

Abstract
Inherited spinocerebellar ataxias (SCAs) are known to be genetically and clinically heterogeneous. Whether severity and survival are variable, however, is not known. We, therefore, studied survival and severity in 446 cases and 509 relatives with known mutations. Survival was 68 years [95% CI: 65–70] in 223 patients with polyglutamine expansions versus 80 years [73–84] in 23 with other mutations (P < 0.0001). Disability was also more severe in the former: at age 60, 30% were wheelchair users versus 3% with other SCAs (P < 0.001). This has implications for genetic counseling and the design of therapeutic trials.
Introduction
Dominantly inherited spinocerebellar ataxias (SCA), a group of neurodegenerative diseases affecting the cerebellum and/or its afferent/efferent tracts, are characterized by genetic and clinical heterogeneity. The mutations remain unknown in 20–60% of cases, depending on their geographical origin.1 Seven SCAs (1–3, 6, 7, 17, DRPLA) share a common mutational mechanism, abnormal Cytosine, Adenine, Guanine (CAG) repeat expansions in genes encoding proteins containing polyglutamine tracts (polyQ-SCAs), but also features such as anticipation and multisystem involvement.2 Natural histories show that polyQ-SCAs have distinguishing features3,4; SCA6, in particular, has later onset and slower progression. In other SCAs, point mutations (SCA5, 11, 13, 14, 15/16, 19/22, 21, 23, 28, 35, and 38), rearrangements (15/16, 20) or non-coding repeat expansions (8, 10, 31, and 36) are found.1,5 The pathological mechanisms are mostly unknown and each subtype is very rare. In this study, we analyzed survival and severity in 446 families with polyQ-SCAs and other mutations.
Patients and Methods
Between 1991 and 2013, we identified 446 index cases with known SCA mutations and 509 affected relatives at the Pitié-Salpêtrière University Hospital in Paris and through the SPATAX network (spatax.wordpress.com). Molecular analyses were performed in the Neurogenetics Unit of the Pitié-Salpêtrière University Hospital, the Brain and Spine Institute (ICM) and collaborating centers. All participants signed informed consent forms (RBM 01-29, RBM 03-48).
We determined age at onset, age when help was needed walking or a wheelchair in 446 index cases and 509 relatives. Age at death, however, was determined in 332 parents of the index cases to limit the bias introduced by index cases still alive, and to include later ages at death. The transmitting parents and their age at death were reported by the index cases; 16% (n = 63) were examined. In 45/114, information concerning the parents was obtained through registers in their place of birth, but remained unknown for 53 transmitting parents. Survival was determined with the Kaplan–Meier estimator.
Causes of death were presumed to be related to the disease, but we cannot exclude that a limited number of other pathologies were responsible. Missing ages when aid walking or wheelchair use where needed were estimated by multiple imputations using the Markov Chain Monte Carlo method. Imputed data were determined as a function of the gene involved and, for the polyQ-SCAs, the length of the CAG repeat, and analyzed using Cox models. The null hypothesis was rejected for P < 0.05. Statistical analysis was performed with SAS software (version 9.3; SAS Institute, Cary, NC). Data are expressed as the mean ± SD [minimum–maximum].
Results
Index cases were divided into two groups: polyQ-SCAs and other known SCAs (Table1); 46% were women in both groups. Among the polyQ group, SCA1 (n = 166), SCA2 (n = 173), SCA3 (n = 301), SCA6 (n = 33) and SCA7 (n = 132) were distinguished. Index cases with polyQ-SCAs (38.4 ± 13.2 [3–78] years, n = 381/392 with information) tended to be older than the others, but not significantly (37.0 ± 16.6 [1–63] years, n = 51/54, P = 0.51); if affected relatives were included in the calculation, however, polyQ-SCA patients were significantly older (36.7 ± 13.6 [0–78] (n = 790/814) versus 33.3 ± 16.3 [1–63] (n = 129/141), P = 0.024) (Table1).
Table 1.
Characteristics of patients (446 index patients and 509 relatives) with autosomal dominant ataxias cases
| Number of patients (index) | Mean age at onset in years | Mean age at examination in years | Mean age when aid walking needed in years | Mean age when wheelchair needed in years | |
|---|---|---|---|---|---|
| POlyQ SCA | |||||
| SCA1 | 166 (72) | 37.3 [14–65] (n = 164) | 44.5 [17–80] (n = 166) | 43.8 [18–75] (n = 57, missing for n = 18) | 48.49 [22–75] (n = 29, missing for n = 7) |
| SCA2 | 173 (82) | 33.7 [3–67] (n = 165) | 43.2 [3–82] (n = 173) | 42.8 [11–70] (n = 41, missing for n = 28) | 45.3 [14–74] (n = 27, missing for n = 11) |
| SCA3 | 301 (161) | 40.2 [10–78] (n = 295) | 49.2 [21–79] (n = 299) | 48.2 [21–75] (n = 79, missing for n = 50) | 51.3 [25–73] (n = 58, missing for n = 23) |
| SCA6 | 33 (18) | 48.2 [24–67] (n = 29) | 53.3 [18–80] (n = 33) | 52.6 [30–70] (n = 11, missing for n = 4) | 59.5 [44–74] (n = 4, missing for n = 2) |
| SCA7 | 132 (51) | 29.4 [0–70] (n = 128) | 38.3 [1.5–85] (n = 128) | 35.1 [1–76] (n = 28, missing for n = 32) | 36.6 [2–70] (n = 28, missing for n = 9) |
| SCA17 | 6 (6) | 38.0 [23–55] (n = 6) | 47.0 [30–63] (n = 6) | 36 (n = 1, missing for n = 3) | 47.7 [39–59] (n = 3) |
| DRPLA | 3 (2) | 27.0 [8–50] (n = 3) | 41.3 [16–71] (n = 3) | 70 (n = 1, missing for n = 2) | 12 (n = 1) |
| Subtotal | 814 (392) | 36.7 [0–78] (n = 791) | 45.3 [1.5–85] (n = 808) | 44.6 [1–76] (n = 218) | 46.8 [2–75] (n = 150) |
| Other SCA | |||||
| SCA5 | 9 (4) | 30.4 [14–60] (n = 9) | 45.0 [24–77] (n = 9) | (missing for n = 2) | n = 0 |
| SCA11 | 2 (1) | 35; 48 (n = 2) | 39; 64 (n = 2) | 61 (n = 1) | n = 0 |
| SCA12 | 1 (1) | 41 (n = 1) | 50 (n = 1) | n = 0 | n = 0 |
| SCA13 | 13 (4) | 8.2 [1–61] (n = 13) | 34.2 [2–67] (n = 13) | 48 (n = 1, missing for 1) | n = 0 |
| SCA14 | 38 (9) | 33.5 [6–60] (n = 38) | 52.9 [15–95] (n = 38) | 54 (n = 1, missing for 2) | n = 0 |
| SCA15 | 14 (6) | 32.0 [18–48] (n = 13) | 44.5 [20–68] (n = 14) | (missing for n = 1) | n = 0 |
| SCA22 | 8 (1) | 36.8 [24–51] (n = 8) | 46.1 [28–62] (n = 8) | 57 (n = 1, missing for 2) | 67 (n = 1) |
| SCA23 | 3 (3) | 49.7 [45–54] (n = 3) | 58 [57–59] (n = 3) | (missing for n = 1) | 57 (n = 1) |
| SCA28 | 24 (8) | 29.6 [6–60] (n = 20) | 44.3 [10–77] (n = 24) | 33; 58 (n = 2, missing for 1)) | 76 (n = 1) |
| SCA31 | 4 (4) | 42.3 [28–55] (n = 4) | 50.0 [34–61] (n = 4) | n = 0 | n = 0 |
| SCA32 | 5 (1) | <1 (n = 5) | 46.8 [34–71] (n = 5) | 33.5 [28–40] (n = 4) | 41 (n = 1, missing for 1) |
| SCA36 | 20 (12) | 49.7 [40–63] (n = 18) | 61.6 [49–83] (n = 20) | 47;53 (n = 2, missing for 3) | 63 (n = 1, missing for 1) |
| Subtotal | 141 (54) | 33.2 [1–63] (n = 129) | 49.0 [2–95] (n = 141) | 45.4 [28–61] (n = 12) | 60.8 [41–75] (n = 65) |
| Total | 955 (446) | ||||
SCA, spinocerebellar ataxias.
Survival in the transmitting parents of the index cases
Death occurred in 223/291 parents with polyQ-SCAs (76%) and 23/41 (57%) parents with other SCAs. Survival was significantly shorter in the parents of polyQ-SCA patients: median age at death was 68 years [95% CI: 65–70] versus 80 years [73–84] in parents with other SCAs (P < 0.0001; Fig.1A). Among the polyQ-SCAs, survival was significantly shorter in SCA1 than SCA 2, 3, 6, and 7 (P < 0.0001) (Fig.1B). Survival was comparable in SCA2, 3, 6, and 7.
Figure 1.
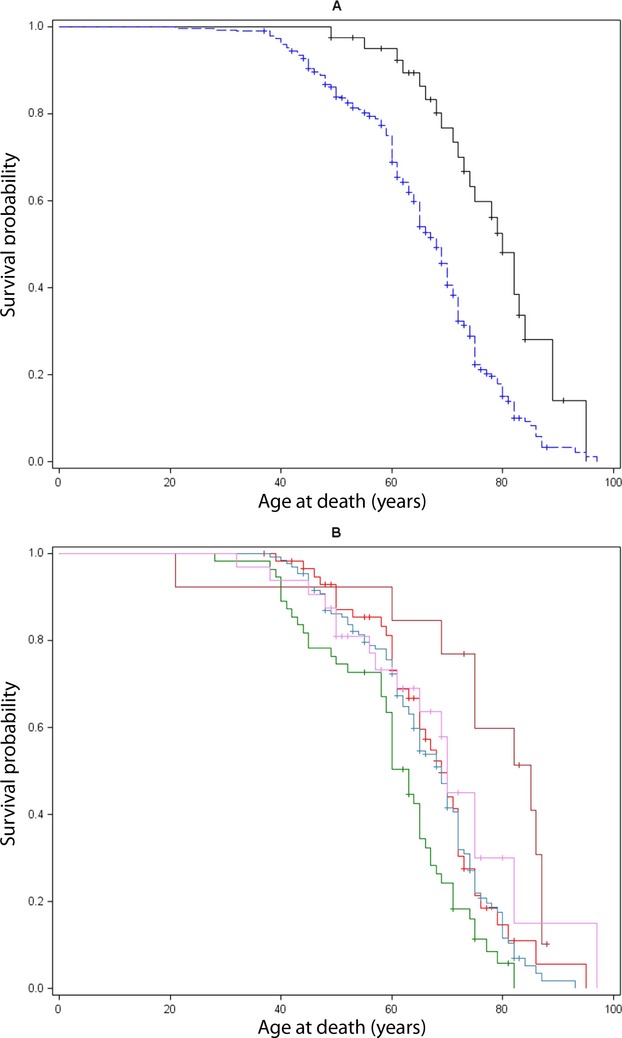
(A) Lifetest in 332 patients with familial autosomal dominant ataxias, according to genotype (polyQ-SCAs in blue vs. SCAs with other mutations in black). Median age at death was 69 years [95% CI: 67–71] but polyQ-SCA patients died significantly earlier than the others (68 vs. 80 years, P < 0.0001). The probability of survival for polyQ-SCA patients was over 95% at age 40, but only 20% at 80, a tendency towards an exponential decrease in life span; most deaths occurred between age 50 and 70 years. (B) Lifetest procedure according to polyQ-SCAs genotype: SCA 1 (green), 2 (red), 3 (steel), 6 (brown), and 7 (violet). Median age at death in SCA 1 was 63 years, significantly earlier than SCA 2, 3, 6 and 7, P < 0.0001. SCA, spinocerebellar ataxias.
Survival was longer when the polyQ-SCA was transmitted by the mother (median 70 vs. 65 years, P = 0.0251). The effect of the gender of the transmitting parent was not significant in the other SCAs (P = 0.47). The patients' gender had no effect (P = 0.23 for other SCAs, 0.06 for polyQ-SCAs). Unfortunately and due to the retrospective nature of the analysis, the effect of CAG repeat length on survival could not be evaluated, since the genotypes of the parents were available for only 8%. Age at onset was available for 41% of the parents. Survival after onset of the disease did not differ significantly among polyQ-SCAs.
Disease severity
At their first visit, after 8.8 ± 7.1 and 16.2 ± 13.3 years of disease duration (P < 0.001), respectively, 48% (354/731, 83 missing) of the polyQ-SCA patients needed aid walking versus 19% (26/134, 3 missing) of the other SCA patients P < 0.001), and 28% (202/728) of the polyQ-SCA patients used a wheelchair versus 5% (7/134) of other SCA patients (P < 0.001). Gender did not influence the age when wheelchairs were used (P = 0.09). PolyQ-SCA patients were more prone to need help walking (HR: 3.3 [95% CI: 2.1–5.3], Fig.2A) or a wheelchair (HR: 6.5 [2.9–14.5], Fig.2B) than other SCA patients. PolyQ-SCA patients with SCA7 were more prone to need help walking than SCA3 (HR: 1.9 [1.3–2.7]) and SCA6 (2.4 [1.3–4.6]) patients, and a higher risk of wheelchair use than SCA1 (HR: 1.7 [1.1–2.7]), SCA3 (HR: 1.7 [1.1–2.5]) and SCA6 patients (3.6 [1.5–8.7]). SCA2 patients were more prone to need help walking than SCA3 patients (1.4 [1.0–1.8]) and had higher risk of wheelchair use than SCA6 patients (HR: 2.5 [1.0–5.9]) (Fig.2C). The age when help walking or a wheelchair were needed was influenced by CAG size in SCA1, 2, 3, 7; the larger the repeat, the earlier the age (P < 0.01).
Figure 2.
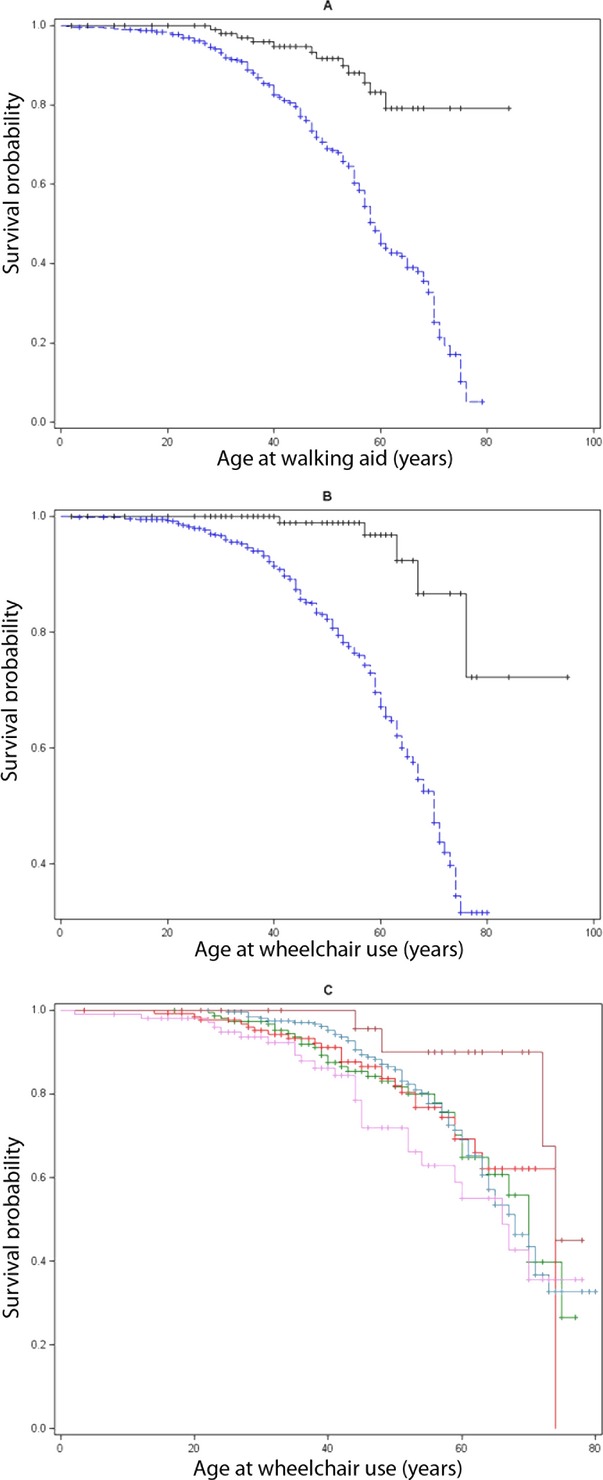
(A) Lifetest procedure for the effect of genotype (polyQ-SCAs in blue vs. other SCAs in black) on age when aid was needed for walking in 862 patients, P < 0.0001 (377 patient walked with aid, 485 were censored at last visit). (B) Lifetest procedure for the effect of genotype (polyQ-SCAs in blue vs. other SCAs in black) on age when a wheelchair was needed in 835 patients, P < 0.0001 (207 patients used wheelchair, 628 were censored at last visit). (C) Lifetest procedure for the effect of polyQ-SCAs genotype: SCA 1 (green), 2 (red), 3 (steel), 6 (brown), and 7 (violet), on age when a wheelchair was needed. SCA, spinocerebellar ataxias.
Discussion
PolyQ-SCAs stand out among the dominant cerebellar ataxias because of their high frequency and highly variable clinical features.1 Based on daily clinical observations, we determined that severity was greater and survival shorter in patients with polyglutamine expansions than in cerebellar ataxias due to other mutations. Survival could not be determined in the other SCAs because of their heterogeneity and rarity. We are aware that distinguishing polyQ-SCAs from the others might appear artificial. Our aim, however, was to determine specifically the characteristics of the polyQ-SCA subgroup.
Survival rates according to the SCA genotype revealed that polyQ-SCA patients have shorter survival, both as a group and individually. We could not correlate survival with CAG repeat number, since it was determined by the age of death of the transmitting parent for whom CAG repeat numbers were not always available. However, since age at onset and severity are dependent on CAG repeat length, survival probably is as well. Because polyQ-SCAs are more frequent than other SCAs in most countries, published survival or mortality rates are often restricted to the former; for example, SCA3.6,7 In our cohort, mean disease duration until death was 19.3 ± 9.7 years in polyQ-SCAs, which is comparable to previous reports.6,7
The mean age at death in our cohort (66 years [SD 65–67]; median: 68 years [CI 95% 65–70]) is earlier than the mean age at death in the Swedish Registry study (75 [SD 64–82] years)8 that included unspecified inherited ataxias. This could be due to a smaller proportion of polyQ-SCA patients in Sweden, as in other Scandinavian countries9; the median age at death in other SCAs in our study was 80 [73–84] years (mean: 79 years).
There was a possible bias in favor of later deaths in that only persons with descendents were analyzed, excluding juvenile cases. However, the median age at death in the generation of polyQ-SCA index patients still being followed was 71 years, i.e., close to the mean age at death of the parents (59 deceased, 119 still alive and 268 censored at last visit, data not shown).
We report, for the first time, that the shortest survival in polyQ-SCAs is in SCA1. Natural histories and clinical characteristics have been well described for SCA1, 2, 3, 6 and 7,3,4,10 as well as SCA1711 and DRPLA.12 CAG repeat size modulates clinical expression, disease duration and phenotype.4 It is difficult to distinguish between the different polyQ-SCAs on clinical grounds, but extreme phenotypes may characterize certain SCAs: motor neuron disease in SCA1,12 chorea in SCA213 or dystonia and spastic ataxia in SCA3,14 notably in cases with early onset.
In addition to shorter survival, polyQ-SCA patients also had a more severe disease than those with other SCA mutations. Severity in polyQ-SCAs was clearly related to repeat size. The influence of repeat size on age at onset in polyQ-SCAs is well known.15,16 The natural history is also significantly influenced by repeat size: the longer the repeat the faster the disease evolves.4 In SCA1, SCA2 and SCA3, the evolution of SARA scores was mainly determined by the length of the repeat on the expanded allele, age at onset and disease duration.4,10 Most polyQ-SCAs are multisystem disorders characterized by prominent brainstem atrophy, even before disease onset.17,18 This also differentiates polyQ-SCAs from other SCAs in which isolated cerebellar atrophy is frequent.
In conclusion, we determined survival and severity in a large group of polyQ-SCA patients and individually in those with SCA1, 2, 3, 6, and 7. Of interest, survival in non-polyQ-SCAs was similar to that of the French population in general, and disease progression slower than in polyQ-SCAs despite an earlier age at onset. This observation has to be confirmed in larger populations of other SCAs. Genetic heterogeneity complicates genetic counseling, since information concerning the more frequent subtypes cannot be extrapolated to rarer forms of the disease. This is important for the understanding of these diseases and could have implications for patient care and the design of treatment trials.
Acknowledgments
Many thanks to the patients and families for their help. We are very grateful to Merle Ruberg for her critical reading of the manuscript. We acknowledge the clinicians of the SPATAX network involved in this study: M. Tchikviladzé, M. Anheim, P. Ribai, I. Leber A. Benomar, S. Medjbeur, E. Ollagnon, V. Meininger, A. Vighetto, J. Pouget, C. Tranchant, H. Aliouche, D. Brassat, A. Destee, A. Toutain, C. Goizet, A. Lannuzel, D. Hannequin, A. Lagueny, P. Damier, C. Delpirou, D. Broglin, G. Ponsot, J. C. Pages, A. Autret, C. Vial, P. Labauge, P. Clavelou, P. Amati-Bonneau, E. Kaphan, R. Bellance, J. M. Leger, C. Desnuelle, P. Coutinho, J. Melki, Y. Boukhriche, V. Paquis, F. Tison, A. Barois, R. Ghnassia, J. Gayon, J. Julien, M. Abada-Bendib, M. Clanet, J. Guimaraes, Laurent, P. Desfontaines, E. Bieth, M. L. Klimek, S. Schaeffer, P. Calvas, H. Chneiweiss, Bataillard, B. Fontaine, J. C. Netter, P. Bejani, G. Fenelon, D. Grid, J. M. Vallat, F. Viallet, J. P. Azulay, N. Pageot, I. Ferrer, P. Edery, Foubert, J. L. Gastaut, W. Camu, P. Sarda, S. Brique, D. Devos, I. Vuillaume, E. Roze, G. Castelnovo, D. Malapert, T. Fioretos, J. Honnorat, J. Koht, J. Zlotogora, P. Pollak, A. Megarbane, A. David, F. Durif, Bertrandeau, D. Cherif, Ménage, D. Rochefort, L. Faivre, E. Salort, Soulages, P. Vandebergh, P. Jonveaux.
Conflict of Interest
Dr. Durr reports non-financial support from INSERM, during the conduct of the study; In addition, has a patent N° 14/308,966 issued.
Dr. Stevanin reports grants from Agence National pour la Recherche, from Verum foundation, during the conduct of the study.
References
- Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. doi: 10.1016/S1474-4422(10)70183-6. [DOI] [PubMed] [Google Scholar]
- Klockgether T. Handbook of ataxia disorders. New York: CRC Press; 2000. [Google Scholar]
- Jacobi H, Bauer P, Giunti P, et al. The natural history of spinocerebellar ataxia type 1, 2, 3, and 6: a 2-year follow-up study. Neurology. 2011;77:1035–1041. doi: 10.1212/WNL.0b013e31822e7ca0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezenas du Montcel S, Charles P, Goizet C, et al. Factors influencing disease progression in autosomal dominant cerebellar ataxia and spastic paraplegia. Arch Neurol. 2012;69:500–508. doi: 10.1001/archneurol.2011.2713. [DOI] [PubMed] [Google Scholar]
- Di Gregorio E, Borroni B, Giorgio E, et al. ELOVL5 mutations cause Spinocerebellar Ataxia 38. Am J Hum Genet. 2014;95:209–217. doi: 10.1016/j.ajhg.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether T, Lüdtke R, Kramer B, et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain J Neurol. 1998;121(Pt 4):589–600. doi: 10.1093/brain/121.4.589. [DOI] [PubMed] [Google Scholar]
- Kieling C, Prestes PR, Saraiva-Pereira ML, Jardim LB. Survival estimates for patients with Machado-Joseph disease (SCA3) Clin Genet. 2007;72:543–545. doi: 10.1111/j.1399-0004.2007.00910.x. [DOI] [PubMed] [Google Scholar]
- Ji J, Sundquist K, Sundquist J. Cancer incidence in patients with polyglutamine diseases: a population-based study in Sweden. Lancet Oncol. 2012;13:642–648. doi: 10.1016/S1470-2045(12)70132-8. [DOI] [PubMed] [Google Scholar]
- Juvonen V, Hietala M, Kairisto V, Savontaus M-L. The occurrence of dominant spinocerebellar ataxias among 251 Finnish ataxia patients and the role of predisposing large normal alleles in a genetically isolated population. Acta Neurol Scand. 2005;111:154–162. doi: 10.1111/j.1600-0404.2005.00349.x. [DOI] [PubMed] [Google Scholar]
- Schmitz-Hübsch T, Coudert M, Bauer P, et al. Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology. 2008;71:982–989. doi: 10.1212/01.wnl.0000325057.33666.72. [DOI] [PubMed] [Google Scholar]
- Reetz K, Lencer R, Hagenah JM, et al. Structural changes associated with progression of motor deficits in spinocerebellar ataxia 17. Cerebellum. 2010;9:210–217. doi: 10.1007/s12311-009-0150-4. [DOI] [PubMed] [Google Scholar]
- Goldfarb LG, Vasconcelos O, Platonov FA, et al. Unstable triplet repeat and phenotypic variability of spinocerebellar ataxia type 1. Ann Neurol. 1996;39:500–506. doi: 10.1002/ana.410390412. [DOI] [PubMed] [Google Scholar]
- Dürr A, Smadja D, Cancel G, et al. Autosomal dominant cerebellar ataxia type I in Martinique (French West Indies). Clinical and neuropathological analysis of 53 patients from three unrelated SCA2 families. Brain J Neurol. 1995;118(Pt 6):1573–1581. doi: 10.1093/brain/118.6.1573. [DOI] [PubMed] [Google Scholar]
- Rosenberg RN. Machado-Joseph disease: an autosomal dominant motor system degeneration. Mov Disord. 1992;7:193–203. doi: 10.1002/mds.870070302. [DOI] [PubMed] [Google Scholar]
- Globas C, du Montcel ST, Baliko L, et al. Early symptoms in spinocerebellar ataxia type 1, 2, 3, and 6. Mov Disord. 2008;23:2232–2238. doi: 10.1002/mds.22288. [DOI] [PubMed] [Google Scholar]
- Van de Warrenburg BPC, Sinke RJ, Verschuuren-Bemelmans CC, et al. Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis. Neurology. 2002;58:702–708. doi: 10.1212/wnl.58.5.702. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Borkert J, Wolf S, et al. Visualization, quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1, 3 and 6. NeuroImage. 2010;49:158–168. doi: 10.1016/j.neuroimage.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Jacobi H, Reetz K, du Montcel ST, et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol. 2013;12:650–658. doi: 10.1016/S1474-4422(13)70104-2. [DOI] [PubMed] [Google Scholar]