

Introduction
Congestive heart failure is the leading cause of morbidity and mortality worldwide, resulting in an extensive economic burden to healthcare systems. Despite recent advances in pharmacological and device therapies, rates of heart disease continue to rise due to an ageing population therefore new treatment options are urgently needed. An understanding of heart disease at a molecular level and the identification of key transporters and proteins affected during pathogenesis has paved the way for new strategies to treat heart disease, namely the calcium handling proteins in the sarcoplasmic reticulum (SR).
As a second messenger, calcium has many signaling roles and is involved in processes ranging from cell death to muscle contraction (Berridge et al., 2000). It is the maintenance of a calcium gradient across endoplasmic or sarcoplasmic reticulum membranes, generated by calcium channels, ATPase pumps, transporters and exchangers working in synergy with calcium binding proteins, which allows these processes to occur. The contractile function of cardiomyocytes is controlled by excitation-contraction (EC) coupling which results in rapid changes in intracellular calcium concentration leading to contraction (systole) and relaxation (diastole) (Figure 1). During systole, an action potential causes the depolarization of the plasma membrane (sarcolemma) which results in the entry of a small amount of extracellular calcium into the cytosol through the voltage-gated L-type calcium channel (LTCC). This calcium binds to receptors on the ryanodine receptor (RyR2), triggering a massive efflux of calcium from the SR into the cytosol; this process is termed calcium-induced calcium release. This approximate tenfold increase in intracellular calcium concentration activates calcium-sensitive contractile proteins (troponin C; TN-C) which then use ATP to produce tension and muscle contraction. For muscle relaxation to occur, calcium is removed from the cytosol – approximately 30% is transported out of the cell (primarily by the sodium-calcium exchanger (NCX) and plasma membrane calcium ATPase (PMCA)) while 70% is pumped back into the SR via the cardiac SR calcium ATPase (SERCA2a) (Bers, 2008).
Figure 1. Excitation-contraction coupling in cardiac myocytes.
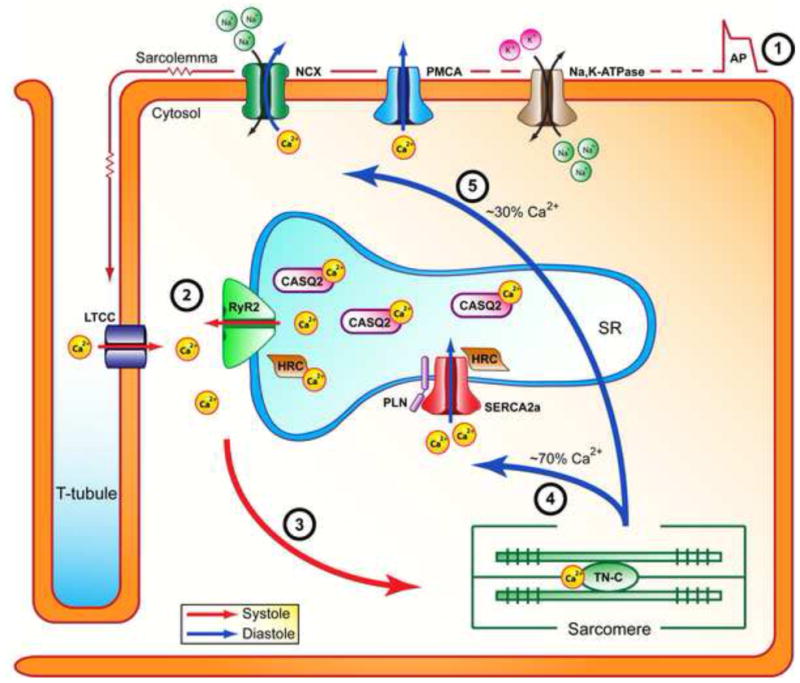
During systole, an action potential depolarizes the sarcolemma and induces opening of the LTCC, allowing a minor amount of extracellular calcium to enter the cytosol (step 1). In turn, this initiates massive calcium release from the SR through the RyR2 channels (step 2). This sudden increase in cytosolic calcium concentration results in binding of calcium to TN-C and initiation of muscle contraction (step 3). The rapid removal of cytosolic calcium during diastole is primarily facilitated by SERCA2a, which returns calcium to the SR (step 4). Some calcium also exits the cell through NCX and PMCA (step 5). This decrease in intracellular calcium leads to dissociation of calcium from TN-C and muscle relaxation. Abbreviations: Na,K-ATPase, sodium-potassium ATPase; PMCA, plasma membrane calcium ATPase; NCX, sodium calcium exchanger; LTCC, voltage dependent L-type calcium channel; RyR2, ryanodine receptor isoform 2; CASQ2, calsequestrin isoform 2; HRC, histidine rich calcium binding protein; SERCA2a, sarco(endo)plasmic reticulum calcium ATPase; PLN, phospholamban; TN-C, troponin-C.
EC coupling is modulated by many signaling pathways, including the β-adrenergic pathway. Activation of the β-adrenergic pathway by β-agonists, such as adrenaline, initiates the production of cyclic AMP (cAMP) by adenylate cyclase which activates protein kinase A (PKA) (Antos et al., 2001). This results in the downstream phosphorylation of multiple targets in the cardiomyocyte that collectively produce an increase in the frequency and strength of contraction (Feldman and Gros, 2007). For example, PKA phosphorylates the SERCA2a modulator, phospholamban (PLN), resulting in relief of inhibition and an increase in the quantity and rate of cytosolic calcium removal back into the SR (Haghighi et al., 2004); phosphorylation of the LTCC increases calcium current and force of contraction (Kamp and Hell, 2000); and troponin I has reduced sensitivity to calcium when phosphorylated, leading to increased calcium removal from the cytosol (Li et al., 2000). Therefore, activation of the β-adrenergic pathway results in both an increase in rate of contraction (positive inotropy) and relaxation (positive lusitropy) (Lohse et al., 2003).
Balanced cardiac energetics are crucial to proper contractile function, as energy producing and utilizing pathways are tightly regulated in the heart. ATP is primarily produced by oxidative phosphorylation in the mitochondria (>95%), with small contributions made by substrate level phosphorylation and the tricarboxylic acid (TCA) cycle (<5%) (Ingwall, 2009). The major ATP-users in the heart are the actomyosin ATPase in the myofibril, SERCA2a in the SR, and PMCA and Na,K-ATPase in the sarcolemma (Figure 2). The concentration of ATP in the heart is kept relatively constant (10mM) despite the relatively high energy demand necessary for cardiac performance (Ingwall, 2009). The energetic state of the heart is also dependent on levels of phosphocreatine (PCr), which is the primary energy reserve source in the heart and is present at levels twice that of ATP (Bittl and Ingwall, 1985).
Figure 2. Structural interactions between the SR and mitochondria and energy metabolism in cardiac myocytes.
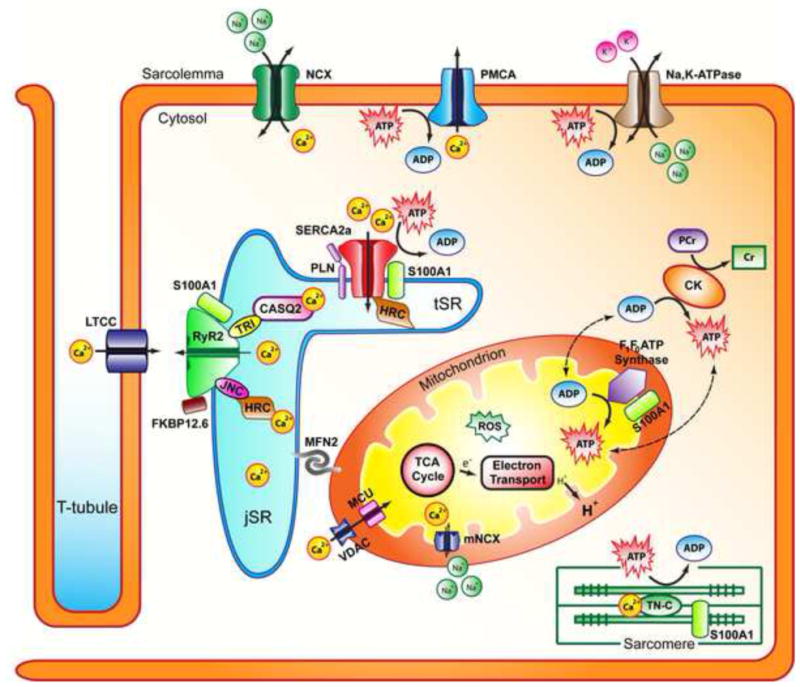
The interfibrillar mitochondria and jSR are in close proximity, tethered by multiple complexes including MFN2. RyR2 and SERCA2a interactomes in the SR are responsible for calcium release and reuptake from the cytosol during muscle contraction and relaxation. These interactomes affect calcium concentration in the cytosol that is available for uptake by VDAC and MCU in the mitochondria. Changes in mitochondrial calcium concentration influence multiple processes related to energy metabolism (ATP synthesis, substrate usage). Mitochondria also produce ROS as a byproduct of energy metabolism, which affects ATP production. The energy state of the cell is defined by ATP and PCr production and consumption. Multiple proteins involved in cardiac function use ATP and their function is affected by the energetic state of the cell. Abbreviations: jSR, junctional sarcoplasmic reticulum; tSR transverse sarcoplasmic reticulum; Na,K-ATPase, sodium-potassium ATPase; PMCA, plasma membrane calcium ATPase; NCX, sodium calcium exchanger; RyR2, ryanodine receptor isoform 2; FKBP12.6, RyR2 accessory binding protein; TRI, triadin; JNC, junctin; CASQ2, calsequestrin isoform 2; HRC, Histidine rich calcium binding protein; SERCA2a, sarco(endo)plasmic reticulum calcium ATPase; PLN, phospholamban; S100A1, S100 calcium binding protein A1; TN-C, troponin-C; MFN2, mitofusin isoform 2; MCU, mitochondrial calcium uniporter; VDAC, voltage dependent anion channel; mNCX, mitochondrial sodium-calcium exchanger; CK, Creatine Kinase; PCr, phosphocreatine; Cr, creatine; ROS, reactive oxygen species.
In patients with cardiac disease, defects in both systolic and diastolic function have been reported. During heart disease, gross physiological changes in the heart, such as increased chamber dimensions and thinning of ventricle walls, are accompanied by myocyte morphological changes, including an increase in length/size, sarcomeric disorganization, and myofibrillar disarray (Harvey and Leinwand, 2011; Kehat and Molkentin, 2010). These abnormalities often stem from changes in calcium homeostasis caused by altered expression or function of calcium transporting or binding proteins. Whether the cause or result of this, the failing heart also has multiple defects in both energy supply and demand which altogether result in an organ that is both energy-starved and ill-functioning. In this Review, we will discuss the role of calcium homeostasis, particularly in terms of SR calcium handling, and how it relates to energy metabolism in heart failure. We will also overview the therapeutic advantages of targeting calcium handling proteins in terms of benefits for both EC coupling and cardiac energetics when pertinent.
Impaired calcium cycling in heart failure
Defects in EC coupling, due to abnormal expression and/or function of calcium handling and transport proteins, are a hallmark of cardiac dysfunction. These defects manifest in changes in the calcium transient: reduced amplitude, increased duration, and prolonged decay – the consequence of which is decreased contractility and reduced cardiac output. In the next subsections, the role of reduced SR calcium load during heart failure will be discussed in terms of excessive calcium entry into the cytosol, decreased SR calcium uptake, decreased SR calcium content, and calcium leak from the SR.
Cytosolic calcium overload
In heart failure, an increase in cytosolic calcium can be caused by excessive calcium entry into the cytosol or reduced calcium efflux from the cytosol. Excessive cytosolic calcium entry is caused by defects in LTCC, NCX and store-operated calcium entry (SOCE). In human cardiomyocytes, cytosolic calcium influx occurs almost entirely through the LTCC and, during heart failure, there is increased phosphorylation of the LTCC (Schroder et al., 1998). This has been shown to result in a compensatory leak of SR calcium through RyR2 (Goonasekera et al., 2012). Modification of channel activity through pharmacological inhibition reduced cardiac remodeling after pressure overload and prevented cardiomyopathy in murine models (Liao et al., 2005; Semsarian et al., 2002) but was detrimental to survival of patients with heart failure (Mahe et al., 2003). SOCE is a mechanism by which calcium-release activated calcium channels (Orai-1) in the sarcolemma sense depletion of intracellular calcium and open to allow an influx of extracellular calcium and this process may be enhanced during pathological remodeling of heart failure (Luo et al., 2012). STIM1 is a SR calcium sensor and has a role in the detection of calcium depletion and the activation of Orai-1 channels. Overexpression of STIM1 in adult rat cardiomyocytes increased SOCE current and activated pathological hypertrophic responses while depletion of STIM1 prevented cardiac hypertrophy and NFAT activation in rats (Hulot et al., 2011). Reduced calcium efflux across the sarcolemma from the cytosol is primarily caused by defective activity of NCX. Sodium overload in the cytosol inhibits calcium efflux from the cell by the NCX as it can cause the exchanger to work in reverse mode. However, more typically during heart failure, NCX is less effective at extruding intracellular calcium due to a diminished transmembrane sodium gradient (Sipido et al., 2002). Some studies have also found that NCX is upregulated during heart failure (Hasenfuss et al., 1999) which could be an initial adaptive response to compensate for SERCA2a downregulation. However, sustained activation of NCX would be maladaptive as it would contribute to a decrease in the SR calcium content due to increased cytosolic calcium removal.
Defects in SR calcium uptake
During heart failure, one of the most pronounced cellular changes is an increase in end-diastolic cytosolic calcium levels and prolongation of the calcium transient during diastole. This is primarily due to a decrease in SR calcium uptake because of SERCA2a dysfunction. In both animal models of and patients with heart failure, there is a reduction in SERCA2a expression (mRNA and protein levels) and activity (Hasenfuss et al., 1994; Kiss et al., 1995). PLN reversibly inhibits SERCA2a in cardiac tissue thereby reducing influx of calcium into the SR. PLN is phosphorylated at Ser16 by PKA or Thr17 by calcium/calmodulin-dependent protein kinase II (CaMKII) or protein kinase B (Akt). When phosphorylated at either or both of these sites, the inhibition of SERCA2a is alleviated and calcium flux into the SR increases. During heart failure, there are no changes in PLN protein expression (Movsesian et al., 1994) but some studies have seen a decrease in PLN mRNA levels in patients with dilated or ischemic cardiomyopathy (Flesch et al., 1996). The reduction in SERCA2a expression in heart failure results in an increase in the PLN-to-SERCA2a ratio, leading to increased inhibition of SERCA2a. Simultaneously, a reduction in phosphorylation of PLN has been shown in animal models of heart failure (Huang et al., 1999) and human failing myocardium (Schwinger et al., 1999). This is in part due to increased activity of protein phosphatase 1 (PP1), which dephosphorylates PLN (Yamada et al., 2006). Defects in PP1 and its modulation by its inhibitor, inhibitor-1, have been found in heart failure and have been targeted by therapeutic modalities, which will be discussed in detail later in this Review.
Several mutations in PLN have been identified in patients and linked to hereditary dilated cardiomyopathy (DCM) (Table 1). The precise cardiac pathology of the different PLN mutations varies but all patients present with cardiac hypertrophy and decreased ejection fraction. The Arg9Cys mutation was identified in an American family and results in DCM with congestive heart failure (Schmitt et al., 2003). The Arg14del mutation was first identified in a Greek patient but has since been found to have a high prevalence in the Netherlands (~15% of DCM cases) (van der Zwaag et al., 2013). This mutation causes “arrhythmogenic cardiomyopathy” but the mechanism isn’t understood. The Leu39stop mutation is a PLN truncation and no detectable PLN activity can be found in homozygous patients (Haghighi et al., 2003). Patients heterozygous for Leu39stop exhibit mild hypertrophy without diminished cardiac performance and patients homozygous for Leu39stop develop DCM and heart failure. The Arg9Leu and Arg9His mutations were recently identified in several patients in Brazil with cardiac hypertrophy and DCM (Medeiros et al., 2011) and initial molecular characterization has revealed defects in SERCA2a inhibition by PLN and/or phosphorylation of PLN by PKA (Ceholski et al., 2012a; Ceholski et al., 2012b).
Table 1.
Disease-causing mutations in SR calcium handling proteins.
| Protein | Mutation | Disease Phenotype | References |
|---|---|---|---|
| CASQ2 | R33Q, Y55C, L167H, E177Q, F189L, K206N, D307H, P308L | CPVT | de La Fuente 2008 Kirchhhefer 2010 |
| HRC | S96A | ventricular arrhythmias, DCM | Arvanitis 2008 |
| PLN | R9C, R9L, R9H R14del L39stop |
DCM arrhythmogenic cardiomyopathy hypertrophy, DCM |
Schmitt 2003, Medeiros 2011 Haghighi 2006 Haghighi 2003 |
| RyR2 | R414L, I419F, S2246L, V2306I, R2311D, P2328S, A2387P, A2403T, R2474S, L2534V, L3778F, G3946S, N4097S, N4104K, E4146K, T4158P, Q4201R, R4497C, F4499C, N4504I, A4510T, A4608P, V4653F, G4671R, V4771I, I4848V, A4860G, I4867M, V4880A, N4895D, P4902L, E4950K R176Q, R420W, L433P, N2386I, Y2392C, T2504M |
CPVT ARVD/C2 |
Yano 2005 |
Abbreviations: ARVD/C2, arrhythmogenic right ventricular cardiomyopathy type 2; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy. For complete list of RyR2 and CASQ2 mutations see http://triad.fsm.it/cardmoc/.
Reduced SR calcium content
Calcium buffering proteins within the lumen of the SR serve an important role during EC coupling. The major calcium buffering protein in cardiac SR is calsequestrin isoform 2 (CASQ2), which has a high capacity and a moderate affinity for calcium, and binds to approximately 50–75% of total SR calcium (Gyorke et al., 2009). As a result, CASQ2 reduces the free luminal calcium concentration in order to facilitate SERCA2a-mediated calcium transport into the SR during diastole as well as leave a sufficient calcium reserve within the SR during systole (Gyorke and Terentyev, 2008). CASQ2 interacts with RyR2 through triadin, which allows the modulation of RyR2 activity by CASQ2. Although the mRNA and protein levels of CASQ2 remain unaltered in heart failure, CASQ2 has been associated with cardiac arrhythmias, such as catecholaminergic polymorphic ventricular tachycardia (CPVT) (Kontula et al., 2005) (Table 1). The CPVT phenotype was confirmed in a transgenic mouse model, where knocking out the CASQ2 gene caused an autosomal-recessive form of CPVT with unaltered calcium transients (Knollmann et al., 2006).
Another SR calcium binding protein which is thought to have a role in calcium uptake and release is the histidine-rich calcium binding protein (HRC). Found in the lumen of the cardiac SR, HRC has been shown to interact with SERCA2a and RyR2 through junctin (an accessory membrane protein), indicating its potentially important role in the crosstalk between SR calcium storage and release (Arvanitis et al., 2007) (Figure 2). Overexpression of HRC in adult rat cardiomyocytes and transgenic mice increased the calcium storage capacity of the SR but decreased SR calcium release, resulting in depressed contractility (Fan et al., 2004; Gregory et al., 2006). The protein levels of HRC have been shown to be significantly reduced in human and animal models of heart failure (Fan et al., 2004), which may be a compensatory mechanism that increases calcium release from the SR in an attempt to improve cardiac contraction. More recently, a HRC mutation (Ser96Ala) was found to cause defects in regulation, as evidenced by depressed calcium transients and prolonged calcium decay, which lead to DCM and ventricular arrhythmias (Arvanitis et al., 2008; Han et al., 2011) (Table 1).
S100A1 is a calcium binding protein which has recently emerged as a key regulator of calcium homeostasis in cardiac muscle. Preferentially expressed in the left ventricle of the heart, S100A1 is a membrane protein which localizes to the SR, sarcomere, and mitochondria (Figure 2). S100A1 has been shown to interact with both RyR2 and SERCA2a in the cardiac SR membrane, thereby facilitating calcium release during systole and calcium reuptake during muscle relaxation, respectively (Kiewitz et al., 2000; Volkers et al., 2007). Therefore, S100A1 directly augments SR calcium uptake and increases SR calcium load. In addition to being expressed in the SR, S100A1 is also present in the mitochondria where it interacts with F1F0 ATP synthase, resulting in an increase in its activity and increased ATP production (Boerries et al., 2007). Decreased expression of S100A1 were shown in animal models of heart failure and human heart samples with dilated and ischemic cardiomyopathy, suggesting that S100A1 plays a crucial role in proper cardiac function (Brinks et al., 2011; Remppis et al., 1996). Indeed, S100A1 knockout mice were shown to develop accelerated congestive heart failure after myocardial infarction and increased mortality (Brinks et al., 2011; Remppis et al., 1996). In contrast, transgenic mice overexpressing S100A1 exhibited improved calcium handling, enhanced contractile response to β-adrenergic stimulation, and survival (Brinks et al., 2011).
SR calcium leak
RyR2-mediated release of calcium from the SR into the cytosol is a key event during EC coupling and is required for cardiac contraction. During muscle relaxation, it is imperative for RyR2 channels to remain tightly closed in order for SR calcium stores to be refilled and primed for subsequent muscle contraction. Aberrant gating of the RyR2 channels has been associated with cardiac arrhythmias and congestive heart failure (Kushnir and Marks, 2010; Wehrens and Marks, 2003). It is important to note that unlike SERCA2a, the expression of RyR2 is unaltered in heart failure (Sainte Beuve et al., 1997). However, several other factors can contribute to defective function of RyR2. It has been reported that increased β-adrenergic signaling results in hyperphosphorylation of RyR2 at Ser2808 by PKA, leading to impaired binding of the channel-stabilizing protein FKBP12.6 (also referred to as calstabin2) and diastolic leakage of calcium from the SR during heart failure (Hain et al., 1995; Marx et al., 2000). Moreover, the levels of protein phosphatase 1 and 2A, both of which associate with RyR2, were shown to be reduced in patients with heart failure, suggesting a slower dephosphorylation rate of RyR2 and dissociation of FKBP12.6 from the channel complex, as well as increased calcium sensitivity of RyR2 (Marx et al., 2000). Nevertheless, the mechanism of PKA-mediated hyperphosphorylation of RyR2 during heart failure is under debate, as several studies reported RyR2 channel activity to be slightly decreased in response to phosphorylation (Valdivia et al., 1995; Xiao et al., 2005). CaMKII-dependent phosphorylation of RyR2 has also emerged as an important modulator of calcium channel activity in heart failure (Chelu et al., 2009). Studies have shown elevated CaMKII levels as well as increased CaMKII-dependent phosphorylation of RyR2 at Ser2008 and Ser2814 in heart failure (Dobrev and Wehrens, 2014), which appear to be an alternative mechanism for increased calcium release from the SR during diastole. Finally, several missense mutations in RyR2 have been identified in patients with cardiac arrhythmias (Table 1), specifically CPVT, resulting in reduced binding affinity for FKBP12.6 and increased permeability of RyR2 to SR calcium (Priori et al., 2001). Although further work is required to elucidate the mechanism of RyR2 regulation in heart failure, stabilization of the RyR2-FKBP12.6 complex appears to be a promising target for the treatment of heart failure.
An energy crisis in the failing heart
The high energy demands of the heart must be maintained in order for it to sustain continuous work. During heart failure, the dynamic metabolic network in the cardiomyocyte is perturbed and these alterations cause energy depletion, directly affecting contractile function. Mitochondrial dysfunction has been linked to cardiac arrhythmias and sudden death (Brown et al., 2010). Heart failure is associated with multiple mitochondrial defects – reduced ATP production, a decline in mitochondrial respiratory activity, and structural defects – but it is unknown as to whether this is due to a reduction in mitochondrial mass or defects in mitochondrial function (Ide et al., 2001; Rosca et al., 2008; Shen et al., 1999). These multiple mitochondrial perturbations are linked to alterations in calcium signaling and EC coupling. In the following subsections, we describe the multifaceted roles of the mitochondria in the heart, the energetic phenotype of the failing heart, and the importance of SR-mitochondria crosstalk in the cardiomyocyte as well as how it is affected in heart failure.
Mitochondria in the human heart
Mitochondria play a key role in cardiac metabolism and energetics, as they occupy as much as 40% of cell volume (Palmer et al., 1977). Mitochondria have multifaceted roles in the cardiac cell including energy homeostasis, signaling, metabolism and mediation of cell death pathways. Consequently, the number, morphology and function of mitochondria are regulated according to cell-specific requirements. This regulation is largely controlled by a network of transcription factors, which mediate replication and transcription of the mitochondrial genome, and respond to changes in energy metabolism, nutrient availability and physiological state of the cell, allowing for adjustments in mitochondrial biogenesis and function (Hock and Kralli, 2009). Mitochondria are dynamic organelles that continuously undergo fission and fusion, and these processes are indispensable to their proper function within the cell (Chan, 2012). Genetic defects in transcription factors or genes involved in mitochondrial biogenesis and dynamics have been linked to multiple pathologies including cancer, heart disease and neurological disorders (Archer, 2013). While energy metabolism is the primary role of mitochondria (discussed in detail in the next subsection), they also have roles in mediating cell signaling and cell death pathways. The primary mediators of cell signaling in mitochondria are calcium and reactive oxygen species (ROS). In cardiac myocytes, there are two subpopulations of mitochondria that differ primarily by their localization - the interfibrillar mitochondria are located adjacent to the junctional SR parallel to the myofibrils and the subsarcolemmal mitochondria are situated underneath the plasma membrane – but also have slightly different biochemical properties and protein and lipid composition (Palmer et al., 1977). Since they are adjacent to the SR, interfibrillar mitochondria are exposed to intracellular calcium transients and are capable of accumulating large amounts of intracellular calcium in response to calcium flux (Carafoli and Lehninger, 1964). The mitochondrial calcium uniporter (MCU) is the primary means for mitochondrial calcium uptake and its activity is dependent on intracellular calcium concentration (Rizzuto et al., 2000) (Figure 2). Calcium extrusion from the matrix is accomplished by the mitochondrial NCX (mNCX), reversal of the MCU or by a permeability transition of the inner mitochondrial membrane (Bernardi and Petronilli, 1996; Montero et al., 2001; Saotome et al., 2005). The accumulation of calcium in mitochondria has been implicated in rate of energy production and activation of cellular apoptosis and necrosis pathways (Nakayama et al., 2007). As a byproduct of ATP production, mitochondria produce ROS which relay stress signals in the cell but can also jeopardize mitochondrial viability and cause cell damage or death (Brand et al., 2004). In the cardiomyocyte, this process has been implicated in ischemia-reperfusion injury (Brand et al., 2004; Yoshida et al., 2000). Cytosolic calcium overload and oxidative stress can lead to changes in the permeability of the mitochondrial inner membrane which is associated with opening of the permeability transition pore, leading to dissipation of the mitochondrial membrane potential, uncoupling of oxidative phosphorylation, loss of ATP and activation of necrotic pathways (Walters et al., 2012). In particular, mice that overexpress cyclophilin D, a regulatory component of the permeability transition pore, show mitochondrial swelling and spontaneous cell death while cyclophilin D knockout mice are protected from ischemia-reperfusion-induced cell death (Baines et al., 2005). In addition, mitochondrial autophagy (mitophagy) plays an early cardioprotective role during ischemia-reperfusion in order to prevent activation of pro-death pathways in favor of adaptation to stress (Kubli and Gustafsson, 2012).
Energetic phenotype of the failing myocardium
While energetic profiles can differ between types of cardiac pathology, it has been well established that the failing heart is energy starved. ATP levels are significantly depleted (~25–30%) during heart failure, as seen in human heart biopsies and animal models (Starling et al., 1998). In the canine rapid pacing heart failure model, a reduction in ATP is observed quickly, soon after the onset of rapid pacing, but occurs at a slow and progressive rate (~0.35% per day) (Shen et al., 1999). This reduction occurs before any left ventricular systolic dysfunction but occurs so gradually that it would be beyond detection until overt heart failure has already occurred. Whether this depletion of ATP is a cause or effect of heart disease is unknown, as a reduction in ATP in a normal heart has never been noted or examined.
While ATP is the major source of energy in the heart, total creatine and PCr levels are better markers of the energetic state of the heart, as they are the primary source of cardiac energy reserve. In the normal heart, approximately two-thirds of the total creatine pool is phosphorylated via creatine kinase (PCr+ADP+H+ → creatine+ATP) so the energy is kinetically trapped and primed as a source of ATP (Figure 2). In animal models and patients with severe heart failure, total creatine levels fall by as much as 60% (Katz, 1998). This reduction results in a concomitant decrease in PCr, the depletion of which is more severe than that of ATP, which results in a decrease in [PCr]/[ATP]. The [PCr]-to-[ATP] ratio is a better predictor of overall and cardiovascular mortality than New York Heart Association Class and LV ejection fraction (Ingwall and Weiss, 2004).
Substrate utilization for energy production is markedly different in heart failure, described as a “metabolic shift” from fatty acid oxidation to glucose use through glycolysis (Siddiqi et al., 2013). This change has been described as a shift to the fetal phenotype, although aerobic metabolism is still dominant. Glucose utilization is inefficient at generating energy compared to fatty acid metabolism but uses less oxygen to generate the same amount of ATP (Camici et al., 1989). Due to limiting oxygen in myocardial ischemia, this may be an adaptive response of the heart. Insulin resistance is also commonly found in patients with heart failure, as either an etiological mechanism with diabetic cardiomyopathy or a consequence of heart failure itself (Ashrafian et al., 2007), and it impairs insulin-mediated glucose uptake and leads to the toxic accumulation of triglycerides (Poornima et al., 2006). In fact, cardiac-specific overexpression of GLUT1 was found to delay the development of heart failure in mice due to pressure-overload by increasing the capacity for glucose uptake and utilization (Liao et al., 2002).
SR-mitochondria crosstalk and its implications for calcium dysregulation
The importance of SR-mitochondria crosstalk is demonstrated by their close physical association and reciprocal functional influence. Electron tomography experiments have shown the distance between the SR and mitochondria to be between 10 and 50nm (Csordas et al., 2006) and evidence of a physical association was confirmed by electron microscopy (Ramesh et al., 1998). This is accomplished by the formation of two types of tethering complexes between the mitochondria and SR: one group is purely structural, providing only a supportive link between the two organelles (ex. Mitofusin-2), and the other is both structural and functional, also providing signaling communication between the mitochondria and the SR (ex. Mitostatin, RyR2, VDAC) (Eisner et al., 2013; Griffiths and Rutter, 2009) (Figure 2). Disruption of SR-mitochondria interactions results in reduced ATP production and triggers stress and apoptotic pathways. In fact, ablation of Mitofusin-2 in mouse cardiac myocytes resulted in accumulation of functionally and morphologically abnormal mitochondria and ultimately led to DCM (Chen and Dorn, 2013). It has been known for some time that intracellular calcium levels influence calcium uptake by mitochondria but the high levels of calcium required to induce these changes were higher than typical maximal physiological relevant calcium levels (Javadov et al., 2003). It was later discovered that mitochondria respond specifically at those sites directly adjacent to the SR, where there are large increases in calcium concentration in transient microdomains (Garcia-Perez et al., 2008). SR calcium release has been shown to be highly regulated by cleft calcium concentration (the region between the junctional SR and the T tubule) and recent developments have allowed the accurate measurement of calcium microdomains in the cardiomyocyte. Despa and colleagues developed calcium sensors to specifically target subcellular microdomains and their results demonstrate the existence of a permanent calcium gradient between the cleft and bulk cytosol during diastole (Despa et al., 2014). By exploiting these standing gradients, mitochondria act as calcium sensors, allowing the propagation of the calcium signal initiated by SR release. Indeed, efficient production of ATP and its delivery to the SR are influenced by cytosolic and mitochondrial calcium levels, as several mitochondrial enzymes are activated by increases in calcium (Denton et al., 1972; Denton et al., 1978; McCormack and Denton, 1979) and defective energy transfer between the SR and mitochondria has been implicated in EC coupling deterioration (Wilding et al., 2006). It has also been proposed that mitochondrial calcium levels can alter the activity of the F1F0 ATP synthase; although shown to bind calcium directly, the F1F0 ATP synthase is likely to be regulated via post-translational modifications and phosphorylation of its γ-subunit, which is sensitive to mitochondrial calcium concentration (Hopper et al., 2006). It has also been shown that S100A1 can bind to the F1F0 ATP synthase in a calcium dependent manner, allowing for an increase in ATP production (Boerries et al., 2007) (Figure 2). In addition, ROS microdomains may impact SR function and calcium microdomains, as oxidation of cysteine thiol groups of the RyR receptor has been shown to result in an increase in channel activity (Zima and Blatter, 2006). In rat cardiac myocytes, mitochondrial ROS production resulted in the production of calcium sparks from the SR (Yan et al., 2008). The integrity of the mutual relationship between the SR and mitochondria is therefore imperative for individual organelle and whole cardiomyocyte function.
Potential therapies for heart failure
Our understanding of normal and pathophysiological calcium transport and signaling has allowed for the development of therapies that target calcium-handling proteins. During heart failure, there is cytosolic calcium overload due to SR malfunction. Recent therapies have been designed to improve SR function – either by increasing SR calcium uptake or preventing SR calcium leak. These therapies will be discussed in the context of cardiac energetics when applicable.
Increasing SR calcium uptake
SERCA2a
During heart failure, the expression and activity of SERCA2a is reduced and gene transfer of SERCA2a into failing human cardiomyocytes improved contractility (del Monte et al., 1999). Similarly, SERCA2a gene transfer improved survival and cardiac contractility in multiple animal models of heart failure, including pressure-overload rat (Muller et al., 2003) and porcine (Kawase et al., 2008) models. Adenoviral-mediated gene expression of SERCA2a was also shown to significantly improve cardiac energetics via an increase in the [PCr]/[ATP] ratio in a rat model of heart failure (Figure 3A) (del Monte et al., 2001). This was unexpected given the increased requirements for ATP and PCr upon SERCA2a overexpression. Indeed, SERCA2a overexpression in a sham rat heart resulted in reduced [PCr]/[ATP] ratio. A later study by the same group showed that short and long term adeno-associated viral (AAV) SERCA2a overexpression in the same rat model of heart failure restored (decreased) the oxygen consumption of cardiomyocytes to normal levels (Figure 3B) (Sakata et al., 2007). The oxygen cost of contractility is thought to be proportional to the energetic cost of contractility, both of which are decreased during heart failure. Of note, a more recent study found that SERCA2a overexpression is limited by the energy supply of the failing heart in a pressure-overload transgenic mouse model (Pinz et al., 2011). In this study, SERCA2a was overexpressed prior to the development of cardiac hypertrophy induced by pressure-overload and despite prevention of downregulation of SERCA2a during the progression of heart failure there were no overt beneficial effects on cardiac reserve. Therefore, increasing SERCA2a levels may be most effective in improving cardiac energetics when heart failure is established.
Figure 3. Gene transfer of SERCA2a improves cardiac metabolism and energetic function in failing hearts.
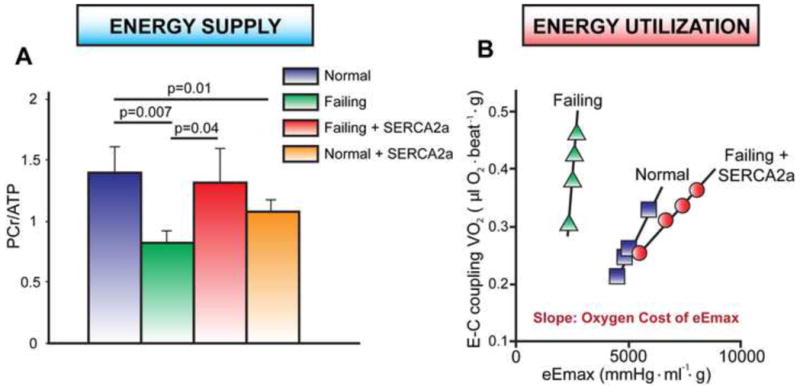
A. The ratio of [PCr]-to-[ATP] and the levels of both PCr and ATP are lower in failing hearts compared to healthy hearts. The overexpression of SERCA2a in failing heart restores the [PCr]-to-[ATP] ratio toward normal (del Monte et al., 2001). B. Linear relationship between the oxygen cost of calcium handling (VO2) and left ventricular contractility (eEmax) in normal (■), failing (▲) and failing+SERCA2a (●) hearts. The slope of these linear relations represents the oxygen cost of left ventricular contractility. Treatment of failing hearts by SERCA2a gene therapy improves the energy cost of left ventricular contractility (Sakata et al., 2007).
Following a multitude of studies in large animal models of heart failure using AAV carrying SERCA2a through intra-coronary delivery, First-in-Man clinical gene therapy trials in patients with advanced heart failure began (Kawase et al., 2008). AAV vectors offer distinct advantages in that they are safe, transduce the myocardium efficiently, afford long-term expression and do not elicit an immune response. CUPID (Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease) is the first clinical trial of AAV1 delivery of SERCA2a (AAV1.SERCA2a) in patients with advanced heart failure. In 2007, phase I of this study began, with the primary objective being to investigate the safety and biological effects of restoring SERCA2a activity in patients with advanced heart failure (Jaski et al., 2009). AAV1.SERCA2a was administered to twelve patients by single coronary artery infusion in a dose escalation manner (1.4×1011–1.0×1013 DNAse-resistant particles (DRP)). This trial revealed no safety concerns and several patients showed improvements in heart failure parameters and cardiac function. Phase II of the CUPID trial was a double-blind, placebo-controlled, randomized study of 39 patients with advanced heart failure where patients received either placebo or low (6×1011 DRP), medium (3×1012 DRP) or high (1×1013 DRP) dose of AAV1.SERCA2a (Jessup et al., 2011). After 6 months, patients who received the high dose displayed an improvement or stabilization of heart failure symptoms and, after 12 months, had fewer cardiovascular events and shorter hospitalizations. Three-year follow-up of these patients showed sustained improvement in clinical event rates in patients receiving high dose AAV1.SERCA2a along with persistence of the vector within the myocardium for up to 31 months (Zsebo et al., 2014). An international Phase IIb/III trial is currently underway to assess the effectiveness of AAV1.SERCA2a in improving clinical outcomes in patients with advanced heart failure (saline vs. AAV1.SERCA2a 1×1013 DRP). Other ongoing trials are evaluating the ability of AAV1.SERCA2a delivery to halt adverse left ventricular remodeling and improve ventricular function in patients with left ventricular assist devices.
Phospholamban
In failing human myocardium, SERCA2a expression and activity is decreased but is not accompanied by a concurrent decrease in PLN expression (Hasenfuss et al., 1994), resulting in SERCA2a superinhibition and prolonged muscle relaxation due to higher PLN-to-SERCA ratios. Multiple antisense RNA or small hairpin RNA (shRNA) studies to specifically target and downregulate PLN have been performed in vitro and in vivo in order to rescue cardiac function (del Monte et al., 2002; Suckau et al., 2009). A recent large animal study involving a shRNA against PLN proved to be toxic due to adverse interference with micro-RNA pathways (Bish et al., 2011). However, use of AAV9-mediated expression of a shRNA to silence PLN resulted in an increase in SERCA2a activity and improved cardiac function in aortic-banded rats (Suckau et al., 2009). Overexpression of dominant-negative and pseudo-phosphorylated (Ser16-to-Glu) forms of PLN have improved cardiac function in animal models of heart failure (Kaye et al., 2007; Ziolo et al., 2005). However, the Ser16-to-Glu PLN mutant cannot be phosphorylated in vivo which may limit its therapeutic potential.
Protein phosphatase 1
PLN is phosphorylated by multiple kinases, including PKA, CaMKII, and Akt, but is only known to be dephosphorylated by protein phosphatase 1 (PP1). When phosphorylated, PLN is non-inhibitory and results in an increase in SR calcium uptake and subsequent increase in force of contraction. One therapeutic approach to increase the proportion of phosphorylated PLN is to inhibit the activity of PP1. It is well-established that there is increased expression of PP1 during heart failure (Wittkopper et al., 2011). In addition, there is decreased expression and phosphorylation of protein phosphatase inhibitor-1 (I-1), a PP1-specific inhibitor, which culminates in decreased phosphorylation of PLN and inhibition of SERCA2a activity (Wittkopper et al., 2011). I-1 is phosphorylated by PKA, upon which it inhibits PP1, and propagates the β-adrenergic response in the heart. Since it has an extensive number of physiological substrates, chronic inhibition of PP1 is not ideal and has been shown to result in hyperphosphorylation of RyR2, leading to cardiac arrhythmogenesis (Wittkopper et al., 2011). Recent studies have examined the inhibition of PP1 through its interaction with I-1 as a potential therapeutic approach to enhance cardiac function in heart failure. A truncated form of I-1 (I-1c) which is constitutively active has been shown to increase phosphorylation of PLN at Ser16 but have no effect on phosphorylation of RyR2, troponin I, or myosin binding protein C in animal models of heart failure (Nicolaou et al., 2009). This suggests this construct causes an increase in calcium uptake into the SR without modification of normal SR calcium release (Kawashima et al., 2009). In addition, I-1c overexpression was reported to improve cardiac performance post-ischemia and was cardioprotective in a mouse model of heart failure (Nicolaou et al., 2009), indicating that PP1 may be a promising therapeutic target.
SUMO1
Recently, the small ubiquitin-related modifier 1 (SUMO1) was shown to play an important role in cardiac contractility (Kho et al., 2011). Through its modification of SERCA2a, SUMO1 has been shown to enhance SERCA2a activity and stability and is thought to have an important role in maintaining proper cardiac function. In human and animal models of heart failure, the expression of SUMO1 and resultant SUMOylation of SERCA2a are reduced. Cardiac-specific overexpression of SUMO1 by AAV9-mediated gene delivery in mouse hearts was found to improve SERCA2a protein stability and activity. More recent studies in porcine models of ischemic heart failure have shown that SUMO1 gene transfer improves cardiac contractility and prevents progression of left ventricular dilatation (Tilemann et al., 2013). In addition, combined delivery of SUMO1 and SERCA2a genes had synergistic effects on improvement of cardiac function. The positive effects of SUMO1 gene delivery were proportional to dosage, with greater benefits seen with higher doses of SUMO1. Moreover, SUMO1 overexpression in porcine heart failure models has been shown to protect SERCA2a from oxidative stress during cardiac hypertrophy and heart failure (Lee et al., 2014). Although further studies are needed to better define the role of SUMO1 in heart failure, current data support the critical role of SERCA2a SUMOylation and its potential as a therapeutic target in heart failure.
Preventing SR calcium leak
Ryanodine receptor 2
Hyperphosphorylated or unstable RyR2 channels result in SR calcium leak, leading to decreased SR calcium content and reduced systolic calcium transients, which in turn cause decreased contractility and reduced cardiac output (Belevych et al., 2011; Wehrens et al., 2006). One therapeutic strategy that has been explored to overcome hyperphosphorylation of RyR2 by PKA or CaMKII is to overexpress FKBP12.6, a RyR2 regulatory protein. Cardiac-specific overexpression in mice (Huang et al., 2006) or adenovirus-mediated delivery in isolated rabbit cardiomyocytes (Loughrey et al., 2004) of FKBP12.6 resulted in stabilization of RyR2, increased SR calcium content and improved myocyte shortening. Pharmacological intervention in reducing SR calcium leak has also been investigated, where SR calcium release is inhibited either by altered RyR2 gating or ion translocation. One such compound is JTV-510 (K201), a benzothiazepine, and it was reported to improve cardiac function by stabilizing the closed state of RyR2 (Ito et al., 2000; Kaneko et al., 1997). More recently, a new class of compounds called Rycals (K201 derivatives) was shown to reduce arrhythmic episodes and act as protective agents against heart failure progression (Bellinger et al., 2009). These drugs are thought to reduce SR calcium leak by stabilizing the interaction between RyR2 and FKBP12.6. However, there are conflicting results on the role of PKA-mediated phosphorylation of RyR2 therefore alternative mechanisms may be important for RyR2 dysfunction in heart failure.
S100A1
Another promising therapeutic target for heart failure is the S100A1 calcium-sensing protein, which interacts with RyR2 and SERCA2a in the SR and is also present in the mitochondria. During heart failure, S100A1 expression is downregulated (Remppis et al., 1996) but in in animal models of heart failure and failing human cardiomyocytes, overexpression of S100A1 significantly improved contractile function, calcium handling and cardiac energetics (Most et al., 2004; Remppis et al., 1996). Due to its interaction with the F1F0 ATP synthase, S100A1 treatment has been shown to improve [PCr]-to-[ATP] and [NADH]-to-[NAD+] ratios in human failing cardiomyocytes (Brinks et al., 2011).
Perspectives
Recent improvements in our understanding of calcium cycling mechanisms in cardiomyocytes have led to novel therapies for heart failure. These approaches mostly include targeting SR calcium handling proteins that have altered activity and/or expression during disease. Current therapies are primarily targeted at increasing SR calcium uptake or preventing SR calcium leak and many are currently being assessed in animal models or patients with heart failure. Cardiac energetics should also be considered in therapies targeting SR calcium proteins, as dysregulation of cardiac calcium and metabolism appear to be symbiotic in heart failure. Some current therapies have progressed in phases of evaluation after having shown early signs of success (SERCA2a) while others are earlier in development (SUMO1, S100A1). An emphasis on SR calcium handling proteins and cardiac energetics will pave the way for future advances in new treatment options for heart failure.
Acknowledgments
We gratefully acknowledge the following funding: R.J.H. is the Arthur & Janet C. Ross Professor of Medicine and the Director of the Cardiovascular Research Center at the Icahn School of Medicine at Mount Sinai Hospital. R.J.H is funded by the National Institutes of Health, American Heart Association and Fondation Leducq.
Footnotes
Author Contributions
P.A.G. and D.K.C. researched the material and co-wrote the manuscript. R.J.H. outlined the content of all material discussed and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res. 2001;89:997–1004. doi: 10.1161/hh2301.100003. [DOI] [PubMed] [Google Scholar]
- Archer SL. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- Arvanitis DA, Sanoudou D, Kolokathis F, Vafiadaki E, Papalouka V, Kontrogianni-Konstantopoulos A, Theodorakis GN, Paraskevaidis IA, Adamopoulos S, Dorn GW, 2nd, et al. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. European heart journal. 2008;29:2514–2525. doi: 10.1093/eurheartj/ehn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitis DA, Vafiadaki E, Fan GC, Mitton BA, Gregory KN, Del Monte F, Kontrogianni-Konstantopoulos A, Sanoudou D, Kranias EG. Histidine-rich Ca-binding protein interacts with sarcoplasmic reticulum Ca-ATPase. American journal of physiology Heart and circulatory physiology. 2007;293:H1581–1589. doi: 10.1152/ajpheart.00278.2007. [DOI] [PubMed] [Google Scholar]
- Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007;116:434–448. doi: 10.1161/CIRCULATIONAHA.107.702795. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Gyorke S. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. 2011;90:493–502. doi: 10.1093/cvr/cvr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr. 1996;28:131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- Bish LT, Sleeper MM, Reynolds C, Gazzara J, Withnall E, Singletary GE, Buchlis G, Hui D, High KA, Gao G, et al. Cardiac gene transfer of short hairpin RNA directed against phospholamban effectively knocks down gene expression but causes cellular toxicity in canines. Hum Gene Ther. 2011;22:969–977. doi: 10.1089/hum.2011.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittl JA, Ingwall JS. Reaction rates of creatine kinase and ATP synthesis in the isolated rat heart. A 31P NMR magnetization transfer study. J Biol Chem. 1985;260:3512–3517. [PubMed] [Google Scholar]
- Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, Aebi U, Schoenenberger CA. Ca2+-dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol. 2007;27:4365–4373. doi: 10.1128/MCB.02045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Buckingham JA, Esteves TC, Green K, Lambert AJ, Miwa S, Murphy MP, Pakay JL, Talbot DA, Echtay KS. Mitochondrial superoxide and aging: uncoupling-protein activity and superoxide production. Biochem Soc Symp. 2004:203–213. doi: 10.1042/bss0710203. [DOI] [PubMed] [Google Scholar]
- Brinks H, Rohde D, Voelkers M, Qiu G, Pleger ST, Herzog N, Rabinowitz J, Ruhparwar A, Silvestry S, Lerchenmuller C, et al. S100A1 genetically targeted therapy reverses dysfunction of human failing cardiomyocytes. J Am Coll Cardiol. 2011;58:966–973. doi: 10.1016/j.jacc.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O’Rourke B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–679. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camici P, Ferrannini E, Opie LH. Myocardial metabolism in ischemic heart disease: basic principles and application to imaging by positron emission tomography. Prog Cardiovasc Dis. 1989;32:217–238. doi: 10.1016/0033-0620(89)90027-3. [DOI] [PubMed] [Google Scholar]
- Carafoli E, Lehninger AL. Binding of adenine nucleotides by mitochondria during active uptake of CA++ Biochem Biophys Res Commun. 1964;16:66–70. doi: 10.1016/0006-291x(64)90212-8. [DOI] [PubMed] [Google Scholar]
- Ceholski DK, Trieber CA, Holmes CF, Young HS. Lethal, hereditary mutants of phospholamban elude phosphorylation by protein kinase A. J Biol Chem. 2012a;287:26596–26605. doi: 10.1074/jbc.M112.382713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceholski DK, Trieber CA, Young HS. Hydrophobic imbalance in the cytoplasmic domain of phospholamban is a determinant for lethal dilated cardiomyopathy. J Biol Chem. 2012b;287:16521–16529. doi: 10.1074/jbc.M112.360859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. The Journal of clinical investigation. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Monte F, Harding SE, Dec GW, Gwathmey JK, Hajjar RJ. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002;105:904–907. doi: 10.1161/hc0802.105564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–2311. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, Lewandowski ED, Hajjar RJ. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation. 2001;104:1424–1429. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM, Randle PJ, Martin BR. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J. 1972;128:161–163. doi: 10.1042/bj1280161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J. 1978;176:899–906. doi: 10.1042/bj1760899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Shui B, Bossuyt J, Lang D, Kotlikoff MI, Bers DM. Junctional cleft [Ca(2)(+)]i measurements using novel cleft-targeted Ca(2)(+) sensors. Circ Res. 2014;115:339–347. doi: 10.1161/CIRCRESAHA.115.303582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D, Wehrens XH. Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. 2014;114:1311–1319. doi: 10.1161/CIRCRESAHA.114.300568. discussion 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner V, Csordas G, Hajnoczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca(2)(+) and reactive oxygen species signaling. J Cell Sci. 2013;126:2965–2978. doi: 10.1242/jcs.093609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan GC, Gregory KN, Zhao W, Park WJ, Kranias EG. Regulation of myocardial function by histidine-rich, calcium-binding protein. American journal of physiology Heart and circulatory physiology. 2004;287:H1705–1711. doi: 10.1152/ajpheart.01211.2003. [DOI] [PubMed] [Google Scholar]
- Feldman RD, Gros R. New insights into the regulation of cAMP synthesis beyond GPCR/G protein activation: implications in cardiovascular regulation. Life Sci. 2007;81:267–271. doi: 10.1016/j.lfs.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Flesch M, Schwinger RH, Schnabel P, Schiffer F, van Gelder I, Bavendiek U, Sudkamp M, Kuhn-Regnier F, Bohm M. Sarcoplasmic reticulum Ca2+ATPase and phospholamban mRNA and protein levels in end-stage heart failure due to ischemic or dilated cardiomyopathy. J Mol Med (Berl) 1996;74:321–332. doi: 10.1007/BF00207509. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez C, Hajnoczky G, Csordas G. Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J Biol Chem. 2008;283:32771–32780. doi: 10.1074/jbc.M803385200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonasekera SA, Hammer K, Auger-Messier M, Bodi I, Chen X, Zhang H, Reiken S, Elrod JW, Correll RN, York AJ, et al. Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J Clin Invest. 2012;122:280–290. doi: 10.1172/JCI58227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KN, Ginsburg KS, Bodi I, Hahn H, Marreez YM, Song Q, Padmanabhan PA, Mitton BA, Waggoner JR, Del Monte F, et al. Histidine-rich Ca binding protein: a regulator of sarcoplasmic reticulum calcium sequestration and cardiac function. Journal of molecular and cellular cardiology. 2006;40:653–665. doi: 10.1016/j.yjmcc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta. 2009;1787:1324–1333. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Gyorke S, Stevens SC, Terentyev D. Cardiac calsequestrin: quest inside the SR. The Journal of physiology. 2009;587:3091–3094. doi: 10.1113/jphysiol.2009.172049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovascular research. 2008;77:245–255. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- Haghighi K, Gregory KN, Kranias EG. Sarcoplasmic reticulum Ca-ATPase-phospholamban interactions and dilated cardiomyopathy. Biochem Biophys Res Commun. 2004;322:1214–1222. doi: 10.1016/j.bbrc.2004.07.164. [DOI] [PubMed] [Google Scholar]
- Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. The Journal of biological chemistry. 1995;270:2074–2081. doi: 10.1074/jbc.270.5.2074. [DOI] [PubMed] [Google Scholar]
- Han P, Cai W, Wang Y, Lam CK, Arvanitis DA, Singh VP, Chen S, Zhang H, Zhang R, Cheng H, et al. Catecholaminergic-induced arrhythmias in failing cardiomyocytes associated with human HRCS96A variant overexpression. American journal of physiology Heart and circulatory physiology. 2011;301:H1588–1595. doi: 10.1152/ajpheart.01153.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011;194:355–365. doi: 10.1083/jcb.201101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G, Schillinger W, Lehnart SE, Preuss M, Pieske B, Maier LS, Prestle J, Minami K, Just H. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–648. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, Witzmann FA, Harris RA, Balaban RS. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry. 2006;45:2524–2536. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang S, Qin D, Boutjdir M, El-Sherif N. Diminished basal phosphorylation level of phospholamban in the postinfarction remodeled rat ventricle: role of beta-adrenergic pathway, G(i) protein, phosphodiesterase, and phosphatases. Circ Res. 1999;85:848–855. doi: 10.1161/01.res.85.9.848. [DOI] [PubMed] [Google Scholar]
- Huang F, Shan J, Reiken S, Wehrens XH, Marks AR. Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: rescue of heart failure by calstabin2 in mice. Proc Natl Acad Sci U S A. 2006;103:3456–3461. doi: 10.1073/pnas.0511282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulot JS, Fauconnier J, Ramanujam D, Chaanine A, Aubart F, Sassi Y, Merkle S, Cazorla O, Ouille A, Dupuis M, et al. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation. 2011;124:796–805. doi: 10.1161/CIRCULATIONAHA.111.031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81:412–419. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- Ito K, Shigematsu S, Sato T, Abe T, Li Y, Arita M. JTV-519, a novel cardioprotective agent, improves the contractile recovery after ischaemia-reperfusion in coronary perfused guinea-pig ventricular muscles. Br J Pharmacol. 2000;130:767–776. doi: 10.1038/sj.bjp.0703373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15:171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549:513–524. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- Kaneko N, Matsuda R, Toda M, Shimamoto K. Inhibition of annexin V-dependent Ca2+ movement in large unilamellar vesicles by K201, a new 1,4-benzothiazepine derivative. Biochim Biophys Acta. 1997;1330:1–7. doi: 10.1016/s0005-2736(97)00132-6. [DOI] [PubMed] [Google Scholar]
- Katz AM. Is the failing heart energy depleted? Cardiol Clin. 1998;16:633–644. viii. doi: 10.1016/s0733-8651(05)70040-0. [DOI] [PubMed] [Google Scholar]
- Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, Hadri L, Yoneyama R, Hoshino K, Takewa Y, et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol. 2008;51:1112–1119. doi: 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Kawashima H, Satoh H, Saotome M, Urushida T, Katoh H, Hayashi H. Protein phosphatase inhibitor-1 augments a protein kinase A-dependent increase in the Ca2+ loading of the sarcoplasmic reticulum without changing its Ca2+ release. Circ J. 2009;73:1133–1140. doi: 10.1253/circj.cj-08-0871. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Preovolos A, Marshall T, Byrne M, Hoshijima M, Hajjar R, Mariani JA, Pepe S, Chien KR, Power JM. Percutaneous cardiac recirculation-mediated gene transfer of an inhibitory phospholamban peptide reverses advanced heart failure in large animals. J Am Coll Cardiol. 2007;50:253–260. doi: 10.1016/j.jacc.2007.03.047. [DOI] [PubMed] [Google Scholar]
- Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122:2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiewitz R, Lyons GE, Schafer BW, Heizmann CW. Transcriptional regulation of S100A1 and expression during mouse heart development. Biochimica et biophysica acta. 2000;1498:207–219. doi: 10.1016/s0167-4889(00)00097-5. [DOI] [PubMed] [Google Scholar]
- Kiss E, Ball NA, Kranias EG, Walsh RA. Differential changes in cardiac phospholamban and sarcoplasmic reticular Ca(2+)-ATPase protein levels. Effects on Ca2+ transport and mechanics in compensated pressure-overload hypertrophy and congestive heart failure. Circ Res. 1995;77:759–764. doi: 10.1161/01.res.77.4.759. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. The Journal of clinical investigation. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontula K, Laitinen PJ, Lehtonen A, Toivonen L, Viitasalo M, Swan H. Catecholaminergic polymorphic ventricular tachycardia: recent mechanistic insights. Cardiovascular research. 2005;67:379–387. doi: 10.1016/j.cardiores.2005.04.027. [DOI] [PubMed] [Google Scholar]
- Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir A, Marks AR. The ryanodine receptor in cardiac physiology and disease. Advances in pharmacology. 2010;59:1–30. doi: 10.1016/S1054-3589(10)59001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Jeong D, Mitsuyama S, Oh JG, Liang L, Ikeda Y, Sadoshima J, Hajjar R, Kho C. The Role of SUMO-1 in Cardiac Oxidative Stress and Hypertrophy. Antioxid Redox Signal. 2014 doi: 10.1089/ars.2014.5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Desantiago J, Chu G, Kranias EG, Bers DM. Phosphorylation of phospholamban and troponin I in beta-adrenergic-induced acceleration of cardiac relaxation. Am J Physiol Heart Circ Physiol. 2000;278:H769–779. doi: 10.1152/ajpheart.2000.278.3.H769. [DOI] [PubMed] [Google Scholar]
- Liao R, Jain M, Cui L, D’Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. 2002;106:2125–2131. doi: 10.1161/01.cir.0000034049.61181.f3. [DOI] [PubMed] [Google Scholar]
- Liao Y, Asakura M, Takashima S, Ogai A, Asano Y, Asanuma H, Minamino T, Tomoike H, Hori M, Kitakaze M. Benidipine, a long-acting calcium channel blocker, inhibits cardiac remodeling in pressure-overloaded mice. Cardiovasc Res. 2005;65:879–888. doi: 10.1016/j.cardiores.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- Loughrey CM, Seidler T, Miller SL, Prestle J, MacEachern KE, Reynolds DF, Hasenfuss G, Smith GL. Over-expression of FK506-binding protein FKBP12.6 alters excitation-contraction coupling in adult rabbit cardiomyocytes. J Physiol. 2004;556:919–934. doi: 10.1113/jphysiol.2003.057166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Hojayev B, Jiang N, Wang ZV, Tandan S, Rakalin A, Rothermel BA, Gillette TG, Hill JA. STIM1-dependent store-operated Ca(2)(+) entry is required for pathological cardiac hypertrophy. J Mol Cell Cardiol. 2012;52:136–147. doi: 10.1016/j.yjmcc.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahe I, Chassany O, Grenard AS, Caulin C, Bergmann JF. Defining the role of calcium channel antagonists in heart failure due to systolic dysfunction. Am J Cardiovasc Drugs. 2003;3:33–41. doi: 10.2165/00129784-200303010-00004. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180:533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros A, Biagi DG, Sobreira TJ, de Oliveira PS, Negrao CE, Mansur AJ, Krieger JE, Brum PC, Pereira AC. Mutations in the human phospholamban gene in patients with heart failure. Am Heart J. 2011;162:1088–1095. e1081. doi: 10.1016/j.ahj.2011.07.028. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Albillos A, Garcia-Sancho J, Alvarez J. Mitochondrial Ca(2+)-induced Ca(2+) release mediated by the Ca(2+) uniporter. Mol Biol Cell. 2001;12:63–71. doi: 10.1091/mbc.12.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, Loffler E, Janssen PM, Eckhart AD, Martini J, et al. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest. 2004;114:1550–1563. doi: 10.1172/JCI21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movsesian MA, Karimi M, Green K, Jones LR. Ca(2+)-transporting ATPase, phospholamban, and calsequestrin levels in nonfailing and failing human myocardium. Circulation. 1994;90:653–657. doi: 10.1161/01.cir.90.2.653. [DOI] [PubMed] [Google Scholar]
- Muller OJ, Lange M, Rattunde H, Lorenzen HP, Muller M, Frey N, Bittner C, Simonides W, Katus HA, Franz WM. Transgenic rat hearts overexpressing SERCA2a show improved contractility under baseline conditions and pressure overload. Cardiovasc Res. 2003;59:380–389. doi: 10.1016/s0008-6363(03)00429-2. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou P, Rodriguez P, Ren X, Zhou X, Qian J, Sadayappan S, Mitton B, Pathak A, Robbins J, Hajjar RJ, et al. Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ Res. 2009;104:1012–1020. doi: 10.1161/CIRCRESAHA.108.189811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
- Pinz I, Tian R, Belke D, Swanson E, Dillmann W, Ingwall JS. Compromised myocardial energetics in hypertrophied mouse hearts diminish the beneficial effect of overexpressing SERCA2a. J Biol Chem. 2011;286:10163–10168. doi: 10.1074/jbc.M110.210757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006;98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- Ramesh V, Sharma VK, Sheu SS, Franzini-Armstrong C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann N Y Acad Sci. 1998;853:341–344. doi: 10.1111/j.1749-6632.1998.tb08295.x. [DOI] [PubMed] [Google Scholar]
- Remppis A, Greten T, Schafer BW, Hunziker P, Erne P, Katus HA, Heizmann CW. Altered expression of the Ca(2+)-binding protein S100A1 in human cardiomyopathy. Biochimica et biophysica acta. 1996;1313:253–257. doi: 10.1016/0167-4889(96)00097-3. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol. 2000;529(Pt 1):37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, Sabbah HN, Hoppel CL. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80:30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainte Beuve C, Allen PD, Dambrin G, Rannou F, Marty I, Trouve P, Bors V, Pavie A, Gandgjbakch I, Charlemagne D. Cardiac calcium release channel (ryanodine receptor) in control and cardiomyopathic human hearts: mRNA and protein contents are differentially regulated. Journal of molecular and cellular cardiology. 1997;29:1237–1246. doi: 10.1006/jmcc.1996.0360. [DOI] [PubMed] [Google Scholar]
- Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, Tsuji T, Takewa Y, del Monte F, Peluso R, et al. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol. 2007;42:852–861. doi: 10.1016/j.yjmcc.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Katoh H, Satoh H, Nagasaka S, Yoshihara S, Terada H, Hayashi H. Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2005;288:H1820–1828. doi: 10.1152/ajpheart.00589.2004. [DOI] [PubMed] [Google Scholar]
- Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- Schroder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, Schwinger RH, Weil J, Herzig S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation. 1998;98:969–976. doi: 10.1161/01.cir.98.10.969. [DOI] [PubMed] [Google Scholar]
- Schwinger RH, Munch G, Bolck B, Karczewski P, Krause EG, Erdmann E. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J Mol Cell Cardiol. 1999;31:479–491. doi: 10.1006/jmcc.1998.0897. [DOI] [PubMed] [Google Scholar]
- Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, et al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Asai K, Uechi M, Mathier MA, Shannon RP, Vatner SF, Ingwall JS. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: a compensatory role for the parallel loss of creatine. Circulation. 1999;100:2113–2118. doi: 10.1161/01.cir.100.20.2113. [DOI] [PubMed] [Google Scholar]
- Siddiqi N, Singh S, Beadle R, Dawson D, Frenneaux M. Cardiac metabolism in hypertrophy and heart failure: implications for therapy. Heart Fail Rev. 2013;18:595–606. doi: 10.1007/s10741-012-9359-2. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Volders PG, Vos MA, Verdonck F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc Res. 2002;53:782–805. doi: 10.1016/s0008-6363(01)00470-9. [DOI] [PubMed] [Google Scholar]
- Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Mol Cell Biochem. 1998;180:171–177. [PubMed] [Google Scholar]
- Suckau L, Fechner H, Chemaly E, Krohn S, Hadri L, Kockskamper J, Westermann D, Bisping E, Ly H, Wang X, et al. Long-term cardiac-targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation. 2009;119:1241–1252. doi: 10.1161/CIRCULATIONAHA.108.783852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilemann L, Lee A, Ishikawa K, Aguero J, Rapti K, Santos-Gallego C, Kohlbrenner E, Fish KM, Kho C, Hajjar RJ. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci Transl Med. 2013;5:211ra159. doi: 10.1126/scitranslmed.3006487. [DOI] [PubMed] [Google Scholar]
- Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zwaag PA, van Rijsingen IA, de Ruiter R, Nannenberg EA, Groeneweg JA, Post JG, Hauer RN, van Gelder IC, van den Berg MP, van der Harst P, et al. Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth Heart J. 2013;21:286–293. doi: 10.1007/s12471-013-0401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkers M, Loughrey CM, Macquaide N, Remppis A, DeGeorge BR, Jr, Wegner FV, Friedrich O, Fink RH, Koch WJ, Smith GL, et al. S100A1 decreases calcium spark frequency and alters their spatial characteristics in permeabilized adult ventricular cardiomyocytes. Cell calcium. 2007;41:135–143. doi: 10.1016/j.ceca.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Walters AM, Porter GA, Jr, Brookes PS. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ Res. 2012;111:1222–1236. doi: 10.1161/CIRCRESAHA.112.265660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Marks AR. Altered function and regulation of cardiac ryanodine receptors in cardiac disease. Trends in biochemical sciences. 2003;28:671–678. doi: 10.1016/j.tibs.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Wilding JR, Joubert F, de Araujo C, Fortin D, Novotova M, Veksler V, Ventura-Clapier R. Altered energy transfer from mitochondria to sarcoplasmic reticulum after cytoarchitectural perturbations in mice hearts. J Physiol. 2006;575:191–200. doi: 10.1113/jphysiol.2006.114116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopper K, Dobrev D, Eschenhagen T, El-Armouche A. Phosphatase-1 inhibitor-1 in physiological and pathological beta-adrenoceptor signalling. Cardiovasc Res. 2011;91:392–401. doi: 10.1093/cvr/cvr058. [DOI] [PubMed] [Google Scholar]
- Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, Walsh MP, Warltier DC, Cheng H, Chen SR. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circulation research. 2005;96:847–855. doi: 10.1161/01.RES.0000163276.26083.e8. [DOI] [PubMed] [Google Scholar]
- Yamada M, Ikeda Y, Yano M, Yoshimura K, Nishino S, Aoyama H, Wang L, Aoki H, Matsuzaki M. Inhibition of protein phosphatase 1 by inhibitor-2 gene delivery ameliorates heart failure progression in genetic cardiomyopathy. FASEB J. 2006;20:1197–1199. doi: 10.1096/fj.05-5299fje. [DOI] [PubMed] [Google Scholar]
- Yan Y, Liu J, Wei C, Li K, Xie W, Wang Y, Cheng H. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc Res. 2008;77:432–441. doi: 10.1093/cvr/cvm047. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Maulik N, Engelman RM, Ho YS, Das DK. Targeted disruption of the mouse Sod I gene makes the hearts vulnerable to ischemic reperfusion injury. Circ Res. 2000;86:264–269. doi: 10.1161/01.res.86.3.264. [DOI] [PubMed] [Google Scholar]
- Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Ziolo MT, Martin JL, Bossuyt J, Bers DM, Pogwizd SM. Adenoviral gene transfer of mutant phospholamban rescues contractile dysfunction in failing rabbit myocytes with relatively preserved SERCA function. Circ Res. 2005;96:815–817. doi: 10.1161/01.RES.0000163981.97262.3b. [DOI] [PubMed] [Google Scholar]
- Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res. 2014;114:101–108. doi: 10.1161/CIRCRESAHA.113.302421. [DOI] [PubMed] [Google Scholar]