

Abstract
The first synthesis of any and all members of the mezzettiaside family of natural products has been achieved. The reported synthesis features the iterative use of the Taylor catalyst in a dual nucleophilic boron/electrophilic palladium catalyzed regioselective glycosylation. In addition, the de novo approach utilizes atomless protecting groups and the minimal use of protecting groups (2 chloroacetates for the synthesis of 10 natural products). These divergent syntheses occurred in a range of 13 to 22 longest linear steps and required only 41 total steps to prepare the entire family of mezzettiasides.
Introduction
The mezzettiasides are a family of partially acetylated oligorhamnose natural products that were isolated from the fruit and bark of several medicinally relevant Mezzettia leptopoda Annonaceae plants located in the Malaysian island of Borneo.1 These anticancer natural products were shown to consist of α-1,3-linked L-rhamno-di-, tri- and tetra-saccharides with differing patterns of acetylation (Fig. 1). Initial testing of the mezzettiasides revealed their μg/mL cytotoxicity against three human cell lines (KB, Col2 and Lu1).2
Figure 1.

The mezzettiaside family of natural products
As part of a program aimed at the synthesis and biological investigation of anticancer/antibiotic oligosaccharide natural products,3 we became interested in the synthesis of mezzettiasides. This interest came out of our synthesis and study of a related class of tri- and tetrasaccharide natural products, the cleistriosides and cleistetrosides, 4 where we found the pattern of acetylation had a significant effect on the biological activity. As a continuation of these studies, we desired access to all ten members of the mezzettiaside natural products. While re-isolation of these materials from the various sources was considered, we instead decided upon a de novo asymmetric synthetic approach, 5 as this approach would nicely position us for further structure activity relationship (SAR) studies.6 We were particularly intrigued at the possibility of efficiently assembling all members of the mezzettiasides via a divergent approach that would minimize the use of protecting groups. Herein, we disclose our successful de novo asymmetric approach to all ten members of the mezzettiaside where all the stereocenters in the represented natural products were derived from achiral acetyl furan (1).
Retrosynthetically, we envisioned that the three tetrasaccharide (5-7) and the four trisaccharide (2-4 & 8) mezzettiasides could be obtained from a common pyranone containing trisaccharide intermediate 13 (Scheme 1). In turn, trisaccharide 13 as well as, the three disaccharide natural products (9-11) could be prepared from pyran containing rhamnoside 12. Finally allylic alcohol 12 could be obtained from achiral 2-acetyl furan (1) using our de novo approach to carbohydrates. 7 Key to the success of this approach is the strategic use of atomless/minimal protecting groups (enone of a pyranone as a masked triol and a chloroacetate as the only protecting group) in combination with the iterative use of a highly stereo- and regio-selective organoboron/Pd-catalyzed glycosylation (i.e., a dual nucleophilic/electrophilic catalysis).8,9
Scheme 1.

Retrosynthetic analysis of mezzettiasides
Results and discussion
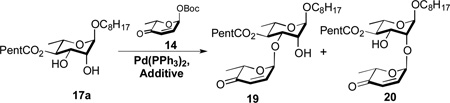
Our synthesis began with pyranone 14, which can be prepared in optically pure form in 3 steps from achiral furan 1.7, 10 Glycosylation of 1-octanol with pyranone 14 using our typical conditions (Pd2(dba)3•CHCl3/4PPh3 in CH2Cl2)6 gave enone 15 (93%). Luche reduction (NaBH4/CeCl3) of enone 15 gave allylic alcohol 16 (80%). The C-4 hexanoate ester was installed by a Steglich esterification (DCC/C5H11CO2H/DMAP) and an Upjohn dihydroxylation (OsO4/NMO(aq)) was used to diastereoselectively install the C-2/3 rhamno-stereochemistry (16 to 17a) (75%, 2 steps).11
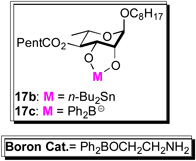
We next investigated on a catalytic regio-/stereoselective glycosylation of the C-3 hydroxyl of 17a. When the Pd-glycosylation was performed on diol 17a, a 1:2 mixture of regioisomer (C-3 vs C-2) was produced. Previously, we have found that the tin acetal of similar diols can react regioselectively (~7:1) under similar Pd-glycosylation conditions.4 The tin acetal 17b formation, however, was difficult to optimize, as conversion to the crude tin acetal must be complete to get good yields and regioselectivities. When this 2-step procedure was performed under optimal conditions, a 5:1 mixture of C-3 to C-2 regioisomers was obtained (68%) (Table 1).
Table 1.
Boron vs Tin mediated regioselective glycosylation
 | ||||
|---|---|---|---|---|
| Additive | solvent(s) | 19:20 | yield | |
 |
none | CH2Cl2 | 1:2 | 61% |
| Bu2Sn=O (1.1 equiv) |
CH2Cl2 | 5:1 | 68% | |
|
Boron cat. (15 mol%) |
CH2Cl2 | 2.5:1 | 61% | |
|
Boron cat. (10 mol%) |
CH3CN/CH2Cl2 (1:0.2) |
3:1 | 65% | |
|
Boron cat. (15 mol%) |
CH3CN/THF (1:0.1) |
6:1 | 74% | |
|
Boron cat. (30 mol%) |
CH3CN/THF (1:0.1) |
7.5:1 | 77% | |
A practical alternative to this difficult to execute procedure emerged when we replaced the stochiometric tin reagent with the catalytic boron reagent developed by Taylor (Ph2BOCH2CH2NH2) resulting in a dual B-nucleophilic/Pd-electrophilic catalyzed glycosylation.12,13 For instance, when the Taylor catalyst was added to our typical glycosylation conditions (2.5% Pd2(dba)3•CHCl3/4PPh3, 15 mol% Ph2BOCH2CH2NH2 in CH2Cl2) a 2.5:1 mixture of C-3 to C-2 regioisomers was obtained. This ratio was significantly increased to 6:1, when the solvent system was changed from CH2Cl2 to CH3CN/THF (10:1). Varying the percentage of the boron catalyst from 10 to 30 percent found the optimal B/Pd ratio to be 6:1 for this glycosylation reaction. Presumably the anionic nature of the borinate complex 17c makes it an ideal coupling partner with the cationic Pd-π-allyl complex (e.g., intermediate 18, Fig. 2), whereas the more polar solvent improves the turnover rate for the formation of borinate 17c. Thus, rendering it significantly more reactive than the neutral diol 17a and as a result requiring on catalytic amounts to impart excellent regiocontrol.
Figure 2.

Plausible transition state for glycosylation via dual catalysis
Under our optimized conditions (2.5 mol% Pd2(dba)3•CHCl3/4PPh3, 30 mol% Ph2BOCH2CH2NH2 in CH3CN/THF(10:1)) a mixture of 17a/14 (1:1.1) was coupled in an 77% yield with good regiocontrol (7.5:1). It is worth noting that when the C-4 ester is smaller (e.g., OAc), significantly better regiocontrol (16:1) was observed in this B-/Pd-dual catalyzed glycosylation (e.g., 23 to 24, Scheme 5). The mixture of regioisomeric products 19 and 20 was difficult to separate at preparative scales. This was easily solved by protecting the remaining hydroxyl group 19 and 20 as a chloroacetate (21) and reducing the enone to give pure allylic alcohol 12 in 85% after silica gel chromatography.
Scheme 5.

Synthesis of mezzettiaside-5
The allylic alcohol portion of 12 could be easily elaborated into the three disaccharide members of the mezzettiasides 9-11 (Scheme 3). Synthesis of mezzettiaside-9 was accomplished in four steps (64%). Specifically, the allylic alcohol 12 underwent chloroacetylation at the C-4 position ((ClAc)2O, 10 mol% DMAP in Py), followed by an Upjohn dihydroxylation (OsO4/NMO(aq)), bis-acetylation (Ac2O/Py) and finally bis-deprotection of the two chloroacetate groups (thiourea, NaHCO3 and n-Bu4NI) 14 to give mezzettiaside-9. When the chloroacetylation step was removed from the above sequence, the resulting three steps sequence provides mezzettiaside-11, in overall good yield (66%). The regioisomeric diacetate mezzettiaside-10 was prepared by a slightly different sequence. The allylic alcohol 12 was C-4 acetylated, dihydroxylated, and regioselectively acetylated via orthoester formation to give the protected disaccharide 22 (68%, 3 steps). Once again, selective removal to the C-2 chloroacetate group gave mezzettiaside-10 (86%). In addition to being an intermediate for the synthesis of mezzettiaside-10, disaccharide 22 proved to be the launching point for the syntheses of the remaining tri- and tetrasaccharide members of the mezzettiasides (Schemes 4-6).
Scheme 3.

Synthesis of mezzettiaside-9, 10 & 11
Scheme 4.

Synthesis of mezzettiaside-2, 3, 4 & 8
Scheme 6.

Synthesis of the tetrasaccharide mezzettiaside-6 & 7
The application of this approach to the trisaccharide mezzettiasides 2-4 and 8, began with the Pd-catalyzed glycosylation of disaccharide 22 with pyranone 14 to give trisaccharide 13 (68%). A four step post glycosylation sequence was used to convert trisaccharide 13 into mezzettiaside 8. This involved a two-step post-glycosylation transformation (NaBH4/CeCl3 and OsO4/NMO(aq)) of the enone functionality of 13 into a rhamno-sugar, an orthoester mediated axial acetylation at C-2 followed by chloroacetate deprotection to give mezzettiaside 8 (53%, 4 steps).
The key tris-rhamno-trisaccharide intermediate 23 for the synthesis of mezzettiaside 2, 3 and 4 was synthesized from trisaccharide 13 by incorporating a C-4 acetylation into the above post-glycosylation sequence (NaBH4/CeCl3, Ac2O/Py and OsO4/NMO(aq)), (Scheme 4). Mezzettiaside 4 was obtained by removing the chloroacetate protecting group (23 to 4, 82%). The required C-2 acetylation in mezzettiaside 2 was achieved via orthoester while the C-3 acetate for mezzettiaside 3 was obtained via Taylor borinate catalyzed acetylation of 23.12 Finally removal of chloroacetate gave mezzettiaside 2 and mezzettiaside 3 in 64% and 58% overall yield respectively (Scheme 4).
As with disaccharide 22, trisaccharide 23 served as a point of divergence for the route to the final three tetrasaccharide mezzettiasides 5-7 (Scheme 5). The route to the tetrasaccharide natural products began with a second regioselective C-3 glycosylation (23 and 14). This was accomplished once again with use of the Taylor catalyst in the dual B-nucleophilic/Pd-electrophilic catalyzed glycosylation (2.5 mol% Pd2(dba)3•CHCl3/4PPh3, 15 mol% Ph2BOCH2CH2NH2 in THF/CH3CN) to give tetrasaccharide enone 24 in excellent yield 76%. In contrast to 17a with a more hindered C-4 hexanoate, for diol 23 with a C-4 acetate, only 15 mol% boron catalyst was needed to achieve near complete regiocontrol (>16:1) in the glycosylation. The resulting enone was converted into a rhamno-sugar 25 by a successive Luche reduction and Upjohn dihydroxylation (73%, 2 steps). As before, a regioselective C-3 acetylation using catalytic borinate followed by chloroacetate deprotection converted compound 25 into mezzettiaside-5 (58%, 2 steps). In contrast, a C-2 acetylation via orthoester and a successive chloroacetate deprotection gave mezzettiaside-6 (25 to 6; 61%, 2 steps). Whereas, a direct chloroacetate deprotection of 25 provided the final tetrasaccharide natural product, mezzettiaside-7 (83%).
Comparison of the spectral data (1H, 13C NMR) for synthetic mezzettiasides with the data reported for the isolated materials was complicated by the choice of mixed solvent systems (CD3OD/C6D6) used in the isolation work. The same features that allow for increase resolution of the 1H and 13C spectra from the use of CD3OD/C6D6 also lead to the increased sensitivity to small changes in solvent ratio and water impurities. In addition, slightly different chemical shifts are obtained depending on which solvent is used as the reference standard. This problem with carbohydrate natural products has been previously observed by us15 and others.16 By exercising with great care in the solvent ratios we were able to generate 13C NMR spectra for nine of the ten mezzettiasides were the shifts fell within (+/−) 0.5 ppm. Unfortunately, for mezzettiaside 8, we were unsuccessful at finding the identical solvent system to reproduce the spectra of the natural material. Using the reported ratio (CD3OD:C6D6 (6:1)) the difference in 13C NMR shifts was as high as (+/−) 0.9 ppm. It should be noted that the multiplicities and coupling constants in 1H NMR were in agreement with that found for the synthetic material and consistent with the structure reported. In order to confirm the 3D-structure of the synthetic material, a detailed NMR studies were conducted on synthetic mezzettiaside-8 (see, SI) as well as mezzettiasides-3 and 4. Thus, we believe that the structure of the synthetic and isolated mezzettiaside-8 are the same. In this regard, we recommend future comparisons to mezzettiaside NMR data should be to the CDCl3 and C6D6 spectra for the mezzettiasides reported herein.
Conclusions
In conclusion, we have successfully completed the first total synthesis of any and all members of the mezzettiaside family of natural products. This divergent synthesis of all 10 mezzettiasides occurred in a range of 13 to 22 longest linear steps. A more notable measure of efficiency is that the entire family of natural products was prepared in only 41 total steps (~ 4 steps/natural product). This highly efficient, regio- and stereo-selective divergent synthesis relied on the strategic use of a novel dual B-/Pd-catalyzed glycosylation, which was used to construct two of the four-glycosidic bonds in progressively more complex settings. Additional efficiencies were achieved by the use of numerous regioselective acetylation as well as an atomless-protecting group strategy which minimized the number of protecting groups required to only two chloroacetate for 10 natural products. The synthesis was amenable for the preparation of significant quantities of each member of the mezzettiasides (5-15 mg) for further biological studies, which will be reported in due course.
Supplementary Material
Scheme 2.

Synthesis of key disaccharide intermediate 12
Acknowledgements
We are grateful to the NIH (GM088839) and NSF (CHE-0749451) for their generous support of our research program.
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures, 1H and 13C NMR spectra and correlation studies.
See DOI: 10.0000/x0xx00000x
Author Contributions
S.O.B conducted all the synthetic work. N.G.A. was responsible for the NMR structure elucidation/confirmation. E.U.S. and G.A.O. provided guidance. All authors contributed to the preparation of the manuscript.
Complete experimental procedures and spectral data for all new compounds. This material is available free of charge via the internet at DOI: 10.1039/x0xx00000x
Notes and references
- 1.(a) Etse JT, Gray AI, Lavaud C, Massiot G, Nuzillard J-M, Waterman PG. J. Chem. Soc., Perkin Trans. 1. 1991:861. [Google Scholar]; (b) Powell DA, York WS, Halbeek HV, Etse JT, Gray AI, Waterman PG. Can. J. Chem. 1990;68:1044–1050. [Google Scholar]
- 2.Cui B, Chai H, Santisuk T, Reutrakul V, Farnsworth NR, Cordell GA, Pezzuto JM, Kinghorn AD. J. Nat. Prod. 1998;61:1535. doi: 10.1021/np980270j. [DOI] [PubMed] [Google Scholar]
- 3.Shi P, Silva MC, Wang HL, Akhmedov NG, Li M, Beuning PJ, O’Doherty GA. ACS Med. Chem. Lett. 2012;3(12):1086. doi: 10.1021/ml300303g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Wu D, Li M, O’Doherty GA. Org. Lett. 2010;12(23):5466. doi: 10.1021/ol1023344. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seidel V, Bailleul F, Waterman PG. J. Nat. Prod. 2000;63:6. doi: 10.1021/np9901478. [DOI] [PubMed] [Google Scholar]
- 5.(a) Ko SY, Lee AW, Masamune S, Reed LA, Sharpless KB. Science. 1983;220:949. doi: 10.1126/science.220.4600.949. [DOI] [PubMed] [Google Scholar]; (b) Northrup AB, MacMillan DWC. Science. 2004;305:1752. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]; (c) Ahmed Md M, Berry BP, Hunter TJ, Tomcik DJ, O'Doherty GA. Org. Lett. 2005;7:745. doi: 10.1021/ol050044i. [DOI] [PubMed] [Google Scholar]; (d) Harris JM, Keranen MD, Nguyen H, Young VG, O’Doherty GA. Carbohydr. Res. 2000;328:17. doi: 10.1016/s0008-6215(00)00031-8. [DOI] [PubMed] [Google Scholar]; (e) Harris JM, Keranen MD, O'Doherty GA. J. Org. Chem. 1999;64:2982–2983. doi: 10.1021/jo990410+. [DOI] [PubMed] [Google Scholar]
- 6.(a) Babu RS, O'Doherty GA. J. Am. Chem. Soc. 2003;125:12406. doi: 10.1021/ja037097k. [DOI] [PubMed] [Google Scholar]; (b) Babu RS, Zhou M, O'Doherty GA. J. Am. Chem. Soc. 2004;126:3428. doi: 10.1021/ja039400n. [DOI] [PubMed] [Google Scholar]; (c) Babu RS, O'Doherty GA. J. Carb. Chem. 2005;24:169. [Google Scholar]; (d) Guo H, O'Doherty GA. Angew. Chem. Int. Ed. 2007;46:5206. doi: 10.1002/anie.200701354. [DOI] [PubMed] [Google Scholar]; (e) Guo H, O'Doherty GA. J. Org. Chem. 2008;73:5211. doi: 10.1021/jo800691v. [DOI] [PubMed] [Google Scholar]
- 7.(a) Li M, Scott JG, O’Doherty GA. Tetrahedron Lett. 2004;45:1005. [Google Scholar]; (b) Guo H, O’Doherty GA. Org. Lett. 2005;7:3921. doi: 10.1021/ol051383e. [DOI] [PubMed] [Google Scholar]
- 8.For an example of a dual catalyzed phosphine/Pd reactions, see: Jellerichs BG, Kong J-R, Krische MJ. J. Am. Chem. Soc. 2003;125:7758. doi: 10.1021/ja0301469.. For an example of Sn/Pd catalysis, see: Kuriyama M, Takeichi T, Ito M, Yamasaki N, Yamamura R, Demizu Y, Onomura O. Chem. Eur. J. 2012;18:2477. doi: 10.1002/chem.201102709.
- 9.For an example of a stepwise nucleophilic phosphine then Pd-catalyzed reactions, see: Fan Y, Kwon O. Org. Lett. 2012;14:3264. doi: 10.1021/ol301154f.
- 10.(a) Shan M, O’Doherty GA. Org. Lett. 2010;12:2986. doi: 10.1021/ol101009q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shan M, O’Doherty GA. Org. Lett. 2006;8:5149. doi: 10.1021/ol062076r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Noyori R, Ohkuma T, Kitamura M. J. Am. Chem. Soc. 1987;109:5856. [Google Scholar]; (d) Achmatowicz O, Bukowski P, Szechner B, Zwierzchowska Z, Zamojski A. Tetrahedron. 1971;27:1973. [Google Scholar]
- 11. In general, we have found the NaBH4 (with and without CeCl3) reduction of α-pyranones (13, 15, 19 and 24) and the OsO4 catalyzed dihydroxylation of the resulting allylic alcohol (e.g., 12, 16) to proceed with virtually complete diastereocontrol, see refs: 4, 6, 7 10.
- 12.(a) Lee D, Taylor MS. J. Am. Chem. Soc. 2011;133:3724. doi: 10.1021/ja110332r. [DOI] [PubMed] [Google Scholar]; (b) Gouliaras C, Lee D, Chan L, Taylor MS. J. Am. Chem. Soc. 2011;133:13926. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]; (c) McClary CA, Taylor MS. Carbohydr. Res. 2013;381:112. doi: 10.1016/j.carres.2013.09.001. [DOI] [PubMed] [Google Scholar]; (d) Dimitrijevic E, Taylor MS. Chem. Sci. 2013;4:3298. [Google Scholar]; (e) Beale TM, Taylor MS. Org. Lett. 2013;15:1358. doi: 10.1021/ol4003042. [DOI] [PubMed] [Google Scholar]
- 13. While the Taylor catalyst has been used in regioselective glycosylations (glycosylbromide with excess Ag2O, ref. 12), this is the first example of its use to catalytically induce regiocontrol in a catalyzed glycosylation.
- 14.(a) Clausen MH, Madsen R. Chem.-Eur. J. 2003;9:3821–3832. doi: 10.1002/chem.200204636. [DOI] [PubMed] [Google Scholar]; (b) Naruto M, Ohno K, Takeuchi N. Tetrahedron Lett. 1979;251 [Google Scholar]
- 15.Sharif EU, Wang HL, Akhmedov NG, O’Doherty GA. Org. Lett. 2014;16:492. doi: 10.1021/ol403369h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Nagano T, Pospísil J, Chollet G, Schulthoff S, Hickmann V, Moulin E, Herrmann J, Müller R, Fürstner A. Chem. Eur. J. 2009;15:9697. doi: 10.1002/chem.200901449. [DOI] [PubMed] [Google Scholar]; (b) Molinaro A, De Castro C, Lanzetta R, Manzo E, Parrilli MJ. J. Am. Chem. Soc. 2001;123:12605. doi: 10.1021/ja016471i. [DOI] [PubMed] [Google Scholar]; (c) Bekiroglu S, Sandström A, Kenne L, Sandström C. Org. Biomol. Chem. 2004;2:200. doi: 10.1039/b311852e. [DOI] [PubMed] [Google Scholar]; (d) Kondoh A, Arlt A, Gabor B, Fürstner A A. Chem. Eur. J. 2013;19:7731. doi: 10.1002/chem.201300827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.