

Abstract
The excessive activities of the serine proteinases neutrophil elastase and proteinase 3 are associated with tissue damage in chronic obstructive pulmonary disease. Reduced concentrations and/or inhibitory efficiency of the main circulating serine proteinase inhibitor α-1-antitrypsin result from point mutations in its gene. In addition, α-2-macroglobulin competes with α-1-antitrypsin for proteinases, and the α-2-macroglobulin-sequestered enzyme can retain its catalytic activity. We have studied how serine proteinases partition between these inhibitors and the effects of α-1-antitrypsin mutations on this partitioning. Subsequently, we have developed a three-dimensional reaction-diffusion model to describe events occurring in the lung interstitium when serine proteinases diffuse from the neutrophil azurophil granule following degranulation and subsequently bind to either α-1-antitrypsin or α-2-macroglobulin. We found that the proteinases remained uninhibited on the order of 0.1 s after release and diffused on the order of 10 μm into the tissue before becoming sequestered. We have shown that proteinases sequestered to α-2-macroglobulin retain their proteolytic activity and that neutrophil elastase complexes with α-2-macroglobulin are able to degrade elastin. Although neutrophil elastase is implicated in the pathophysiology of emphysema, our results highlight a potentially important role for proteinase 3 because of its greater concentration in azurophil granules, its reduced association rate constant with all α-1-antitrypsin variants studied here, its greater diffusion distance, time spent uninhibited following degranulation, and its greater propensity to partition to α-2-macroglobulin where it retains proteolytic activity.
Keywords: α-1-antitrypsin, chronic obstructive pulmonary disease, reaction-diffusion model, enzyme kinetics, serine proteinase
chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and mortality worldwide (40) and creates a significant economic burden (60). Several pulmonary phenotypes have been recognized, including chronic bronchitis, which affects the large airways and is associated with chronic mucus hypersecretion (28a), and emphysema, which is associated with destruction of alveolar walls and enlargement of airspaces distal to the terminal bronchioles (38). Although the main risk factor for the development of COPD is cigarette smoking (10), only around 20% of smokers develop clinically significant disease (49), indicating that other environmental and genetic risk factors are important in the etiology. To date, deficiency of the serine proteinase inhibitor (serpin) α-1-antitrypsin (A1AT) is the only widely recognized genetic predisposition.
A1AT is secreted by hepatocytes (16), enters the lungs primarily by passive diffusion, and inhibits neutrophil serine proteinases (NSPs) irreversibly with 1:1 stoichiometry. Circulating levels of A1AT are in the range of 20–53 μM in individuals with the normal (protease inhibitor MM, PiMM) genotype (5). Over 100 naturally occurring genetic variants of A1AT have been described. The most common allele associated with marked deficiency of A1AT is the Z variant (glutamate 342 → lysine), which is characterized by a reduction in A1AT quantity (∼20% of the serum level of the normal M variant), a reduction in inhibitory efficiency, and a tendency to form polymers that are retained in the synthesizing cell (24). Pathological consequences then result from a reduction in circulating levels of A1AT, increasing the risk of developing early-onset emphysema (12) attributable to the reduced ability to control NSP activity in the tissues. Other variants described in this paper include the S (glutamate 264 → valine), F (arginine 223 → cysteine), and I (arginine 39 → cysteine) variants, which have also been identified in subjects with lung disease although less commonly than the Z variant (2, 15). Population and in vitro studies (7) suggest that, below a minimum serum A1AT threshold of 11 μM, A1AT is markedly less able to protect the lungs from NSPs, leading to an increased risk of developing emphysema.
The NSP family includes neutrophil elastase (NE) and proteinase 3 (PR3), which have been shown directly to cause pathological changes consistent with emphysema in animal models (14, 26). Although many studies to date have focused on the importance of NE in the proteinase/antiproteinase imbalance (45, 50), there is increasing evidence that other NSPs such as PR3 may also play an important role (44). PR3 is the most abundant serine proteinase in the neutrophil with each cell being estimated to store 3.0 and 1.1 pg of PR3 and NE, respectively (8). In addition, inhibition of PR3 in the lungs is likely to be less efficient than for NE because PR3 is not inhibited by the locally produced chelonianin inhibitor, secretory leukoproteinase inhibitor (SLPI) (4), and the remaining airway inhibitors A1AT and elafin bind to NE more readily than to PR3 (59). The third NSP, cathepsin G, has not been shown to produce emphysema-like lesions in animal or in vitro studies to date and was therefore not included in the current study.
Previous studies of the proteinase/antiproteinase imbalance in COPD have not taken into account factors such as competitive binding when more than 1 NSP is released, when NSPs are released in varying amounts, and differences in the association rate constants for each NSP. Furthermore (at least in serum), a second inhibitor, α-2 macroglobulin (A2M) will also compete with A1AT for NSPs, and, at least for NE, the sequestered enzyme may retain its catalytic activity (52). In this study, we have examined how the NSPs NE and PR3 partition between their inhibitors A1AT and A2M following degranulation and the effects of A1AT mutations on this partitioning. We have developed a three-dimensional reaction-diffusion model to describe events likely to occur in the lung interstitium and hence pertinent to the pathophysiology of emphysema. The model describes the diffusion of NE and PR3 from an azurophil granule following external degranulation and their subsequent inhibition by binding to either A2M or A1AT. The results highlight the importance of PR3 in the proteinase/antiproteinase imbalance in emphysema and increase our understanding of the mechanisms involved that may have implications for treatment of A1AT deficiency (A1ATD) (including the role of A1AT augmentation therapy) and usual COPD.
MATERIALS AND METHODS
Subject selection.
Subjects with A1ATD were identified from the United Kingdom national registry (based at Queen Elizabeth Hospital, Birmingham, United Kingdom) and had their diagnosis confirmed by phenotyping and genotyping (Heredilab, Salt Lake City, UT). Healthy controls were partners of patients with A1ATD and had a normal PiMM A1AT phenotype. All subjects provided written informed consent, and ethical approval was obtained for all aspects of this study (South Birmingham Research Ethics Committee LREC 3359).
Sample collection.
Venous blood was collected from all subjects and processed within 30 min of collection. Serum was obtained using Vacuette serum tubes (Greiner Bio-One, Stonehouse, United Kingdom), allowing clot formation. The tubes were centrifuged at 1,800 g for 10 min at room temperature, and serum was harvested and stored in multiple aliquots at −70°C until analysis to minimize freeze-thaw cycles.
Active site titration of enzymes.
Pure PR3 (Merck, Feltham, United Kingdom; EC number 3.4.21.76) and pure NE (Athens Research and Technology, Athens, GA; EC number 3.4.21.37) were active site titrated against pure M variant A1AT (Athens Research and Technology), as described previously (44).
Measurement of A1AT and A2M concentrations in serum samples.
The A1AT concentration was measured in the serum samples using a locally developed ELISA, which has been described elsewhere (44). The A2M concentrations of serum samples were determined using a commercially available A2M ELISA kit (Universal Biologicals, Cambridge, United Kingdom), according to the manufacturer's instructions.
Inhibition assays of NSPs with serum from various A1AT genotypes.
Pure NE of predetermined activity was diluted in NE assay buffer (0.01 M Tris·HCl, 0.5 M NaCl, 0.1% Triton X-100, pH 8.6) to 1.5 μM active enzyme. Serum samples from subjects with various A1AT genotypes were taken, and the A1AT concentration was measured by ELISA. Each serum sample was then diluted in NE assay buffer so that the concentration of A1AT was 1.5 μM. Ten microliters of NE was added to a 96-well plate (Costar, Corning, NY) in triplicate sets, and increasing volumes of serum were then added to each set of wells. Each experimental set had an appropriate control containing no NE but containing all other components including diluted serum. All wells were made up to 110 μl with NE assay buffer, and the plate was covered and incubated with gentle shaking at 37°C for 20 min to allow the NE to form complexes with the serum inhibitors. Residual activity was measured by adding 100 μl of the NE-specific substrate N-succinyl-Ala-Ala-Ala-p-nitroanilide (SlaaapN; Sigma-Aldrich, Gillingham, United Kingdom) at a concentration of 1 mg/ml in NE assay buffer. The absorbance at 410 nm was read at 5-min intervals up to 60 min using a Biotek Synergy HT plate reader at 37°C. Average results were taken for all triplicates, and control values were subtracted. Inhibition slopes were constructed by plotting the percentage of remaining enzyme activity against the molar ratio of A1AT:NE, and each experiment was performed in triplicate.
NE inhibition assays were also performed using 150 μl of the substrate N-Methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide (MSaapvN; Sigma-Aldrich) at a concentration of 0.2 mg/ml in NSP assay buffer [50 mM Hepes, pH 7.4, 150 mM NaCl, 0.05% Igepal CA-630 (vol/vol)] For these experiments, NE was used at a lower concentration of 0.1 μM because of the different kinetic parameters between the substrates (57), and the serum samples were diluted accordingly.
PR3 inhibition assays were performed using a similar method with an active PR3 concentration of 1.5 μM, and the serum samples were diluted accordingly. These experiments were performed in NSP assay buffer and used MSaapvN as the substrate, which is hydrolyzed by both NE and PR3.
Inhibition of NSPs with mixtures of pure A2M and pure A1AT.
With the use of the values obtained from the A2M and A1AT ELISAs for serum samples, mixtures of pure A2M and pure M variant A1AT were created in the same concentrations as would be found in the serum samples. Inhibition assays of NSP activity were then performed as described above.
Inhibition assays of NSPs using elastin-fluorescein as the substrate.
Pure NE or PR3 was diluted in elastin buffer (0.2 M Tris base, pH 8.8) to an active concentration of 1.5 μM, and 20 μl was added to an Eppendorf 1.5-ml tube (Eppendorf, Stevenage, United Kingdom). Serum or pure A1AT was diluted in elastin buffer so that the concentration of A1AT was 1.5 μM, and increasing volumes of serum/A1AT were added to each Eppendorf tube. The total volume in each tube was made up to 300 μl with elastin buffer, and the tubes were incubated for 20 min at 37°C to allow the NE or PR3 to form complexes with inhibitors. The control tube contained buffer alone. After incubation, 200 μl of elastin-fluorescein (Elastin Products, Owensville, MO; purified from bovine neck ligament and labeled with fluorescein-isothiocyanate) was added, and the tubes were incubated on a blood tube rotator at 37°C for 20 h. The tubes were centrifuged, and 100 μl of supernatant was taken and added to a 96-well plate; the absorbance was read at 495 nm in a Biotek Synergy HT plate reader. Control values were subtracted, and inhibition slopes were constructed by plotting the percentage of remaining enzyme activity against the molar ratio of A1AT to NE or PR3.
Treatment of serum with methylamine to inactivate A2M.
To measure the second-order association rate constant (Kass) values for serum A1AT with NE or PR3, it was first necessary to inactivate A2M. This was achieved by treating the serum samples with 0.1 M methylamine (Fisher Scientific, Loughborough, United Kingdom) for 10 min at 37°C (33). The inactivation of A2M was confirmed indirectly by measuring NE and PR3 inhibition by serum samples with and without treatment with methylamine. The substrate used for these experiments was MSaapvN at 0.6 mg/ml in 0.1 M Hepes, pH 7.5, 9.5% dimethyl sulfoxide.
Measurement of Kass for A1AT variants and NE.
Pure NE of predetermined activity was diluted to an active concentration of 100 nM with NSP assay buffer. Methylamine-treated serum samples of various A1AT genotypes were also diluted to an A1AT concentration of 100 nM, and these samples were titrated against NE to determine the percentage of active A1AT. Following this initial step, the time-dependent measurements were performed by incubating equimolar amounts of NE and active A1AT (1 nM each) in a 1-ml polystyrene cuvette with a 1-cm path length at 23°C. Residual NE activity at 1–5 min was measured by the addition of 200 μl of MSaapvN and measurement of the change in absorbance (410 nm) over 10 min using a spectrophotometer (Jasco V550). At each time point, the percentage of NE inhibition was determined. The Kass was calculated by plotting the inverse of NE activity at each time point vs. time. From the linear portion of the slope, the half time (t½) of the reaction was calculated using the formula:
| (1) |
where c is the y-axis intercept and m is the gradient of the line. Kass was calculated using the following equation (34):
| (2) |
Measurement of Kass for A1AT variants and PR3.
Pure PR3 of predetermined activity was diluted to an active concentration of 400 nM with NSP assay buffer. Methylamine-treated serum samples were also diluted to an A1AT concentration of 400 nM, and these samples were titrated against PR3 to determine the percentage of active A1AT. Following this initial step, the time-dependent measurements were performed by incubating equimolar amounts of PR3 and active A1AT (1 nM each) in a black opaque polypropylene low-binding plate (Sigma) at 23°C with a well volume of 150 μl. Residual PR3 activity at 1–5 min was measured by adding 3 μl of the PR3-specific Förster resonance energy transfer substrate Abz-VAD-norV-ADRQ-EDDnp (Alta Biosciences, Birmingham, United Kingdom) at a concentration of 1 mM (18). The change in fluorescence (excitation 320 nm, emission 420 nm) was measured every 2 min for 20 min using a Biotek Synergy 2 Multi-Mode Microplate Reader. At each time point, the percentage of PR3 inhibition was determined. The Kass was calculated using the same method as that used for NE.
Development of a three-dimensional reaction-diffusion model.
The model parameters included the diffusion of PR3 and NE from the azurophil granule following external degranulation and their subsequent binding reactions with inhibitors, modeled by a system of spherically symmetric reaction-diffusion equations and solved numerically by a finite difference method implemented in the software MATLAB version R2013a 8.1.0 (MathWorks; Matlab, Natick, MA). Reactions between NSPs and inhibitors were treated as irreversible mass action equations based on their respective Kass values:
| (3) |
where E is the enzyme (NSP) and S is the inhibitor. Although dissociation of the complex to form inactivated inhibitor may occur, the rate is not sufficient to significantly change the stoichiometry of inhibition or the quantity of inactivated inhibitor (28). Therefore, these reactions based on Kass values are valid approximations.
Kass values or A1AT concentrations for protease inhibitors MZ and SZ (PiMZ and PiSZ) sera were not determined in this study. However, we were able to model these variants by adjusting the model code such that the two A1AT variants in these heterozygous states were treated as separate entities. We assumed that 50% of the total A1AT concentrations in the PiMM, PiSS, and PiZZ homozygotes (see Table 1) contributed equivalent A1AT concentrations to PiMZ and PiSZ heterozygotes and that each A1AT variant had the same Kass value as in the homozygote (see Table 2). Therefore, values of 15.15 μM for M A1AT, 7.2 μM for S A1AT, and 2.15 μM for Z A1AT were used, giving total A1AT concentrations of 17.3 μM for PiMZ and 9.35 μM for PiSZ.
Table 1.
A1AT serum concentrations
| A1AT Genotype | Serum A1AT Concentration, μM |
|---|---|
| PiMM | 30.3 |
| PiSS | 14.4 |
| PiFZ | 21.6 |
| PiIZ | 16.6 |
| PiZZ | 4.3 |
A1AT, α-1-antitrypsin; Pi, protease inhibitor.
Table 2.
Association rate constants for A1AT variants with NE or PR3
| A1AT Variant | Association Rate Constant with NE | Association Rate Constant with PR3 |
|---|---|---|
| M | (1.45 ± 0.02) x 107 | (9.24 ± 0.48) x 105 |
| S | (1.14 ± 0.36) x 107 | (9.51 ± 3.00) x 105 |
| Z | (7.34 ± 0.03) x 106 | (1.10 ± 0.21) x 106 |
| FZ | (5.75 ± 1.43) x 106 | (1.43 ± 0.29) x 106 |
| IZ | (8.64 ± 1.22) x 106 | (1.00 ± 0.12) x 106 |
Values are means ± SE, taken at 23°C, represented as M−1/s. NE, neutrophil elastase; PR3, proteinase 3.
Starting concentrations of PR3 and NE were taken as 13.4 mM (8) and 5.4 mM (22), respectively, and confined to a volume approximating the azurophil granule with radius 0.171 μm and volume of 2.28 × 10−17 l, as published elsewhere (22). A1AT serum concentrations for each variant were calculated as described above, and an A2M concentration of 3 μM was used as published previously (6) and confirmed by measured values in this study. Association rate constants of A2M with NE and PR3 were given as 4.1 × 107 M−1/s and 1.1 × 107 M−1/s, respectively, as previously published (41, 54).
Reaction-diffusion equations.
Reaction-diffusion of NSPs were modeled via partial differential equations, as
| (4) |
with an analogous equation holding for [PR3].
The three-dimensional spherically symmetric Laplacian operator is given by:
| (5) |
The diffusion coefficients (D) for PR3, NE, A1AT, and A2M were estimated as 7.94, 7.90, 6.54, and 2.72 × 10−11 m2/s, respectively, per Eq. 6 from Tyn and Gusek (53) D = 2.44 × 10−9 m2·s−1·g1/3·mol−1/3 × M−1/3, where M is the molecular mass in g mol−1. Diffusivity decreases with molecular mass but not much because D decreases with the inverse of the radius and therefore with the inverse of the cube root of molecular mass. Inhibitors were assumed to be present long before the release of the NSPs and hence spatially uniformly distributed initially. Reaction-diffusion of inhibitors was modeled by
| (6) |
with an analogous equation holding for [A2M].
Finally, NSP-inhibitor complexes were also assumed to diffuse, approximately at the same rate as free inhibitor, and therefore evolve as
| (7) |
with analogous equations holding for [A2M:NE], [A1AT:PR3], and [A2M:PR3].
The system was solved on the domain 0.02 μm < r < 50 μm for a time period of 0 < t < 0.5 s, with no-flux (zero-derivative) boundary conditions at r = 0, 50 μm, the domain being chosen sufficiently large that the choice of condition at the outer boundary does not significantly affect the results. Initial conditions were taken as spatially uniform for inhibitor, zero for complex, and a smoothed step function of radius 0.171 μm for enzyme boluses.
The equations were solved numerically via the Crank-Nicholson method (31) for the diffusion operator and first-order forward time difference for nonlinear reaction terms. The spatial and temporal discretizations of δr = 0.01 μm and δt = 0.4 μs were verified to be sufficiently small by testing the results against those calculated with finer spatial and temporal grids.
Calculation of Rmax, Tmax and partitioning.
Maximum diffusion distances (Rmax) and maximum time free in solution (Tmax) were calculated from the resultant matrices generated by the MATLAB script, giving a change in NE and PR3 concentrations with respect to time and space (as presented in the surface plots, Fig. 7). Rmax was determined as the furthest radial distance from the center at which the concentration exceeded 10−15 M. Tmax was determined as the latest time point at which the concentration at the center exceeded 10−15 M. Rmax and Tmax can therefore be considered upper bounds.
Fig. 7.

Surface plots for NE and PR3 in the presence of PiMM or PiZZ serum. A surface representation of the log10 of the concentration of active NE or PR3 at a given radius from the center of the azurophil granule (μm) and in time (s). M and Z variants of A1AT are detailed, as these show the extremes of the variants simulated in this model. White regions correspond to concentrations below the threshold value of 10−15 M. Plots show detailed results as follows: NE in PiMM serum (A), PR3 in PiMM serum (B), NE in PiZZ serum (C), and PR3 in PiZZ serum (D). The plots show that active PR3 diffuses to a greater distance from the azurophil granule than NE and that both NE and PR3 activity would be above a threshold of 10−15 M at greater distances in the presence of PiZZ serum compared with PiMM serum. This pattern is also observed in time, where active PR3 would be present for longer than NE, especially in the presence of PiZZ serum.
Partitioning of NE and PR3 between A2M and A1AT was calculated using the final concentrations of the complexes at the end of the 0.5 s of simulated time, at which point all reactions were complete.
RESULTS
Active site titration of enzymes.
Purified PR3 and NE were active site titrated against a pure M variant A1AT of known activity (see materials and methods) and found to be 85% and 87% active, respectively. Active enzyme concentrations were used in all subsequent calculations.
A1AT and A2M concentrations in serum samples.
Serum samples were taken from subjects with various A1AT genotypes, and the concentrations of A1AT are shown in Table 1. Serum concentrations of A2M were measured and found to be similar regardless of A1AT genotype (2.8 ± 0.1 μM). Serum concentrations are within the range of previously published values (5, 6).
Inhibition assays of NSPs with serum from various A1AT genotypes using low-molecular-weight substrates.
When serum samples were used to inhibit the activities of NE or PR3 individually, initially the enzyme activity decreased linearly as the ratio of A1AT:proteinase approached 1:1, indicating that inhibition of enzyme activity by serum largely reflected the inhibitory capacity of A1AT (Figs. 1 and 2). However, when the A1AT in the serum samples was in a molar excess of the proteinase, residual proteinase activity was still observed, which was greater as the serum concentration of A1AT decreased relative to A2M. Figure 3 shows that altering the relative concentrations of pure A1AT and pure A2M produced a similar effect; reduced concentrations of A1AT relative to A2M resulted in greater residual proteinase activity even when the A1AT was in a molar excess of the proteinase. The results also demonstrate that pure A1AT alone is capable of completely inhibiting the activities of NSPs with 1:1 stoichiometry. These results demonstrate differences in partitioning of NSPs between their serum inhibitors depending on their relative local concentrations and support previously published data (52) suggesting that NE bound to A2M retains its proteolytic potential, at least toward low-molecular-weight substrates (as used in these experiments). The proteolytic potential of A2M:PR3 complexes has not been studied previously, but the results presented here demonstrate that A2M:PR3 complexes also retain proteolytic activity toward low-molecular-weight substrates. The results of the experiments using purified M variant A1AT and pure A2M in varying concentrations (Fig. 3) did not fully replicate the results from the experiments using serum (Fig. 1). This suggests that the concentrations of inhibitors does not solely account for the differences in partitioning of NSPs in serum samples containing mutant A1AT variants (see discussion).
Fig. 1.
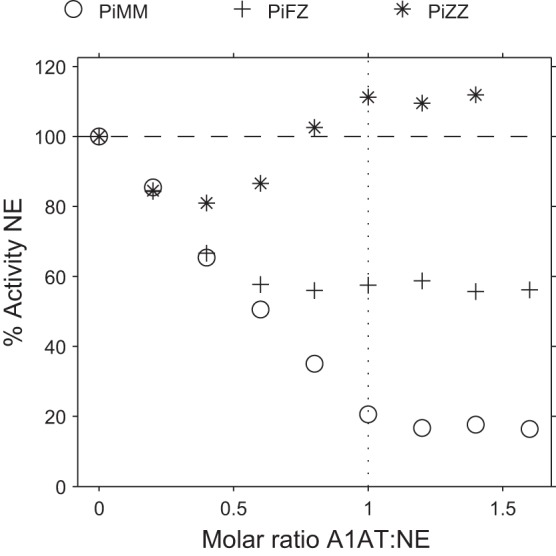
Inhibition of neutrophil elastase (NE) activity by serum from various α-1-antitrypsin (A1AT) genotypes. Inhibition of NE activity by protease inhibitor MM (PiMM), PiFZ, and PiZZ sera using N-succinyl-Ala-Ala-Ala-p-nitroanilide (SlaaapN) as the substrate. The x-axis shows the molar ratio of serum A1AT to active NE. Initially, NE activity decreases linearly as the ratio of A1AT:proteinase approaches 1:1, indicating that NE inhibition largely reflects the inhibitory capacity of A1AT in serum. The residual NE activity observed when serum A1AT is in a molar excess reflects partitioning of the enzyme to α-2 macroglobulin (A2M), where it retains proteolytic activity (and shows enhanced activity in the presence of PiZZ serum). This phenomenon is greater as the serum A1AT concentration reduces relative to A2M. The SE is small and falls within the symbol.
Fig. 2.
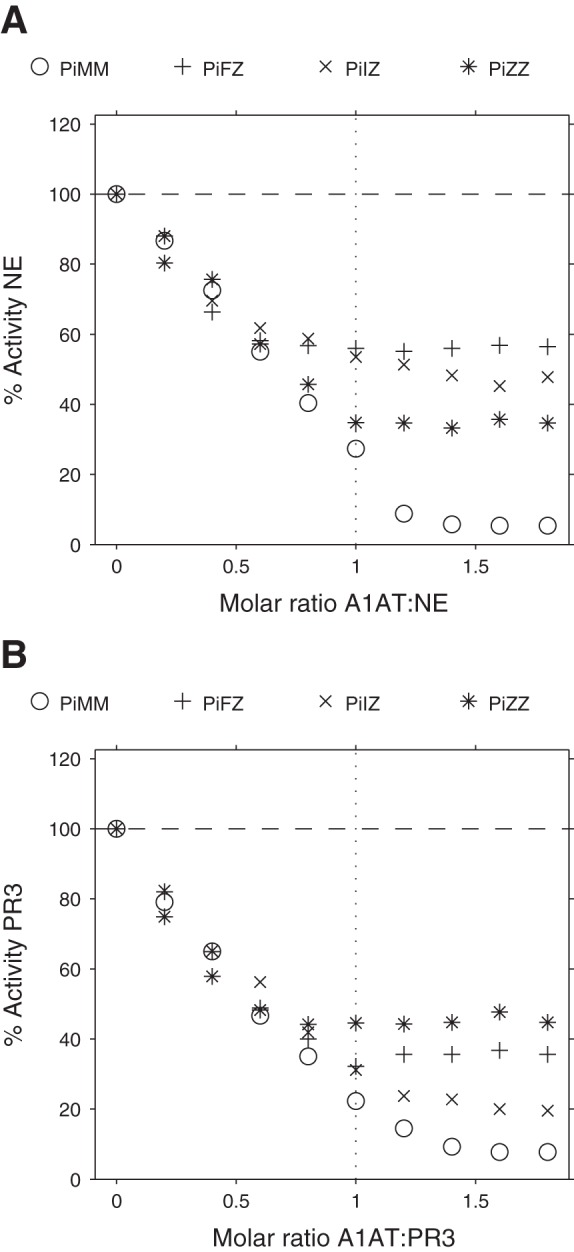
Inhibition of NE or proteinase 3 (PR3) activities by serum from various A1AT genotypes. Inhibition of NE (A) or PR3 (B) activity by PiMM, PiFZ, PiIZ, and PiZZ sera using N-Methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide (MSaapvN) rather than SlaaapN as the substrate. Initially, neutrophil serine proteinase (NSP) activity decreases linearly as the ratio of A1AT:proteinase approaches 1:1, indicating that NSP inhibition largely reflects the inhibitory capacity of A1AT in serum. The residual NSP activity observed when serum A1AT is in a molar excess reflects partitioning of the enzyme to A2M, and this phenomenon is greater in the presence of mutant variants of A1AT. The SE is small and falls within the symbol.
Fig. 3.
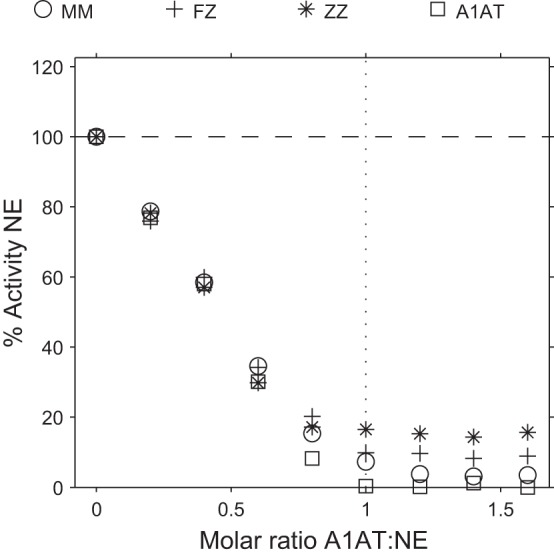
Inhibition of NE with mixtures of pure A2M and pure M variant A1AT in concentrations equivalent to those found in serum from various A1AT genotypes. Inhibition assays of NE using purified inhibitors in concentrations equivalent to those found in PiMM, PiFZ, and PiZZ sera, with SlaaapN as the substrate. The results show that altering the relative concentrations of pure A1AT and pure A2M produces a similar effect to that observed with serum samples, with the greatest residual NE activity being observed when inhibitors are used in proportions equivalent to PiZZ serum. The reduced concentration of A1AT relative to A2M (as found with mutant A1AT variants) results in greater residual NE activity when the A1AT is in a molar excess of NE, suggesting greater partitioning of NE to A2M where the enzyme remains active. Pure A1AT alone (square points) is capable of completely inhibiting NE activity. The SE is small and falls within the symbol.
The inhibition assay of NE by PiZZ serum using SlaaapN as the substrate (Fig. 1) gave different results to those obtained with other serum samples. Although PiZZ serum initially inhibited NE activity, as the ratio of A1AT:NE increased and hence greater volumes of serum (containing A2M) were present, enhanced activity of NE was detected (above that observed with free NE alone). This effect was not observed when MSaapvN was used as the substrate (Fig. 2). Again, this effect is likely to result from NE bound to A2M, and previous authors have reported enhanced activity of NE bound to A2M compared with free NE when using small-peptide substrates, which varies depending on the substrate used from little or no activation to 15-fold activation (52).
The inactivation of A2M in serum by methylamine (Fig. 4) gave further confirmation that the persistent proteinase activity observed when the serum A1AT was in a molar excess of the proteinase was attributable to binding to A2M.
Fig. 4.
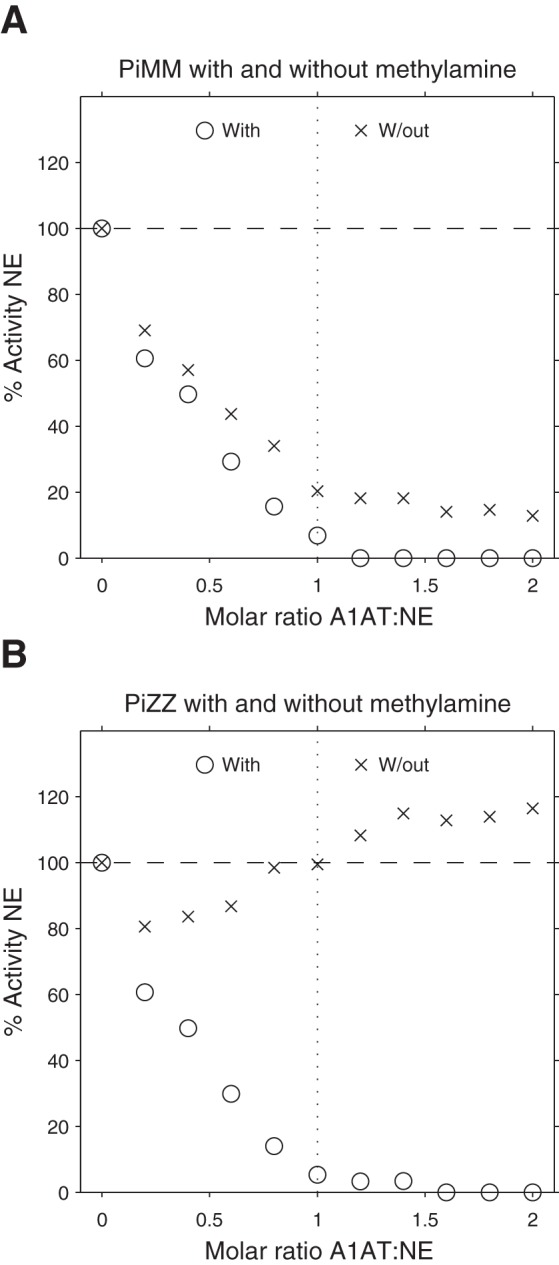
Inhibition of NE activity by serum samples with or without treatment with methylamine. Inhibition of NE using PiMM (A) or PiZZ (B) serum samples with or without treatment with methylamine, using SlaaapN as the substrate. Following inactivation of A2M in the serum samples by methylamine, NE activity is fully inhibited by serum A1AT.
Inhibition assays of NSPs with serum or pure A1AT using elastin-fluorescein as the substrate.
The activity of NE toward elastin could be fully inhibited by stoichiometric amounts of pure M variant A1AT (Fig. 5A). However, when serum was used as the source of inhibitors (PiMM or PiZZ), NE activity against elastin was not completely inhibited even when A1AT was in a molar excess, suggesting that A2M:NE complexes are able to degrade elastin. However, the activity of PR3 against elastin (Fig. 5B) was completely inhibited by serum (PiMM and PiZZ) in the presence of a molar excess of A1AT, suggesting that PR3 complexes with A2M are less likely to play a role in the degradation of mature elastin.
Fig. 5.
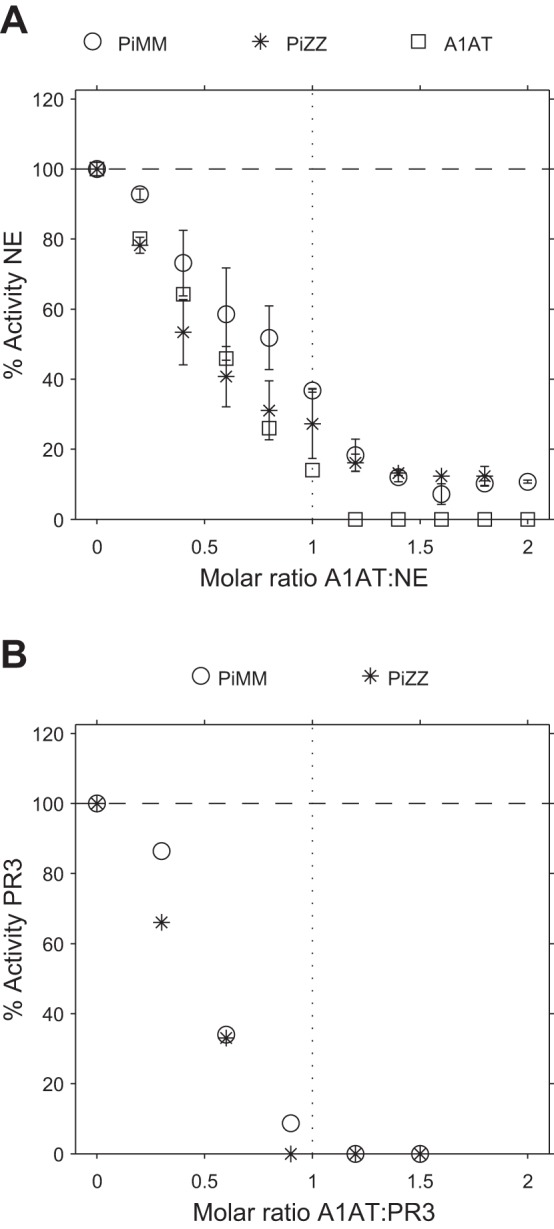
Inhibition of NE or PR3 activities by serum samples or pure A1AT using elastin-fluorescein as the substrate. Inhibition of NE (A) or PR3 (B) activity by PiMM or PiZZ sera or pure A1AT using elastin-fluorescein as the substrate. The results show that pure A1AT was able to completely inhibit NE activity against elastin-fluorescein. However, when serum was used as the source of inhibitors, NE activity against elastin was not completely inhibited even when A1AT was in a molar excess, suggesting that A2M:NE complexes are also able to degrade elastin. The activity of PR3 against elastin could be completely inhibited by PiMM and PiZZ sera. A: mean results of 3 experiments, and error bars indicate the SE. B: results from single experiments.
Association rate constants for A1AT variants and NSPs.
The second-order association rate constant (Kass) values for different variants of A1AT with NE or PR3 are shown in Table 2. The values calculated for NE are comparable to previous results, with the Z variant of A1AT having a Kass value, which is approximately half that of the M variant (34), and the S variant of A1AT having a similar Kass value compared with the M variant (35). For these experiments, it was not possible to use serum from PiFF or PiII homozygotes because no patients had been identified in the United Kingdom. Thus, PiFZ or PiIZ heterozygotes were selected because Z A1AT made a relatively small contribution to the overall concentration of A1AT, and studies have shown that A1AT from PiMZ heterozygotes has a similar Kass with NE to M A1AT alone (9). Furthermore, the results presented here show that FZ A1AT has a Kass value with NE comparable to that obtained with the Z variant and are consistent with results published by Cook et al. (9), who used partially purified F variant A1AT. More recently, we have identified a PiFF homozygote in the United States and were able to confirm our Kass values reported in the current study for F variant A1AT (43). The Kass values calculated in the present study demonstrate that IZ A1AT also had a reduced Kass with NE compared with M A1AT, consistent with a previous study demonstrating that purified I variant A1AT has a Kass value with NE comparable to that obtained with Z A1AT (27).
For all A1AT variants studied here, the Kass with NE was greater than that with PR3. The normal M variant of A1AT has a Kass value for PR3, which is 10 times lower than that for NE, consistent with observations published elsewhere (41). Association rate constants for PR3 and other A1AT variants have not been published previously, but the results presented here suggest that mutants of A1AT have a similar Kass to the M variant. Therefore, the changes in concentration of A1AT in the mutant variants are more likely to influence binding of PR3 to A1AT than changes in association rate constants.
Development of a three-dimensional reaction-diffusion model.
Using the calculated Kass values and serum concentrations for each variant of A1AT along with published values for NE, PR3, and A2M concentrations (see materials and methods), we were able to construct a three-dimensional reaction-diffusion model for the diffusion of NSPs from the azurophil granule following external degranulation and their subsequent inhibition by binding to either A2M or A1AT. The aim of our model was to describe events likely to be occurring in the lung interstitium, which is of particular relevance to the pathophysiology of emphysema.
The summary of the maximum diffusion distance (Rmax) and maximum time free in solution (Tmax) values for NSPs in the presence of different A1AT variants is shown in Fig. 6, and surface plots (depicting diffusion from the azurophil granule in space and time) for NE and PR3 in the presence of either PiMM or PiZZ serum can be seen in Fig. 7. The measures Rmax and Tmax are the distances or times at which the concentrations of the NSPs have fallen below the threshold concentration of 10−15 M, as described in materials and methods. They can therefore be considered upper bounds.
Fig. 6.
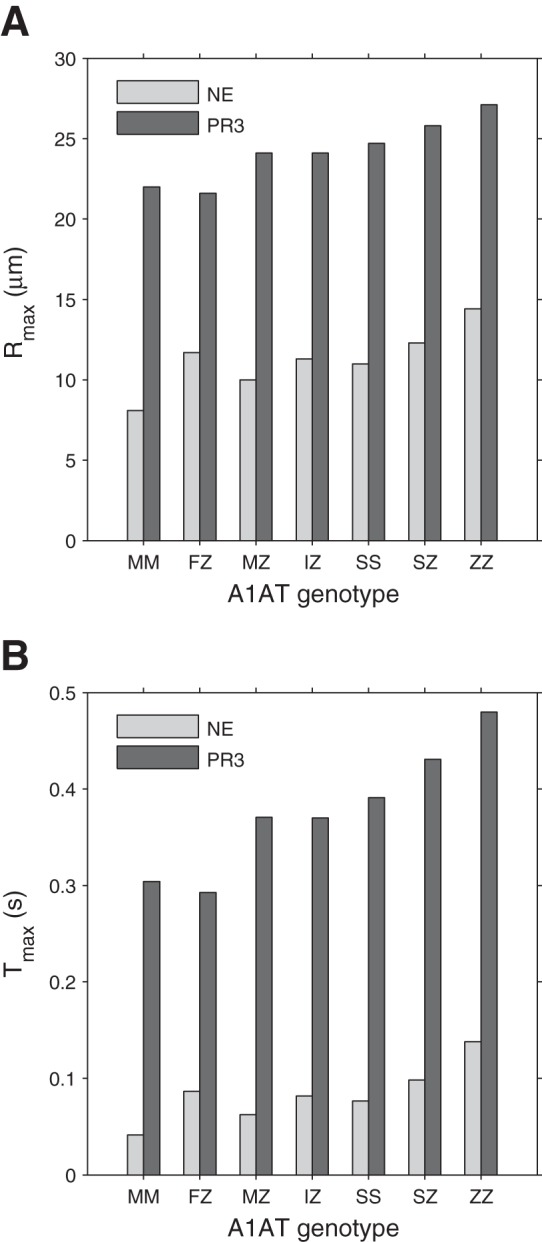
Maximum distance (Rmax) and maximum time (Tmax) at which free NE or PR3 are present in the reaction-diffusion model. Maximum distance (Rmax) (A) and maximum time (Tmax) (B) at which free NE or PR3 are above a threshold of 10−15 M in the reaction-diffusion model using the association rate constant values and serum concentrations of A1AT for each variant. PR3 activity would be present at greater distances, and PR3 would become inhibited later than NE. Mutant A1AT variants result in greater Rmax and Tmax values compared with the M variant.
In the presence of PiMM serum, both NE and PR3 are above the threshold concentration of 10−15 M up to a maximum distance of 8.1 μm and 22 μm, respectively, from the center of the azurophil granule (Fig. 6A), which is also visualized in Fig. 7, A and B, for NE and PR3, respectively. With PiZZ serum, an increase in Rmax to 14.4 μm for NE and to 27.1 μm for PR3 is predicted. An increase in Rmax is predicted for one or both of the NSPs in all mutant A1AT variants (Fig. 6A), with PR3 being free at a greater distance from the center than NE (Fig. 7). The maximum time free in solution (Tmax) of PR3 and NE for each of the A1AT variants is shown in Fig. 6B. The PiZZ genotype exhibits the greatest Tmax values for NE (0.138 s) and PR3 (0.480 s), and the PiMM genotype exhibits the lowest Tmax values (0.041 s for NE and 0.304 s for PR3).
The Rmax and Tmax values for PR3 are predominantly influenced by A1AT concentration because the Kass value of the A1AT:PR3 interaction is similar between A1AT variants (Table 2). However, with NE, differences in Kass are important in addition to A1AT concentrations. For example, PiFZ serum is associated with a concentration of A1AT within the normal range of PiMM serum (5) but has a reduced A1AT:NE Kass value. Therefore, in the presence of PiFZ serum, greater Rmax and Tmax values for NE are observed than with PiMM serum.
Figure 7 represents diffusion from the azurophil granule in space and time for both NE (Fig. 7, A and C) and PR3 (Fig. 7, B and D), respectively, comparing the M variant (Fig. 7, A and B) to the Z variant (Fig. 7, C and D) of A1AT. The results show that active PR3 diffuses to a greater distance from the azurophil granule and persists for longer than NE. In the presence of the Z variant of A1AT, there is increased activity of both NSPs (but particularly PR3) at greater distances from the azurophil granule. The presence of higher concentrations and increased time for activity in solution for both enzymes (especially PR3) will potentially allow greater proteolysis and therefore greater local tissue damage.
Partitioning of NSPs between their inhibitors.
Complex formation through binding of NE or PR3 with the inhibitors A1AT or A2M was calculated from complex concentrations when the system had reached a steady state. The percentage partitioning of PR3 and NE between A2M and variants of A1AT is shown in Fig. 8. This partitioning between A2M and A1AT is important, as NE and PR3 can remain active especially against small substrates when bound to A2M and therefore may continue to contribute to tissue damage until cleared.
Fig. 8.
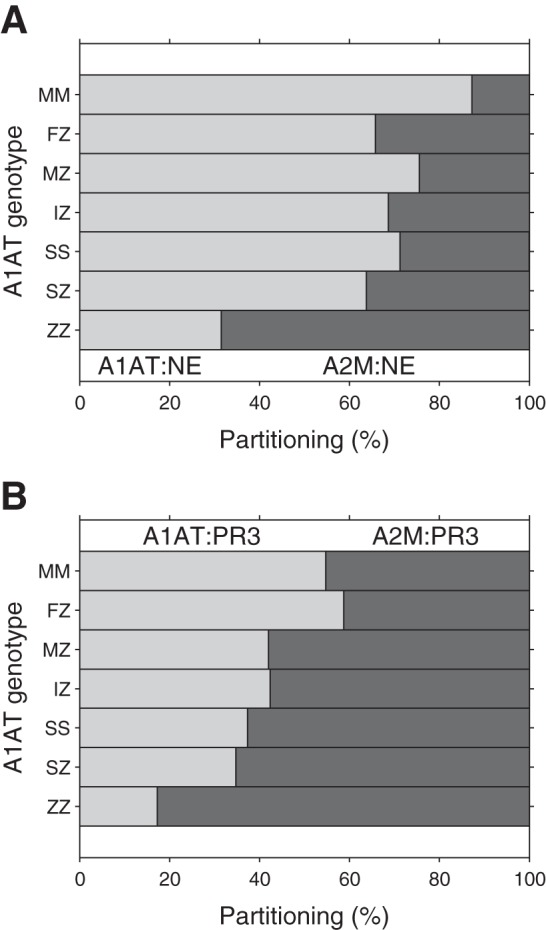
Partitioning of NSPs between inhibitors in the presence of different A1AT variants. Percentage partitioning of NE (A) and PR3 (B) between A2M and variants of A1AT. The PiZZ genotype exhibits the greatest change in partitioning of all the mutant variants, with increased partitioning of NSPs toward A2M.
In the presence of PiMM serum, released NE is mostly inhibited by A1AT with a small percentage bound to A2M, similar to results reported elsewhere (37). Although the Kass value for A2M:NE formation is 4.1 × 107 M−1/s (54) vs. 1.45 × 107 M−1/s for A1AT:NE formation (Table 2), the concentration of A1AT is ∼10-fold higher (30.3 μM against 3 μM), resulting in a greater partitioning of NE toward A1AT. With PR3, the Kass value for A1AT:PR3 formation (9.24 × 105 M−1/s) is ∼10-fold less than the Kass for A2M:PR3 formation (1.1 × 107 M−1/s) (41), and this difference is greater than the difference in NE Kass values. Therefore, PR3 shows an increased partitioning toward A2M, potentially leaving a greater quantity of PR3 active (at least toward low-molecular-weight substrates) than NE.
In the presence of PiZZ serum, a significantly increased partitioning toward A2M is predicted for both NE and PR3. For NE, this is likely to reflect the reduction in Kass with the Z variant of A1AT (Table 2) as well as the reduced A1AT concentration. The Kass for PR3 and A1AT is similar between the M and Z variants and is lower than the Kass with NE. Following release, NSPs (but particularly NE) rapidly deplete free Z A1AT, leaving A2M to inhibit the remainder, and hence PR3 partitions more readily to A2M. A similar pattern is noticed with the other mutant A1AT variants but to a lesser extent because of their higher A1AT serum concentrations compared with the Z variant. This raises the possibility that increased partitioning toward A2M may be partially responsible for any increased tissue damage in these variant A1ATD individuals.
DISCUSSION
The model presented here has highlighted the complex proteinase- inhibitor interactions occurring in COPD and A1ATD, where more than one enzyme is released from the azurophil granules of activated neutrophils and the local inhibitors have different affinities for the enzymes. The results highlight a potentially important role for PR3 in the pathophysiology of emphysema because of its greater concentration in azurophil granules, its reduced Kass with A1AT variants, its greater diffusion distance and time spent uninhibited in solution following release, and its greater propensity to partition to A2M, where it retains proteolytic activity.
NSPs have been the best proven mediators to date replicating the pathological features of COPD and, in particular, emphysema. The interactions between NSPs and their endogenous inhibitors and substrates in vivo are complex and previously poorly understood. Our data provide further information on the potential radius of tissue damage by NSPs following external degranulation from azurophil granules and their partitioning between the major serum inhibitors A1AT and A2M, both of which are found in the lung. Although the size of A2M restricts its diffusion into the airways (and possibly the lung interstitium) in the absence of inflammation (46), partitioning of NSPs to A2M is of biological importance because formation of A2M:NSP complexes does not involve direct interaction with the active site of the enzyme. Our data have shown that A2M:NE complexes retain proteolytic potential, whereas A1AT:NE complexes do not (Fig. 3). Indeed, active A2M:NE complexes have been implicated in the pathogenesis of emphysema in animal models (48), the adult respiratory distress syndrome in humans (56), and the degradation of cartilage matrix in rheumatoid arthritis (30). Our data (Fig. 5) have shown that A2M:NE complexes are able to degrade elastin-fluorescein, and previous studies (29) have also shown that complexes of A2M with porcine pancreatic elastase (PPE) are capable of digesting chemically solubilized elastin. However, other studies have shown that NE complexes with A2M are unable to degrade mature elastin (21) although NE bound to A2M may be able to degrade the elastin precursor tropoelastin (13), which is secreted into the extracellular space before formation of the cross linkages found in elastin (42) and hence could affect the repair process. Further work is therefore required to determine the presence and biological significance of active A2M:NE complexes with particular reference to emphysema.
The proteolytic activity of A2M:PR3 complexes has not been reported previously and may also have clinical relevance, particularly because PR3 has a lower Kass than NE for all variants of A1AT studied here. Therefore, in situations where the local concentration of A1AT is inadequate to inhibit both proteinases, NE would be preferentially inhibited, and PR3 would be more likely to form a complex with A2M. Although A2M:PR3 complexes do not degrade elastin (Fig. 5B), active A2M:PR3 complexes may have indirect consequences for tissue destruction because of their ability to activate proinflammatory cytokines (1). In support of this, Korkmaz et al. (20) confirmed that NE was the main target of A1AT when identical molar amounts of A1AT and NSPs were present together, and PR3 was only inhibited once NE had been totally inhibited. The authors confirmed this for inhibitors present in bronchoalveolar lavage fluid, and they found that PR3 (both free and bound to the neutrophil cell membrane) was the least inhibited NSP.
Our data from the model (Fig. 8) indicate that PR3 would partition more to A2M than NE, especially in the presence of mutant A1AT variants. However, some discrepancies are noted between the data obtained from the model and the in vitro data using serum samples (Fig. 2) in the presence of the F or I variants of A1AT. From the in vitro data, it appears that partitioning of NE to A2M increased in the presence of F or I variant A1AT, which is unlikely to be solely related to the local concentration of A1AT or its Kass because the partitioning is greater than that observed with PiZZ serum. In addition, partitioning of NE to A2M appears to be greater than the partitioning of PR3 to A2M in the presence of the F or I variants of A1AT. The exact reasons for these findings remain uncertain, but there are several possibilities. First, the A1AT:NE complexes formed by F or I variant A1AT could dissociate in the presence of A2M, resulting in the transfer of the enzyme from A1AT to A2M, as described by Ohlsson (36) using trypsin in dog serum and Meyer et al. (29) using PPE in the presence of the two inhibitors. Second, the F or I variant A1AT could be at least partly proteolytically inactivated by NE during the interaction, resulting in inactivated A1AT and free NE (which could then bind to A2M). Further studies into the function of F and I variants of A1AT and the stability of their complexes with NE are therefore necessary.
The inhibition assays using purified inhibitors in varying concentrations (Fig. 3) did not fully replicate the inhibition assays using serum samples (Fig. 1). There are several potential reasons for this discrepancy. First, the experiments using purified inhibitors (Fig. 3) were performed to assess whether alterations in the relative concentrations of A1AT and A2M affected partitioning, and, for this reason, M variant A1AT was used. If purified mutant variants were to be used (such as Z, S, and F variants), further changes in partitioning could be observed due to differences in Kass with NE. Second, mutant variants of A1AT are susceptible to conformational transitions, which may further reduce the levels of functional proteinase inhibitor in vivo. For example, the formation of loop-sheet polymers reduces the antiproteinase capabilities of mutant variants of A1AT beyond that expected for the reduced concentration and Kass (11). Other conformational transitions of mutant variants of A1AT, which may affect their antiproteinase function in vivo, include the formation of unstable intermediates, latent A1AT, or cleaved A1AT following its interaction with NSPs (25). The presence of these conformations of A1AT in the serum samples would influence the partitioning of NSPs more toward binding to A2M.
The inhibition assay of NE by PiZZ serum using SlaaapN as the substrate (Fig. 1) showed enhanced activity of NE (above that observed with free NE alone), as greater volumes of serum (and hence A2M) were added. This phenomenon has been reported previously by Twumasi et al. (52). They reported that the Km, which reflects the affinity of the enzyme for the substrate, was not affected by A2M despite the marked enhancement in the catalytic efficiency of the enzyme as measured by Kcat. They hypothesized that the conformational change in A2M during entrapment of the enzyme uncovers a hydrophobic cavity, which, in conjunction with a positively charged group, orientates the substrate into a position that is more favorable for the enzyme. However, this enhancement of NE activity was not seen when MSaapvN was used as the substrate. The authors demonstrated that the degree of activation of NE bound to A2M varied depending on the synthetic peptide substrate used and varied from little or no activation to 15-fold activation. Interestingly, they also showed that the activity of PPE toward SlaaapN was inhibited by human A2M, thus highlighting species differences in enzyme inhibition and kinetics toward synthetic peptide substrates.
When one compared the results of the inhibition assays using low-molecular-weight substrates (Figs. 1 and 2) with those using elastin as the substrate (Fig. 5), it appears that A2M:NE complexes may be less active toward elastin than against low-molecular-weight peptide substrates. The residual activities of NE (which represent the activities of A2M:NE complexes because the A1AT was in a molar excess) were greater when peptide substrates were used compared with elastin. These findings are consistent with those reported elsewhere (30) and could possibly be due to a proportion of the complexes containing two molecules of NE, which would not allow simultaneous activity of these two NE molecules toward a macromolecular substrate such as elastin.
The results from the three-dimensional reaction-diffusion model (Fig. 7) show that NSPs are able to travel up to 27 μm from the site of release. In the presence of the normal PiMM genotype, the NSPs are confined to a relatively small area of activity. However, in the presence of mutant A1AT variants, the Rmax increases and hence the radius at which NSPs are enzymatically active. This is consistent with the increased tissue destruction and early-onset emphysema observed in PiZZ homozygotes. The use of a radially symmetric model for NE diffusion has previously been tested experimentally (23) and shows high concordance with the current model. The authors demonstrated how degranulation of azurophil granules could significantly damage a substrate surface as a neutrophil passes across it even in the presence of inhibitors. This suggests that, if a neutrophil is close to or adherent to a surface, NSP release from an azurophil granule can cause significant tissue degradation, which would be amplified in the presence of mutant variants of A1AT. In addition, in the presence of infection or increased inflammation in the lung, an influx of neutrophils would result in a large amount of proteinase release, thereby depleting local inhibitors still further.
Our data (Fig. 6) suggest that Rmax and Tmax values for both NE and PR3 are influenced by the local A1AT concentration although Kass values are also important, especially when considering NE activity. This relationship between the radius of potential tissue destruction surrounding the azurophil granules and local A1AT concentration may have significance for one of the treatment methods for A1ATD. In A1ATD individuals, particularly those with the PiZZ phenotype, augmentation therapy with pooled human A1AT reduces the decline in lung density, as measured by CT scanning (47). A serum A1AT concentration of 11 μM has been deemed to be the “protective threshold” below which A1AT augmentation should be considered (51). In the current study, small but significantly increased Rmax values have also been determined for the PiIZ, PiFZ, PiMZ, and PiSS genotypes, which have serum A1AT concentrations above the 11 μM threshold. Whether these increased Rmax values would be clinically significant enough to warrant augmentation therapy is not clear at present. However, importantly, it emphasizes that the potential radius of activity for PR3 exceeds that for NE, and, although A1AT augmentation therapy would beneficially limit activities of both enzymes adding maximum protection, the same would not be true for specific NE inhibitors.
Despite the novel data generated by the model presented here, there are some limitations that may have implications for any conclusions drawn for events taking place in vivo. First, this model has not considered the contribution of the chelonianin inhibitors produced locally in the lungs, SLPI (a reversible inhibitor of NE but not PR3) and elafin (a reversible inhibitor of NE and PR3). Because SLPI does not inhibit PR3 (4), incorporation of this inhibitor into the model may result in an even greater role for PR3 in the pathogenesis of COPD, especially in A1ATD. However, SLPI is predominantly present in the upper airways (55) and may play a less important role in the antiproteinase protection of the distal airways where emphysema occurs although SLPI has been identified in the lung interstitium associated with elastin (58). Second, the model does not incorporate any elimination kinetics of A2M:NSP complexes from the system. Because NSPs remain active when bound to A2M, this may affect extracellular matrix degradation in the lung interstitium attributable to circulation of lymph. Third, the model does not consider any potential binding of NSPs to the neutrophil cell membrane. Membrane-bound NSPs remain catalytically active toward both synthetic peptide and high-molecular-weight biological substrates (8, 39) although this activity can be inhibited by stoichiometric amounts of A1AT (17, 19). The quantity and affinity of cell membrane binding of NSPs remains unknown. However, the model assumes equal affinities of the degranulated NSPs for the cell membrane, which would retain the differential concentrations of the remaining NE and PR3. Finally, the model does not incorporate the third most common NSP, cathepsin G. Although this NSP does not produce emphysema-like lesions in animal and in vitro studies, it would provide a further competing target for A1AT. Importantly, however, the Kass value for cathepsin G and M variant A1AT is more than 100-fold lower than that of NE and about 10-fold lower than that of PR3 (3, 41); therefore, A1AT would be expected to bind cathepsin G only after the other NSPs have been inhibited. Likewise, the Kass value of the cathepsin G-A2M interaction is approximately 10-fold lower than that of the other NSPs (54), and, therefore, its inclusion in the model would be unlikely to affect the predictions reported for the other NSPs. These caveats highlight further areas of research required to gain a greater understanding of the complex proteinase/antiproteinase imbalance occurring in vivo, especially in subjects with emphysema with or without A1AT variants. An increased understanding of the pathogenesis may aid the future development of novel therapies for COPD in general and A1ATD in particular.
In conclusion, the results presented here have provided a greater insight into the proteinase-inhibitor interactions occurring in COPD and A1ATD. The data suggest that PR3 may be more important in the pathophysiology of COPD than previously considered because of the greater quantities stored in azurophil granules, its reduced Kass with A1AT variants, and greater potential radius of activity. In addition, when NSPs form complexes with A2M, they retain their activities, which may have direct (tissue destruction) or indirect (activation of proinflammatory cytokines) effects in vivo. Finally, local A1AT concentrations regulate NSP activities, which may have implications for A1AT augmentation therapy in A1ATD individuals.
DISCLOSURES
N. Sinden has received funding for travel to conferences from GlaxoSmithKline, Boehringer Ingelheim, and Napp Pharmaceuticals. R. Stockley has sat on advisory boards for CSL, Kamada, and Grifols; has received lecture fees from CSL and Grifols; and has received unrestricted research grants from CSL and Grifols. There are no other conflicts of interest, financial or otherwise, declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: N.J.S. performed experiments; N.J.S., M.J.B., D.J.S., J.-U.K., and T.R.D. analyzed data; N.J.S., M.J.B., and R.A.S. interpreted results of experiments; N.J.S. and M.J.B. drafted manuscript; N.J.S., M.J.B., D.J.S., J.-U.K., T.R.D., and R.A.S. approved final version of manuscript; D.J.S. prepared figures; D.J.S., J.-U.K., T.R.D., and R.A.S. edited and revised manuscript; R.A.S. conception and design of research.
ACKNOWLEDGMENTS
The computations described in this paper were performed using the University of Birmingham's BlueBEAR HPC service, which was purchased through HEFCE SRIF-3 funds. See http://www.bear.bham.ac.uk for more details.
REFERENCES
- 1.Bank U, Ansorge S. More than destructive: Neutrophil-derived serine proteases in cytokine bioactivity control. J Leukoc Biol 69: 197–206, 2001. [PubMed] [Google Scholar]
- 2.Baur X, Bencze K. Study of familial α-1-proteinase inhibitor deficiency including a rare proteinase inhibitor phenotype (IZ). I α-1-phenotyping and clinical investigations. Respiration 51: 188–195, 1987. [DOI] [PubMed] [Google Scholar]
- 3.Beatty K, Bieth J, Travis J. Kinetics of association of serine proteinases with native and oxidized α-1-proteinase inhibitor and α-1-antichymotrypsin. J Biol Chem 255: 3931–3934, 1980. [PubMed] [Google Scholar]
- 4.Bergenfeldt M, Axelsson L, Ohlsson K. Release of neutrophil proteinase 4(3) and leukocyte elastase during phagocytosis and their interaction with proteinase inhibitors. Scand J Clin Lab Invest 52: 823–829, 1992. [DOI] [PubMed] [Google Scholar]
- 5.Brantly ML, Wittes JT, Vogelmeier CF, Hubbard RC, Fells GA, Crystal RG. Use of a highly purified α 1-antitrypsin standard to establish ranges for the common normal and deficient α 1-antitrypsin phenotypes. Chest 100: 703–708, 1991. [DOI] [PubMed] [Google Scholar]
- 6.Bristow CL, Di Meo F, Arnold RR. Specific activity of α1proteinase inhibitor and α2macroglobulin in human serum: Application to insulin-dependent diabetes mellitus. Clin Immunol Immunopathol 89: 247–259, 1998. [DOI] [PubMed] [Google Scholar]
- 7.Campbell EJ, Campbell MA, Boukedes SS, Owen CA. Quantum proteolysis by neutrophils: Implications for pulmonary emphysema in α 1-antitrypsin deficiency. J Clin Invest 104: 337–344, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell EJ, Campbell MA, Owen CA. Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibition. J Immunol 165: 3366–3374, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Cook L, Burdon JG, Brenton S, Knight KR, Janus ED. Kinetic characterisation of α-1-antitrypsin F as an inhibitor of human neutrophil elastase. Pathology 28: 242–247, 1996. [DOI] [PubMed] [Google Scholar]
- 10.Doll R, Peto R, Wheatley K, Gray R, Sutherland I. Mortality in relation to smoking: 40 years' observations on male British doctors. BMJ 309: 901–911, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elliott PR, Bilton D, Lomas DA. Lung polymers in Z α1-antitrypsin deficiency-related emphysema. Am J Respir Cell Mol Biol 18: 670–674, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Eriksson S. Pulmonary emphysema and α1-antitrypsin deficiency. Acta Med Scand 175: 197–205, 1964. [DOI] [PubMed] [Google Scholar]
- 13.Galdston M, Levytska V, Liener IE, Twumasi DY. Degradation of tropoelastin and elastin substrates by human neutrophil elastase, free and bound to α2-macroglobulin in serum of the M and Z (Pi) phenotypes for α1-antitrypsin. Am Rev Respir Dis 119: 435–441, 1979. [DOI] [PubMed] [Google Scholar]
- 14.Kao RC, Wehner NG, Skubitz KM, Gray BH, Hoidal JR. Proteinase 3: A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J Clin Invest 82: 1963–1973, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly CP, Tyrrell DN, McDonald GS, Whitehouse DB, Prichard JS. Heterozygous FZ α 1 antitrypsin deficiency associated with severe emphysema and hepatic disease: Case report and family study. Thorax 44: 758–759, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koj A, Regoeczi E, Toews CJ, Leveille R, Gauldie J. Synthesis of antithrombin III and α-1-antitrypsin by the perfused rat liver. Biochim Biophys Acta 539: 496–504, 1978. [DOI] [PubMed] [Google Scholar]
- 17.Korkmaz B, Attucci S, Jourdan ML, Juliano L, Gauthier F. Inhibition of neutrophil elastase by α1-protease inhibitor at the surface of human polymorphonuclear neutrophils. J Immunol 175: 3329–3338, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Korkmaz B, Attucci S, Juliano MA, Kalupov T, Jourdan ML, Juliano L, Gauthier F. Measuring elastase, proteinase 3 and cathepsin G activities at the surface of human neutrophils with fluorescence resonance energy transfer substrates. Nat Protoc 3: 991–1000, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Korkmaz B, Jaillet J, Jourdan ML, Gauthier A, Gauthier F, Attucci S. Catalytic activity and inhibition of Wegener antigen proteinase 3 on the cell surface of human polymorphonuclear neutrophils. J Biol Chem 284: 19896–19902, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korkmaz B, Poutrain P, Hazouard E, de Monte M, Attucci S, Gauthier FL. Competition between elastase and related proteases from human neutrophil for binding to α1-protease inhibitor. Am J Respir Cell Mol Biol 32: 553–559, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Kueppers F, Abrams WR, Weinbaum G, Rosenbloom J. Resistance of tropoelastin and elastin peptides to degradation by α 2-macroglobulin-protease complexes. Arch Biochem Biophys 211: 143–150, 1981. [DOI] [PubMed] [Google Scholar]
- 22.Liou TG, Campbell EJ. Nonisotropic enzyme—inhibitor interactions: A novel nonoxidative mechanism for quantum proteolysis by human neutrophils. Biochemistry 34: 16171–16177, 1995. [DOI] [PubMed] [Google Scholar]
- 23.Liou TG, Campbell EJ. Quantum proteolysis resulting from release of single granules by human neutrophils: A novel, nonoxidative mechanism of extracellular proteolytic activity. J Immunol 157: 2624–2631, 1996. [PubMed] [Google Scholar]
- 24.Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z α 1-antitrypsin accumulation in the liver. Nature 357: 605–607, 1992. [DOI] [PubMed] [Google Scholar]
- 25.Lomas DA, Mahadeva R. α1-Antitrypsin polymerization and the serpinopathies: Pathobiology and prospects for therapy. J Clin Invest 110: 1585–1590, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucey EC, Stone PJ, Breuer R, Christensen TG, Calore JD, Catanese A, Franzblau C, Snider GL. Effect of combined human neutrophil cathepsin G and elastase on induction of secretory cell metaplasia and emphysema in hamsters, with in vitro observations on elastolysis by these enzymes. Am Rev Respir Dis 132: 362–366, 1985. [DOI] [PubMed] [Google Scholar]
- 27.Mahadeva R, Chang WS, Dafforn TR, Oakley DJ, Foreman RC, Calvin J, Wight DG, Lomas DA. Heteropolymerization of S, I, and Z α1-antitrypsin and liver cirrhosis. J Clin Invest 103: 999–1006, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mast AE, Enghild JJ, Pizzo SV, Salvesen G. Analysis of the plasma elimination kinetics and conformational stabilities of native, proteinase-complexed, and reactive site cleaved serpins: Comparison of α 1-proteinase inhibitor, α 1-antichymotrypsin, antithrombin III, α 2-antiplasmin, angiotensinogen, and ovalbumin. Biochemistry 30: 1723–1730, 1991. [DOI] [PubMed] [Google Scholar]
- 28a.Medical Research Council Definition and classification of chronic bronchitis for clinical and epidemiological purposes. A report to the Medical Research Council by their Committee on the Aetiology of Chronic Bronchitis. Lancet 1: 775–779, 1965. [PubMed] [Google Scholar]
- 29.Meyer JF, Bieth J, Metais P. On the inhibition of elastase by serum. Some distinguishing properties of α1-antitrypsin and α2-macroglobulin. Clin Chim Acta 62: 43–53, 1975. [DOI] [PubMed] [Google Scholar]
- 30.Moore AR, Appelboam A, Kawabata K, Da Silva JA, D'Cruz D, Gowland G, Willoughby DA. Destruction of articular cartilage by α 2 macroglobulin elastase complexes: role in rheumatoid arthritis. Ann Rheum Dis 58: 109–113, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morton KW, Mayers DF. Numerical Solution of Partial Differential Equations, second ed.Cambridge University: Cambridge, UK, 2005. [Google Scholar]
- 33.Musumeci V, Vincenti A, Bizzi B. A method for the differential determination of plasma antithrombins. J Clin Pathol 29: 63–68, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogushi F, Fells GA, Hubbard RC, Straus SD, Crystal RG. Z-type α 1-antitrypsin is less competent than M1-type α 1-antitrypsin as an inhibitor of neutrophil elastase. J Clin Invest 80: 1366–1374, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogushi F, Hubbard RC, Fells GA, Casolaro MA, Curiel DT, Brantly ML, Crystal RG. Evaluation of the S-type of α-1-antitrypsin as an in vivo and in vitro inhibitor of neutrophil elastase. Am Rev Respir Dis 137: 364–370, 1988. [DOI] [PubMed] [Google Scholar]
- 36.Ohlsson K. Comparison of affinity of trypsin for two macroglobulin fractions and for 1 antitrypsin in dog serum. Clin Chim Acta 32: 215–220, 1971. [DOI] [PubMed] [Google Scholar]
- 37.Ohlsson K, Olsson I. Neutral proteases of human granulocytes. III. Interaction between human granulocyte elastase and plasma protease inhibitors. Scand J Clin Lab Invest 34: 349–355, 1974. [DOI] [PubMed] [Google Scholar]
- 38.Oldham PD. Measurement of the prevalence of emphysema. Problems of definition and sampling. Proc R Soc Med 69: 127–128, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell surface-bound elastase and cathepsin G on human neutrophils: A novel, nonoxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol 131: 775–789, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 364: 613–620, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Rao NV, Wehner NG, Marshall BC, Gray WR, Gray BH, Hoidal JR. Characterization of proteinase-3 (PR-3), a neutrophil serine proteinase. Structural and functional properties. J Biol Chem 266: 9540–9548, 1991. [PubMed] [Google Scholar]
- 42.Ross R, Bornstein P. Elastic fibers in the body. Sci Am 224: 44–52, 1971. [DOI] [PubMed] [Google Scholar]
- 43.Sinden NJ, Koura F, Stockley RA. The significance of the F variant of α-1-antitrypsin and unique case report of a PiFF homozygote. BMC Pulm Med 14: 132, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinden NJ, Stockley RA. Proteinase 3 activity in sputum from subjects with α-1-antitrypsin deficiency and COPD. Eur Respir J 41: 1042–1050, 2013. [DOI] [PubMed] [Google Scholar]
- 45.Stockley RA. Neutrophils and the pathogenesis of COPD. Chest 121: 151S–155S, 2002. [DOI] [PubMed] [Google Scholar]
- 46.Stockley RA, Mistry M, Bradwell AR, Burnett D. A study of plasma proteins in the sol phase of sputum from patients with chronic bronchitis. Thorax 34: 777–782, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stockley RA, Parr DG, Piitulainen E, Stolk J, Stoel BC, Dirksen A. Therapeutic efficacy of α-1 antitrypsin augmentation therapy on the loss of lung tissue: An integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir Res 11: 136, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stone PJ, Calore JD, Snider GL, Franzblau C. Role of α-macroglobulin-elastase complexes in the pathogenesis of elastase-induced emphysema in hamsters. J Clin Invest 69: 920–931, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tashkin DP, Clark VA, Coulson AH, Simmons M, Bourque LB, Reems C, Detels R, Sayre JW, Rokaw SN. The UCLA population studies of chronic obstructive respiratory disease. VIII. Effects of smoking cessation on lung function: A prospective study of a free-living population. Am Rev Respir Dis 130: 707–715, 1984. [DOI] [PubMed] [Google Scholar]
- 50.Travis J. Structure, function, and control of neutrophil proteinases. Am J Med 84: 37–42, 1988. [DOI] [PubMed] [Google Scholar]
- 51.Turino GM, Barker AF, Brantly ML, Cohen AB, Connelly RP, Crystal RG, Eden E, Schluchter MD, Stoller JK. Clinical features of individuals with PI*SZ phenotype of α 1-antitrypsin deficiency. α 1-Antitrypsin Deficiency Registry Study Group. Am J Respir Crit Care Med 154: 1718–1725, 1996. [DOI] [PubMed] [Google Scholar]
- 52.Twumasi DY, Liener IE, Galdston M, Levytska V. Activation of human leukocyte elastase by human α2-macroglobulin. Nature 267: 61–63, 1977. [DOI] [PubMed] [Google Scholar]
- 53.Tyn MT, Gusek TW. Prediction of diffusion coefficients of proteins. Biotechnol Bioeng 35: 327–338, 1990. [DOI] [PubMed] [Google Scholar]
- 54.Virca GD, Travis J. Kinetics of association of human proteinases with human α 2-macroglobulin. J Biol Chem 259: 8870–8874, 1984. [PubMed] [Google Scholar]
- 55.Vogelmeier C, Hubbard RC, Fells GA, Schnebli HP, Thompson RC, Fritz H, Crystal RG. Anti-neutrophil elastase defense of the normal human respiratory epithelial surface provided by the secretory leukoprotease inhibitor. J Clin Invest 87: 482–488, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wewers MD, Herzyk DJ, Gadek JE. Alveolar fluid neutrophil elastase activity in the adult respiratory distress syndrome is complexed to α-2-macroglobulin. J Clin Invest 82: 1260–1267, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiesner O, Litwiller RD, Hummel AM, Viss MA, McDonald CJ, Jenne DE, Fass DN, Specks U. Differences between human proteinase 3 and neutrophil elastase and their murine homologues are relevant for murine model experiments. FEBS Lett 579: 5305–5312, 2005. [DOI] [PubMed] [Google Scholar]
- 58.Willems LN, Otto-Verberne CJ, Kramps JA, ten Have-Opbroek AA, Dijkman JH. Detection of antileukoprotease in connective tissue of the lung. Histochemistry 86: 165–168, 1986. [DOI] [PubMed] [Google Scholar]
- 59.Ying QL, Simon SR. Elastolysis by proteinase 3 and its inhibition by α(1)-proteinase inhibitor: A mechanism for the incomplete inhibition of ongoing elastolysis. Am J Respir Cell Mol Biol 26: 356–361, 2002. [DOI] [PubMed] [Google Scholar]
- 60.Zaher C, Halbert R, Dubois R, George D, Nonikov D. Smoking-related diseases: The importance of COPD. Int J Tuberc Lung Dis 8: 1423–1428, 2004. [PubMed] [Google Scholar]