

Abstract
Right ventricular (RV) function is a key determinant of survival in patients with both RV and left ventricular (LV) failure, yet the mechanisms of RV failure are poorly understood. Recent studies suggest cardiac metabolism is altered in RV failure in pulmonary hypertension (PH). Accordingly, we assessed mitochondrial content, dynamics, and function in hearts from neonatal calves exposed to hypobaric hypoxia (HH). This model develops severe PH with concomitant RV hypertrophy, dilation, and dysfunction. After 2 wk of HH, pieces of RV and LV were obtained along with samples from age-matched controls. Comparison with control assesses the effect of hypoxia, whereas comparison between the LV and RV in HH assesses the additional impact of RV overload. Mitochondrial DNA was unchanged in HH, as was mitochondrial content as assessed by electron microscopy. Immunoblotting for electron transport chain subunits revealed a small increase in mitochondrial content in HH in both ventricles. Mitochondrial dynamics were largely unchanged. Activity of individual respiratory chain complexes was reduced (complex I) or unchanged (complex V) in HH. Key enzymes in the glycolysis pathway were upregulated in both HH ventricles, alongside upregulation of hypoxia-inducible factor-1α protein. Importantly, none of the changes in expression or activity were different between ventricles, suggesting the changes are in response to HH and not RV overload. Upregulation of glycolytic modulators without chamber-specific mitochondrial dysfunction suggests that mitochondrial capacity and activity are maintained at the onset of PH, and the early RV dysfunction in this model results from mechanisms independent of the mitochondria.
Keywords: pulmonary hypertension, right ventricle, cardiac, mitochondria
heart failure (HF) is characterized by an inability of the ventricles to pump sufficient blood flow to working tissue. Regardless of upstream signaling, the failing heart cannot generate sufficient myofilament force to sustain adequate perfusion pressures. Multiple hypotheses have been put forward to explain this contractile deficit, including myofilament protein dysfunction. However, we have shown in a large animal model of right ventricular (RV) dysfunction that myofilament protein properties are not likely to be solely responsible for the contractile deficit, since isolated skinned cardiac myocytes from hypobaric hypoxia (HH) calves produce similar forces as skinned myocytes from control calves, despite significant organ-level dysfunction (42). Therefore, additional mechanisms must explain the contractile dysfunction observed. Myofilament contraction requires a reliable energy source in the form of ATP for generation of force and maintenance of intracellular calcium gradients. Cardiac hypertrophy and HF are associated with metabolic dysregulation and chronic energy deficiency (15, 29), suggesting energy deficits in the failing heart are associated with and may contribute to the contractile dysfunction. Although a number of metabolic changes have been examined, the outcomes in a number of animal models of ventricular failure have been discordant, with some models showing metabolic adaptation with early stages of dysfunction (18) and others demonstrating additional metabolic deficits with progression to overt decompensated HF (3). However, many of the seminal studies on metabolic profiles during the progression to HF (reviewed in Refs. 4 and 15) have been conducted in the context of left ventricular (LV) failure, and little is known about the metabolic and mitochondrial profile in the context of RV dysfunction.
Recent work suggests mitochondrial dysfunction may be involved in the development of HF and its associated energy deficit and has emerged as a popular hypothesis for both RV and LV dysfunction. In the normal heart, mitochondria are the major site of energy production, providing >95% of the energy supply (ATP) through oxidative phosphorylation. Diminished energy production is associated with loss of myocyte contractility, generation of reactive oxygen species, apoptosis, and myocyte cell death (5, 17, 21, 25). In the heart, as in other tissues, mitochondria exist in a reticulum that is maintained by a dynamic process of fusion and fission, accomplished by mitochondrial fusion proteins mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) and fission proteins such as fission 1 (Fis1) and dynamin-1-like protein (Drp1). Abnormal fission and fusion processes can lead to a disproportionate number of mitochondria, altered intramitochondrial contents, and disorganized mitochondrial shape, all of which have been suggested to play a role in HF (7). Accordingly, HF may be characterized by decreases in cardiac mitochondrial content and oxidative respiration as well as defects in mitochondrial dynamics, all of which might potentially contribute to impaired metabolism and a contractile deficit.
Chronic RV pressure overload in the context of pulmonary arterial banding has been characterized by an increased reliance on glucose as an energy source (41), suggesting altered muscle metabolism may underlie the progression to HF. During ventricular hypertrophy, a metabolic switch occurs altering the fate of glucose and other substrates from oxidative to glycolytic consumption (1, 20). As a result, glycolysis is upregulated alongside downregulated oxidative phosphorylation. Activation of hypoxia-inducible factor-1α (HIF-1α) may facilitate this metabolic switch, resulting in activation of a glycolytic phenotype, including upregulation of vascular endothelial growth factor (VEGF), glucose transporter 4 (GLUT4), phosphofructokinase 1 (PFK1), and pyruvate kinase (PKM), in an effort to increase glucose delivery, transport, and oxidation within the myocardium (19). PKM catalyzes the final rate-limiting step of glycolysis by converting phosphoenolpyruvate and ADP into pyruvate and ATP. Alternative splicing of PKM results in the synthesis of two PKM isoforms: PKM1 and PKM2. Each of these isoforms has differential expression during development and distinct biochemical properties, with PKM1, the adult isoform, promoting oxidative phosphorylation and PKM2, the fetal isoform, promoting aerobic glycolysis (8). Selective upregulation of PKM2 in heart tissue is associated with reduced oxidative metabolism and less efficient ATP production (13). Therefore, important metabolic switches, resulting in increased reliance on glucose metabolism, may also underlie RV dysfunction.
While both humans (38) and animal models (37) of HF show evidence of mitochondrial dysfunction and metabolic abnormalities, only a few studies have examined mitochondrial metabolism in the context of chronic hypoxia (30, 31, 35). Interestingly, a number of these studies have demonstrated changes in LV oxidative capacity with little or no change in the RV (30, 31, 35), and it has been suggested that chronic hypoxia-induced alterations in mitochondria are delayed in the RV, perhaps as an adaptive process in the face of both hypoxia and pressure overload. Therefore, we set out to assess the role of mitochondria and metabolism in the pressure-overloaded RV in a unique large animal model of RV dysfunction, the neonatal calf subjected to HH (12, 39). The neonatal calf model of pulmonary hypertension (PH) induced by a relatively limited-duration (14 days) exposure to acute hypoxia (equivalent to ∼4,500 m) is characterized by elevated pulmonary arterial pressure accompanied by intense inflammatory-fibrotic and structural remodeling of the pulmonary vasculature, pathophysiological features that are recapitulated in established human clinical PH (22, 27). From a cardiac perspective, the animals exhibit overall compensated hemodynamic function and preserved cardiac output in the presence of clear evidence of RV remodeling, including myocardial hypertrophy, chamber expansion, and reexpression of a fetal-hypertrophic contractile gene program (2, 24). Despite significant organ-level dysfunction, isolated skinned myocytes retain contractile properties with only minor contractile changes, suggesting mechanisms other than defects in myofilament force generation may contribute to disease progression. Based on previous literature suggesting a metabolic deficit in the failing left ventricle, we hypothesized that RV from PH animals would show mitochondrial dysfunction and altered metabolic profiles, consistent with the organ-level contractile dysfunction.
MATERIALS AND METHODS
Animal model and hemodynamics measurements.
Details of the model have been previously published (16, 23). Briefly, 1-day-old male Holstein calves (n = 10) were subjected to HH (barometric pressure 445 mmHg) for 2 wk. Age-matched controls (CO; n = 10) were kept at ambient altitude (Denver altitude, barometric pressure 640 mmHg). Standard veterinary care followed institutional guidelines, and the procedures were approved by the Institutional Animal Care and Use Committee; Department of Physiology, School of Veterinary Medicine, Colorado State University. Hemodynamic analysis was performed by right heart catheterization as previously described (16, 23). Briefly, the animals were nonsedated and physically restrained. Catheters were introduced via the jugular vein and advanced into the right atrium, right ventricle, and pulmonary artery for pressure recordings. Following hemodynamic assessments, the animals were killed by pentobarbital sodium (160 mg/kg body wt) and exsanguinated, and the heart was excised. For biochemical analyses, full-thickness biopsies were taken from the RV and LV free wall and flash-frozen in liquid nitrogen.
Immunoblotting.
Approximately 25 mg of sample were homogenized in RIPA buffer (50 mM Tris·HCl, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Nonidet P-40, and 0.5% sodium deoxycholate, pH 7.4) containing 1 mM DTT, 2 mM tributylphosphine, and protease inhibitors. Protein concentration was determined using a modified protein assay (Bio-Rad). Proteins were prepared in Laemmli sample buffer (Bio-Rad) and resolved on either a 12.5 or 10% SDS-PAGE. Proteins were transferred to nitrocellulose and blocked for 1 h at room temperature with 5% nonfat dry milk unless otherwise noted. After being washed with TBS containing 0.5% Tween, membranes were incubated with primary antibody overnight at 4°C. Membranes were washed and incubated with secondary antibody for 1 h at room temperature. Protein bands were visualized using a chemiluminescent substrate and autoradiography. Even loading of proteins was verified by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Ponceau S staining.
Mitochondrial DNA copy number.
DNA was extracted from frozen RV and LV using a QIAamp DNA Mini Kit (Qiagen). Relative mitochondrial DNA (mtDNA) copy number was determined by real-time PCR using primers targeted to mtDNA-encoded cytochrome c oxidase (COX) subunits 1 and 2. Samples were run in duplicate and normalized to the nuclear housekeeping gene desmin. Oligonucleotide sequences were as follows: COX1 forward, ATAGACGTCGACACACGAGC; COX1 reverse, ACCTCCATGAAGTGTTGCCA; COX2 forward, ACTTTCATGACCACACGCTAAT; COX2 reverse, ATGGGTCAGCTTTGTCGTTAGT; desmin forward, CTACATCGAGAAGGTGCGCT; and desmin reverse, TCCTCCTCGTAGATCTCGGC.
RT-qPCR.
RNA was extracted from frozen RV and LV using standard TRIzol-chloroform extraction. RNA was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad). Real-time RT-PCR was performed with the iCycler My iQ using iQ SYBR Green Supermix (Bio-Rad), normalized to the housekeeping gene 18S ribosomal RNA (18S). Oligonucleotide sequences were as follows: PKM1 forward, 5′-CAAGCTGTTTGAAGAACTTG-3′; PKM1 reverse, 5′-AACTATCAAAGCTGCTGCTA-3′; PKM2 forward, 5′-TGCAGCACCTGATTGCCCGT-3′; PKM2 reverse, 5′-ATGGGTGACAGGCGACGGA-3′; 18S forward, 5′-TTTCGATGGTAGTCGCTGTG-3; and 18S reverse, 5′-CTTGGATGTGGTAGCCGTTT-3′.
Isolation of mitochondria.
Mitochondria were isolated using previously published methods (11, 26). Briefly, RV and LV were homogenized in mitochondrial isolation buffer 1 (in mM: 100 KCl, 40 Tris·HCl, 10 Tris base, 5 MgCl2, 1 EDTA, and 1 ATP, pH 7.5). The tissue lysate was spun at 800 g for 10 min, and the supernatant of this spin was centrifuged for 30 min at 9,000 g to pellet mitochondria. The mitochondrial pellet was washed in mitochondrial isolation buffer 2 (100 mM KCl, 10 mM Tris·HCl, 10 mM Tris base, 1 mM MgCl2, 0.1 mM EDTA, and 1.5% BSA, pH 7.4), centrifuged for 10 min at 8,000 g followed by another wash with mitochondrial isolation buffer 2, and spun 10 min at 6,000 g. The mitochondrial pellet was resuspended in PBS for subsequent assessment of mitochondrial complex activity.
Measurement of mitochondrial complex activities.
Complex I and V activity was assessed using commercially available microplate assay kits (MitoSciences) according to the manufacturer's protocol. Briefly, mitochondrial proteins were extracted by adding 1/10 volume lauryl maltoside. Mitochondrial proteins (20 and 5 μg) were added to each well to immunocapture complex I and V, respectively. Complex I activity was determined by the oxidation of NADH to NAD+. Complex V activity was determined by oxidation of NADH to NAD+, which is stoichiometrically coupled to production of ATP. Complex I and V activities are expressed as change in absorbance per minute.
Immunohistochemistry.
Immunohistochemistry was performed on a select subset of samples. Small biopsies of mid, free wall right, and left ventricles were embedded and frozen in optimum cutting temperature buffer. Frozen sections (8 μm) were cut transverse to the ventricular wall on a precooled −20°C cryostat. Slides were washed in TBS containing 0.025% Tween 20 and incubated for 10 min in 1% Triton X-100 in PBS. Slides were washed and blocked for 2 h in 5% BSA in TBS. Sections were incubated with primary antibody [total oxidative phosphorylation (OXPHOS) 1:500] diluted in 1% BSA in TBS overnight. The sections were washed three times with TBS containing 0.025% Triton and incubated with secondary antibodies (1:5,000) for 1 h. Nuclei were visualized with DAPI. Sections were visualized on a Zeiss Laser Scanning Microscope 780. Mitochondrial content was quantified using the Mitophagy plug-in of ImageJ, as previously described (9, 10). Briefly, RGB images were extracted to grayscale and thresholded to optimally resolve mitochondria. The macro outlined mitochondria, and traces were made using the “analyze particles” function. This allowed the calculation of cell area that is occupied by mitochondria using the formula total mitochondrial area/cell area × 100. This calculation of mitochondrial content and the ability of the ImageJ to accurately assess it has been validated (9, 10). Due to technical limitations of this large animal model, we used OXPHOS antibody quantification, since it recapitulates MitoTracker staining of mitochondria (40).
Electron microscopy.
Electron microscopy was performed in conjunction with the Electron Microscopy Center in the Department of Cell and Developmental Biology at the University of Colorado. Select 1-mm punch biopsies were taken from the RV or LV free wall midway between apex and base and rapidly fixed in 4% glutaraldehyde/2.5% paraformaldehyde in 0.1 M cacodylate buffer for a minimum of 48 h. Whole biopsies were dehydrated and were embedded in resin. Slices (50–70 nm thick) were cut on an ultramicrotome and stained with uranyl acetate and lead citrate to enhance contrast. Samples were viewed on an FEI Technai transmission electron microscope.
Materials.
The following antibodies were used: to assess mitochondrial content and integrity: total OXPHOS (1:5,000; Abcam 110413), Mfn1 (1:1,000; Abcam 104274), Mfn2 (1:1,000; Abcam 56889), Fis1 (1:1,000; Abcam epr8412), and Drp1 (1:1,000; Abcam 56788); to assess hypoxia signaling and glycolytic shift: PKM1 (1:2,000; Novus Biologicals NBP2-14833SS), PKM2 (1:2,000; Novus Biologicals NBP1-48308), VEGF (1:500; Abcam 2992–500), GLUT4 (1:1,000; Abcam 25267), PFK1 (1:1,000; Abcam 154804), and HIF-1α (1:500; Novus Biologicals NB100-123). GAPDH (1:500; Santa Cruz sc48166) was used for normalization, and secondary antibodies included 2° anti-mouse and anti-rabbit (1:50,000; Sigma), anti-mouse Alexa488 and anti-rabbit Alexa594 (1:5,000; Invitrogen). The ATP Synthase Enzyme Activity Kit (ab109714) and Complex I Enzyme Activity Kit (ab109721) were purchased from Abcam.
Statistical analyses.
With the exception of immunohistochemistry and electron microscopy, RV and LV tissues from 6–10 CO calves and 6–10 HH calves were examined in all biochemical studies. Furthermore, 6–10 CO and HH calves were examined for hemodynamic measurements. Hemodynamics data were analyzed by one-tailed Student's t-test. Biochemical data were analyzed by two-way ANOVA (ventricle vs. condition). Data are presented as means ± SE. Statistical significance was set a priori for a P value <0.05. Data demonstrating statistical significance are annotated above the respective Figs. 1–6.
Fig. 1.

Two weeks of exposure to hypoxia do not significantly alter mitochondrial shape and content in the right ventricle (RV) and left ventricle (LV). A–D: representative electron micrographs of control (CO) LV (A), CO RV (B), hypobaric hypoxia (HH) LV (C), and HH RV (D). E: expression of mitochondrial electron transport chain complexes was assessed by immunoblotting, using an antibody against one subunit of each complex; n = 10 experiments in each condition. F: mitochondrial DNA copy number was assessed by real-time PCR and expressed relative to a nuclear DNA housekeeping gene; n = 6 in each condition.
Fig. 6.

PH alters the metabolic phenotype in both the RV and the LV. Protein expression of pyruvate kinase (PKM) 1 (A) and PKM2 (B) was unchanged by exposure to hypobaric hypoxia; however, the ratio between 1 and 2 isoforms (C) was significantly diminished as a result of HH. PKM1 and -2 were assessed by immunoblotting and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH); n = 10 in each condition. Real-time PCR was used to assess transcriptional changes in PKM1 and PKM2 isoforms as a result of hypobaric hypoxia. PKM1 (D) and PKM2 (E) were unchanged by HH; however, the ratio between 1 and 2 was attenuated in HH (F), with a significant interaction between the right and left ventricles; n = 6 in each condition. PFK1 (G) and glucose transporter 4 (GLUT4, H) protein expression was upregulated in both ventricles, or unchanged, respectively, as a result of HH; n = 10 in each condition.
RESULTS
Confirmation of PH and RV dysfunction.
Two weeks of HH exposure increased mean pulmonary arterial pressure and total pulmonary resistance, confirming PH. In addition, 2 wk of HH significantly decreased stroke volume and increased RV and right atrial pressures (summarized in Table 1), confirming concomitant RV dysfunction, as previously observed in this model (24, 28).
Table 1.
Hemodynamic measurements in control and HH calves
| Body Wt, kg | HR, beats/min | Cardiac Output, l/min | SV, ml | RVSP, mmHg | RAP, mmHg | MPAP, mmHg | TPR, mmHg·l−1·min−1 | |
|---|---|---|---|---|---|---|---|---|
| CO | 43.7 ± 1.9 | 127 ± 4 | 6.8 ± 0.7 | 48.8 ± 5 | 45. ±2.9 | 2.8 ± 1.3 | 23.8 ± 3.1 | 3.9 ± 0.5 |
| HH | 42.6 ± 4.7 | 155 ± 12 | 5.5 ± 0.7 | 35.6 ± 3.4 | 120.3 ± 13 | 9.7 ± 2.1 | 96.9 ± 6.2 | 20.8 ± 3.1 |
| P value | 0.84 | 0.051 | 0.25 | <0.05* | <0.01* | <0.01* | <0.01* | <0.01* |
Values are presented as means ± SE. HR, heart rate; CO, control; SV, stroke volume; RVSP, right ventricular systolic pressure; RAP, right atrial pressure; MPAP, mean pulmonary arterial pressure; TPR, total pulmonary resistance; HH, hypobaric hypoxia. Hemodynamic measurements were conducted on a subset (n = 8–10) of control and HH calves. Cardiac output was obtained from thermal dilution measurements with a Swann-Ganz catheter and was used to calculate stroke volume and total pulmonary resistance.
Mitochondrial content, dynamics, and electron transport chain activity.
We assessed mitochondrial content by a variety of biochemical and microscopic means. Electron microscopy images indicate there were no gross alterations of mitochondrial structure in the mid ventricle between CO RV and LV (Fig. 1, A and B) and HH RV and LV (Fig. 1, C and D), although increased fibrosis is evident in the HH ventricles. Assessment of individual electron transport complex subunits by total OXPHOS immunoblotting is a surrogate for mitochondrial content. Generally, we found expression of complex subunits to be slightly higher in HH than CO in both the LV and the RV, suggesting minor increases in mitochondrial content (Fig. 1E). mtDNA copy number was unchanged by hypoxia in either ventricle, but COX1 copy number was significantly higher in the RV than the LV (Fig. 1F).
To further verify that mitochondrial abundance and phenotype were unchanged in RV from PH animals, we assessed mitochondrial content by immunohistochemistry and morphometric analysis (Fig. 2A). Staining of transverse sections revealed small decreases in mitochondrial content in both ventricles of the HH animal (Fig. 2B).
Fig. 2.

Mitochondrial content is diminished in both the LV and the RV in response to pulmonary hypertension (PH), with minimal changes in mitochondrial phenotype. A: representative images of midventricle transverse sections stained for total oxidative phosphorylation (OXPHOS), troponin I (TnI), and DAPI for myonuclear identification. B: quantification of mitochondrial content; n = 1/condition, 5 fields/image.
As a means to assess mitochondrial function, we measured electron transport chain activity through spectrophotometric assessment of complex I and V activity. Two weeks of HH exposure significantly attenuated complex I activity in both the LV and the RV (Fig. 3A), whereas no changes occurred in complex V activity in either ventricle in response to HH (Fig. 3B).
Fig. 3.

Exposure to hypobaric hypoxia suppresses complex I activity in both the LV and the RV (A), with no effect on complex V activity (B). Complex I and V activity was assessed spectrophotometrically using commercially available kits and are expressed as a change in absorbance over time; n = 10 in each condition.
Disruption in fission and fusion of mitochondria results in important phenotypic outcomes; thus, we assessed the expression of four proteins involved in mitochondrial fusion and fission (Mfn1, Mfn2, Fis1, and Drp1). Mfn1 was upregulated in both the RV and LV from hypoxic animals (Fig. 4A) and trended toward greater expression in the RV compared with the LV (P = 0.1), whereas Mfn2 (Fig. 4B), Fis1 (Fig. 4C), and Drp1 (Fig. 4D) were unchanged by hypoxia and did not differ between ventricles.
Fig. 4.

Mitochondrial dynamics are generally unchanged in the hypoxic cow heart. Mitofusin 1 (Mfn1, A), mitofusin 2 (Mfn2, B), fission 1 (Fis1, C), and dynamin-1-like protein (Drp1, D) were assessed by immunoblotting; n = 10 in each condition.
Hypoxic and metabolic phenotype.
HIF-1α and VEGF were assessed in the RV and the LV following 2 wk of hypobaric exposure. HIF-1α was significantly upregulated at the protein level in both ventricles (Fig. 5A), whereas VEGF protein expression was unchanged as a result of hypoxia and did not differ between ventricles (Fig. 5B). However, preliminary pathway analysis of microarray data shows increased HIF-1α and VEGF-A mRNAs (HIF-1α, 1.46-fold change, P = 0.029; VEGFA, 1.75-fold change, P = 0.0094) and multiple downstream HIF-1α and VEGF canonical targets are upregulated at the transcript level in hypertensive RV (Brown and Stenmark, unpublished observations).
Fig. 5.
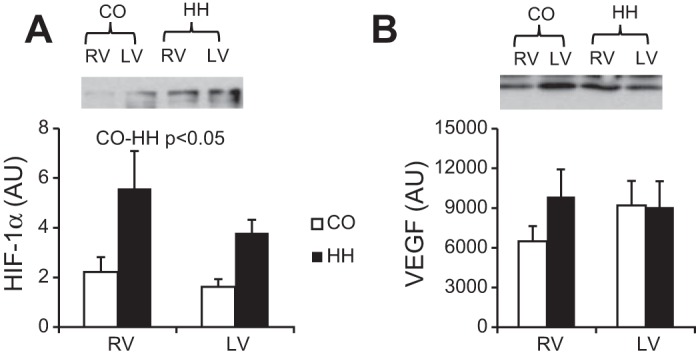
Two weeks of exposure to hypobaric hypoxia significantly increases hypoxia-inducible factor-1α (HIF-1α) protein expression in both the right and left ventricles (A) without altering vascular endothelial growth factor (VEGF) expression (B). HIF-1α and VEGF were assessed by immunoblotting; n = 10 in each condition.
Chronic RV pressure overload in the context of pulmonary arterial banding has been characterized by an increased reliance on glucose as an energy source (41), suggesting altered muscle metabolism may underlie the progression to HF. Accordingly, we assessed the glycolytic mediators PKM, GLUT4, and PFK1 involved in glucose metabolism and uptake. PKM1 (Fig. 6A) and -2 (Fig. 6B) protein expression was unchanged as a result of 2 wk of HH; however, the ratio between the two isoforms was diminished in both the RV and LV in HH animals (Fig. 6C), indicating a small depression of oxidative metabolism. At the transcript level (Fig. 6, D–F), PKM1 and PKM2 were unchanged by HH in either ventricle; however, the ratio between PKM1/2 was significantly depressed only in the RV from HH animals (Fig. 6F). To confirm PKM results, we assessed PFK1 (Fig. 6G) and GLUT4 (Fig. 6H) protein expression and found PFK1 to be upregulated in the HH animals compared with CO and no difference in GLUT4 protein expression between either ventricle or condition. In addition, we assessed GLUT4, lactate dehydrogenase, and hexokinase transcripts for further confirmation of glycolytic metabolic adaptation and found them to be unchanged (lactate dehydrogenase and GLUT4) or upregulated (hexokinase) in both the RV and LV in PH animals (data not shown).
DISCUSSION
It has been suggested that the failing and hypertrophied heart is energy starved, and this energy deficit may explain the contractile dysfunction observed with disease. Mitochondria produce >95% of cardiac ATP and thus are an important source of energy for the energy-sensitive heart. The potential role of impaired metabolism in PH and RV failure has recently been put forth as one mechanism for cardiac disease progression, spurring a metabolic hypothesis of PH that features a global mitochondrial abnormality and metabolic disturbance (33). Accumulating evidence suggests mitochondrial dysfunction may be involved in HF, and mitochondrial dysfunction may explain the energy deficits associated with the failing heart. Therefore, we hypothesized that mitochondrial dysfunction would contribute to the contractile deficit seen in the pressure-overloaded RV in HH. Contrary to our hypothesis, we found little chamber-specific evidence for mitochondrial dysfunction or associated metabolic defects, with RV from PH animals not showing robust decreases in mitochondrial content and activity or alterations in mitochondrial dynamics. We observed small increases in protein expression of mitochondrial electron transport chain subunits with concomitant downregulation of complex I activity. The changes in these mitochondrial parameters and the phenotypic ramifications are summarized in Table 2. Furthermore, we observed small changes in metabolic signaling in the right and left ventricles, indicative of an early switch to glycolytic metabolism. These result are similar to those published by Nouette-Gaulain et al. (31) where it was found that in a rat chronic hypoxia model (21 days) changes in mitochondrial morphology were seen in both ventricles, albeit somewhat delayed in the RV. Importantly, in the current study, all changes were observed in both ventricles, suggesting a response to hypoxic stress, not as a result of the pressure-overloaded right ventricle.
Table 2.
Summary of mitochondrial outcomes as a result of 2 wk of exposure to HH (both ventricles) or the added stress of pressure overload (RV only)
| Assessment | Reflected Phenotype | Response to Hypoxia (Both Ventricles) | Response to PO (RV Only) |
|---|---|---|---|
| Content: OXPHOS, EM, mt/nDNA | %of cell occupied by mitochondria | ↑ | = |
| Function: electron transport chain activity assays | Respiratory chain activity | ↓ | = |
| Fusion: Mfn1, Mfn2 | ΔDynamics: elongation of mitochondrial network; increased mitochondrial mass | ↑ | = (Trend to RV > LV) |
| Fission: Fis1, Drp1 | ΔDynamics: fragmentation of mitochondrial network; increase in no. of smaller mitochondria | = | = |
RV, right ventricle; LV, left ventricle; OXPHOS, oxidative phosphorylation; EM, electron microscopy; mt/nDNA, mitochondrial/nuclear DNA; Mfn1, mitofusin 1; Mfn2, mitofusin 2; Fis1, fission 1; Drp1, dynamin-1-like protein.
Previous investigations of RV dysfunction in PH show that, in rats, a metabolic switch occurs altering the fate of substrates from oxidative to glycolytic consumption (36), suggesting altered muscle metabolism may underlie the contractile deficits in the failing heart. During this metabolic switch, glycolysis is upregulated alongside increased glucose uptake in the energetically starved myocardium. Here we examined a few key signals in the metabolic switch to increased reliance on glucose, including the cardiac glucose transporter GLUT4, the rate-limiting enzyme of glycolysis PFK1, and PKM, the final regulating step of glycolysis, responsible for generating pyruvate to either enter the tricarboxylic acid cycle for oxidative consumption or to be enzymatically converted to lactate. Generally, we found glycolytic signals to be upregulated or unchanged in both the LV and the RV, suggesting exposure to 2 wk of HH increases glycolytic flux in both ventricles. Interestingly, our PKM data suggest that some metabolic adaptations may be specific to the RV, since the ratio between PKM1 and -2 significantly shifts at the transcriptional level and trended toward significance at the protein level (P = 0.1). Mice with enhanced PKM2 expression in skeletal muscle use glucose as a preferential fuel source, over fatty aid oxidation (13). In vitro, preferential splicing of PKM2 shifts muscle cells toward glucose consumption, accompanied by decreased ATP production. This metabolic switch disrupts muscle metabolic homeostasis and results in energy deficits associated with muscle weakness (13). Therefore, it appears that, in rodents, the metabolic switch of increased reliance on glucose uptake and oxidation is insufficient to sustain the failing heart, potentially due to the decreased ATP production per glucose molecule consumed through glycolysis compared with the energy generated through mitochondrial-driven oxidative phosphorylation. In the current study, the majority of changes in glycolytic signals occurred in both ventricles, and we believe this suggests a biventricular response to hypoxia rather than a RV adaptation to pressure overload. Our assessment of HIF-1α supports this, since the oxygen sensor was increased in both ventricles as a result of HH. Therefore, while minimal cardiac metabolic adaptations occurred in response to PH, they do not appear to explain the dysfunction in the RV in this model.
It is well known that many of the physiological and metabolic processes in the heart are regulated by the relative abundance of myosin heavy chain isoforms. In the rodent heart, α-myosin heavy chain predominates (14). α-Myosin heavy chain is associated with a higher ATPase activity, higher cross-bridge turnover rates, and higher resting heart rates. Unlike in rodents, we have previously shown that β-myosin heavy chain predominates in the neonatal calf model, and there is no difference in expression of the α-to-β ratio in the HH calves compared with CO (42). The discrepancies between rodent and calf α/β myosin expression and adaptation to a chronic pressure overload highlights important species differences between the neonatal calf and previously reported mitochondrial and metabolic outcomes in rodent models.
Impaired ATP production may be due to multiple factors within the mitochondria, including decreases in mitochondrial content, attenuated activity of respiratory enzymes, decreased ability to make new mitochondria (biogenesis), or diminished clearance of damaged mitochondria (mitophagy). Mitochondrial biogenic signals and electron transport chain enzymes are known to be impacted during HF. During cardiomyocyte hypertrophy, biogenesis has been shown to be enhanced to meet growing energy demand of the myocardium. However, during pathological decompensation, mitochondrial biogenesis can no longer match energy demand and begins to subsequently decrease (34). Disorders in mitochondrial organization have also been noted in failing hearts, where small fragmented mitochondria predominate over the large mitochondrial reticulum observed in healthy hearts (6). The mitochondria exist in a reticulum that is maintained through constant fission and fusion processes. Fusion serves to mix mitochondrial contents, including metabolites, mtDNA, and mitochondrial proteins, whereas fission enables the segregation of mitochondria to increase distribution and number of the organelle through the cell. Various genetic models of altered fission/fusion show a HF phenotype, including Mfn2-deficient mice, which demonstrate cardiac hypertrophy and depressed cardiac function (32). OPA1, the inner mitochondrial membrane fusion protein, is decreased in both human and rat HF (6), further highlighting the importance of mitochondrial dynamics in regulating mitochondrial and cardiac function. Thus, while it has been suggested that disruptions of mitochondrial dynamics and structure may exist during HF, the mechanisms by which the disruption of these events occurs remains to be elucidated. Here, we show that RV dysfunction in the face of PH is not a result of altered mitochondrial function or dynamics. We found the expression of outer mitochondrial membrane proteins involved in fission and fusion to be unchanged, and immunohistochemical analyses of sectioned RV showed minimal alterations in mitochondrial content. Furthermore, electron microscopy data showed no evidence of mitochondrial structural alterations. Together, the mitochondrial phenotype observed in our large-animal PH model occurred in both the LV and the RV and further support an early adaptive response to hypoxia and the associated energy deficit in the myocardium, not a chamber-specific deficit in the pressure-overloaded dysfunctional RV.
The current study is limited by our inability to measure mitochondrial respiration. The measurement of oxygen consumption requires fresh tissue. Due to sample availability and procurement, we were unable to make these assessments. However, in the face of the current data demonstrating minimal difference in mitochondrial content, dynamics, and activity in the overloaded RV, it is hard to imagine that significant differences in respiration would have been observed between HH and control conditions. Furthermore, even without respiration measurements, we demonstrate for the first time that RV pressure overload is not characterized by mitochondrial dysfunction in the context of HH.
In conclusion, we show that mitochondrial function is largely preserved during early stages of RV remodeling in our PH model. This model of PH represents early adaptation in the RV to the elevated pulmonary arterial pressure and inflammatory-fibrotic and structural remodeling of the pulmonary vasculature. From a cardiac perspective, the animals exhibit overall compensated hemodynamic function and preserved cardiac output in the presence of clear evidence of RV remodeling. In concordance with previous publications of RV hypertrophy, we demonstrate small shifts toward a more glycolytic phenotype in the RV of HH animals. However, we found mitochondrial content, dynamics, and function to be similarly changed in both the RV and LV, which we propose is a compensatory response to the hypoxic stimulus in both ventricles and not due to the selective pressure-overload stress in the RV. It is plausible that, in later stages of RV remodeling when cardiac output can no longer be maintained, mitochondrial deficits may become apparent, a hypothesis that warrants future investigation. However, the abundance of data in the current study demonstrating little mitochondrial abnormality in the pressure-overloaded RV induced by PH suggest that the altered contractile deficit at the level of the RV myocardium cannot be explained by mitochondrially regulated metabolic changes. Taken together with our previous work showing similarly preserved myofilament contractile function in RV myocytes from the hypertensive calf, we suggest that, in this model, additional unexplained mechanisms independent of the mitochondria contribute to RV dysfunction in PH.
GRANTS
This project is supported by National Heart, Lung, and Blood Institute Training Grant 2T32-HL-007822-16 (D. R. Bruns), Program Project Grant P01-HL-014985-36A1, and by the Temple Hoyne Buell Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.R.B. and L.A.W. conception and design of research; D.R.B. and R.D.B. performed experiments; D.R.B. analyzed data; D.R.B. and L.A.W. interpreted results of experiments; D.R.B. prepared figures; D.R.B. drafted manuscript; R.D.B., K.R.S., P.M.B., and L.A.W. edited and revised manuscript; L.A.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Stephen Thoemmes, Hui Zhang, and Yanmei Du for technical assistance.
REFERENCES
- 1.Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol Heart Circ Physiol 267: H742–H750, 1994. [DOI] [PubMed] [Google Scholar]
- 2.Bakerman PR, Stenmark KR, Fisher JH. α-Skeletal actin mRNA increases in acute right ventricular hypertrophy. Am J Physiol Lung Cell Mol Physiol 258: L173–L178, 1990. [DOI] [PubMed] [Google Scholar]
- 3.Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, von Kienlin M, Harre K, Hahn D, Neubauer S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol 40: 1267–1274, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Carley AN, Taegtmeyer H, Lewandowski ED. Matrix revisited: mechanisms linking energy substrate metabolism to the function of the heart. Circ Res 114: 717–729, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol 88: 1880–1889, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res 84: 91–99, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L, Knowlton AA. Mitochondrial dynamics in heart failure. Congest Heart Fail 17: 257–261, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452: 230–233, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Dagda RK, Cherra SJ 3rd Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284: 13843–13855, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson's disease. Autophagy 4: 770–782, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drake JC, Peelor FF 3rd Biela LM, Watkins MK, Miller RA, Hamilton KL, Miller BF. Assessment of mitochondrial biogenesis and mTORC1 signaling during chronic rapamycin feeding in male and female mice. J Gerontol 68: 1493–1501, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frid MG, Li M, Gnanasekharan M, Burke DL, Fragoso M, Strassheim D, Sylman JL, Stenmark KR. Sustained hypoxia leads to the emergence of cells with enhanced growth, migratory, and promitogenic potentials within the distal pulmonary artery wall. Am J Physiol Lung Cell Mol Physiol 297: L1059–L1072, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Z, Cooper TA. Reexpression of pyruvate kinase M2 in type 1 myofibers correlates with altered glucose metabolism in myotonic dystrophy. Proc Natl Acad Sci USA 110: 13570–13575, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haddad F, Bodell PW, Baldwin KM. Pressure-induced regulation of myosin expression in rodent heart. J Appl Physiol 78: 1489–1495, 1995. [DOI] [PubMed] [Google Scholar]
- 15.Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res 81: 412–419, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inscore SC, Stenmark KR, Orton C, Irvin CG. Neonatal calves develop airflow limitation due to chronic hypobaric hypoxia. J Appl Physiol 70: 384–390, 1991. [DOI] [PubMed] [Google Scholar]
- 17.Jung F, Weiland U, Johns RA, Ihling C, Dimmeler S. Chronic hypoxia induces apoptosis in cardiac myocytes: a possible role for Bcl-2-like proteins. Biochem Biophys Res Commun 286: 419–425, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Kagaya Y, Kanno Y, Takeyama D, Ishide N, Maruyama Y, Takahashi T, Ido T, Takishima T. Effects of long-term pressure overload on regional myocardial glucose and free fatty acid uptake in rats. A quantitative autoradiographic study. Circulation 81: 1353–1361, 1990. [DOI] [PubMed] [Google Scholar]
- 19.Kodde IF, van der Stok J, Smolenski RT, de Jong JW. Metabolic and genetic regulation of cardiac energy substrate preference. Comp Biochem Physiol A Mol Integr Physiol 146: 26–39, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Kolwicz SC Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res 90: 194–201, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol 282: L1324–L1329, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Lammers S, Scott D, Hunter K, Tan W, Shandas R, Stenmark KR. Mechanics and function of the pulmonary vasculature: implications for pulmonary vascular disease and right ventricular function. Comprehen Physiol 2: 295–319, 2012. [DOI] [PubMed] [Google Scholar]
- 23.Lammers SR, Kao PH, Qi HJ, Hunter K, Lanning C, Albietz J, Hofmeister S, Mecham R, Stenmark KR, Shandas R. Changes in the structure-function relationship of elastin and its impact on the proximal pulmonary arterial mechanics of hypertensive calves. Am J Physiol Heart Circ Physiol 295: H1451–1459, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemler MS, Bies RD, Frid MG, Sastravaha A, Zisman LS, Bohlmeyer T, Gerdes AM, Reeves JT, Stenmark KR. Myocyte cytoskeletal disorganization and right heart failure in hypoxia-induced neonatal pulmonary hypertension. Am J Physiol Heart Circ Physiol 279: H1365–H1376, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev 13: 137–150, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Miller BF, Robinson MM, Bruss MD, Hellerstein M, Hamilton KL. A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell 11: 150–161, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54: S20–S31, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrell NW, Danilov SM, Satyan KB, Morris KG, Stenmark KR. Right ventricular angiotensin converting enzyme activity and expression is increased during hypoxic pulmonary hypertension. Cardiovasc Res 34: 393–403, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Neubauer S. The failing heart–an engine out of fuel. N Engl J Med 356: 1140–1151, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Nouette-Gaulain K, Forestier F, Malgat M, Marthan R, Mazat JP, Sztark F. Effects of bupivacaine on mitochondrial energy metabolism in heart of rats following exposure to chronic hypoxia. Anesthesiology 97: 1507–1511, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Nouette-Gaulain K, Malgat M, Rocher C, Savineau JP, Marthan R, Mazat JP, Sztark F. Time course of differential mitochondrial energy metabolism adaptation to chronic hypoxia in right and left ventricles. Cardiovasc Res 66: 132–140, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O'Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol 31: 1309–1328, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res 115: 148–164, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Rosca MG, Hoppel CL. Mitochondrial dysfunction in heart failure. Heart Fail Rev 18: 607–622, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rumsey WL, Abbott B, Bertelsen D, Mallamaci M, Hagan K, Nelson D, Erecinska M. Adaptation to hypoxia alters energy metabolism in rat heart. Am J Physiol Heart Circ Physiol 276: H71–H80, 1999. [DOI] [PubMed] [Google Scholar]
- 36.Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res 115: 176–188, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabbah HN, Sharov V, Riddle JM, Kono T, Lesch M, Goldstein S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol 24: 1333–1347, 1992. [DOI] [PubMed] [Google Scholar]
- 38.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol 32: 2361–2367, 2000. [DOI] [PubMed] [Google Scholar]
- 39.Stenmark KR, Aldashev AA, Orton EC, Durmowicz AG, Badesch DB, Parks WC, Mecham RP, Voelkel NF, Reeves JT. Cellular adaptation during chronic neonatal hypoxic pulmonary hypertension. Am J Physiol 261: 97–104, 1991. [DOI] [PubMed] [Google Scholar]
- 40.Taanman JW, Bodnar AG, Cooper JM, Morris AA, Clayton PT, Leonard JV, Schapira AH. Molecular mechanisms in mitochondrial DNA depletion syndrome. Hum Mol Genet 6: 935–942, 1997. [DOI] [PubMed] [Google Scholar]
- 41.Takeyama D, Kagaya Y, Yamane Y, Shiba N, Chida M, Takahashi T, Ido T, Ishide N, Takishima T. Effects of chronic right ventricular pressure overload on myocardial glucose and free fatty acid metabolism in the conscious rat. Cardiovasc Res 29: 763–767, 1995. [PubMed] [Google Scholar]
- 42.Walker LA, Walker JS, Glazier A, Brown DR, Stenmark KR, Buttrick PM. Biochemical and myofilament responses of the right ventricle to severe pulmonary hypertension. Am J Physiol Heart Circ Physiol 301: H832–H840, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]