOR signaling pathway will continue to be a critical area of study in the pathogenesis of pulmonary hypertension, and specific Akt isoforms may help specify therapeutic targets for the treatment of pulmonary hypertension.
Keywords: Akt/mammalian target of rapamycin signaling, hypoxia, pulmonary vascular remodeling, smooth muscle cell proliferation
pulmonary arterial hypertension (PAH) is a progressive and fatal disease characterized by a persistent increase of pulmonary vascular resistance (PVR). Pulmonary vascular remodeling or concentric vascular wall thickening of small pulmonary arteries (PAs) and arterioles, attributable partially to enhanced proliferation and migration of PA smooth muscle cells (PASMC), is a major cause for elevated PVR in patients with PAH and chronic hypoxia-induced pulmonary hypertension (HPH) (32, 54, 74). Multiple cellular and molecular mechanisms have been demonstrated to contribute to the development and progression of pulmonary vascular remodeling through enhanced PASMC proliferation and migration, such as signaling cascades involving intracellular free Ca2+ (12, 37, 50, 75), K+ channels (51, 62, 73), Notch (26, 69), bone morphogenetic protein/transforming growth factor-β (38, 42, 48), and/or Akt/mammalian target of rapamycin (mTOR) (4, 20, 23); however, the specific sequence of events involved in the enhanced PASMC proliferation in pulmonary hypertension remains unclear.
When cells, including PASMC, are activated by extracellular mitogenic factors, such as platelet-derived growth factor (PDGF), insulin-like growth factors (IGF), and endothelin-1, the Akt signaling pathway plays an important role in regulating cell proliferation, migration, and apoptosis, much of which has been linked to subsequent activation of the mTOR, an important downstream signaling protein (45, 52). Inhibition of the Akt/mTOR signaling cascade by phosphatase and tensin homolog deleted on chromosome 10 (PTEN) (a phosphatase that converts phosphatidylinositol 4,5-trisphosphate to phosphatidylinositol 4,5-bisphosphate, inhibiting Akt phosphorylation), rapamycin (which blocks mTOR), or Akt inhibition (specific inhibitors that bind to Akt to prevent its phosphorylation), has been well demonstrated to attenuate cell proliferation in cancer cells (64), embryonic stem cells (2, 61), vascular progenitor cells (25, 27), coronary arterial smooth muscle cells (9, 14), and PASMC (19, 20, 29, 49). Our previous work has also demonstrated the importance of the Akt/mTOR pathway in both thrombin-and PDGF-induced enhancement of store-operated Ca2+ entry (SOCE) and cell proliferation in PASMC (44, 45), whereas downregulation of transient receptor potential canonical (TRPC) channels reduced SOCE and significantly inhibit PASMC proliferation and migration (59, 72).
There are three members in the Akt kinase family, Akt1, Akt2, and Akt3, which share a high degree of homology. Akt1 and Akt2 are broadly expressed in many tissue and cell types, whereas Akt3 is predominantly expressed in brain tissue (10, 67). Each of the Akt isoforms has a distinct but overlapping function in the regulation of cell proliferation, cell apoptosis, protein synthesis, and the cell cycle (17). Numerous clinical studies have indicated differential expression of Akt isoforms in different types of cancer and cells (6, 41, 55, 57, 68). Indeed, many studies have evaluated the distinct roles of Akt isoforms for several types of cancer. The distinct roles of Akt1, as a migration inhibitor, and Akt2, as a promigration kinase, are well defined in breast and ovarian cancer cells (1, 35, 70). In prostate cancer cells, however, Akt1 and Akt2 both function as inhibitors of cell migration and invasion (63). These observations provide compelling evidence that, in cancer cells, Akt1 and Akt2 play a distinctive functional role in the regulation of cell proliferation and migration by their differential effects on receptor tyrosine kinases (RTKs), integrin activity, and microRNAs. Distinct actions of Akt isoforms in vascular tissue have also been noted, such that microvascular permeability and edema formation are attenuated in Akt1−/− mice and by silencing Akt1 in pulmonary vascular endothelial cells, whereas Akt2 does not appear to play a significant role (8).
In human and rodent PASMC, the Akt/mTOR signaling pathway is involved in thrombin- and PDGF-mediated enhancement of SOCE, upregulation of TRPC channels, and cell proliferation (44, 45). Blockade of the Akt/mTOR signaling with rapamycin or an Akt inhibitor can significantly attenuate PASMC proliferation (20). Deletion of PTEN in mouse smooth muscle cells results in vascular remodeling (43), and exposure of PTEN conditional knockout (KO) mice to hypoxia causes severe pulmonary hypertension (19). These studies show the crucial roles of PTEN in the development and progression of pulmonary vascular disease. However, the consequences of suppression of Akt activation by elevating PTEN in pulmonary vascular remodeling and its potential prevention of HPH are still unknown. Furthermore, it remains unclear whether Akt1 and Akt2 both play comparable roles in PASMC proliferation and, ultimately, in the development of pulmonary vascular remodeling leading to pulmonary hypertension.
In this study, we hypothesized that PTEN/Akt/mTOR signaling contributes to the development and progression of pulmonary hypertension, and deletion of different Akt kinase isoforms (e.g., Akt1 or Akt2) may have distinctive effects on the development and progression of pulmonary vascular remodeling in animals with experimental pulmonary hypertension. The aim of this study was to further elucidate the role and significance of the PTEN/Akt/mTOR signaling pathway in the development and progression of pulmonary vascular remodeling and to identify the importance of distinct isoforms of Akt (i.e., Akt1 and Akt2) in the development of pulmonary hypertension.
MATERIALS AND METHODS
Animals.
All animal procedures were conducted with the use of protocols approved by the IACUC of the University of Illinois at Chicago (UIC) and The University of Arizona (UA). All mice used in these studies were of C57BL/6 background and were 6–12 wk old at the time of the experimentation. The Akt1−/− mice were used as commercially available through Jackson Laboratory (stock no. 004912). The Akt2−/− mice are also available commercially through Jackson Laboratory (stock no. 006966). All mice were bred and housed in the UIC Biologic Resources Laboratory and the UA Animal Health Science Center.
Generation of the inducible and conditional mTOR KO mice.
We used Cre-Lox technology to generate an inducible, smooth muscle cell-specific KO mouse line. mTOR floxed mice, herein called mTORflox/flox, were crossed with SMMHC-CreERT2 mice for two generations to create SMMHC-CreERT2+/−/mTORflox/flox mice. In this mouse strain, a tamoxifen-inducible Cre recombinase is under the control of the smooth muscle myosin heavy chain (SMMHC) promoter (66). The reason for employing an inducible recombination strategy was that it allowed for the determination of the consequences of mTOR loss in the adult mouse PASMC.
Tamoxifen administration.
The tamoxifen solution was made freshly before each use by dissolving tamoxifen powder (cat. no. T5648; Sigma Aldrich) in sunflower seed oil to a final concentration of 20 mg/ml. To induce mTOR KO, SMMHC-CreERT2+/−/mTORflox/flox (referred to as mTOR KO) were treated with tamoxifen via intraperitoneal injection once a day for five consecutive days at a dose of 1 mg. Mice were allowed to recover for 2 wk following the tamoxifen administration regimen before any subsequent experimental manipulation.
PCR analysis.
Genomic DNA from a tail biopsy was extracted by standard procedures and subjected to PCR for genotyping. The primer sequences and anticipated band sizes are as follows: Akt1 wild-type (WT): 143 bp, F-GCTCCATAAGCACACCTTCAGG; R-AGCTCTTCTTCCACCTGTCTC; Akt1-Mut: 259 bp, F-GCTCCATAAGCACACCTTCAGG; R-GTGGATGTGGAATGTGTGCGAG; Akt2-WT: 300 bp, F-GAGGTAGAAACAAGAGAATCAT; R-GTTCGCACTGCTGTATGTTG; Akt2-Mut: 800 bp, F-TGTATTTCTACCCCGATGCCA; R-TGCTACTTCCATTTGTCACGTC; SMMHC-CRE: P1-TGACCCCATCTCTTCACTCC; P2-AACTCCACGACCACCTCATC; P3-AGTCCCTCACATCCTCAGGTT. The cycling parameters for the PCR were as follows: 1 cycle at 94°C for 5 min, 33 cycles of 94°C for 45 s, then 45 s of 68°C, and 72°C for 45 s. The PCR ended with 5 min at 72°C.
Western blotting and real-time RT-PCR.
Solubilized protein lysates, isolated from lung tissues and PASMC, were used to detect Akt1, Akt2, mTOR, and PTEN. Cells were lysed in a modified radioimmunoprecipitation assay lysis buffer with a protease inhibitor cocktail (Sigma Aldrich). Total RNA from lung tissues or PASMC was isolated using the RNeasy Plus Mini Kit (cat. no. 74134; Qiagen). Equal amounts (30 μg) of total protein from each sample were loaded onto 4–20% SDS gradient gels (Mini-PROTEAN TGX; Bio-Rad) and separated by gel electrophoresis. Proteins were transferred onto PVDF membranes and stained with Ponceau S. Blots were probed with polyclonal rabbit anti-Akt1 (cat. no. 2938; Cell Signaling), anti-Akt2 (cat. no. 3063; Cell Signaling), and monoclonal β-actin (cat. no. sc-130301; Santa Cruz Biotechnology). Bands were visualized using ImageJ software. To perform the real-time PCR, 2 μg of purified RNA was reverse transcribed to single-stranded cDNA using the TaqMan RNA reverse transcription kit (cat. no. N8080234, Applied Biosystems). Real-time PCR was performed on an ABI 7900HT machine. Specific TaqMan quantitative real-time PCR assays were ordered from Applied Biosystems (specific assay IDs available upon request). The relative mRNA expression levels were normalized to the expression of a housekeeping gene, GAPDH, and determined by calculating the ΔΔCt value, as detailed in the manufacturer's guidelines.
HPH animal model.
In the rodent models of HPH, mice or rats were exposed to room air (normoxia) or 10% oxygen (hypoxia) for 3–5 wk in a BioSpherix A chamber (BioSpherix), and the oxygen concentration (10%) was monitored with a Proox Model P110 oxygen controller (BioSpherix). After chronic exposure to normobaric hypoxia, mice and rats were anesthetized with ketamine/xylazine before hemodynamic measurements or humane death for organ procurement.
Hemodynamic measurements.
Animals were weighed before the experiment and once a week during the experiment. Right ventricular (RV) pressure was measured with a 1.4-French pressure transducer catheter (Millar Instruments) and AcqKnowledge software (Biopac Systems). RV systolic pressure (RVSP) was recorded and used as a surrogate for PA pressure (PAP). The blood was drawn to determine a complete blood cell count, and heart was excised and dissected to determine the ratio of the RV weight to the left ventricle (LV) and septum (S) weight [RV/(LV+S) ratio], i.e., the Fulton index, as a parameter of RV hypertrophy (RVH). Lung tissues were used for Western blotting analysis and real-time RT-PCR analysis as described above, as well as morphometric analysis. Regarding morphometric analysis, tissues were fixed, embedded, and sectioned. Slides were stained with hematoxylin and eosin (H and E) and used to quantitate PA wall thickness.
Isolated perfused/ventilated mouse lung experiments.
The PAP was measured using the isolated perfused/ventilated mouse lung system as previously described (71). Briefly, mice were anesthetized and ventilated with a gas mixture of 21% O2-5% CO2 in N2 via a rodent ventilator (minivent type 845; Harvard Apparatus). A stainless steel catheter was inserted into the main PA after performing a right ventriculotomy, and the PA and ascending aorta were tied together using a 6-0 black silk suture. PAP was measured using a pressure sensor (P75 Type 379, Hugo Sachs Elektronik-Harvard Apparatus), which was connected to the PA catheter. The other end of the catheter was connected to a tube for PA perfusion. The pulmonary circulation was maintained in a closed circuit via a peristaltic pump (ISM 834; ISOMATEC). For data acquisition and data storage, Powerlab 8/30, Quad Bridge Amp, and LabChart (AD Instruments) were used. After basal PAP was stabilized for 40–60 min, the experiments were performed.
Human lung tissues and human PASMC.
Approval for the use of human lung tissues and cells was granted by the UIC Institutional Review Board. Human lung tissues used in this study were from donor lung explants not suitable for lung transplantation and patients with idiopathic PAH (IPAH). Human PASMC were isolated from the lung tissues of normal organ donors and patients with IPAH. In addition, a primary hPASMC cell line from Lonza (CC-2581) was used for cell transfection and proliferation assays. Cells were cultured at 37°C in smooth muscle growth medium (CC-3182; Lonza) and studied at passages 5 to 8.
Mouse PASMC isolation.
PASMC were isolated from mouse lungs, as described previously (65), using a modified method by Marshall et al. (33). A mixture of 5 ml of medium 199 (M199) growth medium containing 5 g/l low-melting-point agarose type VII (Sigma Aldrich), 5 g/l iron beads (diameter <10 μM; Sigma Aldrich), and antibiotics (penicillin and streptomycin) was slowly injected over a period of 60 s through the RV, thereby perfusing the PA. M199 growth medium (1 ml) containing 5 g/l agarose type VII was injected in airways through the trachea. The lungs were plunged in cold PBS to cause the agarose to gel. Because of the rapidly solidifying nature of the agarose and the size of the iron particles, the likelihood of traversing the capillary space is minimized. All the lobes were then isolated and finely minced in a Petri dish. The tissue was further disrupted by passing through a 16-gauge followed by an 18-gauge needle approximately five times. The suspension was then mixed in M199 growth medium containing 80 U/ml type IV collagenase (Sigma Aldrich) and incubated at 37°C for 90 min. With the use of a magnetic column (Invitrogen), the arteries or arterial tissues containing the iron beads were collected. The supernatant was aspirated, and the arteries were washed and suspended in 5 ml M199 containing 20% FBS. Aliquots of the suspension were transferred to T25 culture flasks. Cells from the hypoxic group were incubated at 3% O2, whereas cells from the normoxic control were cultured in air. Smooth muscle cell purity was determined by immunostaining with smooth muscle-specific α-actin antibody.
Bromodeoxyuridine cell proliferation assay.
Briefly, PASMC were plated into a 96-well plate at a density of 5 × 103/100 μl per well and incubated overnight. Bromodeoxyuridine (BrdU) label was added to the culture medium with a dilution of 1:10,000 on the next day. Cells were cultured for another 16 h, and cell proliferation activities were then detected using a BrdU cell proliferation assay kit (Calbiochem) on GloMax-96 Microplate luminometer (Promega) at the wavelength of 450 nm according to the user's manual.
Statistics.
The composite data are expressed as means ± SE. Statistical analysis was performed using paired or unpaired Student's t-test or ANOVA and post hoc tests (Student-Newman-Keuls) where appropriate. Differences were considered to be significant at P < 0.05.
RESULTS
Akt is highly phosphorylated in lung tissues and PAs isolated from patients with IPAH and animals with experimental pulmonary hypertension.
We first examined whether Akt phosphorylation was increased in lung tissues and PASMC isolated from rats and mice with chronic HPH and PASMC isolated from patients with IPAH. As shown in Fig. 1, the level of phosphorylated Akt (p-Akt) in lung tissues isolated from both mice and rats with HPH was significantly higher than lung tissues from normoxic control animals (Fig. 1, A and B), whereas the protein expression level of total Akt (Akt) was comparable between normoxic control and HPH rats. These data correlate with the development of HPH and RVH in both mice (Fig. 1, C and D) and rats (Fig. 1, E and F), which is demonstrated by a rise in RVSP and an increase in Fulton index [RV/(LV+S)] following chronic hypoxia exposure for 4 wk. Additionally, PASMC isolated from patients with IPAH are demonstrated to have increased p-Akt/Akt ratio compared with normal PASMC (Fig. 1D), implying that activation of Akt signaling (enhanced phosphorylation of Akt) is present in PAs isolated from patients with IPAH and lung tissues from animals with experimental pulmonary hypertension and may contribute to pulmonary vascular remodeling in the development and progression of disease.
Fig. 1.
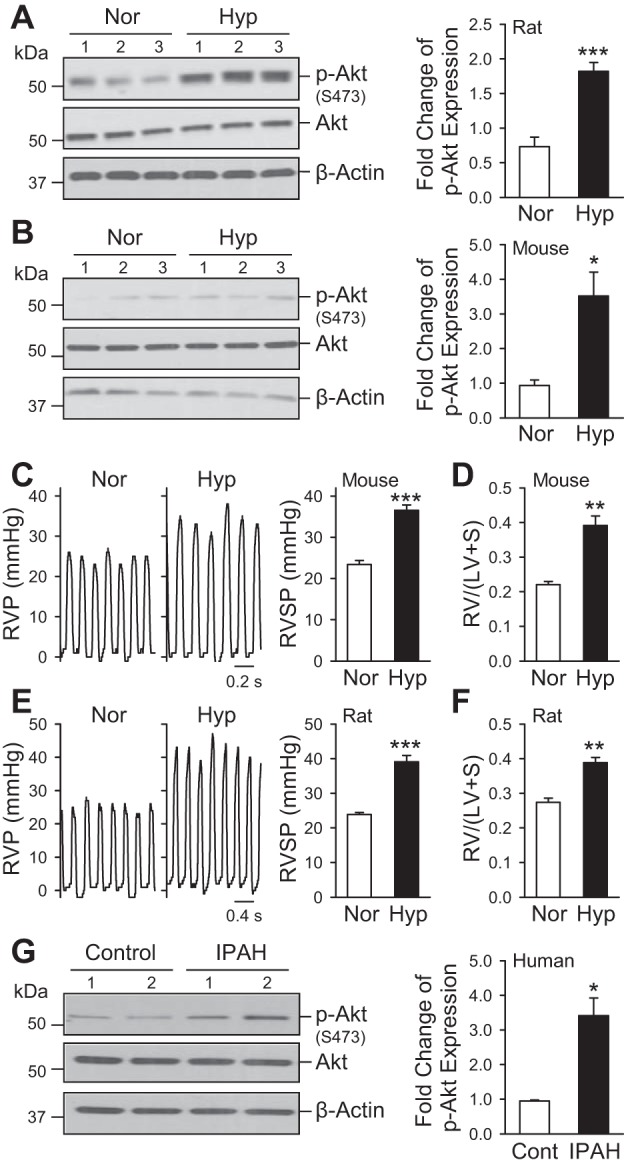
Phosphorylation of Akt1/2 is upregulated in rat and mouse lungs with hypoxia-induced pulmonary hypertension and in pulmonary artery smooth muscle cells (PASMC) from patients with idiopathic PA hypertension (IPAH). A and B: representative Western blots and quantifications of the Akt phosphorylation in lungs from rats (A) and mice (B) exposed to normoxia (Nor) or chronic hypoxia (Hyp). Values are means ± SE (n = 3–5, right). *P < 0.05; ***P < 0.001 vs. Nor. Representative pressure tracings and bar graphs quantifying hemodynamic measurements, right ventricular pressure (RVP), and RV systolic pressure (RVSP) from normoxic and chronic hypoxic mice (C) and rats (E), as well as quantification of RV hypertrophy from normoxic and chronic hypoxic mice (D) and rats (F) measured by the ratio of the RV weight to the left ventricle (LV) and septum (S) weight, called the Fulton index [RV/(LV+S)]. *P < 0.05; **P < 0.01; ***P < 0.001 vs. Nor. G: representative Western blots and quantifications of Akt phosphorylation in PASMC isolated from normal control subjects (Cont) and patients with IPAH. *P < 0.05 vs. Cont.
Deletion of specific Akt isoforms does not affect expression of the other isoform and negligibly affects basal systemic arterial pressure and PAP.
To examine whether Akt, and in particular which Akt isoform (Akt1 or Akt2), is involved in the development and progression of pulmonary hypertension, we first used heterozygous (Akt1+/−, Akt2+/−) and homozygous (Akt1−/−, Akt2−/−) KO mice to compare the basal protein and mRNA expression levels of Akt1 and Akt2, as well as systemic hemodynamics compared with WT mice. The mice were genotyped using the standard PCR procedure and confirmed downregulation and absence of Akt1 mRNA expression in the Akt1+/− and Akt1−/− mice, respectively (Fig. 2, A and B). Similar changes were identified for Akt2 mRNA expression in Akt2+/− and Akt2−/− mice (Fig. 2, A and B). The mRNA expression level of Akt1 in Akt2 +/− mice was not significantly changed compared with the WT littermates, nor was the expression level of Akt2 in Akt1+/− mice. Protein expression of Akt1 was decreased in Akt1+/− and undetectable in Akt1−/− mice with no significant change in Akt2 protein expression compared with WT littermates (Fig. 2C). In Akt2−/− mice, protein expression of Akt2 was not detectable (ND) in lung tissues with no significant change in Akt1 protein expression (Fig. 2D).
Fig. 2.

Deletion of Akt1 does not affect Akt2 expression in lung tissue, and LV hemodynamics are unaffected in Akt1−/− and Akt2−/− mice. A: genotyping characterization of Akt1−/−, Akt2−/−, and wild-type (WT) mice demonstrates the presence of only knockout (KO) alleles in the homozygous mice, whereas heterozygotes have both KO and WT alleles. Lung tissues were harvested from Akt1−/−, Akt2−/−, and WT mice. B: real-time quantitative RT-PCR detecting mRNA expression of Akt1 and Akt2 in the Akt1−/− and Akt2−/− mice. C and D: Western immunoblotting assessed Akt1 and Akt2 protein expression in lung tissue isolated from WT, Akt1+/−, and Akt+/− mice (C) as well as from WT, Akt1−/−, and Akt2−/− mice (D). *P < 0.05; ***P < 0.001 vs. WT. ND indicates not detectable. E and F: representative record of LV systolic pressures (LVSP, E) and systemic arterial pressures (SAP, F) with summarized data (means ± SE, left) in WT mice (n = 6), Akt1−/− mice (n = 3), and Akt2−/− mice (n = 3).
The systemic arterial pressure, measured by a catheter in the carotid artery, and LV systolic pressure, determined by a catheter advanced into the LV, were both comparable among WT, Akt1+/−, and Akt2+/− mice (Fig. 2, E and F). These results have characterized the KO mice used for subsequent experiments, demonstrating no appreciable changes in the mRNA or protein expression of accompanying Akt isoforms in each specific genetic deletion. Additionally, baseline systemic hemodynamics is unaffected in heterozygous KOs.
Deletion of Akt1 (Akt1−/−) significantly attenuates the development and progression of pulmonary hypertension.
No significant difference in the basal pulmonary hemodynamics was observed between WT and Akt1−/− mice (Fig. 3, A and B). Exposure of WT mice to normobaric hypoxia (10% O2) for 4 wk significantly increased PAP, indicated by a significant increase in RVSP (from 24.2 ± 2.6 to 41.9 ± 1.6 mmHg, P < 0.001) (Fig. 3, A and B), which was accompanied by marked RVH, indicated by a significant increase in the Fulton index [the ratio of RV/(LV+S)] (from 0.29 ± 0.01 to 0.46 ± 0.08, P < 0.05) (Fig. 3C). The hypoxia-induced increase in RVSP and RVH in WT mice was associated with significant PA remodeling, indicated by a significant increase in PA wall thickness from both small (<50 μM) and medium-sized (50–100 μm) arteries (from 0.47 ± 0.03 to 0.64 ± 0.06 μm in vessels with diameter <50 μm, P < 0.05, and from 0.43 ± 0.03 to 0.73 ± 0.04 μm in vessels with diameter between 50 and 100 μm, P < 0.01) (Fig. 3, D and E). In Akt1−/− mice, the hypoxia-mediated increases in RVSP, RV/(LV+S) ratio, and PA wall thickness were all significantly less than in WT mice (Fig. 3). The significantly lower RVSP in hypoxic Akt1−/− mice than in hypoxic WT mice (Fig. 3, A and B) correlated with a significantly reduced RVH measured by the Fulton index (Fig. 3C).
Fig. 3.
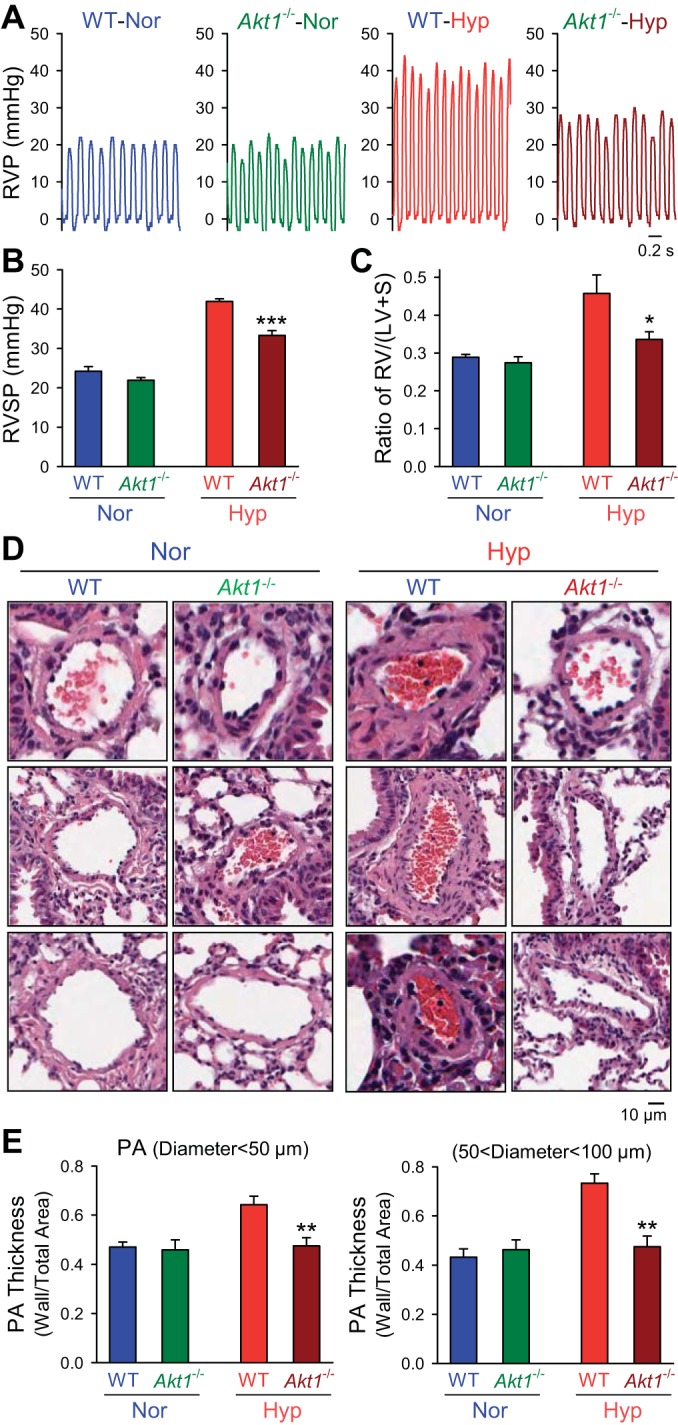
Deletion of Akt1 attenuates hypoxia-induced pulmonary hypertension and pulmonary vascular remodeling. A and B: representative record of RVP (A) and summarized data (means ± SE, B) of peak RVSP (B) in normoxic (n = 6) and hypoxic (n = 6) Akt1−/− mice. ***P < 0.001 vs. WT-Hyp. C: averaged Fulton index (means ± SE) in normoxia and hypoxic WT mice or Akt1−/− mice. *P < 0.05 vs. WT-Hyp. D and E: hematoxylin and eosin (H and E) images (D) of small PAs and summarized data (means ± SE, E) of the medial thickness of PAs with a diameter <50 μm (left) or between 50 and 100 μm (right) from WT mice or Akt1−/− mice under normoxic or hypoxic exposure. All significance markers are vs. hypoxic WT control animals. **P < 0.01 vs. WT-Hyp.
In addition to RVSP and the Fulton index, the wall thickness of small (diameter <50 μm) and medium-sized (diameter 50–100 μm) PAs in hypoxic Akt1−/− mice was significantly lower than in hypoxic WT mice. As shown in Fig. 3, D and E, deletion of Akt1 nearly eliminated hypoxia-mediated PA wall remodeling. These results indicate that Akt1−/− mice are protected from the development of HPH, and the predominant effect appears to be through attenuation of pulmonary vascular remodeling.
Deletion of Akt2 (Akt2−/−) negligibly affects the development and progression of pulmonary hypertension.
It has been well demonstrated that different isoforms of Akt, such as Akt1 and Akt2, play distinctive roles in regulating cell growth, proliferation, and apoptosis (17). The next set of experiments was designed to determine whether deletion of Akt2 (Akt2−/−) would have the same protective effect on the development and progression of pulmonary hypertension.
Similar to findings with Akt1−/− mice, baseline RVSP and Fulton index were similar among Akt2−/− mice and WT littermates (Fig. 4, A–C). In contrast to Akt1−/− mice, Akt2−/− mice exhibited a similar increase in RVSP and RVH following 4 wk of normobaric hypoxia compared with WT mice. As shown in Fig. 4, there was no significant difference in RVSP measurement and the ratio of RV/(LV+S) between hypoxic WT mice and hypoxic Akt2−/− mice (Fig. 4, A–C). Furthermore, Akt2−/− failed to prevent hypoxia-mediated pulmonary vascular remodeling, demonstrated by increases in PA wall thickness (Fig. 4, D and E). These results indicate that deletion of Akt2 (Akt2−/−) does not protect against the development and progression of HPH in mice.
Fig. 4.
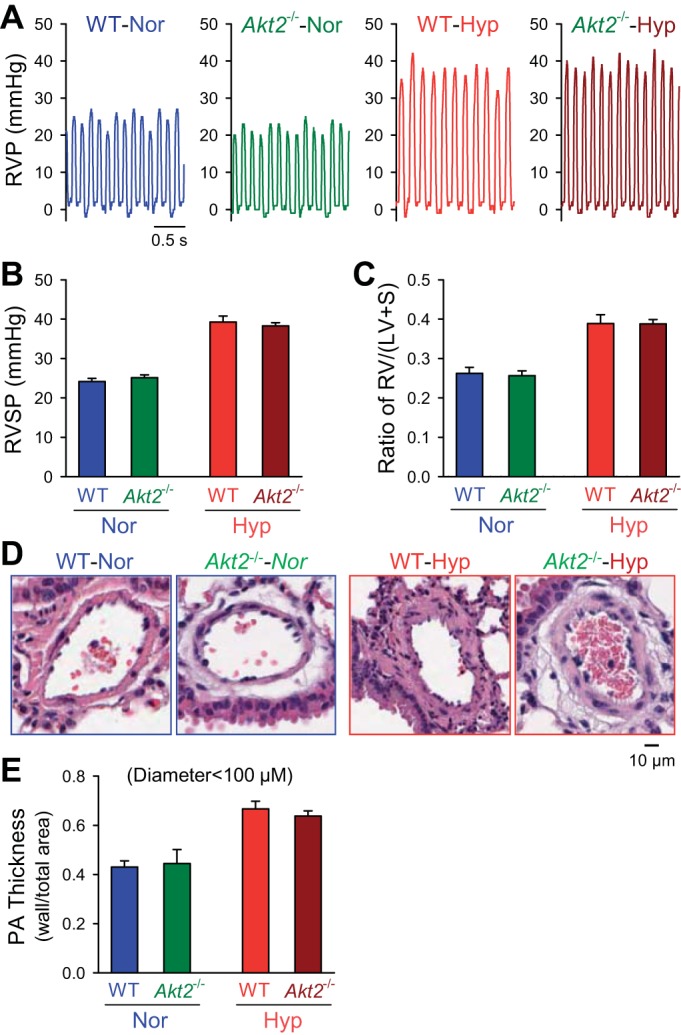
Akt2-deficient mice are not protected against hypoxia-mediated pulmonary hypertension. A and B: representative record (A) of RVP and summarized data (means ± SE, B) of peak RVSP in normoxic (n = 6) and hypoxic (n = 6) Akt2−/− mice. C: averaged Fulton index [RV/(LV+S), means ± SE] in hypoxic WT mice or Akt2−/− mice. D and E: H and E images (D) of small PAs from WT mice or Akt2−/− mice under normoxic or hypoxic exposure with summarized data (means ± SE, E) of medial thickness measurements in the small and medium-sized PAs. No significant differences seen between WT and Akt2−/− mice under normoxic or hypoxic conditions.
Conditional and inducible deletion of mTOR in vascular smooth muscle cells significantly attenuates the development and progression of pulmonary hypertension.
Upon activation of membrane tyrosine kinase receptors (e.g., PDGFR and IGFR) by growth factors, the activated phosphatidylinositol 3-kinase (PI3K)-Akt pathway leads to phosphorylation of mTOR and subsequently activates the downstream signaling cascades, which result in cell proliferation and migration (15). Many of these growth factors are shown to be increased in both animal models of HPH and patients with PAH (24, 47, 56). To examine whether the Akt1/mTOR pathway is the signaling cascade responsible for promoting PASMC proliferation and migration and for the development and progression of HPH, we generated smooth muscle-specific mTOR conditional and inducible KO mice (Fig. 5A), as outlined in materials and methods. The smooth muscle-specific deletion of mTOR was confirmed by Western blot analysis 2 wk after tamoxifen treatment in freshly isolated PA tissues (Fig. 5B). Subsequently, these animals were then exposed to normobaric hypoxia for 3 wk before hemodynamic measurement and histological analysis (Fig. 5C).
Fig. 5.
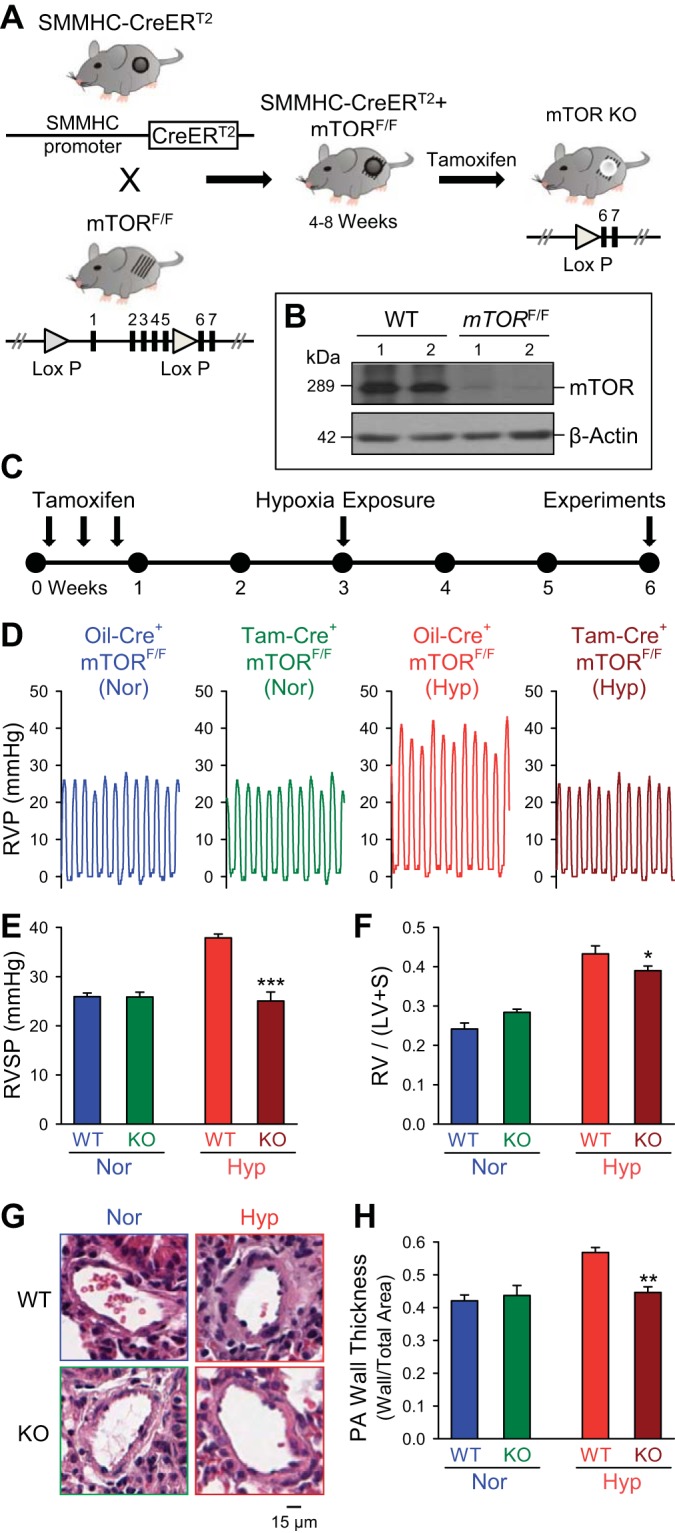
Conditional and inducible KO of mammalian target of rapamycin (mTOR) in PASMC attenuates hypoxia-induced pulmonary hypertension. A: generation of conditional and inducible mTOR KO mice by crossbreeding smooth muscle myosin heavy chain (SMMHC)-CreERT2 mice with mTORflox/flox mice to create SMMHC-CreERT2+/−/mTORflox/flox mice. These mice were treated with tamoxifen (5 consecutive daily doses) 3 wk before hypoxic exposure to induce KO of mTOR in smooth muscle cells. B: representative Western blot demonstrates downregulation of mTOR protein expression in the PA from conditional and inducible mTOR KO mice after treatment with tamoxifen compared with WT littermates treated with oil (WT). C: timeline for induction of conditional KO with tamoxifen and subsequent chronic hypoxia treatment followed by experimental analysis. D and E: representative record of RVP (D) and summarized data (means ± SE, E) showing RVSP in control (Oil-Cre+ mTORF/F) and mTOR-KO (Tam-Cre+ mTORF/F) mice in normoxia and hypoxia. F: summarized data (means ± SE) showing Fulton index [RV/(LV+S)] for control (Oil-Cre+ mTORF/F) and mTOR-KO (Tam-Cre+ mTORF/F) mice in Nor and Hyp. G: representative image of distal PAs from control and mTOR-KO mice in Nor and Hyp. H: summarized data (means ± SE) showing quantification of PA wall thickness, as measured by ratio of wall area to total vessel area, of control and mTOR-KO mice in Nor and Hyp. *P < 0.05; **P < 0.01; ***P < 0.001 vs. WT/control-Nor.
Baseline pulmonary hemodynamic measurements, Fulton index, and PA wall thickness were unchanged between Cre+/mTORF/F mice treated with vehicle control (oil) or tamoxifen (Fig. 5, D–F). Exposure of Cre+/mTORF/F mice treated with vehicle control (oil) to normobaric hypoxia (10% O2) for 4 wk resulted in significantly increased RVSP (Fig. 5, D and E), Fulton index [RV/(LV+S)] (Fig. 5F), and PA wall thickness (Fig. 5, G and H). In Cre+/mTORF/F mice treated with tamoxifen (Tam-Cre+/mTORF/F, or the smooth muscle-specific mTOR KO mice), the hypoxia-mediated increase in RVSP was significantly attenuated compared with vehicle control Cre+/mTORF/F mice treated with oil (Oil-Cre+/mTORF/F) (Fig. 5, D and E). The significantly lower RVSP in hypoxic Tam-Cre+/mTORF/F mice was associated with a significantly lower ratio of RV/(LV+S) compared with vehicle control (Fig. 5F). Additionally, the increased PA wall remodeling was significantly attenuated in the Tam-Cre+/mTORF/F mice compared with control Oil-Cre+/mTORF/F mice (Fig. 5, G and H). These results indicate that smooth muscle-specific KO of mTOR protects against the pulmonary vascular remodeling during the development of HPH.
Upregulation of PTEN in transgenic mice attenuates the development of HPH.
PTEN-transgenic (TG) mice were derived and gifted from the laboratory of Dr. Pandolfi in methods previously described (11). Western blotting of lungs from WT mice showed a significant decrease in PTEN expression when exposed to 4 wk of hypoxia and a significant elevation of PTEN in TG mice compared with hypoxic WT lungs (Fig. 6A). Under normoxic conditions, RVSP and RVH indices are similar among WT and PTEN-TG mice (Fig. 6, B–D). When exposed to 4 wk of normobaric hypoxia, PTEN-TG mice show a reduced RVSP elevation (Fig. 6, B and C) and significantly decreased RVH (Fig. 6D). Figure 6E shows representative cross-sectional views of small PAs from both WT and PTEN-TG mice under normoxic and hypoxic conditions. Quantifying the PA wall thickness as the ratio of wall area to total vessel area, we observe that. in both medium-sized (diameter 50 to 100 μM) and small (diameter <50 μM) PAs, the degree of pulmonary vascular remodeling after hypoxia is significantly attenuated in PTEN-TG mice (Fig. 6F). These results show that upregulation of PTEN, a repressor of Akt activity, protects mice from pulmonary vascular remodeling and development of HPH.
Fig. 6.
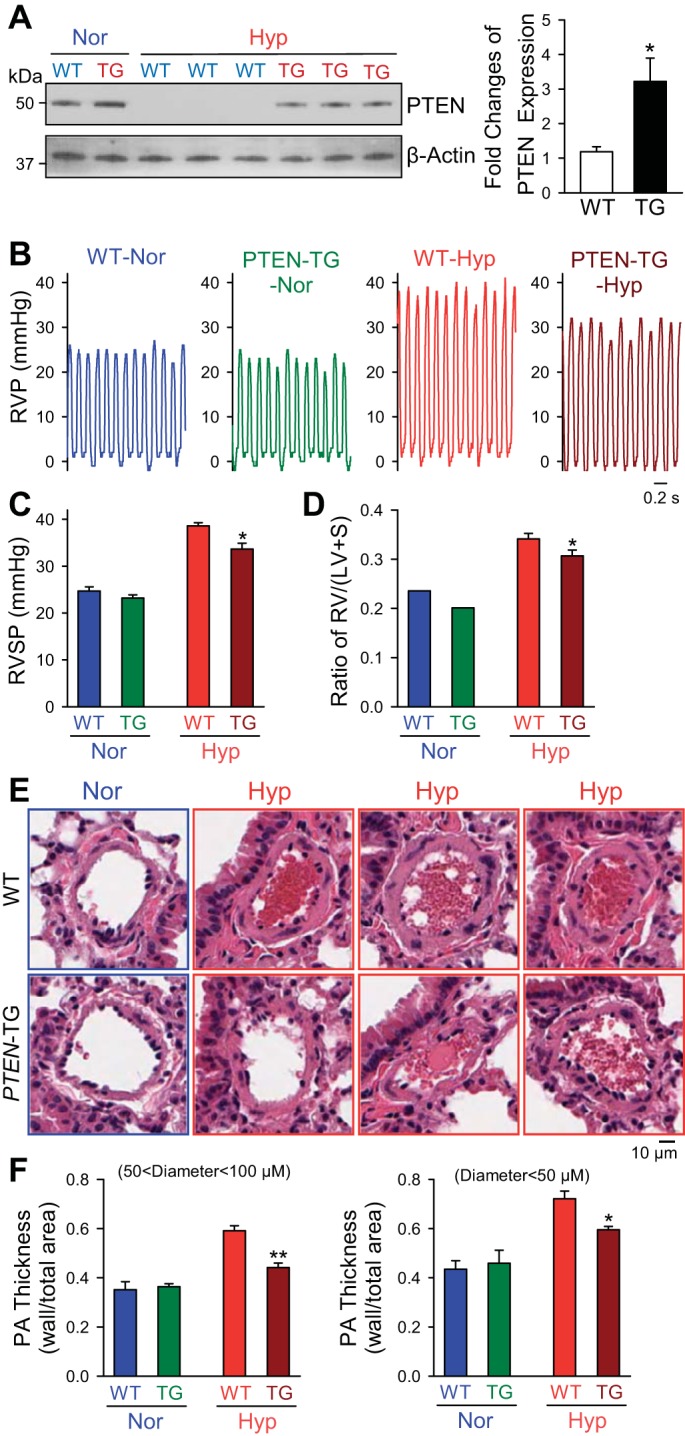
Upregulation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) in PTEN-transgenic (TG) mice attenuates the development and progression of hypoxia-induced pulmonary hypertension. A: representative Western blots (left) and quantifications (means ± SE, n = 3–5, right) of the PTEN expression in lungs from WT and PTEN-TG mice exposed to normoxia or chronic hypoxia. B and C: representative record of RVP (B) and summarized data (means ± SE, C) showing RVSP in PTEN-TG mice and WT littermates during Nor and Hyp conditions. D: summarized data (means ± SE) showing Fulton index [RV/(LV+S)] in PTEN-TG mice and WT littermates during Nor and Hyp conditions. E: representative H and E image of distal PAs from PTEN-TG mice and WT littermates during Nor and Hyp conditions. F: summarized data (means ± SE) showing quantification of PA wall thickness, as measured by ratio of wall area to total vessel area, for both medium-sized (50 μm < diameter <100 μM) and small (diameter <50 μM) PAs in PTEN-TG mice and WT littermates in Nor and Hyp. *P < 0.05; **P < 0.01 vs. WT mice in Hyp.
Smooth muscle-specific deletion of mTOR has the greatest protective effect on HPH and PASMC cell proliferation.
The Akt pathway remains complex, and therefore we sought to determine the relative effect of the various proteins in this pathway. Comparing the effect of chronic hypoxia on Akt1−/− mice, Akt2−/− mice smooth-muscle specific mTOR KO mice, and PTEN-TG mice, we observe that smooth muscle-specific mTOR KO shows the most robust protection from RVSP increases seen after chronic hypoxia (Fig. 7A). Indeed, whereas both Akt1−/− and PTEN-TG mice have nearly 40% protection, the smooth muscle-specific mTOR KO is completely protective and predominately attenuated the RVSP increase after chronic hypoxia (Fig. 7A). RVH and PA remodeling are dramatically protected in the Akt1−/− and smooth muscle-specific mTOR KO mice, whereas the PTEN-TG mice show less protection (Fig. 7, B and C). In fact, PA wall remodeling is eliminated in both Akt1−/− and smooth muscle-specific mTOR KO mice, illustrating the importance of the Akt1/mTOR pathway in PASMC proliferation and pulmonary vascular remodeling.
Fig. 7.
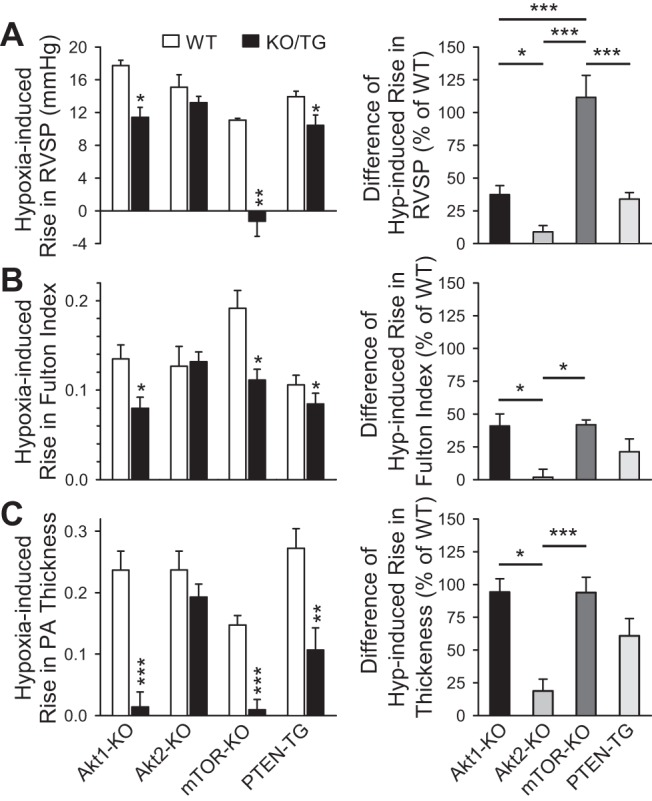
Smooth muscle cell-specific KO of mTOR has the greatest inhibitory effect on hypoxia-induced pulmonary hypertension. A–C: summarized data (means ± SE, left) demonstrating the chronic hypoxia-induced increases in RVSP (A), Fulton index [RV/(LV+S)] (B), and PA wall thickness (C) in WT, Akt1-KO mice (Akt1−/−), Akt2-KO mice (Akt2−/−), smooth muscle-specific conditional and inducible mTOR-KO mice, and PTEN-TG mice. *P < 0.05, **P < 0.01, ***P < 0.001 vs. WT. Right: summarized data (means ± SE) showing the differences of chronic hypoxia-mediated increases in RVSP (A), Fulton index (B) and PA wall thickness (C) between WT mice and Akt1-KO, Akt2-KO, mTOR-KO, or PTEN-TG mice. The difference (% of WT) was calculated by the ratio of the hypoxia-induced increases in KO/TG mice to the hypoxia-induced increases in WT mice. *P < 0.05, **P < 0.01, ***P < 0.001 between different strains indicated by horizontal bars.
To confirm these findings, we performed in vitro studies to determine the relative influence of different signaling proteins in the Akt pathway on PASMC proliferation. Proliferation of human PASMC, measured by BrdU uptake, after knockdown with siRNA of Akt1, Akt2, mTOR, and β-catenin (BCTN), was compared with scrambled siRNA control under both normoxia and hypoxia (for 24 h).
Treatment of human PASMC with specific siRNA for Akt1 significantly reduced protein expression level of Akt1 but had no effect on Akt2 expression level (Fig. 8, A and B). In this experiment, the scrambled siRNA for Akt1 or Akt2 were used as controls. Furthermore, treatment of human PASMC with specific siRNA for mTOR (Fig. 8, C, top, and D, left) or BCTN (Fig. 8, C, bottom, and D, right) significantly decreased protein expression level of mTOR or BCTN, respectively, compared with the cells treated with scrambled siRNA for mTOR or BCTN. These results indicate that the siRNAs for Akt1, Akt2, mTOR, or BCTN we used in this study are specific and effective.
Fig. 8.

Cell proliferation is significantly inhibited by knockdown of Akt1 and mTOR in human PASMC and in PASMC isolated from Akt1−/− mice under both normoxic and hypoxic conditions. A: Western blot analysis of Akt1 and Akt2 in human PASMC transfected with scrambled siRNA (Scramble), siRNA for Akt1, or siRNA for Akt2. β-Actin was used as a loading control. B: summarized data (means ± SE) showing the protein expression level of Akt1 and Akt2 in PASMC transfected with scrambled (Scram) siRNA, siRNA for Akt1 (si-Akt1), or siRNA for Akt2 (si-Akt2). **P < 0.01, ***P < 0.001 vs. Scram. C: Western blot analysis of mTOR and β-catenin (BCTN) in human PASMC transfected with scrambled siRNA (Scram), siRNA for mTOR (top), or siRNA for BCTN (bottom). β-Actin was used as a loading control. D: summarized data (means ± SE) showing the protein expression level of mTOR (left) and BCTN (right) in PASMC transfected with Scram-siRNA, siRNA for mTOR (si-mTOR) or siRNA for β-catenin (si-BCTN). **P < 0.01, ***P < 0.001 vs. Scram. E: cell proliferation measured by bromodeoxyuridine (BrdU) uptake under normoxic (left) and hypoxic (right, for 24 h) treatment in control human PASMC (control, not transfected with siRNA) and human PASMC transfected with scrambled siRNA (si-Scram), si-Akt1, siAkt2, si-mTOR, or si-BCTN. *P < 0.05, **P < 0.01, ***P < 0.001 vs. si-Scram and control. F: cell proliferation measured by BrdU uptake in normoxia and hypoxia (24 h) in PASMC isolated from WT, Akt1−/−, and Akt2−/− mice (left). The bar graph (right) shows the hypoxia-induced increase in BrdU uptake (means ± SE) in PASMC isolated from WT, Akt1−/−, and Akt2−/− mice. ***P < 0.001 vs. WT, ###P < 0.001 vs. Akt1−/−.
Knockdown of either Akt1 or mTOR using siRNA significantly reduced PASMC proliferation under normoxic as well as hypoxic conditions (Fig. 8E), and there is no effect of BCTN knockdown on PASMC proliferation under both normoxic and hypoxic conditions (Fig. 8E). Whereas Akt2 knockdown also significantly reduced PASMC proliferation in hypoxic conditions, the inhibitory effect was significantly less than that of Akt1 knockdown (Fig. 8E). To confirm that these differences are attributable to differences in the Akt1/2 isoforms, PASMC were isolated from WT, Akt1−/−, and Akt2−/− mice for cell proliferation experiments. Despite difficulty in culturing these cells because of the significantly decreased proliferation, we were able to isolate an adequate number of cells for both normoxic and hypoxic experiments. Although PASMC from both Akt1−/− and Akt2−/− mice showed decreased proliferation compared with WT mice under normoxic conditions, there was a trend toward Akt1−/− having a greater effect (Fig. 8F). This effect became evident under hypoxic conditions in which Akt2−/− had little to no effect, whereas proliferation of PASMC isolated from Akt1−/− mice had been significantly attenuated under hypoxic conditions compared with both WT and Akt2−/−. Additionally, it is well known that hypoxia leads to increased proliferation of PASMC, yet we show this increase to be more pronounced in PASMC isolated from Akt2−/− mice (Fig. 8F, right). These findings suggest an important difference in the function of the Akt isoforms under hypoxic conditions, in which Akt1 likely plays a more important role in cell proliferation.
Acute hypoxia rapidly causes Akt phosphorylation in PASMC, and Akt1 may partially contribute to acute hypoxic pulmonary vasoconstriction.
Acute hypoxic pulmonary vasoconstriction (HPV) is an important physiological mechanism that maximizes ventilation/perfusion (V/Q) matching and gas exchange in the lung. Persistent hypoxia, however, causes sustained pulmonary vasoconstriction and contributes to the development of pulmonary hypertension in patients with obstructive lung diseases and chronic high-altitude diseases (58, 60).
Acute alveolar hypoxia, by reducing inspired fractional O2 concentration (FiO2) in the ventilated gas mixture from 21% to 1%, caused a reversible increase in PAP attributable to pulmonary vasoconstriction in the isolated perfused/ventilated lung from WT mice (Fig. 9, A–C). The acute HPV was slightly, but significantly, reduced in the isolated perfused/ventilated lungs from Akt−/− mice compared with the WT littermates (Fig. 9, A–C). Furthermore, in vitro experiments using primary cultured PASMC demonstrate that acute exposure of PASMC to hypoxia (3% O2) ranging from 1 to 10 min increased phosphorylation of Akt1/2 and mTOR. As shown in Fig. 9, D–G, the hypoxia-induced increase in phosphorylated Akt1/2 (p-Akt1/2) and phosphorylated mTOR (p-mTOR) in PASMC took place at 1 min and maximized at 3 min. Therefore, the acute effect of Akt1/2 phosphorylation and activation on the pulmonary vasculature may influence hypoxic vasoconstriction, in addition to its effect on PASMC proliferation described above.
Fig. 9.

Deletion of Akt1 attenuates acute hypoxia-induced pulmonary vasoconstriction. A: representative record of PA pressure (PAP) in isolated perfused and ventilated lungs from WT and Akt−/− mice. B: summarized data (means ± SE) showing the increases in PAP induced by repeated hypoxia challenges (6 times) in isolated and ventilated lungs from WT (open bars) and Akt−/− (solid bars) mice. C: averaged data (means ± SE) showing the averaged increases in PAP induced by all hypoxic challenges in WT (n = 3) and Akt−/− (n = 3) mice. *P < 0.05 vs. WT. D–G: representative Western blot data (D and E) and time course (F and G) of hypoxia-induced increase in phosphorylation of Akt1/2 (p-Akt1/2) and mTOR (p-mTOR) in PASMC. *P < 0.05 vs. 0 min hypoxia.
DISCUSSION
In patients with PAH and HPH, the elevated PAP is principally the result of increased total PVR. Activation of G protein-coupled receptors (GPCRs) and RTKs by mitogenic and angiogenic factors, cytokines, and hormones is one of the important mechanisms leading to cell growth, proliferation, and migration. Upregulated expression and increased function of RTK ligands (e.g., PDGF and VEGF) (47) and RTKs (e.g., VEGFR and PDGFRα/PDGFRβ) (7, 21, 31) in the pulmonary vascular cells (including fibroblasts, smooth muscle cells, and endothelial cells) are believed to play an important role in the initiation and progression of PA wall thickening, as well as the neointimal lesions and intraluminal obliteration observed in patients with PAH and animals with experimental pulmonary hypertension (4, 36, 46). Blockade of various RTKs and GPCRs in the plasma membrane of pulmonary vascular smooth muscle cells and endothelial cells using nonselective and selective receptor antagonists (e.g., imatinib) has been proven to have therapeutic effects on pulmonary hypertension (18, 20, 46, 53, 56).
The PI3K-Akt-mTOR pathway is one of the fundamental intracellular signaling cascades that communicate extracellular mitogenic signals to the nuclear transcriptional machinery that induces cell proliferation and protein synthesis (64). The results from this study indicate several things. 1) Phosphorylated Akt1 and 2 (pAkt1/2) are significantly increased in the lung tissues isolated from mice and rats with HPH compared with normoxic controls and in PASMC isolated from patients with PAH compared with normal controls, whereas mRNA and protein expression levels of total Akt1/2 are comparable in lung tissues isolated from normoxic control mice and HPH mice and rats and in PASMC isolated from normal controls and patients with PAH. 2) Chronic hypoxia-induced increases in RVSP, PA thickening, and RVH were all significantly attenuated in Akt1−/− mice compared with their WT littermates, whereas HPH in Akt2−/− mice was similar to that in WT controls. 3) Conditional and inducible KO of mTOR in vascular smooth muscle cells almost abolished the development and progression of HPH compared with control mice. 4) Upregulation of PTEN in PTEN-TG mice significantly attenuated HPH compared with WT controls. 5) Akt1 and mTOR have the greatest effect on PASMC proliferation under hypoxic conditions. 6) Acute exposure of PASMC to hypoxia rapidly increased phosphorylation of Akt1/2 and mTOR, whereas acute HPV in the isolated perfused/ventilated lung from Akt1−/− mice was slightly, but significantly, less than the hypoxic response in WT mice. These observations imply that 1) the PI3K/Akt/mTOR signaling in PASMC is activated in pulmonary hypertension, and activated Akt1/mTOR signaling cascade is required for or involved in the development and progression of pulmonary hypertension; and 2) the activated Akt1/mTOR signaling specifically involved in the development and progression of pulmonary vascular remodeling is due to enhanced phosphorylation of Akt1 but not Akt2.
Blockade of mTOR with rapamycin has been well documented to inhibit cell proliferation in a variety of cell types, including cancer cells (13, 28), coronary arterial smooth muscle cells (22, 34, 39), embryonic stem cells (40, 76), and vascular progenitor cells (3, 16). Additionally, experimental manipulation of upstream molecules in the Akt pathway has been shown to exert beneficial effects on cell proliferation in cancer (30). Our previous studies show that PDGF-mediated activation of the Akt/mTOR pathway is involved in growth factor-mediated upregulation of TRPC channels and enhancement of SOCE in human PASMC (44, 45). Enhanced Ca2+ entry in PASMC is a critical pathogenic factor of both sustained pulmonary vasoconstriction and pulmonary vascular remodeling characteristic of PAH. Here we confirm the effects of the PTEN/Akt1/mTOR pathway on pulmonary vascular remodeling in an experimental animal model of pulmonary hypertension. The novel approach of manipulating multiple steps both upstream and downstream of Akt, as well as employing smooth muscle-specific and inducible KO animals, allows us to confidently display the importance of this signaling pathway on HPH and pulmonary vascular remodeling. While demonstrating this importance, we were also able to identify differences in the contribution of different Akt isoforms, as well as parallel signaling pathways, to the development of HPH.
In a variety of cancer cells, Akt1 and Akt2 play distinct roles in regulating cell proliferation, migration, and protein synthesis (1, 5, 6, 35, 63, 68). In our study, we attempted to discern the specific effects of Akt isoforms on the development of experimental pulmonary hypertension. Although neither the Akt1−/− mice nor the Akt2−/− mice had any significant differences in hemodynamic measurements at baseline, we were able to note some interesting differences after 4 wk of normobaric hypoxia exposure. We were able to show that deletion of Akt1 in mice caused ∼40% inhibition of hypoxia-mediated increase in RVSP and nearly eliminated hypoxia-mediated PA wall thickening. This would suggest that Akt1 plays a dominant role in pulmonary vascular remodeling, a process in which PASMC proliferation is key. While taking a similar approach with Akt2−/− mice, we were unable to show any significant difference in the development of HPH between Akt2−/− mice and their littermate controls. PA wall thickening was also unaffected in Akt2−/− mice, leading us to believe that Akt2 plays a much less dominant role in the development of HPH and pulmonary vascular remodeling. Both siRNA knockdown of Akt1 and Akt2 and PASMCs isolated from Akt1−/− and Akt2−/− mice confirm that cell proliferation under hypoxic conditions is predominately effected by Akt1. In addition to the chronic effects of Akt1, we have also demonstrated acute effects in which HPV is attenuated in Akt1−/− mice. The rapid increase in Akt/mTOR and rapid increase in PAP during hypoxia imply that hypoxia-mediated activation (phosphorylation) of Akt/mTOR signaling may be involved in acute HPV.
We have strengthened these findings on the importance of the Akt1 signaling pathway in the development of murine HPH by examining both upstream and downstream molecules in this signaling cascade including mTOR and PTEN. It has been clearly demonstrated that Akt activation leads to activation of downstream mTOR and then exerts a proproliferative effect on PASMC, resulting in pulmonary vascular remodeling (23). In this study, we have shown that conditional and inducible deletion of mTOR in PASMC almost abolished chronic hypoxia-induced increase in RVSP (Fig. 5, C and E) and PA wall thickening (Fig. 5, G and H). Knockdown of mTOR with siRNA, along with knockdown of Akt1, causes a significant inhibitory effect on PASMC proliferation (Fig. 8, E and F). These data indicate that the Akt1/mTOR pathway in PASMC is an important signaling pathway for the development and progression of pulmonary vascular remodeling and pulmonary hypertension.
PTEN is an upstream phosphatase that prevents activation of Akt indirectly, and previous work has shown that inactivated PTEN is associated with pulmonary hypertension in humans as well as increased severity of HPH in animal models (19). Our TG PTEN mice have previously been demonstrated to have significantly upregulated PTEN gene and protein expression, which can prevent Akt phosphorylation (11). Indeed, our results from this study show that upregulation of PTEN, and thus inactivation of Akt signaling, is protective against HPH, lending further support to our hypothesis.
In conclusion, our study has demonstrated that unique Akt isoforms have distinctive effects in the development of HPH in mice and that Akt1 and not Akt2 is essential for pulmonary vascular remodeling and the development of HPH. We have also shown the importance of the Akt1/mTOR pathway in which increased levels of PTEN, an upstream repressor, or specific KO of downstream mTOR in PASMC, can significantly attenuate the development and progression of HPH. The Akt/mTOR pathway will continue to be a focus of research aimed at determining factors that affect pulmonary vascular remodeling, and focusing on specific isoforms or targets of this pathway may lead to novel therapeutic targets for pulmonary hypertension.
GRANTS
This work was supported in parts by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL 115014, HL066012, HL125208, and HL098053). D. Fraidenburg was supported by a training grant from the NHLBI of the National Institutes of Health (HL082547). J. Chen was supported by a Proof-of-Concept Research Grant from the Pulmonary Hypertension Association.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.T., D.R.F., S.S., J.R.S., S.O., R.D.Y., M.G.B., R.D.M., J.G.G., R.F.M., A.M., and J.X.-J.Y. conception and design of research; H.T., J.C., D.R.F., S.S., J.R.S., and A.M. performed experiments; H.T., J.C., D.R.F., S.S., J.R.S., A.R.D., S.O., R.D.Y., M.G.B., R.D.M., R.F.M., and J.X.-J.Y. analyzed data; H.T., J.C., D.R.F., S.S., J.R.S., A.R.D., J.G.G., R.F.M., A.M., and J.X.-J.Y. interpreted results of experiments; H.T., D.R.F., and J.X.-J.Y. prepared figures; H.T., D.R.F., and J.X.-J.Y. drafted manuscript; H.T., J.C., D.R.F., S.S., J.R.S., A.R.D., S.O., R.D.Y., M.G.B., R.D.M., J.G.G., R.F.M., A.M., and J.X.-J.Y. edited and revised manuscript; H.T., J.C., D.R.F., S.S., J.R.S., A.R.D., S.O., R.D.Y., M.G.B., R.D.M., J.G.G., R.F.M., A.M., and J.X.-J.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Pier Paolo Pandolfi for providing the Super-PTEN mice used in this study and Ms. Annisa Westcott for critical review of the manuscript.
REFERENCES
- 1.Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, Danino M, Karlan BY, Slamon DJ. Overexpression of AKT2/protein kinase Bβ leads to up-regulation of β1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res 63: 196–206, 2003. [PubMed] [Google Scholar]
- 2.Armstrong L, Hughes O, Yung S, Hyslop L, Stewart R, Wappler I, Peters H, Walter T, Stojkovic P, Evans J, Stojkovic M, Lako M. The role of PI3K/AKT, MAPK/ERK and NFκB signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum Mol Genet 15: 1894–1913, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Butzal M, Loges S, Schweizer M, Fischer U, Gehling UM, Hossfeld DK, Fiedler W. Rapamycin inhibits proliferation and differentiation of human endothelial progenitor cells in vitro. Exp Cell Res 300: 65–71, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Cantoni S, Galletti M, Zambelli F, Valente S, Ponti F, Tassinari R, Pasquinelli G, Galie N, Ventura C. Sodium butyrate inhibits platelet-derived growth factor-induced proliferation and migration in pulmonary artery smooth muscle cells through Akt inhibition. FEBS J 280: 2042–2055, 2013. [DOI] [PubMed] [Google Scholar]
- 5.Chen ML, Xu PZ, Peng XD, Chen WS, Guzman G, Yang X, Di Cristofano A, Pandolfi PP, Hay N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev 20: 1569–1574, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR. Amplification of Akt2 in human pancreatic cells and inhibition of Akt2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA 93: 3636–3641, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dahal BK, Heuchel R, Pullamsetti SS, Wilhelm J, Ghofrani HA, Weissmann N, Seeger W, Grimminger F, Schermuly RT. Hypoxic pulmonary hypertension in mice with constitutively active platelet-derived growth factor receptor-β. Pulm Circ 1: 259–268, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Lorenzo A, Fernandez-Hernando C, Cirino G, Sessa WC. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc Natl Acad Sci USA 106: 14552–14557, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duran-Prado M, Morell M, Delgado-Maroto V, Castano JP, Aneiros-Fernandez J, de Lecea L, Culler MD, Hernandez-Cortes P, O'Valle F, Delgado M. Cortistatin inhibits migration and proliferation of human vascular smooth muscle cells and decreases neointimal formation on carotid artery ligation. Circ Res 112: 1444–1455, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol 25: 1869–1878, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, Anastasiou D, Ito K, Sasaki AT, Rameh L, Carracedo A, Vander Heiden MG, Cantley LC, Pinton P, Haigis MC, Pandolfi PP. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 149: 49–62, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 280: H746–H755, 2001. [DOI] [PubMed] [Google Scholar]
- 13.Granville CA, Warfel N, Tsurutani J, Hollander MC, Robertson M, Fox SD, Veenstra TD, Issaq HJ, Linnoila RI, Dennis PA. Identification of a highly effective rapamycin schedule that markedly reduces the size, multiplicity, and phenotypic progression of tobacco carcinogen-induced murine lung tumors. Clin Cancer Res 13: 2281–2289, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Han G, Ma H, Chintala R, Miyake K, Fulton DJ, Barman SA, White RE. Nongenomic, endothelium-independent effects of estrogen on human coronary smooth muscle are mediated by type I (neuronal) NOS and PI3-kinase-Akt signaling. Am J Physiol Heart Circ Physiol 293: H314–H321, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 18: 1926–1945, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Hegner B, Lange M, Kusch A, Essin K, Sezer O, Schulze-Lohoff E, Luft FC, Gollasch M, Dragun D. mTOR regulates vascular smooth muscle cell differentiation from human bone marrow-derived mesenchymal progenitors. Arterioscler Thromb Vasc Biol 29: 232–238, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal 23: 1515–1527, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galie N, Gomez-Sanchez MA, Grimminger F, Grunig E, Hassoun PM, Morrell NW, Peacock AJ, Satoh T, Simonneau G, Tapson VF, Torres F, Lawrence D, Quinn DA, Ghofrani HA. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation 127: 1128–1138, 2013. [DOI] [PubMed] [Google Scholar]
- 19.Horita H, Furgeson SB, Ostriker A, Olszewski KA, Sullivan T, Villegas LR, Levine M, Parr JE, Cool CD, Nemenoff RA, and Weiser-Evans MC. Selective inactivation of PTEN in smooth muscle cells synergizes with hypoxia to induce severe pulmonary hypertension. J Am Heart Assoc 2: e000188, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, Saker M, Marcos E, Tissot CM, Dubois-Rande JL, Amsellem V, Adnot S. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am J Respir Cell Mol Biol 48: 568–577, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karoor V, Oka M, Walchak SJ, Hersh LB, Miller YE, Dempsey EC. Neprilysin regulates pulmonary artery smooth muscle cell phenotype through a platelet-derived growth factor receptor-dependent mechanism. Hypertension 61: 921–930, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konig S, Browne S, Doleschal B, Schernthaner M, Poteser M, Machler H, Wittchow E, Braune M, Muik M, Romanin C, Groschner K. Inhibition of Orai1-mediated Ca2+ entry is a key mechanism of the antiproliferative action of sirolimus in human arterial smooth muscle. Am J Physiol Heart Circ Physiol 305: H1646–H1657, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Krymskaya VP, Snow J, Cesarone G, Khavin I, Goncharov DA, Lim PN, Veasey SC, Ihida-Stansbury K, Jones PL, Goncharova EA. mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J 25: 1922–1933, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li P, Oparil S, Sun JZ, Thompson JA, Chen YF. Fibroblast growth factor mediates hypoxia-induced endothelin-A receptor expression in lung artery smooth muscle cells. J Appl Physiol 95: 643–651; discussion 863, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Li W, Wang H, Kuang CY, Zhu JK, Yu Y, Qin ZX, Liu J, Huang L. An essential role for the Id1/PI3K/Akt/NFκB/survivin signalling pathway in promoting the proliferation of endothelial progenitor cells in vitro. Mol Cell Biochem 363: 135–145, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Macias J, Yuan JX, Jamieson SW, Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med 15: 1289–1297, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lien SC, Wei SY, Chang SF, Chang MD, Chang JY, Chiu JJ. Activation of PPAR-α induces cell cycle arrest and inhibits transforming growth factor-beta1 induction of smooth muscle cell phenotype in 10T1/2 mesenchymal cells. Cell Signal 25: 1252–1263, 2013. [DOI] [PubMed] [Google Scholar]
- 28.LoRusso PM. Mammalian target of rapamycin as a rational therapeutic target for breast cancer treatment. Oncology 84: 43–56, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Luo C, Yi B, Bai L, Xia Y, Wang G, Qian G, Feng H. Suppression of Akt1 phosphorylation by adenoviral transfer of the PTEN gene inhibits hypoxia-induced proliferation of rat pulmonary arterial smooth muscle cells. Biochem Biophys Res Commun 397: 486–492, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Madhunapantula SV, Robertson GP. The PTEN-Akt3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res 22: 400–419, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malhotra R, Paskin-Flerlage S, Zamanian RT, Zimmerman P, Schmidt JW, Deng DY, Southwood M, Spencer R, Lai CS, Parker W, Channick RN, Morrell NW, Elliott CG, Yu PB. Circulating angiogenic modulatory factors predict survival and functional class in pulmonary arterial hypertension. Pulm Circ 3: 369–380, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandegar M, Remillard CV, Yuan JX. Ion channels in pulmonary arterial hypertension. Prog Cardiovasc Dis 45: 81–114, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Marshall C, Mamary AJ, Verhoeven AJ, Marshall BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol 15: 633–644, 1996. [DOI] [PubMed] [Google Scholar]
- 34.Martin KA, Merenick BL, Ding M, Fetalvero KM, Rzucidlo EM, Kozul CD, Brown DJ, Chiu HY, Shyu M, Drapeau BL, Wagner RJ, Powell RJ. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem 282: 36112–36120, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Meng Q, Xia C, Fang J, Rojanasakul Y, Jiang BH. Role of PI3K and Akt specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal 18: 2262–2271, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Mizuno S, Farkas L, Al Husseini A, Farkas D, Gomez-Arroyo J, Kraskauskas D, Nicolls MR, Cool CD, Bogaard HJ, Voelkel NF. Severe pulmonary arterial hypertension induced by SU5416 and ovalbumin immunization. Am J Respir Cell Mol Biol 47: 679–687, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54: S20–S31, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β1 and bone morphogenetic proteins. Circulation 104: 790–795, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Moses JW, Leon MB, Popma JJ, Fitzgerald PJ, Holmes DR, O'Shaughnessy C, Caputo RP, Kereiakes DJ, Williams DO, Teirstein PS, Jaeger JL, Kuntz RE, Investigators S Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N Engl J Med 349: 1315–1323, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, Kiyama H, Yonezawa K, Yamanaka S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol 24: 6710–6718, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakatani K, Thompson DA, Barthel A, Sakaue H, Liu W, Weigel RJ, Roth RA. Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J Biol Chem 274: 21528–21532, 1999. [DOI] [PubMed] [Google Scholar]
- 42.Nasim MT, Ogo T, Chowdhury HM, Zhao L, Chen CN, Rhodes C, Trembath RC. BMPR-II deficiency elicits pro-proliferative and anti-apoptotic responses through the activation of TGF β-TAK1-MAPK pathways in PAH. Hum Mol Genet 21: 2548–2558, 2012. [DOI] [PubMed] [Google Scholar]
- 43.Nemenoff RA, Simpson PA, Furgeson SB, Kaplan-Albuquerque N, Crossno J, Garl PJ, Cooper J, Weiser-Evans MC. Targeted deletion of PTEN in smooth muscle cells results in vascular remodeling and recruitment of progenitor cells through induction of stromal cell-derived factor-1α. Circ Res 102: 1036–1045, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Ogawa A, Firth AL, Ariyasu S, Yamadori I, Matsubara H, Song S, Fraidenburg DR, Yuan JX. Thrombin-mediated activation of Akt signaling contributes to pulmonary vascular remodeling in pulmonary hypertension. Physiol Rep 1: e00190, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogawa A, Firth AL, Smith KA, Maliakal MV, Yuan JX. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 302: C405–C411, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pankey EA, Thammasiboon S, Lasker GF, Baber S, Lasky JA, Kadowitz PJ. Imatinib attenuates monocrotaline pulmonary hypertension and has potent vasodilator activity in pulmonary and systemic vascular beds in the rat. Am J Physiol Heart Circ Physiol 305: H1288–H1296, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perros F, Montani D, Dorfmuller P, Durand-Gasselin I, Tcherakian C, Le Pavec J, Mazmanian M, Fadel E, Mussot S, Mercier O, Herve P, Emilie D, Eddahibi S, Simonneau G, Souza R, Humbert M. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 178: 81–88, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Pi W, Guo X, Su L, Xu W. BMP-2 up-regulates PTEN expression and induces apoptosis of pulmonary artery smooth muscle cells under hypoxia. PLoS One 7: e35283, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pi WF, Guo XJ, Su LP, Xu WG. Troglitazone upregulates PTEN expression and induces the apoptosis of pulmonary artery smooth muscle cells under hypoxic conditions. Int J Mol Med 32: 1101–1109, 2013. [DOI] [PubMed] [Google Scholar]
- 50.Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ, Yuan JX. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 279: C1540–C1549, 2000. [DOI] [PubMed] [Google Scholar]
- 51.Platoshyn O, Remillard CV, Fantozzi I, Mandegar M, Sison TT, Zhang S, Burg E, Yuan JX. Diversity of voltage-dependent K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 287: L226–L238, 2004. [DOI] [PubMed] [Google Scholar]
- 52.Pluteanu F, Cribbs LL. Regulation and function of Cav3.1 T-type calcium channels in IGF-I-stimulated pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 300: C517–C525, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, Jansa P, Jing ZC, Le Brun FO, Mehta S, Mittelholzer CM, Perchenet L, Sastry BK, Sitbon O, Souza R, Torbicki A, Zeng X, Rubin LJ, Simonneau G, Investigators S Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 369: 809–818, 2013. [DOI] [PubMed] [Google Scholar]
- 54.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 122: 4306–4313, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roy HK, Olusola BF, Clemens DL, Karolski WJ, Ratashak A, Lynch HT, Smyrk TC. Akt proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 23: 201–205, 2002. [DOI] [PubMed] [Google Scholar]
- 56.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 115: 2811–2821, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Staal SP. Molecular cloning of the Akt oncogene and its human homologues Akt1 and Akt2: Amplification of Akt1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci USA 84: 5034–5037, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steiner MK. World Health Organization Class III COPD-associated pulmonary hypertension: Are we there yet in understanding the pathobiology of the disease? Chest 136: 658–659, 2009. [DOI] [PubMed] [Google Scholar]
- 59.Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 283: L144–L155, 2002. [DOI] [PubMed] [Google Scholar]
- 60.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 92: 367–520, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takahashi K, Murakami M, Yamanaka S. Role of the phosphoinositide 3-kinase pathway in mouse embryonic stem (ES) cells. Biochem Soc Trans 33: 1522–1525, 2005. [DOI] [PubMed] [Google Scholar]
- 62.Tang T, Gao MH, Lai NC, Firth AL, Takahashi T, Guo T, Yuan JX, Roth DM, Hammond HK. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation 117: 61–69, 2008. [DOI] [PubMed] [Google Scholar]
- 63.Virtakoivu R, Pellinen T, Rantala JK, Perala M, Ivaska J. Distinct roles of Akt isoforms in regulating β1-integrin activity, migration, and invasion in prostate cancer. Mol Biol Cell 23: 3357–3369, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase Akt pathway in human cancer. Nat Rev Cancer 2: 489–501, 2002. [DOI] [PubMed] [Google Scholar]
- 65.Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H, Ayon RJ, Choudhury MS, Ko EA, Wang J, Wang C, Makino A, Yuan JX. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav32. Am J Physiol Lung Cell Mol Physiol 305: L154–L164, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12–G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14: 64–68, 2008. [DOI] [PubMed] [Google Scholar]
- 67.Xie R, Cheng M, Li M, Xiong X, Daadi M, Sapolsky RM, Zhao H. Akt isoforms differentially protect against stroke-induced neuronal injury by regulating mTOR activities. J Cereb Blood Flow Metab 33: 1875–1885, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu X, Sakon M, Nagano H, Hiraoka N, Yamamoto H, Hayashi N, Dono K, Nakamori S, Umeshita K, Ito Y, Matsuura N, Monden M. Akt2 expression correlates with prognosis of human hepatocellular carcinoma. Oncol Rep 11: 25–32, 2004. [PubMed] [Google Scholar]
- 69.Yamamura H, Yamamura A, Ko EA, Pohl NM, Smith KA, Zeifman A, Powell FL, Thistlethwaite PA, Yuan JX. Activation of notch signaling by short-term treatment with jagged-1 enhances store-operated Ca2+ entry in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 306: C871–C878, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol Cell 20: 539–550, 2005. [DOI] [PubMed] [Google Scholar]
- 71.Yoo HY, Zeifman A, Ko EA, Smith KA, Chen J, Machado RF, Zhao YY, Minshall RD, Yuan JX. Optimization of isolated perfused/ventilated mouse lung to study hypoxic pulmonary vasoconstriction. Pulm Circ 3: 396–405, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 284: C316–C330, 2003. [DOI] [PubMed] [Google Scholar]
- 73.Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. Hypoxic and metabolic regulation of voltage-gated K+ channels in rat pulmonary artery smooth muscle cells. Exp Physiol 80: 803–813, 1995. [DOI] [PubMed] [Google Scholar]
- 74.Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. Inhibition of cytochrome P-450 reduces voltage-gated K+ currents in pulmonary arterial myocytes. Am J Physiol Cell Physiol 268: C259–C270, 1995. [DOI] [PubMed] [Google Scholar]
- 75.Zhang S, Dong H, Rubin LJ, Yuan JX. Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292: C2297–C2305, 2007. [DOI] [PubMed] [Google Scholar]
- 76.Zhou J, Su P, Wang L, Chen J, Zimmermann M, Genbacev O, Afonja O, Horne MC, Tanaka T, Duan E, Fisher SJ, Liao J, Chen J, Wang F. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc Natl Acad Sci USA 106: 7840–7845, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]