

Abstract
Objective
Pathogenesis in facioscapulohumeral muscular dystrophy (FSHD) appears to be due to aberrant expression, particularly in skeletal muscle nuclei, of the full-length isoform of DUX4 (DUX4-FL). Expression of DUX4-FL is known to alter gene expression and to be cytotoxic, but cell responses to DUX4-FL are not fully understood. Our study was designed to identify cellular mechanisms of pathogenesis caused by DUX4-FL expression.
Methods
We used human myogenic cell cultures to analyze the effects of DUX4-FL when it was expressed either from its endogenous promoter in FSHD cells or by exogenous expression using BacMam vectors. We focused on determining the effects of DUX4-FL on protein ubiquitination and turnover and on aggregation of TDP-43.
Results
Human FSHD myotubes with endogenous DUX4-FL expression showed both altered nuclear and cytoplasmic distributions of ubiquitinated proteins and aggregation of TDP-43 in DUX4-FL-expressing nuclei. Similar changes were found upon exogenous expression of DUX4-FL, but were not seen upon expression of the non-toxic short isoform DUX4-S. DUX4-FL expression also inhibited protein turnover in a model system and increased the amounts of insoluble ubiquitinated proteins and insoluble TDP-43. Finally, inhibition of the ubiquitin–proteasome system with MG132 produced TDP-43 aggregation similar to DUX4-FL expression.
Interpretations
Our results identify DUX4-FL-induced inhibition of protein turnover and aggregation of TDP-43, which are pathological changes also found in diseases such as amyotrophic lateral sclerosis and inclusion body myopathy, as potential pathological mechanisms in FSHD.
Introduction
In this study, we examined pathological consequences of DUX4-FL expression in human myogenic cells in vitro. Aberrant expression of the cytotoxic DUX4-FL protein in skeletal muscles is proposed to cause facioscapulohumeral muscular dystrophy (FSHD).1–3 With a prevalence of ∽1 in 20,000, FSHD is a progressive, autosomal dominant disease with a variable age of onset, though often in adolescence and young adulthood, that usually presents with weakness in the face, arms, and shoulder girdle.4 In FSHD1, which accounts for ∽95% of FSHD cases, the disease is genetically linked to 4q35 near the telomere, a region which contains an array of 3.3 kb D4Z4 repeats.5,6 In FSHD1 patients, the D4Z4 repeat number ranges from one to ten. In contrast, unaffected individuals with normal muscle function typically have unshortened arrays with >10 D4Z4 repeats, though individuals with contracted D4Z4 arrays but without FSHD clinical symptoms are found.7 The rarer FSHD2 form of the disease is due to mutations in SMCHD1 in which patients have normal length D4Z4 arrays with >10 repeats.8 Both FSHD1 and FSHD2, however, are associated with DNA hypomethylation in the D4Z4 region suggesting an epigenetic component to FSHD pathogenesis.9–11
The unifying model of FSHD1,3 proposes that pathogenesis is due to aberrant expression of DUX4-FL, a long isoform of the double homeobox protein DUX4. Though a DUX4 open reading frame is found in each of the 3.3 kb repeats of the 4q35 D4Z4 array, a stable poly-adenylated mRNA for DUX4-FL is produced only from the most telomeric repeat and only when that repeat is adjacent to a 4qA telomeric allele which allows poly-adenylation of the transcript. The resulting DUX4-FL protein has a C-terminal transcription activating domain and is typically highly cytotoxic when overexpressed in many cell types.12–15 A shorter DUX4-S isoform that lacks the C-terminal domain can be produced from an alternatively spliced DUX4 mRNA, but DUX4-S is typically non-toxic and may act as a dominant-negative inhibitor of DUX4-FL.16 DUX4-FL is expressed from its endogenous promoter in a very small fraction, typically <0.5%, of the nuclei in cultures of human myogenic cells.2,17 Expression of DUX4-FL can lead to induction of cell death, activation of caspase-3, and aberrant expression of DUX4-FL target genes.12,13,16,18 The DUX4-FL model for FSHD pathogenesis is consistent with a considerable body of experimental work, including the finding, based on analysis of a large library of myogenic cells and biopsies, that DUX4-fl mRNA and DUX4-FL protein are expressed at a much higher level in FSHD1 than in healthy controls.17
In this study, we extended analyses of how myogenic cells respond to DUX4-FL expression. We found that DUX4-FL expression, either from its endogenous promoter or exogenously, led both to altered localization of ubiquitinated proteins and to aggregation of TDP-43 in nuclei. TDP-43 aggregation in the nucleus and/or the cytoplasm has previously been associated with multiple neuromuscular diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration with ubiquitin positive inclusion (FTLD-U), and inclusion body myopathy.19–21 Our further experiments showed that exogenous DUX4-FL expression impaired protein turnover in a model system and that inhibition of the ubiquitin–proteasome system (UPS) by MG132 produced changes similar to DUX4-FL expression. Our study thus identifies DUX4-FL-induced inhibition of protein turnover and nuclear aggregation of TDP-43 as potential mechanisms of pathogenesis in FSHD and raises that possibility that therapies targeted at these abnormalities could prove useful in multiple diseases including FSHD.
Materials and Methods
Cells and culture
The primary myogenic cells used in these studies were prepared from biceps biopsies of FSHD and unaffected donors as described previously17,22 and summarized in Table1. All human cells were obtained from the Wellstone FSHD biobank at the University of Massachusetts, School of Medicine and cells were anonymized prior to receipt with no personal identifying information available to us. The cells had been produced prior to our study from muscle biopsies collected under protocols approved by the appropriate institution that included informed donor consent and approval to publish results in accordance with standards of the Helsinki Declaration. Because our studies were of human cells that were obtained from a cell bank and for which personal identification data were not obtainable by us, the studies were classified as exempt from Human Studies review by the Boston University Institutional Review Board in accordance with U.S.A. Department of Health and Human Services policy.
Table 1.
| Cells1 | Disease status2 | Gender, age, family relation | EcoRI/BlnI sizes (telomere allele) | #DUX4-FL+ve nuclei per 1000 nuclei in myosin+ve cells Ave ± SE (n) |
|---|---|---|---|---|
| 09Abic | FSHD | F, 31 years, proband | 25 kb (4qA, paternal), >112 kb (4qA) | 0.79 ± 0.21 143 |
| 09Ubic | Unaffected | F, 57 years, mother of 09A | 47 kb (4qB), >112 kb (4qA) | 0.12 ± 0.08 (143 |
| 17Abic | FSHD | M, 23 years, proband | 19 kb (4qA, maternal), 87 kb (4qA) | 3.71 ± 0.63 143 |
| 17Ubic | Unaffected | M, 21 years, brother of 17A | 97 kb (4qB), >112 kb (4qA) | 0.021 ± 0.015 123 |
FSHD, facioscapulohumeral muscular dystrophy.
Cell designations include cohort (family) number (09 or 17) followed by A for the FSHD affected subjects or U for the unaffected first degree relative and ending with bic indicating that the biopsy site was in the biceps muscle.
FSHD was confirmed by presence of both clinically apparent muscle weakness and a shortened 4q D4Z4 repeat array identified by an EcoRI/BlnI restriction fragment of <35 kb coupled with a 4qA telomere allele.17,68 Shortened repeat arrays with 4qA telomere alleles are shown in bold.
P < 0.01 by t-test for FSHD versus unaffected within the indicated family.
Myogenic cells from each donor were FACS-purified based on CD56 expression and the resulting myogenic cell populations were >90% desmin-positive. As previously,17,23 (1) DUX4-FL expression was detectable by immunostaining in only a small fraction of nuclei (<0.5%), (2) was found in a much higher proportion of nuclei in FSHD than unaffected cells and (3) was found only in differentiated, myosin-expressing myocytes (Table1). The human primary myogenic cells were grown on gelatin-coated dishes in high-serum medium for proliferation and switched when near confluence to low-serum medium for differentiation as described.17,22 For some experiments as indicated, we used HEK293 cells which were obtained from the ATCC (Manassas, VA). HEK293 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum, 4 mmol/L l-glutamine, and 1% penicillin-streptomycin (Life Technologies, Grand Island, NY). As indicated, proteasome function was inhibited with MG132 (EMD Millipore, Billerica, MA) used at a final concentration of 10 μmol/L and added 5 h before cell harvest.
Immunostaining
Myogenic cells were grown on gelatin-coated 4-well chamber slides (Thermo Fisher Scientific, Waltham, MA) until >90% confluent, then switched to differentiation medium for 4–6 days. Differentiated cultures were washed twice with PBS, fixed with ice-cold 100% methanol or 2% Paraformaldehyde (PFA) for 10 min, washed three times with PBS, permeabilized with 0.5% Triton X-100 for 10 min at room temperature for PFA-fixed specimens and incubated for 60 min at room temperature in a blocking solution consisting of 4% horse serum, 4% goat serum (Life Technologies, Thermo Fisher Scientific), and 4% BSA (EMD Millipore, Billerica, MA) in PBS + 0.1% Triton X-100. Fixed cultures were incubated overnight at 4°C with a primary antibody diluted in blocking solution. The following day cells were rinsed three times with PBS and incubated for 1 h with the appropriate secondary antibody diluted 1:500 in blocking solution. For double immunostaining, cultures were subsequently incubated as above with the appropriate second primary antibody, followed by washing, and incubation with the second secondary antibody.
Antibodies
DUX4-FL was detected with rabbit anti-DUX4-FL mAb E5524 used at 1:200 dilution (Epitomics, Burlingame, CA). Myosin heavy chain (MyHC) isoforms were detected with mouse mAbs F59 or MF20 (Developmental Studies Hybridoma Bank, Iowa City, IA) used at 1:10 dilution of hybridoma supernatant. Activated caspase-3 was detected with a rabbit pAb (Cell Signaling Technologies, Beverly, MA) used at 1:100. TDP-43 was detected with either rabbit anti-TARDBP pAb (cat. 10782-2-AP; Proteintech, Chicago, IL) or mouse anti-TDP-43 mAb (cat. 60019-2; Proteintech). Ubiquitinated proteins were detected with mouse mAb FK2 (MBL, Woburn, MA) which reacts with K29-, K48-, and K63 mono- and poly-ubiquitinated proteins, but not free ubiquitin. V5 epitope tag was detected using either mouse anti-V5 mAb (Life Technologies, Grand Island, NY) or a rabbit pAb (EMD Millipore). GFP was detected with a rabbit pAb (Santa Cruz Biotechnology, Dallas, TX). Primary antibody binding was visualized with appropriate species-specific secondary antibodies (Life Technologies) conjugated to either Alexa Fluor 488 or Alexa Fluor 594 and used at 1:500. Nuclei were stained with bisbenzimide.
Microscopy
Immunostaining was analyzed by manually scanning the entire culture area (e.g. to identify DUX4-FL-positive nuclei or MyHC-positive cells), followed by imaging and manual quantitation. Standard microscope images were acquired using a Nikon E800 microscope with Spot camera and software version 4.6 (Diagnostic Instruments Inc., Sterling Heights, MI). Confocal images were acquired using a Leica SP5 instrument (Leica Microsystems, Buffalo Grove, IL).
Immunoblotting
Human primary myogenic cells or HEK293 cells were homogenized in RIPA buffer (50 mmol/L Tris–HCl pH 7.4, 150 mmol/L NaCl, 1% NP-40, 0.5% sodium deoxycholic acid, and 0.1% SDS) with protease inhibitors (Calbiochem Protease Inhibitor Cocktail III; EMD Millipore), sonicated, and then centrifuged at 15,000g for 15 min. The RIPA-soluble protein in the resulting supernatant was quantified with a Bradford assay (Bio-Rad, Hercules, CA), and the supernatant was mixed with an equal volume of 2× SDS–PAGE sample buffer and boiled for 5 min. 25 μg protein was subjected to SDS–PAGE (Mini-protean TGX gels, Bio-Rad) under reducing conditions and transferred to Polyvinylidene difluoride (PVDF) membranes for immunoblotting (Bio-Rad). The pellets resulting from centrifugation after homogenization in RIPA buffer were resuspended in urea buffer (7 mol/L urea, 2 mol/L thiourea, 4% CHAPS), sonicated, and centrifuged. The supernatant, which contained RIPA-insoluble/urea-soluble protein, was subjected to SDS–PAGE and transferred to PVDF membranes. Blots were blocked with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) and then incubated with primary antibodies. Signals were detected with Alexa-680 (Life Technologies) or IRDye 800 (LI-COR Biosciences)-conjugated secondary antibodies with appropriate species specificity. Immunoblots were visualized by the Image Studio software version 2.1.10 (LI-COR Biosciences) that accompanies the LI-COR Odyssey infrared system (LI-COR Biosciences).
Caspase
Caspase enzymatic activity was measured using the luminescence-based Caspase-Glo 3/7 Assay System (Promega, Madison, WI) according to the manufacturer's instructions and with signal detection on an Infinite M1000 microplate reader (Tecan, Morrisville, NC).
Plasmids and transfection
Plasmids used for expression of V5-tagged DUX4-FL and DUX4-S, which we termed pCMV-DUX4-fl-V5 and pCMV-DUX4-s-V5, were described previously.15 The simian CMV IE4 promoter in these plasmids was derived from pCS2+. The plasmid for expression of full-length TDP-43 fused with GFP under control of a CMV promoter derived from pcDNA3 was described previously.25 These plasmids were transfected into human myoblasts cells using the X-tremeGene HP DNA transfection reagent (Roche Diagnostic Corporation, Indianapolis, IN) following the manufacturer's instructions.
EGFP-CL1 degron assay
The assay was described previously.26 In brief, the nucleotide sequence encoding the CL1 degron peptide (ACKNWFSSLSHFVIHL) was inserted into the XhoI/EcoRI site of plasmid of pEGFP-C3.26 The resulting plasmid was designated pEGFP-CL1 and was used to express the GFP-CL1 degron fusion protein. DUX4-FL and pEGFP-CL1 vectors were co-transfected into HEK293 cells using Polyethylenimine “Max” (Polysciences Inc., Warrington, PA) and cells were collected 48 h after transfection.
BacMam vectors
Though plasmid transfection of myoblasts followed by switch to differentiation medium can be used to overcome the poor transfection efficiency of myotubes, the high toxicity of DUX4-FL expressed from the CMV promoter fragment made it impossible to use this strategy as myoblasts were killed before myotube formation was sufficiently extensive. Though lentivirus vectors can directly infect myotubes, we were unable to generate sufficient titers of lentivirus vectors carrying the simian CMV IE4 promoter-DUX4-fl construct due to DUX4-FL-induced death of the packaging cell lines. These obstacles were overcome by use of baculovirus-derived BacMam vectors.27 BacMam vectors were derived from pCMV-DUX4-fl-V5 and pCMV-DUX4-s-V5 by EcoRI/XbaI restriction digest and cloning of the corresponding DUX4 inserts into pENTR1A (Life Technologies) which was prepared by digestion with EcoRI and XbaI. The resulting entry clones were recombined into the BacMam destination vector pJiF2 using LR ClonaseII (Life Technologies) and recombinant baculovirus generated using the Tn7 transposition system.28 In the BacMam vectors, expression was driven by a human CMV-IE1 promoter. P1 viral supernatants were used in all experiments without further purification and expression was analyzed at 24–48 h after addition as noted.
Results
To identify potential pathological consequences of DUX4-FL expression in human myogenic cells, we identified and compared potentially pathological changes induced by exogenous and endogenous expression of DUX4-FL. In myoblasts, we used both plasmid transfection and BacMam vectors, as indicated, to express DUX4-FL or, as a control, DUX4-S, whereas in myotubes we used BacMam vectors for all studies of exogenous DUX4-FL expression. Expression was driven from constitutively active CMV promoters in both plasmids and BacMam vectors.
Exogenous DUX4-FL expression in myoblasts
Previous studies had shown that exogenous expression of transfected DUX4-FL is accompanied by increased caspase-3 activation, a marker for incipient cell death, in many different types of cells including Hep2, mouse C2C12, fibroblasts, and human primary myogenic cells.12,13,16,18 We first confirmed that exogenous DUX4-FL overexpression increased caspase-3 activation in primary human myoblasts. We used plasmid transfection in cultures of proliferating human healthy control and FSHD myoblasts to induce DUX4-FL expression and found that ∽10–20% of the cells in transfected cultures were positive for DUX4-FL immunostaining at 24–48 h after transfection. In these cultures, the primary human myoblasts did not express MyHC and thus had not differentiated. At 32–48 h after transfection, immunostaining for activated caspase-3 was detectable in a fraction of the cells with DUX4-FL-positive nuclei, whereas DUX4-FL-negative nuclei in the same or parallel cultures showed no increase in caspase-3 immunostaining (Fig.1A–D). Exogenous DUX4-FL expression-induced caspase-3 immunostaining similarly in both healthy control and FSHD myoblasts. To confirm the immunostaining results, we also measured caspase-3 enzymatic activity after BacMam-mediated expression in myoblasts, which resulted in >90% of the nuclei-expressing DUX4-FL or, as a control, DUX4-S at 32–48 h after BacMam addition. We found that caspase-3 enzymatic activity was increased significantly in myoblasts upon exogenous DUX4-FL expression (Fig.1I, proliferating). Activation was specific to DUX4-FL, as caspase-3 was not activated upon BacMam-mediated expression of the short DUX4-S isoform that lacks the C-terminal region of DUX4-FL (Fig.1I). Our results with human primary myoblasts were thus consistent with the previous studies noted above that showed caspase-3 activation by exogenous DUX4-FL in multiple cell types.
Figure 1.
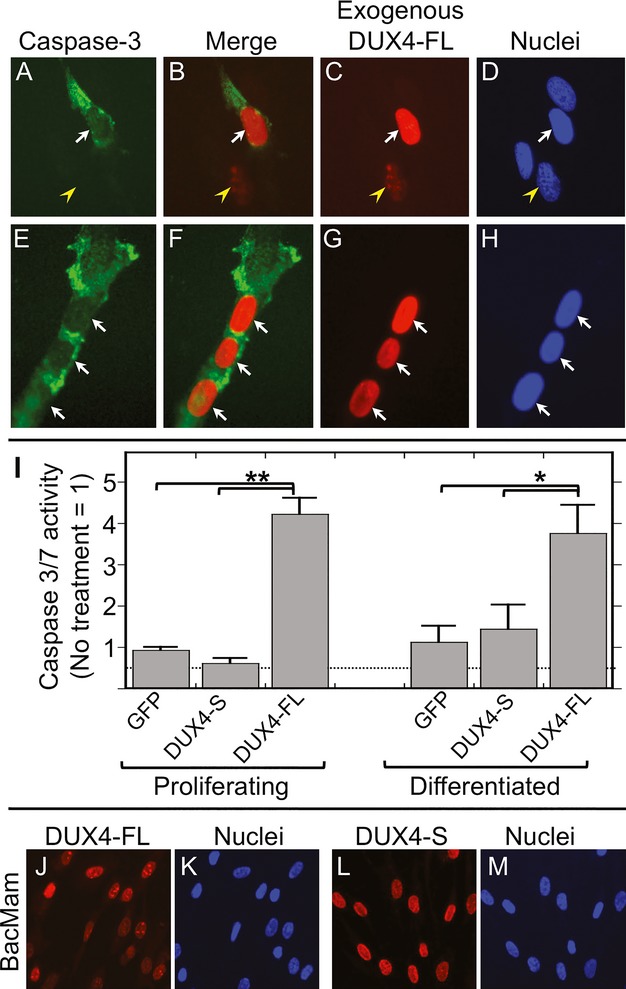
Exogenous DUX4-FL increased caspase activation. (A–H) DUX4-FL (red) was expressed in proliferating human myoblasts (A–D) at 24 h after plasmid transfection and in differentiated myotubes (E–H) at 48 h after BacMam vector addition. Cultures were established with myoblasts from a healthy donor. Immunostaining for activated caspase-3 (green) was found in a fraction of myoblasts and myotubes that expressed exogenous DUX4-FL (arrows). Yellow arrowhead in (A–D) indicates a DUX4-FL-positive myoblast without caspase-3 staining. (I) Caspase 3/7 enzymatic activity was increased by BacMam-mediated expression of DUX4-FL, but not DUX4-S, in both proliferating (myoblast) and differentiated (myotube) cultures of human myogenic cells. Error bars = SE, n = 4. *P < 0.05; **P < 0.01.
We next found that exogenous expression of DUX4-FL altered the intracellular distribution of ubiquitin-conjugated proteins in human myoblasts. We chose to examine ubiquitinated proteins (Ub-proteins) for several reasons. First, impaired functioning of protein turnover systems that process ubiquitinated proteins is often associated with increased cell death, including in myogenic cells29; and ubiquitinated cytoplasmic inclusions are found in at least two myopathies, inclusion body myositis and oculopharyngeal muscular dystrophy.30,31 Second, DUX4-FL expression had been linked previously to altered expression of ubiquitin handling proteins, including increases in ubiquitin ligases.16,32 Finally, impaired protein turnover often leads to protein aggregation,33 and we (this study) and others2,34, have observed that DUX4-FL immunostaining is sometimes punctate, suggesting that perhaps DUX-FL itself can form aggregates within the nucleus.
We used immunostaining with the mouse mAb FK2, which reacts with mono- and poly-ubiquitinated proteins (Ub proteins) but not free ubiquitin, to show that the distribution of Ub-proteins in human myoblasts was altered by exogenous DUX4-FL expression (Fig.2). In the absence of detectable DUX4-FL expression, the most common pattern of Ub-protein immunostaining in human myoblasts included moderate levels of mostly uniformly distributed signal in both nuclei and cytosol (Fig.2A–C). Myoblasts that expressed exogenous DUX4-FL, however, had decreased immunostaining in nuclei (arrows, Fig.2A–C). A subset of both DUX4-FL-negative and DUX4-FL-positive cells also showed regions of punctate staining in the cytoplasm and/or nucleus. To more quantitatively analyze Ub-protein distribution in response to DUX4-FL expression, we examined mAb FK2 immunostaining in DUX4-FL-positive and -negative myoblasts and separately classified nuclear and cytoplasmic staining in each cell as “low,” “mid,” or “high” (examples are given in Fig.2A). This analysis confirmed that the predominant effect of exogenous expression of DUX4-FL in human myoblasts was to decrease nuclear immunostaining for Ub-proteins (Fig.2D).
Figure 2.
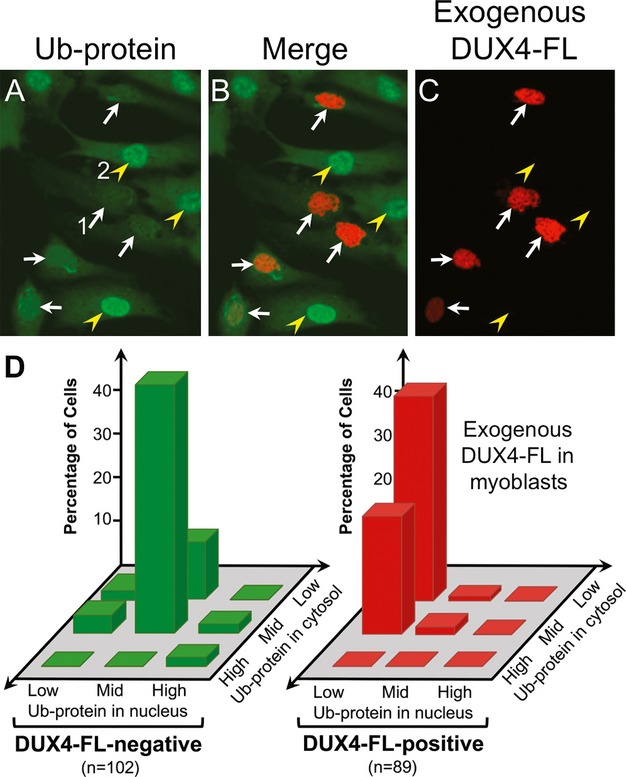
Exogenous DUX4-FL altered ubquitinated protein distribution in myoblasts. (A–C) After plasmid transfection, DUX4-FL (red) was expressed in a subset of myoblasts; and confocal images of immunostaining for ubiquitinated proteins (Ub-protein, green) showed decreased staining in the nuclei of DUX4-FL-positive (arrows) compared to DUX4-FL-negative (arrowheads) myoblasts. Numbered cells give examples of the classifications of Ub-protein staining patterns that are graphed in (D); with the (DUX4-FL-positive) myoblast labeled “1” classified as low nuclear and low cytoplasmic; and the (DUX4-FL-negative) myoblast labeled “2” classified as mid nuclear and mid cytoplasmic. Additional classification examples are in Figure4. (D) Nuclear and cytoplasmic Ub-protein staining patterns were independently classified as low, mid, or high, as described under (A) and in the text, and the number of myoblasts with each pattern was counted. The predominant effect of exogenous DUX4-FL expression on Ub-protein distribution in myoblasts was to decrease nuclear staining with little effect on cytoplasmic staining.
We next found that exogenous expression of DUX4-FL in human myoblasts also altered the intracellular distribution of TDP-43 (transactive response DNA-binding protein 43 kDa, encoded by the TARDBP gene). We examined TDP-43 because TDP-43 aggregates have been reported in inclusion body myositis, where ubiquitin aggregates are also found.20,30,35,36 In addition, TDP-43 functions in alternative mRNA splicing37,38; and proper splicing is required for normal skeletal muscle function,39 may regulate DUX4-fl mRNA production,2 and may be aberrant in myopathies including FSHD.40
As is common in many cell types, human myoblasts that did not express DUX4-FL showed immunostaining for endogenous TDP-43 that was distributed throughout the nuclei and was typically undetectable in the cytoplasm (Fig.3A–C). In contrast, upon exogenous expression of DUX4-FL, TDP-43 immunostaining in myoblasts was no longer uniformly distributed as in DUX4-negative nuclei, but rather appeared to include highly fluorescent puncta suggesting aggregation (Fig.3A–C). This punctate pattern was found in most, but not all, nuclei that expressed exogenous DUX4-FL. In one experiment, for example, we found one or more regions of punctate TDP-43 immunostaining in 22 of 28 (78%) of DUX4-FL-positive nuclei, whereas this punctate pattern was found in only one 51 (2%) of nearby DUX4-FL-negative nuclei in the same cultures (P < 0.01 by Fisher's exact test for observed versus expected number of nuclei with punctate pattern). As found previously in HeLa cells,38 we found that a short 1 h heat shock at 42°C also induced a punctate pattern of TDP-43 immunostaining (Fig.3K and L), indicating aggregation, in the nuclei of human myogenic cells, though the puncta induced after a short (1 h) heat shock were typically smaller than the puncta found after the much longer term (32–48 h) expression of DUX4-FL.
Figure 3.
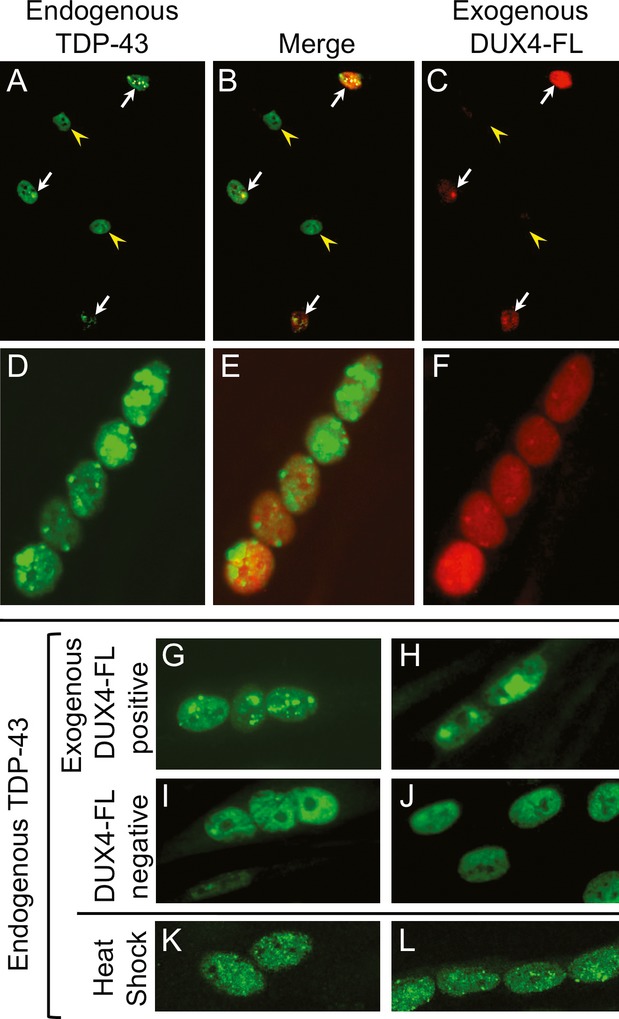
Exogenous DUX4-FL induced aggregation of TDP-43 in myonuclei. (A–C) DUX4-FL (red) was expressed in proliferating human myoblasts using plasmid transfection. Arrows indicate DUX4-FL-positive myoblasts and arrowheads indicate DUX4-FL-negative myoblasts. (D–H) DUX4-FL was expressed in differentiated myotubes using a BacMam vector. Cultures were established with myoblasts from a healthy donor and examined at 32–48 h after addition of plasmids or BacMam vector. TDP-43 immunostaining (green) in DUX4-FL-negative myoblasts (A–C) and myotubes (I and J) was diffusely distributed in nuclei and absent from the cytoplasm, whereas DUX4-FL-positive myoblasts (A–C) and myotubes (D–H) showed a different, punctate pattern of TDP-43 staining indicative of aggregation. (K and L) TDP-43 immunostaining also showed a punctate pattern after a short heat shock (1 h at 42°C).
Exogenous DUX4-FL expression in differentiated myotubes
We next tested whether exogenous expression of DUX4-FL in differentiated, multinucleate myotubes induced changes similar to those we found in myoblasts. Our previous work showed that, under our culture conditions, DUX4-FL was expressed from its endogenous promoter only in differentiated (MyHC-expressing) myogenic cells,23 so it was important to determine if differentiated cells responded differently to DUX4-FL than myoblasts. To express DUX4-FL or, as a control, DUX4-S in human myotubes under control of a constitutively active CMV promoter fragment as in our plasmid transfections of myoblasts, we used the baculovirus-based BacMam vector system.27 At 32–48 h after transduction of differentiated cultures with the BacMam vectors, >90% of the myotube nuclei showed immunostaining for DUX4-FL or DUX4-S. Though found in almost all nuclei, the BacMam-mediated DUX4-FL immunostaining was of highly variable intensity and often of non-uniform punctate distribution, whereas the DUX4-S immunostaining was typically of uniform intensity and distribution within all nuclei in the culture (Fig.1J–M).
In myotubes, exogenous, BacMam-mediated expression of DUX4-FL was accompanied both by increased immunostaining for active caspase-3 (Fig.1E–H) and by a severalfold increase in caspase 3/7 enzymatic activity (Fig.1I). In contrast, parallel cultures with BacMam DUX4-S did not show a significant increase in caspase 3/7 enzymatic activity when compared to controls with BacMam GFP (Fig.1I). Thus, exogenous expression from a constitutive CMV promoter of DUX4-FL, but not DUX4-S, increased caspase activation in both myoblasts and myotubes.
In myotubes, BacMam-mediated DUX4-FL expression also altered the patterns of Ub-protein immunostaining. Double immunostained images showed an increased cytoplasmic immunostaining for Ub-proteins in DUX4-FL-expressing cells compared to DUX4-FL-negative cells in the same cultures (Fig.4A–C). In addition, many of the nuclei in DUX4-FL-positive myotubes showed relatively large aggregates of Ub-proteins (Fig.4A–C). Such large Ub-protein aggregates were seldom seen in DUX4-FL-negative myotube nuclei.
Figure 4.
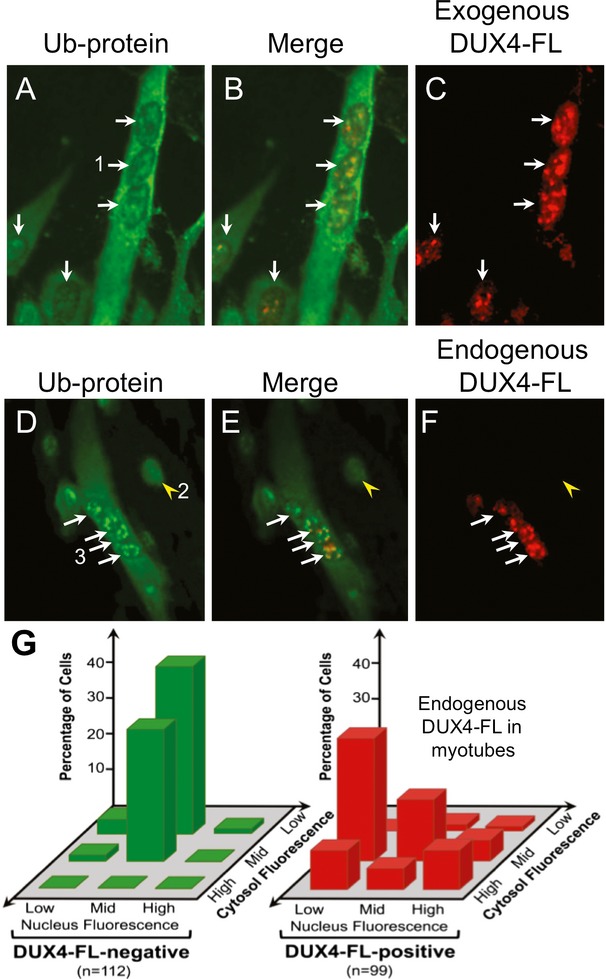
Exogenous and endogenous DUX4-FL altered ubiquitinated protein distribution in myotubes. (A–C) A BacMam vector was used to express exogenous DUX4-FL (red) in myotubes. When compared to the low cytoplasmic and uniform nuclear staining for Ub-proteins in DUX4-FL-negative cells (e.g. Fig.2A and arrowhead in D), DUX4-FL-positive myotubes show increased immunostaining for Ub-proteins (green) in the cytoplasm and variable staining, often with aggregates, in nuclei. (D–F) Expression of DUX4-FL from its endogenous promoter in myotubes formed from FSHD myoblasts (17Abic) also led to increased cytoplasmic staining and variable, often punctate, staining for Ub-proteins. Arrows in all panels indicate cells with DUX4-FL-positive nuclei and the arrowheads in (D–F) indicate a cell with a DUX4-FL-negative nucleus. Additional examples of Ub-protein staining in DUX4-FL-negative cells are shown in Figure7E and F. Numbered cells give examples of the classifications of Ub-protein staining patterns graphed in (G); with the (DUX4-FL-positive) myotube labeled “1” in (A) classified as mid nuclear and high cytoplasmic; the (DUX4-FL-negative) myocyte labeled “2” in (D) classified as mid nuclear and low cytoplasmic; and the (DUX4-FL-positive) myotube labeled “3” in (D) classified as high nuclear and mid cytoplasmic. Note that mid and high-nuclear staining can be punctate, rather than uniform across the nuclei. Additional examples are in Figure2. (G) Nuclear and cytoplasmic Ub-protein staining patterns were independently classified as low, mid, or high, and the number of cells with each pattern was counted. The results showed that endogenous DUX4-FL expression in myotubes increased cytoplasmic staining and produced variable changes in nuclear staining for Ub-proteins.
As in myoblasts, exogenous DUX4-FL also altered the pattern of TDP-43 immunostaining in myotubes. In DUX4-negative myotubes there was a relatively uniform distribution of TDP-43 immunostaining in nuclei with the exception of nucleoli that were unstained (Fig.3I and J). In contrast, there was a distinctly punctate pattern of TDP-43 staining in the nuclei of DUX4-FL-positive myotubes (Fig.3D–H). Thus, in both myoblasts (Fig.3A–C) and myotubes (Fig.3D–H), exogenous expression of DUX4-FL from a constitutive CMV promoter-induced aggregation of TDP-43.
Endogenous DUX4-FL expression
We next examined whether DUX4-FL expression from its endogenous promoter in differentiated cells caused the same changes as exogenous expression. Unlike exogenously expressed DUX4-FL, which was detectable in most nuclei when expressed from the BacMam vector, endogenous expression of DUX4-FL was detectable by immunostaining in only a very small fraction, <0.5%, of the nuclei in differentiated, myosin-expressing cells (Table1), much as seen in previous studies.2,17,23,24 Because the downstream effects of DUX4-FL depend on expression level,13 it was important to determine which of the changes induced by exogenous DUX4-FL from the strong CMV promoter were also induced by DUX4-FL expression from its endogenous promoter.
Our results showed that endogenous DUX4-FL expression was accompanied by altered Ub-protein localization and TDP-43 aggregation, but not caspase-3 activation. In particular, caspase-3 activation, which was detected by immunostaining upon exogenous expression of DUX4-FL in myotubes (Fig.1E–H), was not detected in myotubes that expressed DUX4-FL from the endogenous promoter. The morphology of the endogenous DUX4-FL-positive nuclei also appeared to be normal. Thus, in contrast to the caspase activation that was detectable by immunostaining upon exogenous expression of DUX4-FL in myoblasts or myotubes (Fig.1), there was no detectable activation of caspase-3 upon expression of DUX4-FL from its endogenous promoter.
Endogenous expression of DUX4-FL was, however, accompanied by altered patterns of immunostaining for both Ub-proteins and TDP-43, and these alterations were similar to those seen upon exogenous DUX4-FL expression in myotubes. For Ub-proteins, double immunostaining suggested that endogenous expression of DUX4-FL in myotubes was often accompanied by increased cytoplasmic staining and increased numbers of nuclear aggregates when compared to DUX4-FL-negative cells in the same differentiated cultures (Fig.4). To more quantitatively analyze the Ub-protein staining patterns, we classified nuclear and cytoplasmic Ub-protein staining intensity in myotubes with and without endogenous DUX4-FL expression as “low,” “mid,” or “high,” as we did for exogenous expression in myoblasts (see examples in Figure4A and D). This classification assay showed that one consistent effect of endogenous DUX4-FL in human myotubes was to increase cytoplasmic staining intensity for Ub-proteins (Fig.4G). This increased cytoplasmic staining was also seen upon exogenous expression of DUX4-FL in myotubes (Fig.4A–C), but not myoblasts (Fig.2).
In addition to the increased cytoplasmic stain, most myotube nuclei with endogenous DUX4-FL expression showed altered Ub-protein staining compared to DUX4-FL-negative nuclei in the same culture. Though the Ub-protein and DUX4-FL staining patterns in myotube nuclei were often both punctate (Fig.4) or both uniform, we also found examples of myotube nuclei with uniform DUX4-FL and punctate Ub-protein staining and vice versa. In addition, though some DUX4-FL-positive myotube nuclei appeared to have decreased immunostaining for Ub-proteins, whereas others showing increased, often punctate, staining; the myotube nuclei with the most intense staining for endogenous DUX4-FL did not necessarily have the most intense stain for Ub-protein (Fig.4). The results of the quantitative assay (Fig.4G) suggested that Ub-protein staining patterns were sufficiently distinct in DUX4-FL-positive compared to DUX4-FL-negative cells to allow identification of about two-thirds of the DUX4-FL cells based on Ub-protein staining alone.
To further assess this result, we carried out a blind test to confirm that the Ub-protein staining in endogenous DUX4-FL-positive and -negative nuclei were distinctive. For this test, an observer was provided with 25 unlabeled pairs of images that had been individually stained for Ub-proteins and for nuclei. Each image contained many cells (range 13–60, ave = 28.3, total = 708), and the observer was asked to identify any cells that appeared to have a Ub-protein staining pattern that was distinct from the pattern in the majority of cells. After the observer identified candidate cells, the Ub-protein and nuclear images were compared to companion images that had been immunostained for DUX4-FL but withheld from the observer. In total, the observer correctly identified 18 of the 25 (72%) DUX4-FL-positive cells in the blind image set, whereas only one or two would have been expected to be correctly identified by chance (P < 0.001 by Fisher's exact test, observed vs. expected). Only 16 of 683 DUX4-FL-negative cells were identified as having an unusual ubiquitin pattern. These results implied that the Ub-protein staining pattern in endogenous DUX4-FL-expressing cells was sufficiently different from the pattern in DUX4-FL-negative cells to be accurately identified in a majority (∽70%) of the cases, whereas only a very small percentage (<3%) of DUX4-FL-negative cells had a similar pattern. This finding is similar to the results from our classification assay (Fig.4G), in which about two-thirds of the DUX4-FL-positive cells appeared to have a distinct Ub-protein staining pattern. Thus, as with exogenous expression, expression of DUX4-FL in myotubes from its endogenous promoter altered the intracellular localization of ubiquitinated proteins.
Endogenous expression of DUX4-FL also altered the TDP-43 immunostaining pattern. Most nuclei with endogenous expression of DUX4-FL showed a distinctly more punctate pattern of TDP-43 immunostaining than was observed in DUX4-FL-negative nuclei (Fig.5). As above, we carried out a blind test to assess the extent of altered TDP-43 staining upon expression of endogenous DUX4-FL. For this assessment, the observer was provided with 65 pairs of images that had been stained for TDP-43 and for nuclei and was asked to identify cells with atypical patterns of TDP-43 staining. Of the 425 cells in the image collection, the observer identified 31 of the 45 (69%) endogenous DUX4-FL-positive cells as having an atypical TDP-43 staining pattern (P < 0.001 by Fisher's exact test, observed vs. expected by chance). In contrast, only 11 of the 380 (2.9%) DUX4-FL-negative cells were identified by the observer as having an altered TDP-43 staining pattern. Thus, approximately two-thirds of the endogenous DUX4-FL-positive cells had a TDP-43 immunostaining pattern that was sufficiently distinct for identification when compared to DUX4-FL-negative cells.
Figure 5.
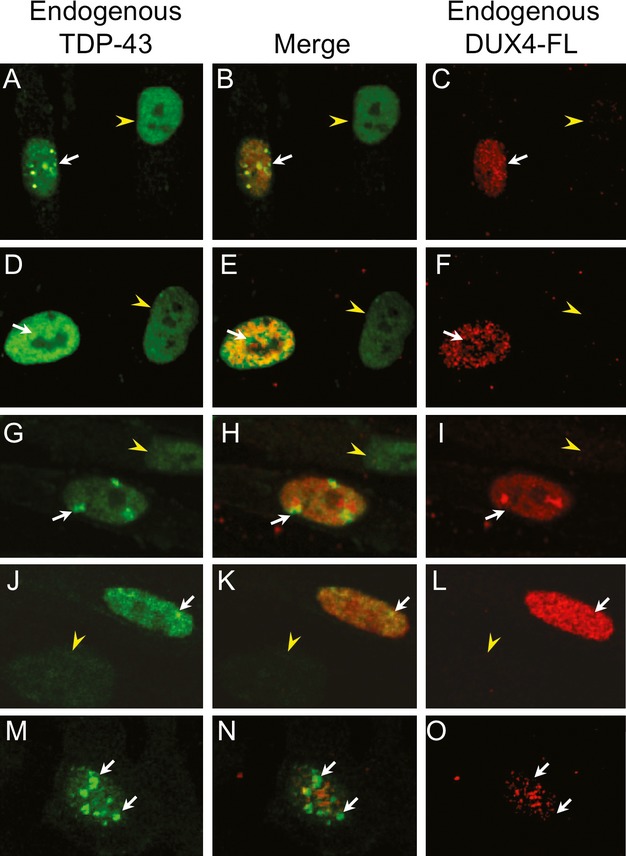
Endogenous DUX4-FL-induced aggregation of TDP-43 in myotube nuclei. Double immunostaining of differentiated cultures for expression of endogenous DUX4-FL (red) and TDP-43 (green) showed that TDP-43 staining was punctate, indicative of aggregation, in nuclei that expressed endogenous DUX4-FL (arrows), whereas nearby nuclei that did not express DUX4-FL (arrowheads) showed a more uniform distribution of TDP-43 immunostaining. Each row shows a different set of nuclei. In the nuclei shown here, DUX4-FL staining was also punctate, however, merged confocal images showed little or no overlap of the distinctly punctate areas of staining for DUX4-FL and Ub-proteins (e.g. H).
UPS inhibition
Based on our findings that exogenous and endogenous DUX4-FL expression in human myoblasts and myotubes led to altered intracellular distribution of ubiquitinated proteins and apparent aggregation of TDP-43, we hypothesized that DUX4-FL expression was accompanied by impaired protein turnover. As one test of this possibility, we used a co-transfection assay to determine if DUX4-FL would inhibit turnover of a modified GFP protein that was made unstable by addition of the 16 amino acid CL-1 degron sequence.41,26,42 In this assay, conditions that impair protein turnover lead to an increased level of the GFP-degron protein. Indeed, by immunoblotting, we found more GFP in HEK293 cell cultures that expressed both the GFP-degron and DUX4-FL than in cultures that expressed only the GFP-degron and not DUX4-FL (Fig.6A), a result that we replicated in three similar experiments. As a positive control, treatment with MG132, an inhibitor of protein turnover through the UPS, also increased the amount of GFP-degron on immunoblots compared to untreated cultures (Fig.6A). In myoblasts, a fluorescent signal from the stabilized GFP-degron was detectable upon plasmid co-transfection and co-expression with DUX4-FL (Fig.6B). These results suggested that exogenous expression of DUX4-FL impairs protein turnover as measured by stabilization of the GFP-degron reporter.
Figure 6.
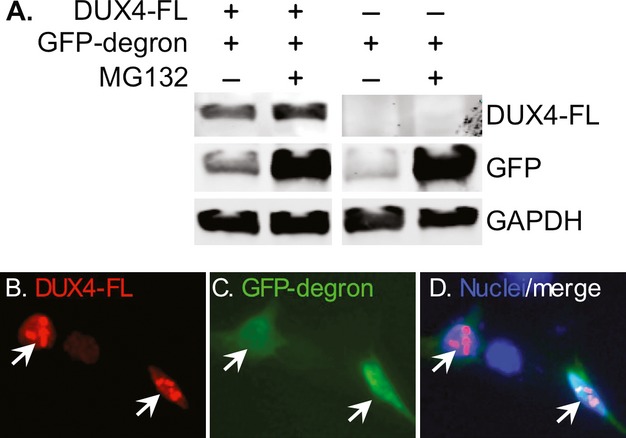
Exogenous DUX4-FL slowed protein turnover. (A) GFP protein made unstable by addition of the 16 amino acid CL1 degron sequence (GFP-degron) was expressed by plasmid transfection in HEK293 cells with or without co-expression of exogenous DUX4-FL and with or without MG132 treatment to inhibit protein turnover through the ubiquitin–proteasome system. Immunoblots showed that more of the GFP-degron protein accumulated in cultures that co-expressed DUX4-FL (compare GFP in lanes 1 and 3) indicating that DUX4-FL expression led to stabilization and slowed turnover of the GFP-degron protein. As a positive control, MG132 treatment also led to accumulation of GFP-degron protein (lanes 2 and 4). GAPDH was used a loading control. (B–D) When co-expressed by plasmid transfection in human myoblasts, GFP-degron protein (green) accumulated when co-expressed with DUX4-FL (red).
To further examine a possible role for UPS inhibition in DUX4-FL-mediated pathology, we next determined if treatment with MG132 would lead to changes in Ub-protein distribution or TDP-43 aggregation similar to those we found in cells that expressed DUX4-FL. After a 5 h treatment with 10 μmol/L MG132, MG132-treated myoblasts and myotubes showed increased cytoplasmic and decreased nuclear staining for Ub-proteins; and these changes did not appear to be additionally altered by exogenous DUX4-FL expression (Fig.7). The MG132-induced changes in Ub-protein staining were in some respects similar to the changes induced by exogenous or endogenous DUX4-FL expression, though the similarities between MG132-induced and DUX4-FL-induced changes were different for myoblasts and myotubes. Myoblasts that expressed exogenous DUX4-FL from a plasmid showed a decrease in nuclear Ub-protein staining similar to that induced by MG132 treatment, but did not show the increase in cytoplasmic staining (Fig.2). In contrast, myotubes that expressed exogenous or endogenous DUX4-FL did show an increase in cytoplasmic Ub-protein staining, but did not show the relatively uniform reduction in nuclear Ub-protein stain seen upon MG132 treatment (Fig.4). Rather, DUX4-FL-expressing nuclei often had a punctate Ub-staining pattern (Fig.4). Furthermore, we found that MG132 treatment induced the accumulation of punctate TDP-43 staining in the nuclei, but not in the cytoplasmic regions, of myotubes (Fig.7I–L); and this MG132-induced change in TDP-43 distribution within nuclei was similar to that induced by exogenous or endogenous DUX4-FL expression (cf. Figs.3, 5).
Figure 7.
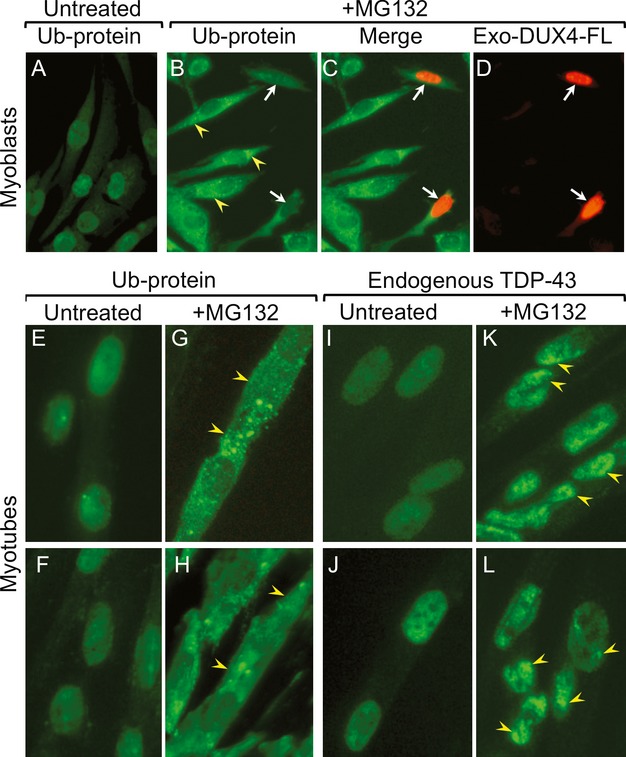
MG132 treatment changed the distributions of ubiquitinated proteins and TDP-43 in human myoblasts and myotubes. (A–D) In untreated myoblasts, staining for Ub-proteins was diffuse in nuclei and low in the cytoplasm (A), whereas, after MG132 treatment, cytoplasmic staining was high and partially punctate in the cytoplasm and nuclear staining was very low (B). Expression of exogenous DUX4-FL by plasmid did not further change the Ub-staining pattern in myoblasts (C and D). (E–H) Staining for Ub-proteins was uniformly diffuse in nuclei and low in the cytoplasm of untreated myotubes (E and F), whereas MG132 treatment led to decreased nuclear staining and increased, often punctate, cytoplasmic staining (G and H). (I–L) Staining for TDP-43 was uniformly diffuse in nuclei and low in the cytoplasm of untreated myotubes (I and J), whereas MG132 treatment led to a punctate, aggregated pattern of TDP-43 staining in nuclei (arrowheads; K and L).
DUX4-FL and protein aggregation
Because DUX4-FL-expressing nuclei often showed punctate patterns of immunostaining for ubiquitinated proteins (e.g. Fig.4D), TDP-43 (e.g. Fig.3D) and DUX4-FL itself (e.g. Fig.4F), we next used double immunostaining to determine if the punctate staining patterns were overlapping or distinct (Fig.8). First, in the subset of nuclei in which we observed punctate staining for both DUX4-FL and Ub-proteins, we found that the DUX4-FL and Ub-protein immunostaining patterns were largely distinct, though with some overlap (Fig.8A–C). There was also little or no overlap of nuclear punctate immunostaining for Ub-proteins and TDP-43 (Fig.8D–F). These results indicate that the DUX4-FL and TDP-43 proteins which generated the punctate immunostaining in these DUX4-FL-positive nuclei were unlikely to be extensively ubiquitinated. Finally, in those nuclei where both the DUX4-FL and TDP-43 showed punctate staining, there was also little or no overlap of staining (Fig.8G–I). Taken together, these results indicate that the Ub-proteins, TDP-43, and DUX4-FL that generated the punctate staining patterns likely did so independently in spatially separated domains.
Figure 8.
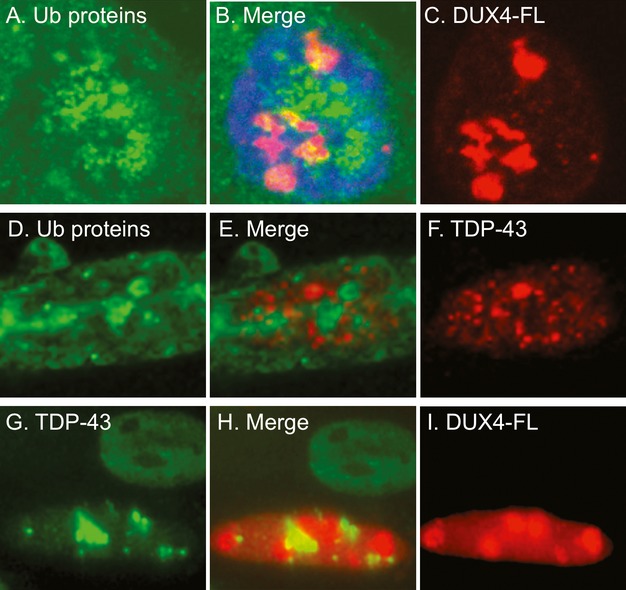
The punctate immunostaining patterns for ubiquitinated proteins, TDP-43, and DUX4-FL in nuclei showed little co-localization. (A–C) Confocal microscope images for ubiquitinated proteins (green, A) and DUX4-FL (red, C) were merged in (B) to show the relatively small regions of overlap (yellow). (B) Includes blue DNA stain for the nucleus. (D–F) Standard microscope images for ubiquitinated proteins (green, D) and TDP-43 (red, F) were merged in (E) and showed little overlap of the punctate areas of immunostaining in the nucleus. (G–I) Standard microscope images for TDP-43 (green, G) and DUX4-FL (red, I) were merged in (H) and showed little overlap of the punctate areas immunostaining seen in the lower DUX4-FL-positive nucleus. Additional examples are in Figure5. The upper DUX4-FL-negative nucleus in (G–I) shows the nearly uniform nuclear immunostaining for TDP-43 that is seen in the absence of DUX4-FL.
Finally, because punctate immunostaining patterns suggested that proteins might be forming insoluble aggregates, we determined if the amounts of insoluble proteins were affected by exogenous expression of DUX4-FL or, as a control, DUX4-S (Fig.9). BacMam DUX4-FL or DUX4-S vectors were added to myogenic cell cultures after 4 days of differentiation, and 48 h later each culture was harvested and separated into a RIPA-soluble fraction and a RIPA-insoluble/urea soluble fraction. In the RIPA-soluble fraction, the amounts of ubiquitinated proteins and TDP-43 were not affected by expression of either DUX4-FL or DUX4-S. In contrast, the RIPA-insoluble/urea soluble fraction had increased amounts of both ubiquitinated proteins and TDP-43 (including both the 43 kDa full-length form and an ∽35 kDa form) upon expression of DUX4-FL, but DUX4-S. Consistent with previous observations of non-muscle cells,43–45 we found some TDP-43 in the insoluble fraction of control cells, however, we also found that the amount of insoluble TDP-43 was consistently increased upon DUX4-FL expression. Furthermore, a large amount of the exogenous DUX4-FL, but little of the DUX4-S, was found in the RIPA-insoluble/urea soluble fraction indicating that a proportion of the DUX4-FL itself became RIPA insoluble.
Figure 9.
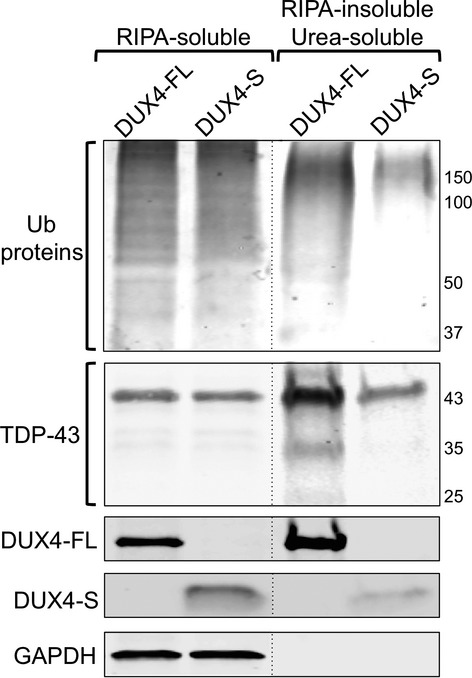
Exogenous expression of DUX4-FL, but not DUX4-S, was followed by increased amounts of insoluble ubiquitinated proteins, TDP-43, and DUX4-FL itself. BacMam DUX4-FL or DUX4-S vectors were added to myogenic cell cultures that had been in differentiation medium for 4 days, and 48 h later the cultures were harvested and separated into a RIPA-soluble fraction and a RIPA-insoluble/urea soluble fraction. Fractions were separated by SDS-PAGE and immunoblotted to identify, as indicated, ubiquitinated proteins (Ub proteins); TDP-43; DUX4-FL and DUX4-S (by V5 epitope tag); and GAPDH (as a marker for the RIPA-soluble fraction). The amounts of ubiquitinated proteins and TDP-43 in the RIPA-soluble fraction was not affected by expression of either DUX4-FL or DUX4-S. In contrast, the RIPA-insoluble/urea soluble fraction had increased amounts of both ubiquitinated proteins and TDP-43 (both the 43 kDa form and the ∽35 kDa form) upon expression of DUX4-FL, but DUX4-S. In addition, much of the DUX4-FL, but little of the DUX4-S, was found in the RIPA-insoluble/urea soluble fraction. All samples were analyzed on the same immunoblot with some lanes re-arranged for presentation as denoted by the vertical dotted lines. The experiment was repeated twice with similar results.
Discussion
In this study, we found that expression of DUX4-FL in human myogenic cells, either from its endogenous promoter or exogenously, was accompanied both by altered intracellular distribution and accumulation of ubiquitinated proteins and by formation of TDP-43 aggregates in nuclei. In addition, inhibition of the UPS by MG132 led both to increased cytoplasmic accumulation of ubiquitinated proteins and to nuclear TDP-43 aggregates similar to those seen upon DUX4-FL expression. Exogenous DUX4-FL expression also slowed turnover of destabilized GFP in a model system and increased the amounts of insoluble ubiquitinated proteins, TDP-43, and DUX4-FL itself. Because aberrant DUX4-FL expression, particularly in skeletal muscle, appears to be causative in FSHD, our results suggest that DUX4-FL-induced inhibition of the normal pathways of protein turnover and nuclear aggregation of TDP-43 may contribute to FSHD pathogenesis.
A current view of pathogenesis in FSHD is that the combination of D4Z4 array contraction and epigenetic changes including DNA hypomethylation leads to a relaxed chromatin state which, in the presence of a functional poly-adenylation locus, allows aberrant transcription in skeletal muscle of a stable DUX4-fl mRNA.3,6,23 The resulting DUX4-FL protein can act as a transcription factor and has been found to alter transcription of many genes and to be cytotoxic when overexpressed in several systems both in vitro and in vivo.12–15,18,46,47 Furthermore, genetic and/or epigenetic changes in FSHD may also lead to dysregulated transcription of non-coding RNAs, such as the long non-coding RNA from the D4Z4 region that is preferentially produced in FSHD48 or the multiple miRNAs that appear to be differentially expressed in FSHD.49,50
In our study, we found that DUX4-FL, when expressed from its endogenous promoter in myotubes, produced both an alteration in ubiquitinated protein distribution and nuclear aggregation of TDP-43 in the absence of significant caspase-3 activation or overt cytotoxicity. In contrast, when DUX4-FL was exogenously expressed in myotubes with a BacMam vector, we did find caspase-3 activation and cytotoxicity. It is likely that this difference in cytotoxicity arose because more DUX4-FL was produced by exogenous expression under the CMV promoter than by expression from the endogenous promoter. This possibility is consistent with a previous study, using the mouse C2C12 myogenic cell line, which showed that low levels of DUX4-FL can interfere with muscle gene regulation in the absence of cytotoxicity, whereas high levels of DUX4-FL are cytotoxic.13 Recently, it was found that endogenous DUX4-FL expression could lead to caspase activation and cytotoxicity under culture conditions which both differed from ours and produced multinucleate cells with aggregations of large numbers of nuclei in close proximity to each other.18 The myotubes formed under our culture conditions, however, contained moderate numbers of nuclei arranged in single file as found in newly forming myotubes in vivo.51 Endogenous DUX4-fl mRNA usually appears to be produced by only one of the nuclei within a myotube,18 whereas the DUX4-FL protein is able to move to and accumulate in multiple nearby nuclei.52 It may be, therefore, that the number and proximity of nearby nuclei, as well as the level of expression, determine whether endogenous DUX4-FL reaches a cytotoxic threshold. It is clear, however, that sub-cytotoxic levels of DUX4-FL are sufficient to induce potentially pathological changes in the host myotube (13, this work).
Additional work is needed to identify the molecular mechanism(s) linking DUX4-FL expression to abnormal protein turnover. Overexpression of DUX4-FL causes abnormal expression of genes in several pathways, including apoptosis, immune mediators, protein turnover, stress response, ubiquitin, and unfolded protein response, as well as alteration in expression of additional transcription factors such as PITX1.13,16,47,53 Thus, inhibition of protein processing could be due to DUX4-FL-induced gene expression changes that alter the composition or level of protein processing systems. Such gene expression changes could be due either to DUX4-FL directly or secondarily through DUX4-FL-dependent induction of additional transcription factors, particularly PITX1 which can itself induce myopathy when ectopically expressed.14,54 An additional possible linkage between DUX4-FL and impaired protein turnover could be through oxidative stress. FSHD muscles can show signs of mitochondrial dysfunction and increased oxidative stress, for example, as measured by lipofuscin accumulation.55 Furthermore, ectopic DUX4-FL expression has recently been linked to pathology through possibly increased oxidative stress56; and increased oxidative stress through multiple mechanisms, including mitochondrial dysfunction, is known to lead to impaired protein turnover.57,58
Dysregulation of protein homeostasis and TDP-43 aggregation have now been linked to multiple nerve and muscle diseases.57,59,60 For example, nuclear, cytoplasmic, and neuritic inclusions that contain TDP-43 are found in neurons and glia in Frontotemporal Lobar Degeneration with ubiquitin-positive inclusions (FTLD-U) and amyotrophic lateral sclerosis (ALS), though TDP-43 is ubiquitinated only in cytoplasmic and not in nuclear aggregates.61 TDP-43 aggregates are also usually not ubiquitinated in multiple protein aggregate myopathies.62 Similarly, TDP-43 also does not appear to be ubiquitinated when in nuclear aggregates induced by heat shock38 or upon DUX4-FL expression or MG132 treatment (this work). Consistent with previous studies, we did not find cytoplasmic accumulation of endogenous TDP-43 after a short treatment (5 h) with MG132, though, based on work in non-muscle cells, cytoplasmic aggregates of TDP-43 would be expected after longer treatment (e.g. >24 h).43 In our studies, we found that both exogenous and endogenous DUX4-FL induced only nuclear aggregates of the endogenously expressed TDP-43 protein. Because endogenous DUX4-FL expression did not induce detectable activation of caspase-3, it appears that this nuclear aggregation of TDP-43 was not dependent on caspase-3-mediated cleavage of TDP-43 itself.45 Nuclear aggregation of TDP-43 may be a downstream result of DUX4-FL-induced impairment in proteasome function, because MG132 treatment also produced nuclear TDP-43 aggregates. The nuclear ubiquitin–proteasome system63 might be particularly sensitive to DUX4-FL expression. The extent of DUX4-FL-induced nuclear TDP-43 aggregation, as well as balance in formation of nuclear and cytoplasmic aggregates, is likely to evolve over time and to be determined by a combination of DUX4-FL and TDP-43 levels, ubiquitination, interactions with binding partners, and extent of impairment of the ubiquitin–proteasome system.
Our study adds a new facet to the potential pathological consequences of aberrant DUX4-FL expression in FSHD, but much remains to be learned. In particular, it will be important to examine FSHD muscle biopsies for signs of proteasome dysfunction, changes in ubiquitinated protein distribution, and nuclear aggregates of TDP-43 and, if found, to determine if such changes correspond to sites of concurrent DUX4-FL expression or are more widespread. It will also be informative to identify any additional proteins that may co-aggregate with TDP-43 in muscle cells, such as, for example, alpha-synuclein or stress granule components,64,65 to identify if a specific subset of proteins is abnormally ubiquitinated and mis-localized in DUX4-FL-expressing myogenic cells, and to determine if the TDP-43 in DUX4-FL-induced nuclear aggregates is phosphorylated or cleaved.66 Because TDP-43 normally regulates multiple aspects of mRNA and non-coding RNA, studies are also needed to determine if DUX4-FL expression leads to loss of TDP-43 function, thereby possibly contributing to pathology through a loss-of-function mechanism.67 Multiple diseases show proteasome and TDP-43 pathologies and therapies that target these types of pathologies are under rapid development. For development of FSHD therapies, one promising focus has been to develop techniques to inhibit the expression or function of DUX4-FL.56,68 Inhibition of DUX4-FL expression or function, if successful in patients, might also be expected to normalize proteasome function and TDP-43 aggregation.
Acknowledgments
This work was supported by grants to J. B. M. from the National Institutes of Health (R01AR060328), the Muscular Dystrophy Association (216422), the FSH Society, the Association Française contre les Myopathies, and the Boston University Clinical and Translational Science Institute which is supported by the National Institutes of Health (UL1-TR000157); by a subcontract to J. B. M. on an award from the National Institutes of Health to Peter L. Jones (R01AR062578); by a grant to S. H. from the Thoracic Foundation (Boston MA); and by a grant from the National Institutes of Health that supported the Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center for FSHD Research (U54HD060848). We thank Kathryn Wagner and Genila Bibat (Kennedy-Krieger Institute and Johns Hopkins School of Medicine) for obtaining the initial biopsies; the Wellstone FSHD Center (University of Massachusetts Medical School) for providing human myogenic cells prepared from biopsies; Stephen Tapscott and Linda N. Geng (Fred Hutchinson Cancer Center) for samples of DUX4-FL mAbs; Daniel Perez of the FSH Society; Shinichi Takayama (Boston University School of Medicine) for plasmids and much useful advice; Hiroaki Mitsuhashi and Louis M. Kunkel (Boston Children's Hospital) for DUX4 plasmids; and Peter L. Jones and Takako I. Jones (University of Masschusetts Medical School) for plasmids and key discussions.
Conflict of Interest
Dr. Boyce holds relevant patents and receives royalties from commercial use of BacMam vectors. Dr. Homma reports grants from Thoracic Foundation, during the conduct of the study. Dr. Miller reports grants from NIH, Muscular Dystrophy Association, Association française contre les myopathies, FSH Society, Boston University CTSI, during the conduct of the study.
References
- Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider L, Geng LN, Lemmers RJ, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawil R, van der Maarel SM, Tapscott SJ. Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet Muscle. 2014;4:12. doi: 10.1186/2044-5040-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawil R. Facioscapulohumeral muscular dystrophy. Neurotherapeutics. 2008;5:601–606. doi: 10.1016/j.nurt.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabianca DS, Gabellini D. The cell biology of disease: FSHD: copy number variations on the theme of muscular dystrophy. J Cell Biol. 2010;191:1049–1060. doi: 10.1083/jcb.201007028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maarel SM, Tawil R, Tapscott SJ. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med. 2010;17:252–258. doi: 10.1016/j.molmed.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scionti I, Greco F, Ricci G, et al. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am J Hum Genet. 2012;90:628–635. doi: 10.1016/j.ajhg.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maarel SM, Miller DG, Tawil R, et al. Facioscapulohumeral muscular dystrophy: consequences of chromatin relaxation. Curr Opin Neurol. 2010;25:614–620. doi: 10.1097/WCO.0b013e328357f22d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartweck LM, Anderson LJ, Lemmers RJ, et al. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology. 2013;80:392–399. doi: 10.1212/WNL.0b013e31827f075c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard MC, Roche S, Dion C, et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology. 2014;83:733–742. doi: 10.1212/WNL.0000000000000708. [DOI] [PubMed] [Google Scholar]
- Kowaljow V, Marcowycz A, Ansseau E, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord. 2007;17:611–623. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Bosnakovski D, Xu Z, Gang EJ, et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008;27:2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuebbles RD, Long SW, Hanel ML, Jones PL. Testing the effects of FSHD candidate gene expression in vertebrate muscle development. Int J Clin Exp Pathol. 2010;3:386–400. [PMC free article] [PubMed] [Google Scholar]
- Mitsuhashi H, Mitsuhashi S, Lynn-Jones T, et al. Expression of DUX4 in zebrafish development recapitulates facioscapulohumeral muscular dystrophy. Hum Mol Genet. 2013;22:568–577. doi: 10.1093/hmg/dds467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng LN, Yao Z, Snider L, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell. 2010;22:38–51. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TI, Chen JCJ, Rahimov F, et al. DUX4-fl expression in both FSHD subjects and unaffected relatives provides evidence for modifiers and a quantitative model of pathogenesis. Hum Mol Genet. 2010;21:4419–4430. doi: 10.1093/hmg/dds284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block GJ, Narayanan D, Amell AM, et al. Wnt/β-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum Mol Genet. 2013;22:4661–4672. doi: 10.1093/hmg/ddt314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17:659–667. doi: 10.1016/j.molmed.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SA. Inclusion body myositis. Curr Opin Rheumatol. 2011;23:574–578. doi: 10.1097/BOR.0b013e32834b53cc. [DOI] [PubMed] [Google Scholar]
- Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34:812–826. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- Homma S, Chen JCJ, Rahimov F, et al. A unique library of myogenic cells from facioscapulohumeral muscular dystrophy subjects and unaffected relatives: family, disease, & cell function. Eur J Hum Genet. 2012;20:404–410. doi: 10.1038/ejhg.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeda C, Debarnot C, Homma S, et al. Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy associated DUX4 gene. Mol Cell Biol. 2014;34:1942–1955. doi: 10.1128/MCB.00149-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng LN, Tyler AE, Tapscott SJ. Immunodetection of human double homeobox 4. Hybridoma (Larchmt) 2011;30:125–130. doi: 10.1089/hyb.2010.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T, Kametani F, Arai T, et al. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet. 2009;18:524–530. doi: 10.1093/hmg/ddp275. [DOI] [PubMed] [Google Scholar]
- Hishiya A, Iemura S, Natsume T, et al. A novel ubiquitin-binding protein ZNF216 functioning in muscle atrophy. EMBO J. 2006;25:554–564. doi: 10.1038/sj.emboj.7600945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce FM, Bucher NL. Baculovirus-mediated gene transfer into mammalian cells. Proc Natl Acad Sci USA. 1996;93:2348–2352. doi: 10.1073/pnas.93.6.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckow VA, Lee SC, Barry GF, Olins PO. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J Virol. 1993;67:4566–4579. doi: 10.1128/jvi.67.8.4566-4579.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone J, Ciammola A, Tiloca C, et al. Apoptosis induced by proteasome inhibition in human myoblast cultures. Eur J Histochem. 2006;50:109–118. [PubMed] [Google Scholar]
- Askanas V, Serdaroglu P, Engel WK, Alvarez RB. Immunolocalization of ubiquitin in muscle biopsies of patients with inclusion body myositis and oculopharyngeal muscular dystrophy. Neurosci Lett. 1991;130:73–76. doi: 10.1016/0304-3940(91)90230-q. [DOI] [PubMed] [Google Scholar]
- Küsters B, van Hoeve BJ, Schelhaas HJ, et al. TDP-43 accumulation is common in myopathies with rimmed vacuoles. Acta Neuropathol. 2009;117:209–211. doi: 10.1007/s00401-008-0471-2. [DOI] [PubMed] [Google Scholar]
- Vanderplanck C, Ansseau E, Charron S, et al. The FSHD atrophic myotube phenotype is caused by DUX4 expression. PLoS One. 2011;6:e26820. doi: 10.1371/journal.pone.0026820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastle M, Grune T. Protein oxidative modification in the aging organism and the role of the ubiquitin proteasomal system. Curr Pharm Des. 2011;17:4007–4022. doi: 10.2174/138161211798764898. [DOI] [PubMed] [Google Scholar]
- Tassin A, Laoudj-Chenivesse D, Vanderplanck C, et al. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? J Cell Mol Med. 2013;17:76–89. doi: 10.1111/j.1582-4934.2012.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Pestronk A. Sporadic inclusion body myositis: possible pathogenesis inferred from biomarkers. Curr Opin Neurol. 2010;23:482–488. doi: 10.1097/WCO.0b013e32833d3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubourg O, Wanschitz J, Maisonobe T, et al. Diagnostic value of markers of muscle degeneration in sporadic inclusion body myositis. Acta Myol. 2011;30:103–108. [PMC free article] [PubMed] [Google Scholar]
- Arnold ES, Ling SC, Huelga SC, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci USA. 2013;110:E736–E745. doi: 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udan-Johns M, Bengoechea R, Bell S, et al. Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum Mol Genet. 2014;23:157–170. doi: 10.1093/hmg/ddt408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spletter ML, Schnorrer F. Transcriptional regulation and alternative splicing cooperate in muscle fiber-type specification in flies and mammals. Exp Cell Res. 2014;321:90–98. doi: 10.1016/j.yexcr.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistoni M, Ghigna C, Gabellini D. Alternative splicing and muscular dystrophy. RNA Biol. 2010;7:441–452. doi: 10.4161/rna.7.4.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- Tydlacka S, Wang CE, Wang X, et al. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci. 2008;28:13285–13295. doi: 10.1523/JNEUROSCI.4393-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eersel J, Ke YD, Gladbach A, et al. Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS One. 2011;6:e22850. doi: 10.1371/journal.pone.0022850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Hou SC, Way TD, et al. Heat-shock protein dysregulation is associated with functional and pathological TDP-43 aggregation. Nat Commun. 2013;4:2757. doi: 10.1038/ncomms3757. [DOI] [PubMed] [Google Scholar]
- Huang CC, Bose JK, Majumder P, et al. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J Cell Sci. 2014;127:3024–3038. doi: 10.1242/jcs.136150. [DOI] [PubMed] [Google Scholar]
- Wallace LM, Garwick SE, Mei W, et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol. 2011;69:540–552. doi: 10.1002/ana.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Snider L, Balog J, et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum Mol Genet. 2014;23:5342–5352. doi: 10.1093/hmg/ddu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabianca DS, Casa V, Bodega B, et al. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell. 2012;149:819–831. doi: 10.1016/j.cell.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev P, Stankevicins L, Ansseau E, et al. Defective regulation of microRNA target genes in myoblasts from facioscapulohumeral dystrophy patients. J Biol Chem. 2013;288:34989–35002. doi: 10.1074/jbc.M113.504522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harafuji N, Schneiderat P, Walter MC, Chen YW. miR-411 is up-regulated in FSHD myoblasts and suppresses myogenic factors. Orphanet J Rare Dis. 2013;8:55. doi: 10.1186/1750-1172-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrin S, Latil M, Soave S, et al. Normal muscle regeneration requires tight control of muscle cell fusion by tetraspanins CD9 and CD81. Nat Commun. 2013;4:1674. doi: 10.1038/ncomms2675. [DOI] [PubMed] [Google Scholar]
- Ferreboeuf M, Mariot V, Furling D, et al. Nuclear protein spreading: implication for pathophysiology of neuromuscular diseases. Hum Mol Genet. 2014;23:4125–4133. doi: 10.1093/hmg/ddu129. [DOI] [PubMed] [Google Scholar]
- Dixit M, Ansseau E, Tassin A, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci USA. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SN, Cabotage J, Shi R, et al. Conditional over-expression of PITX1 causes skeletal muscle dystrophy in mice. Biol Open. 2012;1:629–639. doi: 10.1242/bio.20121305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turki A, Hayot M, Carnac G, et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic Biol Med. 2012;53:1068–1079. doi: 10.1016/j.freeradbiomed.2012.06.041. [DOI] [PubMed] [Google Scholar]
- Bosnakovski D, Choi SH, Strasser JM, et al. High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skelet Muscle. 2014;4:4. doi: 10.1186/2044-5040-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höhn TJ, Grune T. The proteasome and the degradation of oxidized proteins: part III-Redox regulation of the proteasomal system. Redox Biol. 2014;2:388–394. doi: 10.1016/j.redox.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segref A, Kevei E, Pokrzywa W, et al. Pathogenesis of human mitochondrial diseases is modulated by reduced activity of the ubiquitin/proteasome system. Cell Metab. 2014;19:642–652. doi: 10.1016/j.cmet.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta. 2014;1843:13–25. doi: 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Olivé M, Janué A, Moreno D, et al. TAR DNA-binding protein 43 accumulation in protein aggregate myopathies. J Neuropathol Exp Neurol. 2009;68:262–273. doi: 10.1097/NEN.0b013e3181996d8f. [DOI] [PubMed] [Google Scholar]
- von Mikecz A. The nuclear ubiquitin-proteasome system. J Cell Sci. 2006;119:1977–1984. doi: 10.1242/jcs.03008. [DOI] [PubMed] [Google Scholar]
- Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol. 2012;71:680–693. doi: 10.1097/NEN.0b013e31826183c8. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Raphael AR, LaDow ES, et al. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L. Rodent models of TDP-43 proteinopathy: investigating the mechanisms of TDP-43-mediated neurodegeneration. J Mol Neurosci. 2011;45:486–499. doi: 10.1007/s12031-011-9610-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Wang H, Qiao T, et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2014;111:E1121–E1129. doi: 10.1073/pnas.1322641111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace LM, Liu J, Domire JS, et al. RNA interference inhibits DUX4-induced muscle toxicity in vivo: implications for a targeted FSHD therapy. Mol Ther. 2012;20:1417–1423. doi: 10.1038/mt.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]