

Abstract
During the maturation phase of mammalian erythroid differentiation, highly proliferative cells committed to the erythroid lineage undergo dramatic changes in morphology and function to produce circulating, enucleated erythrocytes. These changes are caused by equally dramatic alterations in gene expression, which in turn are driven by changes in the abundance and binding patterns of transcription factors such as GATA1. We have studied the dynamics of GATA1 binding by ChIP-seq and the global expression responses by RNA-seq in a GATA1-dependent mouse cell line model for erythroid maturation, in both cases examining seven progressive stages during differentiation. Analyses of these data should provide insights both into mechanisms of regulation (early versus late targets) and the consequences in cell physiology (e.g., distinctive categories of genes regulated at progressive stages of differentiation). The data are deposited in the Gene Expression Omnibus, series GSE36029, GSE40522, GSE49847, and GSE51338.
Keywords: Gene regulation, ChIP-seq, Transcription factor, DNA binding, RNA-seq
| Specifications | |
|---|---|
| Organism/cell line | Mus musculus cell lines G1E and G1E-ER4 |
| Strain | 129 |
| Sex | Male |
| Sequencer or array type | Illumina Genome Analyzer IIx, Illumina HiSeq 2000 |
| Data format | Sequence reads: fastq; mapped reads: bam, bai (bam index file); peaks calls: broadPeak; signal tracks: bigwig |
| Experimental factors | Mouse cell line (G1E) with a genetic knockout of the Gata1 gene and a daughter cell line (G1E-ER4) transduced with an estrogen-activated Gata1-estrogen receptor transgene that can rescue erythroid maturation in an estrogen-dependent manner. |
| Experimental features | G1E cells (untreated) and G1E-ER4 cells treated with 10 nM estradiol for six time points (0, 3, 7, 14, 24, and 30 h) were (a) analyzed genome-wide for binding by GATA1 using ChIP-seq (including control samples of input DNA at each time point) and (b) mapped for transcription genome-wide by strand-specific, paired-end RNA-seq of poly A + RNA, including biological replicates. |
| Consent | Not applicable |
| Sample source location | Not applicable |
Direct link to deposited data
All data are available through ENCODE data portals:
The GATA1-ChIP-seq data sets are available in three GEO Series: GSE51338, GSE36029, and GSE49847.
http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE51338
http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36029
http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE49847
The RNA-seq data sets are available in three GEO Series: GSE40522, GSE51338, and GSE49847.
http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40522
http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE51338
http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE49847
The individual data sets and links are listed in Table 1.
Table 1.
Genomic data sets and URLs for access.
Number.
Experimental design, materials, and methods
Cell lines used
G1E cells are an immortalized Gata1 null cell line derived from embryonic stem cells [1], and the daughter cell line G1E-ER4 has been stably rescued by transduction with a virus expressing a hybrid gene encoding the GATA1-ER protein [2], [3]. Both G1E and untreated G1E-ER4 cells proliferate and show many properties of immature erythroid progenitor cells [2], [4]. Upon treatment with an estrogen such as estradiol (E2), G1E-ER4 cells mature synchronously and rapidly, recapitulating many aspects of normal erythroid differentation in a manner dependent on activation of GATA1-ER [2], [4], [5], [6]. Among the changes during differentation are a loss of proliferative capacity, a reduction in cell size, condensation of the nucleus, increase and then decrease in CD44, and an increase in TER119 [7] (Fig. 1, left).
Fig. 1.

Illustrative changes in morphology, cell surface markers, RNA, and GATA1 occupancy during GATA1-dependent erythroid maturation. G1E cells and G1E-ER4 cells either untreated (0 h) or treated with E2 for increasing lengths of time (top to bottom) were examined by (left to right) microscopy after staining cytospins with May–Grunwald–Giemsa (MGG), and FACS after fluorescent staining with antibody against CD44 and TER119. Results of RNA-seq on polyA + RNA and ChIP-seq with antibody against GATA1 are shown on the right for the locus encoding beta-globins and olfactory receptors, specifically a 145,400 bp region at chr7:110,928,295-111,073,694 (NCBI37/mm9 assembly of the mouse genome). In the locus map, genes transcribed from left to right are above the line and those in opposite orientation are below the line. RNA-seq reads map to bottom (minus) strand of the assembly, and thus the quantitation of the RNA is plotted as negative numbers. The locus control region (LCR) is a complex enhancer regulating the Hbb genes encoding beta-globins. Additional DNase hypersensitive sites (HSs) even more distal from the Hbb genes are also shown.
Cell culture methods
G1E and G1E-ER4 cells were grown in IMDM media with 15% fetal calf serum 2 U/ml erythropoietin (EpoGen from Amgen) and 50 ng/ml stem cell factor [1], [2]. To induce erythroid maturation, G1E-ER4 cells were treated with 10− 8 mol/L β-estradiol for 3, 7, 14, 24, and 30 h. Cells were harvested by centrifugation at 500 ×g for 5 min at 4 °C and washed once in 1 × PBS.
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed as previously described [2]. Briefly, 75 million cells in 1 × PBS were cross-linked for 10 min by adding formaldehyde at a final concentration of 0.4%, and glycine was added at a final concentration of 125 mM to quench cross-linking. Cells were then lysed (10 mM Tris–HCl, pH 8.0, 10 nM NaCl, 0.2% NP40) for 10 min on ice, washed once in 1 × PBS, followed by nuclear lysis (50 mM Tris–HCl 8.0, 10 mM EDTA, 1% SDS) for 10 min on ice. Chromatin was then diluted further with Immunoprecipitation Buffer (20 mM Tris–HCl, pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.01% SDS) and a 1 × Protease Inhibitor Cocktail set V, EDTA-free (Calibiochem, La Jolla, CA). A Misonix S-4000 sonicator was used to shear samples in 8 repeats of 30 cycles of 1 s on, 1 s off sonication at 30% output power 30 on ice. Fragments in the size range of 200-400 base pairs were obtained. Sonicated chromatin was pre-cleared overnight at 4 °C with 20 μg rat non-immune sera (IgG) on protein G agarose beads. Ten micrograms of the rat anti-GATA1 (sc-265, Santa Cruz Biotechnology, Santa Cruz, CA; lot number L1609) antibody were also pre-bound to protein G agarose beads overnight at 4 °C. For binding, pre-cleared chromatin was added to the antibody–bead complex and incubated with rotation at 4 °C for 4 h; 200 μL of pre-cleared chromatin was saved for use as input. After binding, the beads were washed with Wash Buffer I (20 mM Tris–HCl, pH 8.0, 2 mM EDTA, 50 mM NaCl, 1% Triton X-100, 0.1% SDS), High-Salt Wash Buffer (20 mM Tris–HCl, pH 8.0, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.1% SDS), Wash Buffer II (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, 250 mM LiCl, 1% NP40, 1% deoxycholate), and 1 × TE. DNA:protein complexes were then eluted from beads with Elution Buffer (1% SDS, 100 mM NaHCO3). Reverse cross-linking of immunoprecipitated chromatin was accomplished by the addition of NaCl to ChIP and input samples, followed by incubation overnight at 65 °C with 1 μg RNase A. To remove proteins, each sample was treated with 6 μg Proteinase K for 2 h at 45 °C. Immunoprecipitated DNA was finally purified using the Qiagen PCR Purification Kit.
Illumina library preparation for ChIP-Seq
All samples including input were processed for library construction for Illumina sequencing using Illumina's ChIP-seq Sample Preparation Kit. In brief, DNA fragments were repaired to generate blunt ends, and a single ‘A’ nucleotide was added to each end. Double-stranded Illumina adaptors were ligated to the fragments. Ligation products were amplified by 18 cycles of PCR, and the DNA between 250 and 350 base pairs was gel purified. Completed libraries were quantified with Quant-iT dsDNA HS Assay Kit. The DNA libraries were sequenced on the Illumina Genome Analyzer IIx or HiSeq 2000 as indicated (Table 2) using Illumina's kits and reagents as appropriate.
Table 2.
Characteristics and quality metrics for ChIP-Seq data sets.
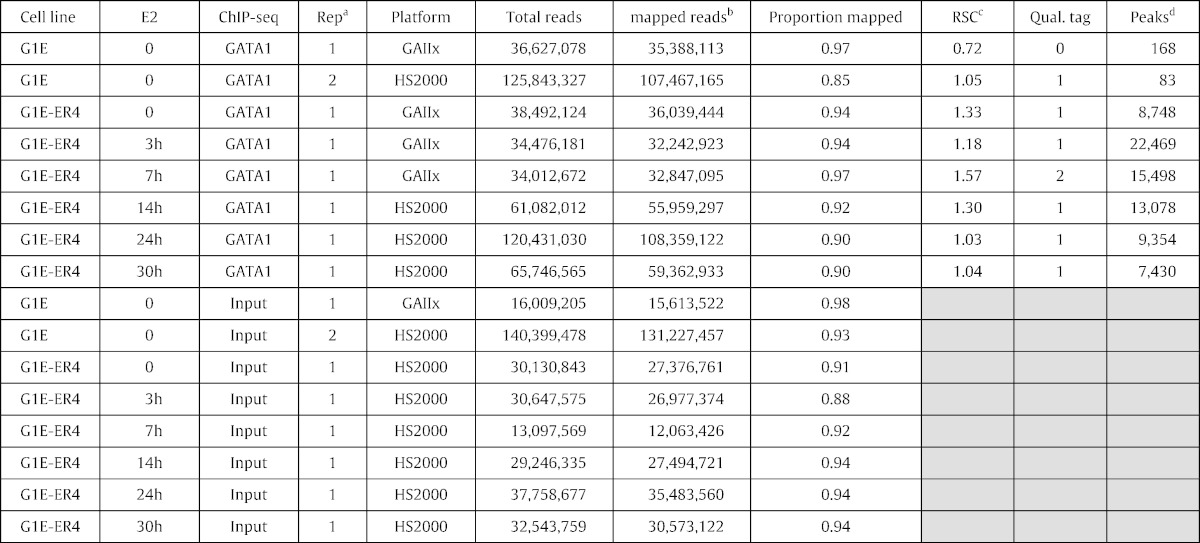
aNumber of the replicate sample. Most samples have one determination.
bIncludes reads that mapped at multiple locations.
cDetermined from uniquely mapping reads.
dNumber of peaks after removing those overlapping blacklisted regions.
Mapping for ChIP-Seq
Raw ChIP-seq reads were first groomed using FASTQ Groomer on Galaxy [8], [9], [10].This program verifies that each base call has a corresponding quality value, and that the quality value is in the Sanger, Phred + 33 format. Groomed reads were then mapped to mouse mm9 genome using Bowtie [11] using the parameters –m = − 1 (no limit), −k = 1, − y, and − best, thus allowing reads to map to multiple locations, but reporting only the single, best alignment. This option was chosen to allow reads to map in duplicated regions.
Peak calling for ChIP-seq
The mapped reads for each time point in G1E-ER4 cells and untreated G1E cells were then passed to MACS [12] with the matched control (input) data set for peak calling using an mfold of 12, p-value threshold of 1e − 05 and bw (bandwidth) set to 120. We filtered ChIP-seq peaks which had an overlap of at least one base pair with blacklisted regions described in either Pimkin et al. [13] or mm9 blacklisted regions identified by ENCODE [14].The number of peaks in each data set (after removing the ones overlapping blacklisted regions) is given in Table 2. The ENCODE blacklisted regions were obtained from https://sites.google.com/site/anshulkundaje/projects/blacklists.
Quality assessment of ChIP-seq
GATA1 ChIP-seq and input samples were sequenced to a high depth, ranging from 12 million to 138 million mapped reads (Table 2), with mean and median sequencing depths of 48 million and 34 million mapped reads, respectively. All ChIP-seq data sets had a very high proportion of sequence reads mapping to the genome; all but one were above 90% with a mean of 93%.
High-quality ChIP-seq data sets should show substantial clustering of the mapped reads, which can be measured by cross-correlation analysis of reads mapping to the two strands of DNA [15]. The values for these metrics are sensitive to the method of mapping, and thus to generate quality scores we remapped the sequencing reads and only accepted uniquely mapped reads. We report the Relative Strand Cross-correlation Coefficient (RSC) values (Table 2); values greater than 1 are indicative of high quality [16]. Almost all the RSC scores for samples in the GATA1 ChIP-seq time course are above 1, and they fit well within the distribution of RSC scores for all ChIP-seq data sets from mouse ENCODE [17], shown in Fig. 2. Quality score tags were derived from defined ranges of RSC scores, with − 2 corresponding to minimal read clustering and + 2 corresponding to a highly clustered library [16]. Quality score tags were 1 or 2 for the GATA1 ChIP-seq samples in cell types containing GATA1. We also performed cross-correlation analysis for GATA1 in the G1E cell line, which has no GATA1. As expected, replicate 1 for this sample had an RSC score less than 1 and a quality score tag of 0. However, replicate 2 actually passed the quality score thresholds, even though no antigen was present in the cell line. The “signal” track is basically just noise (Fig. 1), and almost no peaks were called by MACS [12]. This illustrates some of the limitations in applying quality score metrics, as discussed by Landt et al. [15] and Marinov et al. [16].
Fig. 2.

Distribution of RSC scores for mouse ENCODE ChIP-seq data sets. The RSC scores were computed by the mouseENCODE consortium [17] on the uniformly processed alignments of the GATA1 ChIP-seq data sets. The results are available from (https://docs.google.com/spreadsheet/ccc?key=0Ao3-Or4FCMJEdFpPY2lwWnlZTV92MUNLOHYxbEl4Vnc&usp=drive_web#gid=0). The QC tags are discrete categories based on RSC values as described in Marinov et.al [16].
RNA extraction and cDNA synthesis
Total RNA was extracted from ~ 5 to 10 million cells using Invitrogen's TRIzol reagent and the Ambion PureLink RNA Extraction Mini Kit (Life Technologies #12183018A). Invitrogen's Dynabeads mRNA Purification Kit (#610-06) was used isolate mRNA in two rounds of selection. Isolated mRNA was subjected to fragmentation at 94 °C for 2 min 30 s in a high-salt 1 × fragmentation buffer (200 mM Tris acetate pH 8.2, 500 mM potassium acetate, and 150 mM magnesium acetate) [18], and fragmentation ions were removed using a Sephadex G-50 column (USA Scientific). First-strand cDNA was synthesized from 100 ng mRNA primed with 3 μg random hexamers [18] and the four conventional dNTPs (dATP, dTTP, dGTP, dCTP) using Invitrogen's ThermoScript RT-PCR System (#11146-024). ActinomycinD was added to prevent leaky second-strand synthesis. Second-strand cDNA was synthesized at 16 °C for 2.5 h in 11 × SSB (500 mM Tris–HCl pH 7.5, 100 mM MgCl2 and 10 mM DTT) with all the four dNTPs, except using dUTP in place of dTTP to label the second-strand cDNA [19]. Uracil-labeled second-strand cDNA was then further processed for sequencing library preparation using the Illumina ChIP-seq DNA Sample Prep Kit, including end repair, A-tailing, and adaptor ligation. Prior to PCR amplification, the dUTP-labeled second strand was selectively digested so as to amplify only first-strand cDNA. The adaptor-ligated double-stranded cDNA library was treated with 1 μL Uracil N-glycosylase (Applied Biosystems GeneAmp AmpErase #11146-024) for 15 min at 37 °C, followed by high heat (95 °C for 5 min) to remove uracil from nucleotides and to promote abasic scission of the second strand. For PCR, betaine was added to the PCR buffer at a final concentration of 1.8 M to improve amplification of GC-rich sequences [20]. Strand-specific libraries were sequenced on the Illumina HiSeq 2000 to obtain 2 × 99 nt paired-end reads. All samples were determined as biological replicates.
Mapping and estimation of transcript abundance
Mapping and estimation of transcript abundance were performed as previously described in [21], with a few modifications. As with ChIP-seq, RNA-seq reads were first groomed using the FASTQ Groomer tool on Galaxy. Groomed reads were mapped to the mm9 genome using the splice-junction mapper TopHat [11], [22] in reference-assisted mode (− G, using a custom gene model annotation file, other options—library-type fr—first strand, − j using a custom junctions file). The custom junctions file was obtained by combining splice junctions from TopHat output across all transcriptomes for a comprehensive splice junction annotation. The custom gene model annotation file was generated so as to represent each gene by a single canonical transcript. Starting with an Illumina iGenomes RefSeq mm9 GTF, we obtained canonical transcripts for each gene from the “knownCanonical” table using the UCSC Table Browser to represent that gene. For genes without any record of a canonical transcript, we chose the representative transcript based on transcript length (longest), CDS length (longest) and number of exons (greater). We excluded genes positioned on chrN_random or chrUn_random. Genes encoding small RNAs (snoRNAs matching the pattern "Snora") were excluded because estimation of their expression levels is not reliable. This resulted in 22,977 genes, each with a single transcript. We used Cufflinks and Cuffdiff [22], [23], [24], [25] to obtain expression levels for individual replicates and pooled samples, respectively, using this custom GTF. However, regions on mouse chr11 and chr7 containing alpha and beta globin transcripts were masked from both tools, using option − M (see section on "Globin expression estimation" below). Other Cuffdiff options used include dispersion-method = per-condition, library-type = fr-first strand, max-bundle-frags = 20000000, min-reps-for-js-test = 2, and − b for bias correction. Transcript abundance levels pooled across replicates were expressed in terms of log2-transformed fragments per kilobase of exon model (FPKMs) per million mapped fragments, after addition of a value of 1.1 as noise. Noise addition was done to avoid log-transforms of zero values and divide-by-zero issues. Thus, genes with FPKM of 0 are log2-transformed to an expression level of 0.1375.
Globin expression estimation
Globins are expressed in enormous amounts in erythroid cells. Despite the availability of high-performance compute clusters, estimating abundances for globins and performing differential expression tests at these loci is time and memory-intensive (programs often do not complete running), depending upon the number of reads. To avoid these issues, we (bioinformatically) masked the alpha and beta globin loci on chr11 and chr7, while estimating expression levels. The option − M/− maskfile was used with a custom GTF covering the globin loci to achieve this. As a result, all alpha and beta globin genes, including fetal globins had an FPKM of 0. To obtain some measure of expression for globins, we extrapolated the expected FPKM of globins from their read counts, by comparing read counts and FPKMs of the top 20 highly expressed genes. The median ratio between the read counts and FPKMs for these genes, 2, was used to obtain expression levels for globin genes.
Quality assessment of RNA-seq data
Table 3 provides a summary of raw and mapped alignments for the fourteen RNA-seq data sets. As expected for high-quality RNA-seq data, the two replicates for each cell line and time point were highly correlated (Fig. 3), with a Spearman's correlation coefficient ranging from 0.90 to 0.96 across samples. The Spearman's correlation coefficient was chosen since it is less affected by extreme values than the Pearson's correlation coefficient. The lowess line (red) closely follows the 45° diagonal, also indicating that the two replicates agree with each other quite well.
Table 3.
Raw reads and mapped alignment statistics for RNA-Seq.
| Cell line | Treatment | Raw reads Rep1 | Raw reads Rep2 | No. Alignments Rep1 | No. Alignments Rep2 |
|---|---|---|---|---|---|
| G1E | untreated | 2 x 136,192,858 | 2 x 123,009,356 | 151,366,580 | 181,528,211 |
| G1E-ER4 | untreated | 2 x 137,017,614 | 2 x 120,209,833 | 167,089,002 | 127,431,78 |
| G1E-ER4 | 3 h E2 | 2 x 95,549,285 | 2 x 125,119,593 | 81,973,316 | 143,130,692 |
| G1E-ER4 | 7 h E2 | 2 x 116,159,379 | 2 x 122,712,837 | 142,846,469 | 115,629,100 |
| G1E-ER4 | 14 h E2 | 2 x 112,218,860 | 2 x 121,004,971 | 102,138,513 | 146,590,846 |
| G1E-ER4 | 24 h E2 | 2 x 139,565,377 | 2 x 106,547,243 | 169,441,332 | 46,871,607 |
| G1E-ER4 | 30 h E2 | 2 x 157,853,165 | 2 x 118,793,140 | 187,803,929 | 137,994,877 |
Fig. 3.

Scatterplots showing reproducibility of measurements of gene expression in replicates of RNA-seq. Each dot is the expression level of a gene in replicate 1 (Rep1) versus the expression level in replicate 2 (Rep2). Red dotted lines indicate the lowess fit between the measurements in replicates, and grey dotted lines indicate the 45° diagonal. Spearman correlations between replicates are given in the graphs.
Recapitulation of prior literature
High-quality data sets should also recapitulate observations made in the earlier literature. Examination of the distribution of expression levels confirmed that most genes are not expressed in either G1E cells or G1E-ER4 cells treated with E2 (Fig. 4). Importantly, non-erythroid genes such as Vwf and Pf4 [26] were among those measured as silent in the RNA-seq data from both cell types, whereas well-known erythroid genes are expressed. Transcripts from late erythroid marker genes such as Alas2 and Epb4.9 were hardly detectable in G1E cells but were expressed at high levels in maturing G1E-ER4 treated with E2 for 30 h. Conversely, genes known to be downregulated during erythroid differentiation, such as Kit [27] and Myb [28], were strongly expressed in G1E cells, but they were repressed in maturing G1E-ER4 cells.
Fig. 4.

Distribution of gene expression levels before and after GATA1-induced erythroid maturation. The number of genes in each bin of expression level is plotted for the erythroid progenitor model G1E cells (left) and the model for maturing erythroblasts, G1E-ER4 cells treated with E2 for 30 h (right). Specific examples of genes expressed at low levels are indicated by gene names in blue, and examples of more highly expressed genes are indicated by names in red. Colored tick marks corresponding to these genes on the top x-axis indicate expression levels on the bottom x-axis.
The quality of the GATA1 ChIP-seq data sets was also evaluated by examining a locus at which the dynamics of GATA1 occupancy has been studied, the Hbb locus encoding beta-like globins. As reported previously [2], [29], we found that the locus control region (LCR) upstream of the globin locus was occupied by GATA1 at the earliest time points, whereas GATA1 bound to the Hbb-b1 promoter at later times (starting at 7 h), before accumulation of transcripts from Hbb-b1 transcripts at 14 h (Fig. 1, right). Moreover, several sites appear to lose GATA1 occupancy during maturation; this is an example of a phenomenon worthy of further analysis with these data sets.
Thus, both the RNA-seq and the ChIP-Seq data sets agree well with published literature, and we infer that they are high quality.
Discussion
The dynamics of GATA1 binding and effects on transcription have been studied at individual genetic loci [2], [29]. The data sets reported here provide genome-wide information on these relationships. Further investigation should reveal many new insights, especially by combining the analysis of binding with the expression of candidate target genes. We expect that many of the sites bound later in the time course could be directing activation of erythroid-specific genes, but some of the late binding could also implicated in repression. GATA1 binding only at early stages of the examined course of differentiation was unexpected; it has not been examined extensively in prior studies. It is important to investigate whether specific categories of genes are enriched as candidate targets of early-bound GATA1, and if so, determine whether they represent genes characteristic of alternative cell fates.
While these data sets are valuable, users should be aware that they are generated on material from an immortalized mouse cell line that recapitulates many but not all aspects of erythroid maturation during differentiation. The cells do not fully mature, and enucleated reticulocytes and erythrocytes are not produced. Furthermore, the genetic knockout of Gata1 in G1E cells and the rescue with a hormone-activatable form of GATA1-ER in G1E-ER4 cells generates a large and immediate shift in the concentrations of GATA2 and GATA1, whereas in normal erythropoietic maturation, the concentration of GATA2 decreases and that of GATA1 increases gradually [1], [2]. These caveats should be kept in mind while mining these important new data sets.
Acknowledgments
We thank Daniela I. Drautz for sequencing libraries on Illumina Genome sequencer (GAIIx) and Anshul Kundaje for generating the cross-correlation scores for the mouse ENCODE ChIP-seq samples. This work was supported by the National Institutes of Health grants R01DK065806 (RCH, MJW, GAB), U01HL099656 and P30DK090969 (MJW), RC2HG005573, R56DK065806, and U54HG006998 (RCH). This work was supported in part through instrumentation funded by the National Science Foundation through grant OCI-0821527 (the Penn State CyberSTAR and BioSTAR computers).
References
- 1.Weiss M.J., Yu C., Orkin S.H. Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol. Cell. Biol. 1997;17:1642–1651. doi: 10.1128/mcb.17.3.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Welch J.J., Watts J.A., Vakoc C.R., Yao Y., Wang H., Hardison R.C. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood. 2004;104:3136–3147. doi: 10.1182/blood-2004-04-1603. [DOI] [PubMed] [Google Scholar]
- 3.Tsang A.P., Visvader J.E., Turner C.A., Fujiwara Y., Yu C., Weiss M.J. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell. 1997;90:109–119. doi: 10.1016/s0092-8674(00)80318-9. [DOI] [PubMed] [Google Scholar]
- 4.Cheng Y., Wu W., Kumar S.A., Yu D., Deng W., Tripic T. Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 2009;19:2172–2184. doi: 10.1101/gr.098921.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregory T., Yu C., Ma A., Orkin S.H., Blobel G.A., Weiss M.J. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood. 1999;94:87–96. [PubMed] [Google Scholar]
- 6.Rylski M., Welch J.J., Chen Y.-Y., Letting D.L., Diehl J.A., Chodosh L.A. GATA-1-mediated proliferation arrest during erythroid maturation. Mol. Cell. Biol. 2003;23:5031–5042. doi: 10.1128/MCB.23.14.5031-5042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen K., Liu J., Heck S., Chasis J.A., An X., Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc. Natl. Acad. Sci. U. S. A. 2009;106:17413–17418. doi: 10.1073/pnas.0909296106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giardine B., Riemer C., Hardison R.C., Burhans R., Elnitski L., Shah P. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blankenberg D., Von Kuster G., Coraor N., Ananda G., Lazarus R., Mangan M. Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 2010:1–21. doi: 10.1002/0471142727.mb1910s89. (Chapter 19, Unit 19.10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goecks J., Nekrutenko A., Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pimkin M., Kossenkov A.V., Mishra T., Morrissey C.S., Wu W., Keller C.A. Divergent functions of hematopoietic transcription factors in lineage priming and differentiation during erythro-megakaryopoiesis. Genome Res. 2014;24:1932–1944. doi: 10.1101/gr.164178.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marinov G.K., Kundaje A., Park P.J., Wold B.J. Vol. 4. 2014. Large-scale quality analysis of published ChIP-seq data, G3 (Bethesda) pp. 209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 19.Parkhomchuk D., Borodina T., Amstislavskiy V., Banaru M., Hallen L., Krobitsch S. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin J.Z., Yassour M., Adiconis X., Nusbaum C., Thompson D.A., Friedman N. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods. 2010;7:709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu W., Cheng Y., Keller C.A., Ernst J., Kumar S.A., Mishra T. 2011. Dynamics of the epigenetic landscape during erythroid differentiation after GATA1 restoration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts A., Pimentel H., Trapnell C., Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- 24.Roberts A., Trapnell C., Donaghey J., Rinn J.L., Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szalai G., LaRue A.C., Watson D.K. Molecular mechanisms of megakaryopoiesis. Cell. Mol. Life Sci. 2006;63:2460–2476. doi: 10.1007/s00018-006-6190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jing H., Vakoc C.R., Ying L., Mandat S., Wang H., Zheng X. Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Mol. Cell. 2008;29:232–242. doi: 10.1016/j.molcel.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartůnek P., Králová J., Blendinger G., Dvorák M., Zenke M. GATA-1 and c-myb crosstalk during red blood cell differentiation through GATA-1 binding sites in the c-myb promoter. Oncogene. 2003;22:1927–1935. doi: 10.1038/sj.onc.1206281. [DOI] [PubMed] [Google Scholar]
- 29.Im H., Grass J.A., Johnson K.D., Kim S.-I., Boyer M.E., Imbalzano A.N. Chromatin domain activation via GATA-1 utilization of a small subset of dispersed GATA motifs within a broad chromosomal region. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17065–17070. doi: 10.1073/pnas.0506164102. [DOI] [PMC free article] [PubMed] [Google Scholar]