

Abstract
Foxp3+ regulatory T cells are abundant in the intestine where they prevent dysregulated inflammatory responses to self and environmental stimuli. It is now appreciated that Treg cells acquire tissue-specific adaptations that facilitate their survival and function1; however, key host factors controlling the Treg response in the intestine are poorly understood. IL-1 family member IL-33 is constitutively expressed in epithelial cells at barrier sites2 where it functions as an endogenous danger signal or alarmin following tissue damage3. Recent studies in humans have described high levels of IL-33 in inflamed lesions of inflammatory bowel disease (IBD) patients4-7 suggesting a role for this cytokine in the pathogenesis of IBD. In the intestine, both protective and pathologic roles for IL-33 have been described in murine models of acute colitis8-11 but its contribution to chronic inflammation remains ill defined. Here we show that the IL-33 receptor ST2 is preferentially expressed on colonic Treg (cTreg) cells, where it promotes Treg function and adaptation to the inflammatory environment. IL-33 signaling into T cells stimulates Treg responses in several ways. Firstly, it enhances transforming growth factor-β1 (TGF-β1) mediated differentiation of Treg cells and secondly, it provides a necessary signal for Treg accumulation and maintenance in inflamed tissues. Strikingly, IL-23, a key pro-inflammatory cytokine in the pathogenesis of IBD, restrained Treg responses through inhibition of IL-33 responsiveness. These results demonstrate a hitherto unrecognized link between an endogenous mediator of tissue damage and a major anti-inflammatory pathway, and suggest that the balance between IL-33 and IL-23 may be a key controller of intestinal immune responses.
To identify potential tissue-specific modulators of cTreg cells, we compared the mRNA expression profiles of mesenteric lymph node (MLN) and cTreg cells. We identified St2, the transcript coding for the IL-33 receptor12, as one of the top differentially upregulated genes in cTreg cells (Fig. 1a,b). Flow-cytometric analysis confirmed selective enrichment of ST2+ Treg cells in the colon (Fig. 1c) and these cells expressed high levels of the activation markers KLRG1, CD103 and OX40 (Fig. 1d). Analysis of Helios expression revealed that ST2+ Treg cells are a heterogeneous population containing thymus-derived T regulatory (tTreg) cells as well as peripherally generated Helios− Treg cells (Fig. 1e)13. A significant proportion of intestinal Foxp3+ Treg cells co-express the transcription factor GATA314-16 and GATA3 is known to regulate ST2 expression in Th2 cells17. Indeed, ST2 expression was largely restricted to GATA3-expressing cTreg cells (Fig. 1e) and selective ablation of GATA3 in Foxp3-expressing cells, using Gata3f/f-Foxp3-Cre mice15, caused a marked reduction of ST2 protein levels (Fig. 1f).
Figure 1. ST2-expressing Treg cells are enriched in the colon.

a, Change in gene expression in cTreg cells vs. MLN Treg cells (n=3 per group) presented as volcano plot. b, Top differentially upregulated transcripts in cTreg vs. MLN Treg cells. c, ST2 protein expression on Treg cells from indicated organs. d, Phenotypic analysis of ST2− or ST2+ cTreg cells. e, Expression of transcription factors in cTreg cells. f, Representative histograms gated on cTreg cells from control or Gata3f/f-Foxp3-Cre mice.
Given that ST2+ Treg cells are prominent in the colon we postulated that IL-33 may modulate in vitro-induced Treg-cell (iTreg) differentiation. To test this, we sort-purified naïve CD4+ T cells from Foxp3gfp reporter mice and activated them in the presence of TGF-β1. Notably, both Gata3 and St2 expression were induced under iTreg-differentiation conditions (Extended Data Fig. 1). Addition of IL-33 to iTreg cultures significantly increased both the percent and total number of Foxp3-expressing cells but had no effect on Foxp3 expression in the absence of TGF-β1 (Fig. 2a). The presence of IL-33 in iTreg cultures did not affect induction of Th2 cytokines or expression of Th1 and Th17-associated transcription factors Tbx21 and Rorc (Extended Data Fig. 1), suggesting that IL-33 preferentially regulates Foxp3 expression. Thus, our data indicate that the alarmin IL-33 is a novel co-factor in TGF-β1-mediated iTreg generation.
Figure 2. Effects of IL-33 on iTreg and tTreg cells.

a, Naïve CD4+ T cells were cultured with anti-CD3/CD28 plus indicated cytokines and the frequencies and absolute numbers of Foxp3+ T cells determined 3 days later (mean ± s.e.m. of three independent experiments). b, Naïve CD4+ T cells were cultured for 48h with anti-CD3/CD28 plus TGF-β1 followed by stimulation with IL-33 for 45 minutes. Blots are representative of two independent experiments. c-d, Cells were cultured and stimulated as in (b) and recruitment of GATA3 or RNA polymerase II (Pol II) to the indicated regions assessed by ChIP-qPCR. Data are from one experiment representative of two (mean ± s.d.). e, Representative plots of Treg cells cultured with anti-CD3/CD28 plus indicated cytokines and analyzed after 3 days. Data are representative of three independent experiments. f, Treg cells were cultured with anti-CD3/CD28 for 24h followed by stimulation with IL-33. Blots are representative of three independent experiments. g, Mixed chimaeras were generated containing WT and St2−/− bone marrow cells. Reconstituted mice were analyzed at steady state or two weeks after infection with Helicobacter hepaticus and anti-IL-10R treatment (inflamed). Absolute numbers of WT or St2−/− Treg cells in steady state (n = 3) and inflamed (n = 6) hosts (mean ± s.e.m). h, Analysis of Foxp3 expression in Treg cells from inflamed chimaeric hosts presented as geometric mean fluorescence intensity (gMFI). *P<0.05, **P<0.01 *** P<0.001 as calculated by 1way-ANOVA with Bonferroni post-test or paired Students’ t-test.
GATA3 is highly expressed in ST2+ Treg cells (Fig. 1e) and IL-33 has been shown to activate GATA3 in Th2 cells18 as well as innate lymphoid cells19. Consistent with this notion, we observed serine phosphorylation of GATA3 upon acute stimulation of iTreg cells with IL-33 (Fig. 2b). The Foxp3 locus contains putative GATA3-binding sites within its promoter and intragenic conserved noncoding sequences (CNS) 1-314. To investigate whether IL-33 influences the binding of GATA3 to any of these elements in iTreg cells, we performed chromatin immunoprecipitation (ChIP) followed by quantitative PCR. Acute stimulation of iTreg cells with IL-33 induced GATA3 recruitment to the Foxp3 promoter but not CNS1, 2 or 3 (Fig. 2c). In addition, RNA polymerase II (Pol II) was recruited to the Foxp3 promoter upon IL-33 stimulation (Fig. 2d) suggesting that IL-33 directly regulates Foxp3 expression through activation and recruitment of GATA3 to the Foxp3 promoter. In Th2 cells, GATA3 has been shown to promote St2 gene expression by binding to an enhancer element located 12-kb upstream of the St2 transcription start site17. Consistent with this we detected recruitment of GATA3 to the St2 enhancer upon acute stimulation of iTreg cells with IL-33 and this correlated with RNA Pol II enrichment at the St2 promoter (Extended Data Fig. 2). Thus, in addition to its role in Foxp3 induction, IL-33 also promoted its own receptor expression in iTreg cells through direct transcriptional regulation of the St2 locus providing an amplification loop for further enhancement of iTreg-cell differentiation.
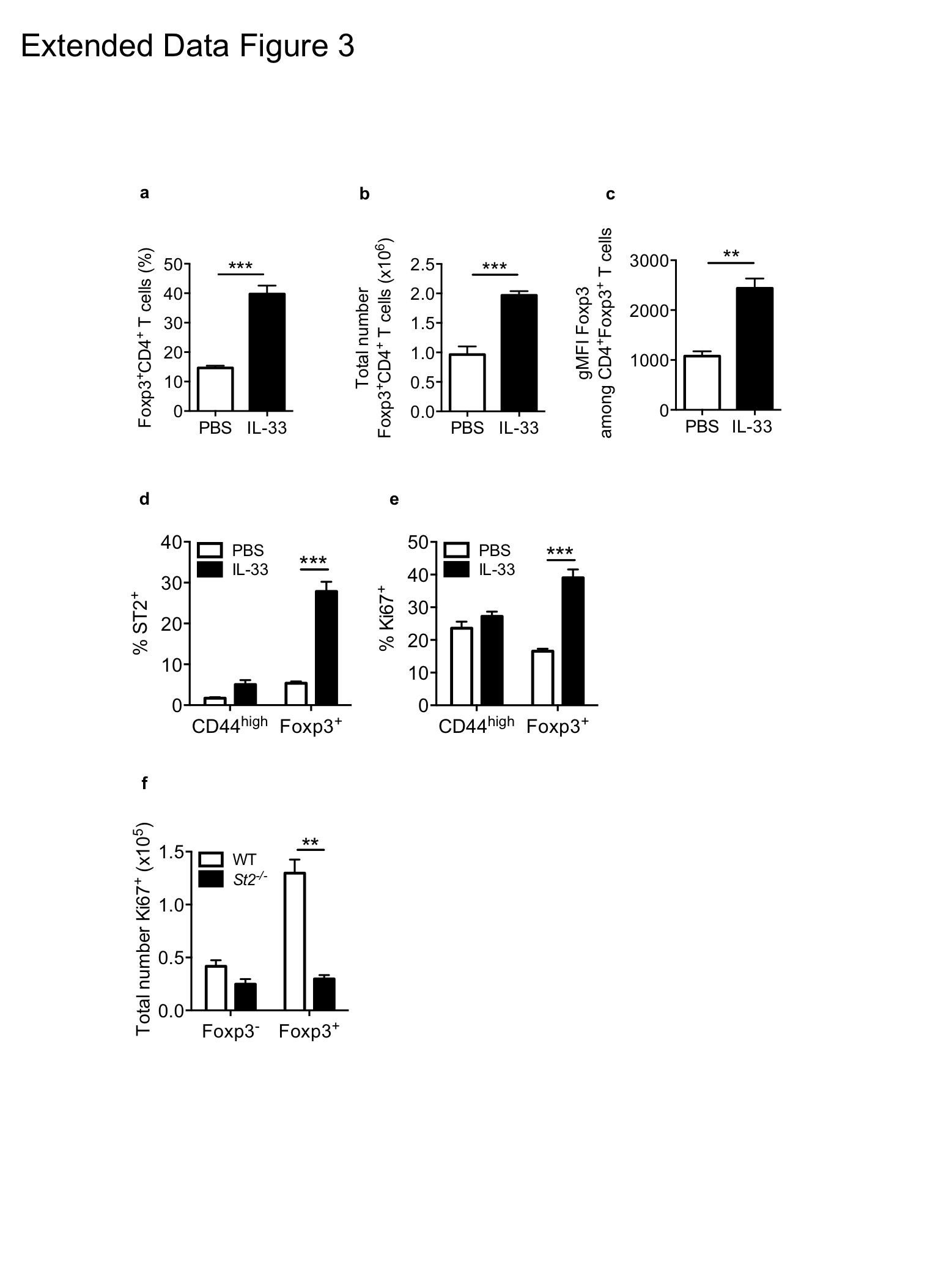
Next we focused on thymus-derived Treg cells, which constitute a significant proportion of ST2+ cTreg cells (Fig. 1e). In line with published reports20,21 administration of recombinant IL-33 led to a significant increase in the frequency and total number of splenic Treg cells (Extended Data Fig. 3a,b) and these IL-33-elicted Treg cells expressed higher levels of Foxp3 and ST2 (Extended Data Fig. 3c,d). Further analysis of proliferation marker Ki67 showed IL-33 induced proliferation in splenic Treg cells but not T effector cells (Extended Data Fig. 3e). To examine if IL-33 acts directly on Treg cells we injected IL-33 into chimeric mice containing a mixture of WT and St2−/− hematopoietic cells. In this setting, the proliferative capacity of St2−/− Treg cells was significantly impaired (Extended Data Fig. 3f), suggesting that IL-33 acts directly on tTreg cells to promote their proliferation and accumulation in vivo. This is further supported by the finding that sort-purified splenic Treg cells cultured in the presence of IL-33 expressed higher levels of ST2, showed a more activated phenotype and expressed increased amounts of Foxp3 protein (Fig. 2e). In addition, acute stimulation of TCR-activated splenic Treg cells with IL-33 induced serine phosphorylation of GATA3 (Fig. 2f) further demonstrating that IL-33 acts directly on tTreg cells.
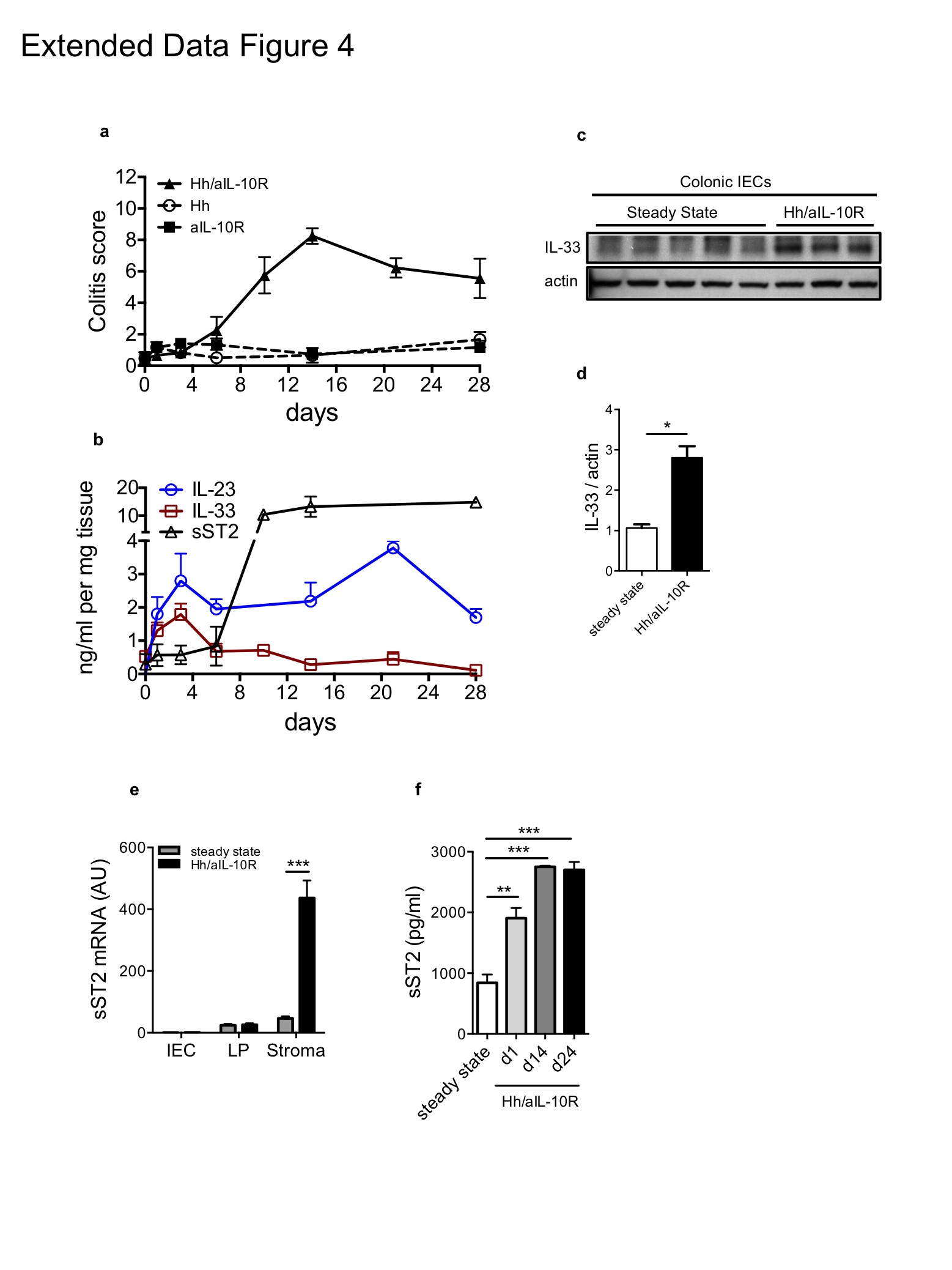
To assess the impact of IL-33 on the Treg response during intestinal inflammation we induced chronic colitis by infection with Helicobacter hepaticus and administration of an IL-10R blocking antibody22 (Extended Data Fig. 4a). We detected an increase in IL-33 protein levels in colon explant cultures and its expression kinetics mirrored that of IL-23, which is essential for the development of intestinal inflammation in this model (Extended Data Fig. 4b). Consistent with its pattern of expression, IL-33 protein levels were elevated in colonic intestinal epithelial cells isolated from the inflamed gut (Extended Data Fig. 4c-d). Interestingly, the onset of intestinal pathology correlated with a marked increase of soluble ST2 (sST2), which is produced primarily by colonic stromal cells (Extended Data Fig. 4b, e-f). sST2 is thought to limit IL-33 bioavailability by acting as a decoy receptor23 and is increased in patients with active IBD6,24 suggesting that the chronic inflammatory tissue environment may antagonize IL-33 activity. Despite high levels of sST2, analysis of chimeric mice showed that accumulation of St2−/− Treg cells in the colon but not the spleen was significantly impaired during the peak of intestinal inflammation (Fig. 2g). In addition, colonic St2−/− Treg cells expressed lower levels of Foxp3 protein on a per cell basis as compared to their WT counterparts (Fig. 2h). Together, these observations indicate that the alarmin IL-33 acts in a cell-intrinsic manner to promote the tissue-specific accumulation and stability of the Treg phenotype in the intestine under inflammatory conditions. Furthermore, high levels of sST2 during chronic intestinal inflammation may represent a mechanism to further perpetuate pathogenic responses by limiting IL-33-driven Treg accumulation.
We next sought to compare the suppressive capacity of WT and St2−/− Treg cells. St2−/− Treg cells inhibited T cell proliferation to the same extent as WT Treg cells in vitro (Extended Data Fig. 5) and addition of IL-33 did not enhance WT Treg suppressor function. We then tested the ability of St2−/− Treg to protect from colitis induced by adoptive transfer of naïve CD4+ T cells. Interestingly, ST2 was highly expressed on WT Treg cells upon T cell transfer pointing toward a potential role of ST2 in modulating Treg function in this model (Extended Data Fig. 6a). Indeed, St2−/− Treg cells were significantly impaired in their ability to prevent colonic inflammation and cellular infiltration (Fig. 3a,b), demonstrating that IL-33 signaling into Treg cells is important for their suppressive function in vivo. Analysis of WT or St2−/− Treg cells two weeks post transfer, prior to the onset of intestinal pathology, showed similar proliferative capacity and Foxp3 expression between groups (Fig. 3c). The ratio of T effector cells (CD45.1+ RBhi progeny) to Treg cells (CD45.1−Foxp3+ Treg progeny) was also similar (Fig. 3d) suggesting that IL-33 signaling in Treg cells is dispensable for their ability to expand and index with effector cells in the lymphopenic host at this time point. By contrast, analysis at 8 weeks post-transfer showed that progeny of St2−/− Treg cells contained a significantly lower proportion of Foxp3+ cells and expressed significantly less Foxp3 on a per cell basis suggesting they had lost Foxp3 expression (Fig. 3e,h). Under these circumstances the ratio of T effector/Treg cells and the total number of T effector cells (CD45.1+ RBhi progeny) was markedly increased in recipients of St2−/− Treg cells (Fig. 3f,g). Importantly, ST2-deficient Treg cells did not themselves acquire the capacity to produce inflammatory cytokines (Extended Data Fig. 6c). Perturbations of Foxp3 expression have been shown to affect Treg cell function25-27 and our data indicate that IL-33 signaling in Treg cells contributes to the maintenance of Foxp3 expression under inflammatory stress, enabling Treg cells to compete in the inflammatory niche and control the intestinal effector T cell response.
Figure 3. IL-33 promotes Treg cell stability and function in vivo.

a, C57BL/6 Rag1−/− mice were injected with CD45.1+ naïve T cells alone (RBhi, n = 4) or in combination with WT (n = 4) or St2−/− (n = 6) CD45.1− Treg cells. Mice were sacrificed after 6-8 weeks post transfer and colitis scores are shown. b, Absolute numbers of colon lamina propria (LP) cells from mice in (a). c, C57BL/6 Rag1−/− mice were injected as in (a) and sacrificed at 2 weeks post injection. Representative plots are gated on colonic Treg cell progeny (CD45.1−). d, Ratio of RBhi T cell progeny (CD45.1+) to WT or St2−/− Foxp3+ Treg cell progeny (CD45.1−) in the colon (n = 5 per group) from mice in (c), (mean ± s.e.m). e, C57BL/6 Rag1−/− mice were injected as in (a) and sacrificed at 8 weeks post injection. Representative plots are gated on colonic Treg cell progeny (CD45.1−). f, Ratio of RBhi T cell progeny (CD45.1+) to WT or St2−/− Treg cell progeny (CD45.1−) in the colon from mice in (e), (mean ± s.e.m). g, Absolute numbers of RBhi T cell progeny (CD45.1+) in the colon from mice in (e), (mean ± s.e.m). h, Analysis of Foxp3 expression in colonic Foxp3+CD45.1− Treg cells presented as gMFI (mean ± s.e.m).
Results are representative of two independent experiments. *P<0.05 **P<0.01 *** P<0.001 **** P<0.0001 as calculated by 1way-ANOVA with Bonferroni post-test or Students’ t-test.
Next we sought to integrate our observations with existing pro-inflammatory pathways. IL-23 is a pivotal mediator of intestinal inflammation and polymorphisms in the IL23R locus are associated with increased susceptibility to IBD in humans28. We previously showed that IL-23 promotes intestinal inflammation in part through inhibition of iTreg cell differentiation29,30. However, the mechanism by which IL-23 blocked iTreg generation remained undefined. Interestingly, whole transcriptome analysis of IL-23 target genes in colonic effector CD4+ T cells revealed that IL-23 inhibits expression of Gata3 and St2 (Extended Data Fig. 7). Based on this observation we hypothesized that IL-23 might limit T cell responsiveness to IL-33. Indeed, the co-factor activity of IL-33 on TGF-β1-mediated Foxp3 induction in vitro was completely abrogated in the presence of IL-23 (Fig. 4a). Notably, addition of IL-23 prevented induction of Gata3 and St2 mRNA under iTreg differentiation conditions (Fig. 4b) resulting in reduced ST2 protein expression (Fig. 4c). Consequently, acute stimulation of IL-23-exposed iTreg cells with IL-33 did not lead to recruitment of GATA3 to the ST2 enhancer (Extended Data Fig. 8). We observed a similar role for IL-23 in limiting Treg ST2 expression during bacterially driven intestinal inflammation (Extended Data Fig. 9). Collectively our data indicate that IL-23 inhibits iTreg differentiation by regulating T cell responsiveness to IL-33.
Figure 4. IL-23 inhibits the effects of IL-33 on Treg cells.

a, Naïve CD4+ T cells were cultured with anti-CD3/CD28 plus TGF-β1 as well as indicated cytokines and the frequencies of Foxp3+ T cells determined after 3 days later (mean ± s.e.m. of three independent experiments). b, Naïve CD4+ T cells were cultured with anti-CD3/CD28 plus indicated cytokines for 48h. Data are from one experiment representative of two (mean ± s.d.). c, Naïve CD4+ T cells were cultured as indicated. Representative blots of two independent experiments are shown. d, C57BL/6 Il23a−/−Rag1−/− mice were injected with CD45RBhi WT (n = 9) or St2−/− (n = 10) T cells. Mice were sacrificed after 6-8 weeks post transfer and frequencies of Foxp3+CD4+ T cells in colon are shown. e, Colitis scores for mice in (d). f, Expression of indicated cytokines by colonic CD4+ T cells from mice in (d). g, Treg cells were cultured with anti-CD3/CD28 for 24h followed by stimulation with IL-33 in the presence or absence of IL-23. Representative blots of two independent experiments are shown. h, Treg cells were cultured in the presence of anti-CD3/CD28 for 24h and mRNA expression of indicated genes measured following stimulation with IL-33 for 45 minutes in the presence or absence of IL-23 (mean ± s.e.m. of three independent experiments). i, Treg cells were cultured with anti-CD3/CD28 for 24h and representative blots of two independent experiments are shown. *P<0.05, **P<0.01 *** P<0.001 as calculated by 1way-ANOVA with Bonferroni post-test or Students’ t-test.
We previously showed that IL-23 restrains Treg cells in vivo because naïve T cell transfer into Il23a−/−Rag1−/− recipients resulted in increased Treg differentiation29. Therefore, we hypothesized that enhanced responsiveness to IL-33 may contribute to increased iTreg differentiation in Il23a−/−Rag1−/− hosts. To test this we transferred WT or St2−/− naïve T cells into Il23a−/−Rag1−/− hosts and monitored iTreg generation. Indeed, ST2-deficient T cells were significantly impaired in their ability to differentiate into Treg cells (Fig. 4d) and this correlated with a significant increase in intestinal pathology (Fig. 4e). Importantly, ST2-deficiency had minor effects on in vivo differentiation of Th1, Th2 or Th17 cells (Fig. 4f) further supporting the notion that the increased colitogenic potential of St2−/− CD4+ T cells is a consequence of deficient iTreg differentiation rather than dysregulated effector T cell responses. In summary, our data strongly suggest that IL-33 is a major factor responsible for driving iTreg differentiation in the absence of IL-23.
Finally, we investigated whether IL-23 can interfere with ST2 signaling in Treg cells. Interestingly, sort-purified ST2+ Treg cells from the colon expressed detectable levels of Il23r (Extended Data Fig. 10). Indeed, exposure of TCR-activated tTreg cells to IL-23 completely abolished IL-33-mediated GATA3 phosphorylation (Fig. 4g). Furthermore, IL-33 preferentially induced genes co-regulated by Foxp3 and GATA316 and this was completely abrogated in the presence of IL-23 (Fig. 4h). Addition of a specific inhibitor of STAT3, the main transcription factor downstream of IL-23 signaling, reversed this inhibitory effect of IL-23 (Fig. 4i). Together our data suggest that IL-23 inhibits ST2 signal transduction and expression of a distinct set of GATA3 regulated genes in tTreg cells.
Our results identify a new function for IL-33 as an important link between inflammation-driven tissue damage and the local intestinal Treg cell response. We show that colonic Treg cells are poised to respond to the release of IL-33 upon tissue damage through selective expression of ST2 and that signaling through this pathway plays an essential role in their capacity to adapt to the inflammatory tissue environment and restrain intestinal inflammation. The ability of IL-33 to amplify regulatory networks in response to tissue injury may represent a more general mechanism by which alarmins limit immune-mediated damage to self at barrier tissues. Strikingly, IL-23 limits this regulatory mechanism through inhibition of Treg cell responsiveness to IL-33 suggesting that the balance between IL-23 and IL-33 may be a major determinant of the outcome of intestinal immune responses.
Method Summary
A description of the in vitro assays and details of in vivo models can be found in the full Methods.
Materials and Methods
Mice
Wild-type C57BL/6, congenic B6.SJL-Cd45.1, C57BL/6 Il23r−/−, C57BL/6 Rag1−/−, C57BL/6 Il23a−/−Rag1−/− and Foxp3gfp reporter mice31,32 were bred and maintained under specific pathogen-free conditions in accredited animal facilities at the University of Oxford. Where indicated, mice were i.p. injected with recombinant IL-33 (1 μg / injection; Biolegend) for 5 consecutive days and sacrificed 24 h after the last injection. Gata3f/f-Foxp3Cre mice were kept at the NIH and experiments were performed at the NIH. Spleen, MLN and bone marrow from C57BL/6 St2−/− mice were obtained from Padraic Fallon (Trinity College Dublin, Ireland) or Max Loehning (Charité – University Medicine Berlin, Germany). All procedures were conducted in accordance with the UK Scientific Procedures Act of 1986. Mice were negative for Helicobacter spp. and other known intestinal pathogens, were age and sex-matched and more than 6 weeks old when first used. Both female and male mice were used in experiments. Wherever possible preliminary experiments were performed to determine requirements for sample size, taking into account resources available and ethical, reductionist animal use. Generally, each mouse of the different experimental groups is reported. Exclusions criteria such as, inadequate staining or low cell yield due to technical problems, were pre-determined. Animals were assigned randomly to experimental groups. Each cage contained animals of all the different experimental groups.
Generation of mixed bone marrow chimaeras
Bone marrow isolated from WT (CD45.2+), Il23r−/− or St2−/− mice was mixed at a 1:1 ratio with bone marrow taken from B6.SJL-Cd45.1 mice and injected intravenously into gamma-irradiated (5.5 Gy, 550 rad) C57BL/6 Rag1−/− recipients and chimaeras were used in experiments >8 weeks after injection.
T cell transfer colitis
For naïve T cell transfer colitis 4×105 CD4+CD25−CD45RBhi T cells were injected i.p. into Rag1−/− or Il23a−/−Rag1−/− recipients. In co-transfer experiments 2×105 CD4+CD25−CD45RBhi T cells from each source were mixed and injected i.p. into Rag1−/− hosts. For Treg mediated protection from colitis 4×105 CD4+CD25−CD45RBhi T cells and 2×105 CD4+CD25+ Treg cells were mixed and injected i.p. into Rag1−/− hosts33. Mice were sacrificed at indicated time points or sacrificed when weight loss approached 20% of the original body weight at the start of the experiment.
Induction of Helicobacter hepaticus/anti-IL-10R colitis
Mice were fed 1×108 cfu Helicobacter hepaticus by oral gavage with a 22G curved needle on day 0, day 1 and day 2 of the experiment. In addition mice received 1mg of an anti-IL-10R blocking antibody by i.p. injection once weekly starting at the day of Helicobacter hepaticus infection.
Histological assessment of intestinal inflammation
Proximal, mid, and distal colon samples were fixed in buffered 10% formalin solution. Paraffin embedded sections were cut (5 mm) and stained with hematoxylin and eosin, and inflammation was scored in a blinded fashion using a previously described scoring system34.
Isolation of leukocyte subpopulations and flow cytometry
Cell suspensions from spleen, MLN, and the lamina propria were prepared as described previously35 and first incubated with anti-CD16/CD32 (eBioscience) to prevent nonspecific binding. Single cell suspensions were stained with antibodies against CD4, CD25, TCR-β, CD45.1, CD45.2, CD103, OX40, IL-17A, IFN-γ, IL-13, Foxp3, GATA3, Helios, (all from eBioscience), RORγt, Ki67, (BD Biosciences) and CD45RB, KLRG1 (all from Biolegend) and anti-ST2 conjugated to biotin (mdBioproducts). Intracellular staining was performed as follows: cells were restimulated for 4 h as previously described36, washed and incubated with anti-CD16/CD32. Cells were washed and stained for surface markers indicated above and fixed in eBioscience Fix/Perm buffer, followed by permeabilization in eBioscience permeabilization buffer for 1 h in the presence of antibodies. Cells were acquired with a BD LSRII and analysis was performed with FlowJo (Tree Star) software.
Treg cell cultures
CD25−CD62L+CD44lo GFP− naïve CD4+ T cells were sort-purified from Foxp3gfp reporter mice. Cells were plated at 2×105 cells/well in flat bottomed 96 well plates coated with anti-CD3 (5 μg/ml) in RPMI (Invitrogen) containing 10% FCS, 2 mM l-glutamine, 0.05 mM 2-mercaptoethanol, and 100 U/ml each of penicillin and streptomycin (complete media). Antibodies and cytokines were added at the following concentrations, anti-CD28 (2 μg/ml), TGF-β1 (R&D, 500pg/ml), IL-2 (Peprotech, 1U/ml), IL-23 (R&D, 20ng/ml), IL-33 (Biolegend, 1 ng/ml). Cells were harvested at indicated times, lysed for use for qPCR, western blot or analyzed by flow cytometry. GFP+CD4+ Treg cells were sort-purified from Foxp3gfp reporter mice and cells were plated at 2×105 cells/well in flat bottomed 96 well plates coated with anti-CD3 (5 μg/ml) in complete media and anti-CD28 (2 μg/ml), IL-2 (Peprotech, 1U/ml) or IL-33 (Biolegend, 1 ng/ml) were added at the start of the culture. Cells were analyzed after 3 days by flow cytometry. For acute stimulation, 2×105 cells/well were plated in 96 well plates coated with anti-CD3 (5 μg/ml) in complete media and soluble anti-CD28 (2 μg/ml) and cultured for 24 h. Cells were then stimulated with IL-33 (10 ng/ml) for the indicated time and used for qPCR or western blot.
Treg cell suppression assay
CD25+CD4+ Treg cells were sorted by flow cytometry from WT and St2−/− mice. CD25− CD62L+CD44−CD4+ T cells (responder cells) were sorted from St2−/− mice. Antigen presenting cells (APCs) were isolated by magnetic bead separation from splenocytes and LN cells from St2−/− mice. T responder cells were labeled with Violet cell trace and plated together with the sorted Tregs at ratios of 1:1 and 1:3 together with irradiated APCs in the presence of anti-CD3 (1μg/ml). IL-33 was added at 30 ng/ml. After 4 days proliferation of T responder cells was measured by flow cytometry.
Colon explant cultures
Organ explants were prepared as previously described36 and cultured overnight in complete media. Cytokine levels in the supernatants were determined by ELISA IL-23 and IL-33 (eBioscience) and soluble ST2 (R&D), and concentrations were normalized to the weight of the explants.
Isolation and culture of colonic stromal cells
Colonic tissue was digested and cells from the 30:40% Percoll gradient interface harvested and washed twice in PBS containing 2% BSA. Cells were plated at 10×106 cells/ml in complete DMEM containing 40μg/ml Gentamycin (Sigma). After 24-48 hours, non-adherent cells were removed by vigorous washing, and media replaced. Adherent intestinal stromal cells were cultured for 10 days until confluent. Cells were then plated at 1×106 cells/ml for 48 hours and the concentration of sST2 in supernatants was determined by ELISA (R&D Systems).
qPCR and microarray preparation
RNA was extracted according to the manufacturers’ protocol (RNeasy, Qiagen) and cDNA synthesis was performed using Superscript III reverse transcription and Oligo dT primers (both from Invitrogen). qPCR reactions were performed using TaqMan Gene Expression Assays and normalized to HPRT (all from Applied Biosystems). Alternatively, qPCR reactions were performed using SYBR green PCR SensiMix (Quantace) with primers from37. For soluble ST2 the following primers were used: sST2 forward 5′-tcgaaatgaaagttccagca -3′ and sST2 reverse 5′-tgtgtgagggacactccttac -3′ Samples were assayed in duplicate on a Bio-Rad CFX96 RT-qPCR machine and differences were calculated using the 2ΔC(t) method.
For microarray gene expression analysis RNA was extracted using the RNAqueous-Micro total RNA Isolation Kit or RiboPure RNA Purification Kit (both Ambion). For RNA amplification and labelling the TargetAmp2-Round Biotin-aRNA Amplification Kit 3.0 (Epicentre, Illumina) or the Illumina TotalPrep RNA Amplification Kit (Ambion) were used and RNA quality was assessed using the Agilent 2100 Bioanalyzer and only samples with RNA integrity number (RIN) values above 7 were used for further processing. Biotinylated cRNA was hybridised to MouseWG-6 v2.0 Expression BeadChip (Illumina). Hybridisations to BeadChips and data acquisition were performed by the microarray core facility at the Wellcome Trust Center for Human Genetics, Oxford. Microarray analysis was performed using GeneSpring GX12 software (Agilent). Data was normalised using 75% percentile shift normalisation algorithm and baseline transformed to the median of all samples. Statistical significance was determined using an unpaired t-test followed by Benjamini Hochberg false discovery rate multiple testing correction. P value cut-off was set to 0.05.
Total protein extracts and immunoblot analysis
Total protein extracts were prepared as described38. Equal amounts of protein were resolved by SDS-PAGE and analyzed with anti-pGATA3 (ab61052, Abcam), anti-total GATA3 (L50-823, BD Pharmingen), anti-ST2 (101001B, mdBioproducts), anti-actin (A5541; Sigma) or anti-IL-33 (396118, R&D). For inhibition of STAT3 activity in vitro, STAT3 VI (10 mM, Santa Cruz) was added 30min before cytokine stimulation.
Chromatin immunoprecipitation
ChIP assays were performed as described38 with antibody to GATA3 (L50-823, BD Pharmingen) or RNA polymerase II (sc-899; Santa Cruz). The immunoprecipitated DNA fragments were then analyzed by real-time PCR with SYBR Premix Ex Taq II master mix (Takara Bio) and the following primers: locus encoding St2 enhancer, 5′-gccaaccacaacagcagatggggaaa -3′ and 5′-actgagatcctgccctggcttccct -3′; locus encoding St2 promoter, 5′-tggcctccttggaaaggcttggt -3′ and 5′-agtgcaggaggggcatggagatga. Primers for analyzing binding to CNS1, CNS2, CNS3 and promoter locus of Foxp3 were from25. Data were analyzed using Rotogene 6000 software (Corbett Research Ltd). Results are normalized to input DNA and presented as fold enrichment relative to unstimulated cells.
Statistical analysis
Where appropriate Student’s t-test was used. For comparison of more than two groups a one-way ANOVA followed by a Bonferroni multiple comparison test was performed. All statistical analysis was calculated in Prism (GraphPad). Differences were considered to be statistically significant when P ≤ 0.05.
Supplementary Material
Extended Data Figure 1: Effects of IL-33 on gene expression during iTreg differentiation
a, Sort-purified CD25−CD62L+CD44lo GFP− naïve CD4+ T cells from Foxp3gfp reporter mice were stimulated with anti-CD3/anti-CD28 in the presence of indicated cytokines. mRNA expression of indicated genes was measured at 24h, 48h and 72h and presented as fold change over time 0. Data are representative of two independent experiments and show the mean ± s.d.
{kind=link}
Extended Data Figure 2: IL-33 directly regulates ST2 expression
Sort-purified CD25−CD62L+CD44lo GFP− naïve CD4+ T cells from Foxp3gfp reporter mice were cultured with anti-CD3/anti-CD28 in the presence of TGF-β1 followed by acute stimulation with IL-33 for 45 minutes. Shown are the recruitment of GATA3 or RNA polymerase II (Pol II) to the indicated regions of the gene encoding ST2 assessed by ChIP followed by qPCR. Results are normalized to those obtained with genomic DNA (input) and presented as fold enrichment relative to unstimulated cells. Data are from one experiment representative of two (error bars, s.d.).
{kind=link}
Extended Data Figure 3: IL-33 acts directly on tTreg cells to promote their proliferation and accumulation in vivo
a-e, Foxp3gfp reporter mice were injected i.p with recombinant IL-33 (1μg/mouse/day) for 5 days. a, Frequencies of TCRβ+CD4+Foxp3+ splenic tTreg cells. b, absolute numbers of TCRβ+CD4+Foxp3+ splenic Treg cells. c, gMFI of Foxp3 among TCRβ+CD4+Foxp3+ splenic Treg cells (n = 10 for PBS and n = 14 for IL-33). d, Frequencies of ST2+ cells among splenic CD44high T cells or Foxp3+ Treg cells. e, Frequencies of Ki67+ cells among splenic CD44high T cells or Foxp3+ Treg cells (n = 5 for PBS and n = 8 for IL-33). f, Mixed bone marrow chimaeras were generated by lethal irradiation of C57BL/6 Rag1−/− mice followed by i.v. injection of 2.5×106 WT (CD45.1+) and 2.5×106 St2−/− (CD45.1−) bone marrow cells (n = 5). Following reconstitution, mixed chimaeras were injected with recombinant IL-33 (1μg/mouse/day) for 5 days and the absolute number of Ki67+ among TCRβ+CD4+Foxp3− or TCRβ+CD4+Foxp3+ cells are shown. **P<0.01 *** P<0.001 as calculated by unpaired (WT mice) or paired (chimaeric mice) Students’ t-test.
{kind=link}
Extended Data Figure 4: Kinetics of IL-33 and IL-23 during Helicobacter hepaticus/anti-IL-10R colitis
C57BL/6 mice were infected by oral gavage with ~108 cfu Helicobacter hepaticus on three consecutive days with concomitant i.p. injections of a blocking anti-IL-10R mAb (1mg/week) starting on the day of the first infection. a, Colitis scores for the indicated groups and b, IL-33, IL-23 and soluble ST2 protein production in colon explant cultures from Hh/aIL-10R treated mice were determined at the indicated time points (n = 4, data show the mean ± s.e.m.). c, Colonic intestinal epithelial cells (IECs) were isolated from steady state or colitic (day 10) mice. Total protein extracts from each time point were analyzed by immunoblot and biological replicates are shown. d, Densitometric quantification of immunoblot in (c). e, mRNA expression analysis of intestinal epithelial cells (IEC), lamina propria cells (LP) and stromal cells isolated from the colon at steady state or 10 days after induction of colitis. f, Colonic stromal cells were isolated at the indicated time points, cultured and passaged and spontaneous sST2 protein production determined in supernatants from 48hr cultures. Stromal cells were uniformly CD45− EpCAM−. Data represent mean ± s.e.m. *P<0.05, **P<0.01 *** P<0.001 as calculated by 1way-ANOVA with Bonferroni post-test or Students’ t-test.
{kind=link}
Extended Data Figure 5: The in vitro suppressive function of St2−/− Treg cells is not impaired.
CD25+CD4+ Treg cells were sorted by flow cytometry from WT and St2−/− mice. CD25− CD62L+CD44−CD4+ T cells (responder cells) and antigen presenting cells (APCs) were sorted from St2−/− mice. T responder cells were labeled with Violet cell trace and plated together with the sorted Treg cells at ratios of 1:1 and 1:3 together with irradiated APCs in the presence of anti-CD3 (1μg/ml). IL-33 was added at 30 ng/ml. After 4 days proliferation of Tresp was measured by flow cytometry. Data are representative of two independent experiments.
{kind=link}
Extended Data Figure 6: ST2 is preferentially expressed on Treg cell progeny
a, C57BL/6 Rag1−/− mice were injected i.p. with 4×105 CD4+CD25−CD45RBhi CD45.1+ naive T cells in combination with 2×105 WT CD4+CD25+ CD45.1− Treg cells. Mice were sacrificed at 6-8 weeks post transfer. Representative plots of the colon gated on progeny of CD4+CD45RBhi cells (CD45.1+) or WT Treg cells (CD45.1−) cells are shown. b, Histogram overlay of Foxp3 expression by WT (solid line) or St2−/− (dotted line) CD4+CD25+ Treg cells prior to injection into C57BL/6 Rag1−/− mice. c, C57BL/6 Rag1−/− mice were injected as in (a) and frequencies of cytokine producing colonic Treg progeny (CD45.1−) among Foxp3− or Foxp3+ cells are shown. WT (n = 4) and St2−/− (n = 6), bars represent mean ± s.e.m..
{kind=link}
Extended Data Figure 7: St2 is an IL-23 target gene in intestinal CD4+ T cells.
C57BL/6 Rag1−/− mice were injected i.p. with a 1:1 mixture of 2×105 WT (CD45.1+) and 2×105 Il23r−/− (CD45.2+) CD4+CD25−CD45RBhi T cells or 2×105 WT (CD45.1+) and 2×105 WT (CD45.2+) CD4+CD25−CD45RBhi T cells. Mice were sacrificed upon development of clinical signs of inflammation (~8 weeks). Viable TCRβ+CD4+CD45.2+ WT or Il23r−/− T cells were sort-purified from the colon and RNA extracted without further manipulation. Whole transcriptome gene expression analysis was performed using the Illumina Bead Array platform. Two-dimensional cluster analysis (hierarchical, Pearson uncentered, average linkage, unsupervised) of the 168 transcripts whose expression was significantly affected in the comparison of Il23r−/− versus WT CD4+ T cells. Each row corresponds to a gene and each column to a sample (n = 6 for WT and n= 5 for Il23r−/−). The 168 transcripts represent 147 unique genes including genes with unknown function.
{kind=link}
Extended Data Figure 8: IL-23 inhibits IL-33-induced GATA3 recruitment to the ST2 locus
Sort-purified CD25−CD62L+CD44lo GFP− naïve CD4+ T cells from Foxp3gfp reporter mice were cultured with anti-CD3/anti-CD28/TGF-β1 followed by acute stimulation with IL-33 for 45 minutes in the presence or absence of IL-23. Shown are the enrichment of GATA3 or RNA polymerase II (Pol II) to the indicated regions of the gene encoding ST2 assessed by ChIP followed by qPCR. Results are normalized to those obtained with genomic DNA (input) and presented as fold recruitment relative to unstimulated cells. Data are from one experiment representative of two (error bars, s.d.)
{kind=link}
Extended Data Figure 9: IL-23 is a negative regulator of ST2 expression in vivo
Mixed bone marrow chimaeras were generated by lethal irradiation of C57BL/6 Rag1−/− mice followed by i.v. injection of 2.5×106 WT (CD45.1+) and 2.5×106 Il23r−/− (CD45.1−) bone marrow cells (n = 5). Reconstituted mice were infected by oral gavage with ~108 cfu Helicobacter hepaticus on three consecutive days with concomitant i.p. injections of a blocking anti-IL-10R mAb (1mg/week) starting on the day of the first infection. Mice were sacrificed at 2 weeks post infection. Frequencies of ST2+ and gMFI of ST2 among TCRβ+CD4+Foxp3+ cTreg cells are shown. *P<0.05, *** P<0.001 as calculated by paired Students’ t-test.
{kind=link}
Extended Data Figure 10: Il23r mRNA is expressed by ST2+Foxp3+ Treg cells
mRNA expression of indicated genes in Foxp3−ST2−, Foxp3+ST2− or Foxp3+ST2+ populations sort-purified from the steady state cLP of Foxp3gfp reporter mice. Bars represent the mean ± s.e.m. from three independent experiments.
{kind=link}
Acknowledgements
C. S, K. A, A. C and F. P are supported by the Wellcome Trust. FP is also supported by Fondation Louis Jeantet. A.N.H is supported by a European Molecular Biology Organization (EMBO) long-term fellowship (ALTF 116-2012). PGF is supported by Science Foundation Ireland. M.L. and A.F. are supported by Volkswagen Foundation (Lichtenberg Program) and BMBF (e:Bio/T-Sys). B.M.J.O is supported by an Oxford-UCB Pharma Postdoctoral Fellowship.
We thank all members of the Oxford Translational Gastroenterology Unit for assistance and support. We are grateful to Helen Ferry and Kate Alford for essential flow cytometry support and the staff of the University of Oxford for excellent animal care. We are also grateful to Dilair Baban (Wellcome Trust Center for Human Genetics, Oxford, UK) for conducting microarray hybridizations.
Footnotes
The authors declare no competing financial interests.
References
- 1.Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nature immunology. 2013;14:1007–1013. doi: 10.1038/ni.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pichery M, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. Journal of immunology. 2012;188:3488–3495. doi: 10.4049/jimmunol.1101977. [DOI] [PubMed] [Google Scholar]
- 3.Palmer G, Gabay C. Interleukin-33 biology with potential insights into human diseases. Nature reviews. Rheumatology. 2011;7:321–329. doi: 10.1038/nrrheum.2011.53. [DOI] [PubMed] [Google Scholar]
- 4.Beltran CJ, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflammatory bowel diseases. 2010;16:1097–1107. doi: 10.1002/ibd.21175. [DOI] [PubMed] [Google Scholar]
- 5.Kobori A, et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol. 2010;45:999–1007. doi: 10.1007/s00535-010-0245-1. [DOI] [PubMed] [Google Scholar]
- 6.Pastorelli L, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8017–8022. doi: 10.1073/pnas.0912678107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seidelin JB, et al. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol Lett. 2010;128:80–85. doi: 10.1016/j.imlet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Oboki K, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18581–18586. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duan L, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Molecular medicine. 2012;18:753–761. doi: 10.2119/molmed.2011.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grobeta P, Doser K, Falk W, Obermeier F, Hofmann C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflammatory bowel diseases. 2012;18:1900–1909. doi: 10.1002/ibd.22900. [DOI] [PubMed] [Google Scholar]
- 11.Sedhom MA, et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut. 2013;62:1714–1723. doi: 10.1136/gutjnl-2011-301785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmitz J, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 13.Thornton AM, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. Journal of immunology. 2010;184:3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity. 2011;35:337–348. doi: 10.1016/j.immuni.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wohlfert EA, et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest. 2011;121:4503–4515. doi: 10.1172/JCI57456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudra D, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nature immunology. 2012;13:1010–1019. doi: 10.1038/ni.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo L, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13463–13468. doi: 10.1073/pnas.0906988106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maneechotesuwan K, et al. Regulation of Th2 cytokine genes by p38 MAPK-mediated phosphorylation of GATA-3. Journal of immunology. 2007;178:2491–2498. doi: 10.4049/jimmunol.178.4.2491. [DOI] [PubMed] [Google Scholar]
- 19.Furusawa JI, et al. Critical Role of p38 and GATA3 in Natural Helper Cell Function. Journal of immunology. 2013;191:1818–1826. doi: 10.4049/jimmunol.1300379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turnquist HR, et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. Journal of immunology. 2011;187:4598–4610. doi: 10.4049/jimmunol.1100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wasserman A, et al. Interleukin-33 augments Treg cell levels: a flaw mechanism in atherosclerosis. The Israel Medical Association journal: IMAJ. 2012;14:620–623. [PubMed] [Google Scholar]
- 22.Kullberg MC, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. The Journal of experimental medicine. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. The Journal of biological chemistry. 2007;282:26369–26380. doi: 10.1074/jbc.M704916200. [DOI] [PubMed] [Google Scholar]
- 24.Diaz-Jimenez D, et al. Soluble ST2: a new and promising activity marker in ulcerative colitis. World journal of gastroenterology: WJG. 2011;17:2181–2190. doi: 10.3748/wjg.v17.i17.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Y, et al. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 27.Kitoh A, et al. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity. 2009;31:609–620. doi: 10.1016/j.immuni.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 29.Izcue A, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahern PP, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–288. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fontenot JD, et al. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 32.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 33.Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. The Journal of experimental medicine. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Izcue A, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uhlig HH, et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. Journal of immunology. 2006;177:5852–5860. doi: 10.4049/jimmunol.177.9.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hue S, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. The Journal of experimental medicine. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rudra D, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nature immunology. 2012;13:1010–1019. doi: 10.1038/ni.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krausgruber T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nature immunology. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended Data Figure 1: Effects of IL-33 on gene expression during iTreg differentiation
a, Sort-purified CD25−CD62L+CD44lo GFP− naïve CD4+ T cells from Foxp3gfp reporter mice were stimulated with anti-CD3/anti-CD28 in the presence of indicated cytokines. mRNA expression of indicated genes was measured at 24h, 48h and 72h and presented as fold change over time 0. Data are representative of two independent experiments and show the mean ± s.d.
Extended Data Figure 2: IL-33 directly regulates ST2 expression
Sort-purified CD25−CD62L+CD44lo GFP− naïve CD4+ T cells from Foxp3gfp reporter mice were cultured with anti-CD3/anti-CD28 in the presence of TGF-β1 followed by acute stimulation with IL-33 for 45 minutes. Shown are the recruitment of GATA3 or RNA polymerase II (Pol II) to the indicated regions of the gene encoding ST2 assessed by ChIP followed by qPCR. Results are normalized to those obtained with genomic DNA (input) and presented as fold enrichment relative to unstimulated cells. Data are from one experiment representative of two (error bars, s.d.).
Extended Data Figure 3: IL-33 acts directly on tTreg cells to promote their proliferation and accumulation in vivo
a-e, Foxp3gfp reporter mice were injected i.p with recombinant IL-33 (1μg/mouse/day) for 5 days. a, Frequencies of TCRβ+CD4+Foxp3+ splenic tTreg cells. b, absolute numbers of TCRβ+CD4+Foxp3+ splenic Treg cells. c, gMFI of Foxp3 among TCRβ+CD4+Foxp3+ splenic Treg cells (n = 10 for PBS and n = 14 for IL-33). d, Frequencies of ST2+ cells among splenic CD44high T cells or Foxp3+ Treg cells. e, Frequencies of Ki67+ cells among splenic CD44high T cells or Foxp3+ Treg cells (n = 5 for PBS and n = 8 for IL-33). f, Mixed bone marrow chimaeras were generated by lethal irradiation of C57BL/6 Rag1−/− mice followed by i.v. injection of 2.5×106 WT (CD45.1+) and 2.5×106 St2−/− (CD45.1−) bone marrow cells (n = 5). Following reconstitution, mixed chimaeras were injected with recombinant IL-33 (1μg/mouse/day) for 5 days and the absolute number of Ki67+ among TCRβ+CD4+Foxp3− or TCRβ+CD4+Foxp3+ cells are shown. **P<0.01 *** P<0.001 as calculated by unpaired (WT mice) or paired (chimaeric mice) Students’ t-test.
Extended Data Figure 4: Kinetics of IL-33 and IL-23 during Helicobacter hepaticus/anti-IL-10R colitis
C57BL/6 mice were infected by oral gavage with ~108 cfu Helicobacter hepaticus on three consecutive days with concomitant i.p. injections of a blocking anti-IL-10R mAb (1mg/week) starting on the day of the first infection. a, Colitis scores for the indicated groups and b, IL-33, IL-23 and soluble ST2 protein production in colon explant cultures from Hh/aIL-10R treated mice were determined at the indicated time points (n = 4, data show the mean ± s.e.m.). c, Colonic intestinal epithelial cells (IECs) were isolated from steady state or colitic (day 10) mice. Total protein extracts from each time point were analyzed by immunoblot and biological replicates are shown. d, Densitometric quantification of immunoblot in (c). e, mRNA expression analysis of intestinal epithelial cells (IEC), lamina propria cells (LP) and stromal cells isolated from the colon at steady state or 10 days after induction of colitis. f, Colonic stromal cells were isolated at the indicated time points, cultured and passaged and spontaneous sST2 protein production determined in supernatants from 48hr cultures. Stromal cells were uniformly CD45− EpCAM−. Data represent mean ± s.e.m. *P<0.05, **P<0.01 *** P<0.001 as calculated by 1way-ANOVA with Bonferroni post-test or Students’ t-test.
Extended Data Figure 5: The in vitro suppressive function of St2−/− Treg cells is not impaired.
CD25+CD4+ Treg cells were sorted by flow cytometry from WT and St2−/− mice. CD25− CD62L+CD44−CD4+ T cells (responder cells) and antigen presenting cells (APCs) were sorted from St2−/− mice. T responder cells were labeled with Violet cell trace and plated together with the sorted Treg cells at ratios of 1:1 and 1:3 together with irradiated APCs in the presence of anti-CD3 (1μg/ml). IL-33 was added at 30 ng/ml. After 4 days proliferation of Tresp was measured by flow cytometry. Data are representative of two independent experiments.
Extended Data Figure 6: ST2 is preferentially expressed on Treg cell progeny
a, C57BL/6 Rag1−/− mice were injected i.p. with 4×105 CD4+CD25−CD45RBhi CD45.1+ naive T cells in combination with 2×105 WT CD4+CD25+ CD45.1− Treg cells. Mice were sacrificed at 6-8 weeks post transfer. Representative plots of the colon gated on progeny of CD4+CD45RBhi cells (CD45.1+) or WT Treg cells (CD45.1−) cells are shown. b, Histogram overlay of Foxp3 expression by WT (solid line) or St2−/− (dotted line) CD4+CD25+ Treg cells prior to injection into C57BL/6 Rag1−/− mice. c, C57BL/6 Rag1−/− mice were injected as in (a) and frequencies of cytokine producing colonic Treg progeny (CD45.1−) among Foxp3− or Foxp3+ cells are shown. WT (n = 4) and St2−/− (n = 6), bars represent mean ± s.e.m..
Extended Data Figure 7: St2 is an IL-23 target gene in intestinal CD4+ T cells.
C57BL/6 Rag1−/− mice were injected i.p. with a 1:1 mixture of 2×105 WT (CD45.1+) and 2×105 Il23r−/− (CD45.2+) CD4+CD25−CD45RBhi T cells or 2×105 WT (CD45.1+) and 2×105 WT (CD45.2+) CD4+CD25−CD45RBhi T cells. Mice were sacrificed upon development of clinical signs of inflammation (~8 weeks). Viable TCRβ+CD4+CD45.2+ WT or Il23r−/− T cells were sort-purified from the colon and RNA extracted without further manipulation. Whole transcriptome gene expression analysis was performed using the Illumina Bead Array platform. Two-dimensional cluster analysis (hierarchical, Pearson uncentered, average linkage, unsupervised) of the 168 transcripts whose expression was significantly affected in the comparison of Il23r−/− versus WT CD4+ T cells. Each row corresponds to a gene and each column to a sample (n = 6 for WT and n= 5 for Il23r−/−). The 168 transcripts represent 147 unique genes including genes with unknown function.
Extended Data Figure 8: IL-23 inhibits IL-33-induced GATA3 recruitment to the ST2 locus
Sort-purified CD25−CD62L+CD44lo GFP− naïve CD4+ T cells from Foxp3gfp reporter mice were cultured with anti-CD3/anti-CD28/TGF-β1 followed by acute stimulation with IL-33 for 45 minutes in the presence or absence of IL-23. Shown are the enrichment of GATA3 or RNA polymerase II (Pol II) to the indicated regions of the gene encoding ST2 assessed by ChIP followed by qPCR. Results are normalized to those obtained with genomic DNA (input) and presented as fold recruitment relative to unstimulated cells. Data are from one experiment representative of two (error bars, s.d.)
Extended Data Figure 9: IL-23 is a negative regulator of ST2 expression in vivo
Mixed bone marrow chimaeras were generated by lethal irradiation of C57BL/6 Rag1−/− mice followed by i.v. injection of 2.5×106 WT (CD45.1+) and 2.5×106 Il23r−/− (CD45.1−) bone marrow cells (n = 5). Reconstituted mice were infected by oral gavage with ~108 cfu Helicobacter hepaticus on three consecutive days with concomitant i.p. injections of a blocking anti-IL-10R mAb (1mg/week) starting on the day of the first infection. Mice were sacrificed at 2 weeks post infection. Frequencies of ST2+ and gMFI of ST2 among TCRβ+CD4+Foxp3+ cTreg cells are shown. *P<0.05, *** P<0.001 as calculated by paired Students’ t-test.
Extended Data Figure 10: Il23r mRNA is expressed by ST2+Foxp3+ Treg cells
mRNA expression of indicated genes in Foxp3−ST2−, Foxp3+ST2− or Foxp3+ST2+ populations sort-purified from the steady state cLP of Foxp3gfp reporter mice. Bars represent the mean ± s.e.m. from three independent experiments.