

Abstract
Maintenance of cellular homeostasis depends upon several pathways that allow a cell to respond and adapt to both environmental stress and changes in metabolic status. New work in this issue of The EMBO Journal reveals a mechanism of cross talk between heat shock factor 1 (HSF1), the primary regulator of the proteotoxic stress response, and AMP-activated protein kinase (AMPK), the primary sensor in the metabolic stress response.
See also: S Dai et al (February 2015)
Organisms are exposed to various protein-damaging or proteotoxic stresses from sources such as environmental temperature, microbial infections, heavy metals, toxins or caloric restriction, which induce biological responses to minimize cellular damage and maintain protein homeostasis (proteostasis) and to ensure cell survival under stressful conditions. The heat shock or proteotoxic stress response (PSR) represents a particularly well-characterized mechanism, which up-regulates molecular chaperones to prevent protein aggregation and aid refolding of damaged proteins. In vertebrates, PSR activation occurs primarily through heat shock transcription factor 1 (HSF1), a trimeric protein that upon stress is rapidly recruited to the promoters of chaperone genes and induces their expression (Anckar & Sistonen, 2011). Stress-inducible HSF1 activation is controlled on multiple levels including chromatin environment, promoter heat shock response element composition, interacting proteins as well as extensive post-translational modifications, in particular differential HSF1 multi-site phosphorylation dependent on the nature and severity of a stress. While the role of HSF1 in protecting cells and organisms against severe stress stimuli is well established, many aspects of how HSF1 senses different forms and quantities of stresses have remained poorly understood.
Recent genome-wide studies identifying many additional HSF1 targets beyond PSR-linked heat shock proteins have implicated HSF1 in many cellular processes including cell cycle regulation, metabolism, gametogenesis, disease and aging (reviewed in Vihervaara & Sistonen, 2014). HSF1-dependent proteostasis maintenance is fundamental for the survival of cancer cells. Cellular aging as well as aging-associated pathological conditions (such as diabetes, cancer, neurodegenerative and cardiac disease) are associated with impaired proteostasis, and loss-of-function experiments in the C. elegans model system support a causal involvement of HSF1 in lifespan regulation and prevention of neurodegeneration-linked protein aggregate formation (reviewed in Anckar & Sistonen, 2011). In aged human cells, HSF1 DNA binding activity is reduced due to increased HSF1 acetylation, caused by lowered levels of the NAD-dependent deacetylase SIRT1 (Westerheide et al, 2009).
HSF1 is also regulated by metabolic processes. The insulin signaling pathway, which monitors the organismal nutrient status, inhibits HSF1 activation and is able to reduce lifespan by silencing HSF1. In contrast, dietary restrictions and nutrient deprivation, resulting in low insulin levels, are known to increase lifespan and reduce the progression of age-related diseases. Increased AMP:ATP ratios upon nutrient deprivation also stimulate AMP-activated protein kinase (AMPK), the master regulator of the metabolic stress response (MSR). AMPK-mediated phosphorylation in turn triggers numerous downstream events aimed at enhancing ATP production and decreasing ATP expenditure, which are fundamental for the survival of cells exposed to metabolic stress. AMPK can be activated by various cellular stresses, including reactive oxygen species (ROS), and a hepatocellular carcinoma mouse model already provided some indications for HSF1 as a conduit for cross talk between PSR and MSR. In this model, insulin stimulation in hsf1 null mice paradoxically both enhanced insulin signal transduction and increased AMPK activation in response to lowered glucose levels, two pathways that usually oppose each other. Since progression of carcinogen-induced hepatocellular carcinoma was also impaired in hsf1 null mice, HSF1 was suggested as a potential target to control hepatic metabolic diseases (Jin et al, 2011). However, few studies have directly investigated the relationship of the PSR and MSR pathways.
The new work by Dai et al (2015) describes a previously unknown mechanism of mutual antagonism between these two pathways for the purpose of maintaining cellular homeostasis. Using a novel ELISA-based DNA binding assay and an adapted proximal ligation assay, the authors investigated the effects of proteotoxic and metabolic stresses on HSF1 DNA binding activity. Induction of metabolic stress through nutrient deprivation or the anti-diabetic drug metformin suppressed HSF1 DNA binding activity and the PSR, via AMPK-mediated phosphorylation of HSF1 on serine-121. Intriguingly, heat shock-induced PSR in turn suppressed the MSR, by inhibiting AMPK serine-172 phosphorylation and activation (Fig1).
Figure 1. HSF1 integrates multiple stress responses.
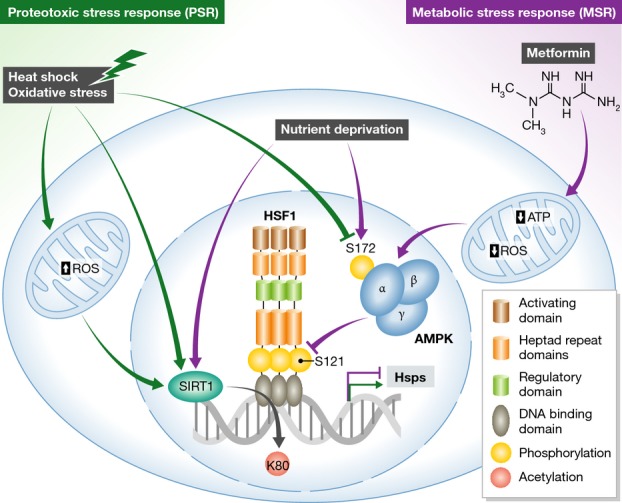
AMPK, a classic sensor of metabolic stresses such as impaired gluconeogenesis resulting from nutrient deprivation or decreased ATP levels induced by the pharmacological agent metformin, functions as an upstream regulator of HSF1 activity. Inhibition of mitochondrial electron transport chain by metformin also reduces mitochondrial ROS levels and oxidative stress. Phosphorylated AMPK inhibits HSF1 activity by preventing HSF1 Ser121 phosphorylation and HSF1-dependent transcription. Proteotoxic stresses, such as heat shock and oxidative stress, inhibit AMPK phosphorylation and are thus permissive for HSF1-activating Ser121 phosphorylation. Both proteotoxic and metabolic stresses recruit SIRT1 to DNA, where it deacetylates HSF1 at Lys80 and maintains binding of HSF1 to target gene promoters. See text for details.
Several studies on the role of HSF1 in maintaining cancer cell proteostasis have followed the groundbreaking original findings by Dai et al (2007) that HSF1-deficient mice are protected from skin tumorigenesis upon p53 mutation and that human cancer cell lines depend on HSF1 to maintain their proliferative and survival capacity. The mechanism for increased cancer cell survival was initially attributed solely to elevated levels of heat shock proteins assisting with protein refolding and maintenance of proteostasis. However, the recent observations of HSF1 recruitment to many non-chaperone genes strongly suggest that HSF1 may promote cell survival through multiple mechanisms (Mendillo et al, 2012), a notion that is further supported by the identification of distinct HSF1 and HSF2 targets in mitotic versus freely cycling human cells (Vihervaara et al, 2013). Importantly, the new study by Dai et al (2015) finds that MSR induction by metformin decreases HSF1 activity also in tumors and increases their susceptibility to proteotoxic stress, suggesting a unifying mechanism by which metabolic stressors in malignancy can inhibit HSF1 and thereby disrupt proteostasis.
Together with the recent implication of HSF1 as a target for cancer therapy, the observed metformin-mediated inhibition of HSF1 activity adds to the mounting body of evidence supporting metformin as a candidate anti-cancer drug, with more than 350 new studies on its potential use in 2014 alone (Foretz et al, 2014). However, the mechanisms by which metformin retards cancer cell growth are still poorly understood. According to this new study, HSF1 inactivation downstream of metformin-induced AMPK and MSR activation provides a novel mechanism that could be explored for anti-cancer drug development. Still, Dai et al caution that prolonged use of metformin might contribute to neurodegeneration, since pronounced AMPK activation in neural cells from patients with Huntington's disease and Alzheimer's disease has been observed, suggesting that AMPK activation may exacerbate aggregation of poly-glutamine tract proteins, a common cause of neurodegenerative disorders. This is further supported by a recent study investigating AMPK activation upon increased levels of amyloid beta peptides in Alzheimer's disease patients (Mairet-Coello et al, 2013). In a transgenic mouse model expressing mutant human amyloid precursor protein, increased intracellular calcium levels activated AMPK, which in turn phosphorylated the Tau protein, ultimately resulting in dendritic spine loss. Therefore, the valid caveat raised by the authors should be remembered when considering metformin as an anti-cancer drug.
Another possible mechanism by which the MSR pathway may affect the PSR and cancer is through ROS production in mitochondria. HSF1 knockdown affects mitochondrial function and ROS production, and ROS production upon oxidative stress can activate both HSF1 and (albeit through unclear mechanisms) AMPK (Ahn & Thiele, 2003; Mairet-Coello et al, 2013; Zhu et al, 2014). Feedback regulation of ROS and AMPK has been reported to control lifespan in C. elegans (Hwang et al, 2014). On the other hand, metformin reduces ROS production and activates AMPK through inhibition of the mitochondrial respiratory chain (Foretz et al, 2014). It will thus be important to determine whether proteotoxic stresses other than heat shock, such as ROS, inflammation or heavy metals, exhibit a similar antagonistic relationship with the MSR pathway. The finding that heavy metals cause dephosphorylation of AMPK at serine-172 and subsequent inhibitory phosphorylation of HSF1 at serine-303 suggests that antagonism between the PSR and the MSR may be a general phenomenon for proteotoxic stresses, but some stress-specific mechanisms may exist, such as differential effects on specific HSF1 phosphorylation sites (Zhu et al, 2014).
Dai et al (2015) found AMPK to be an upstream kinase regulating the DNA binding and transcriptional activity of HSF1, which in turn is dependent upon the metabolic status of the cell. HSF1's capacity to remain bound at its chromatin target sites is also known to be regulated by SIRT1, which deacetylates lysine-80 within the HSF1 DNA binding domain (Westerheide et al, 2009). Increased SIRT1 levels upon caloric restriction prolong lifespan, and since SIRT1 levels decrease with age, the aging-related PSR decline may be linked to SIRT1-mediated control of HSF1 activity. Together, this suggests that HSF1 acts as regulatory hub in a network linking cell nutrition, stress and lifespan.
Kennedy et al (2014) recently defined seven ‘pillars of aging’: adaptation to stress, epigenetics, inflammation, metabolism, macromolecular damage, stem cells and regeneration, and proteostasis. Intriguingly, HSF1 and AMPK have both been found to play key roles in most, if not all, of these processes. Biological systems change with age, and the lynchpin is that aging itself is the predominant risk factor for most diseases and conditions that limit lifespan. Moreover, interventions such as dietary restrictions or pharmaceuticals such as rapamycin, acarbose and metformin all have been shown to extend lifespan as well as to delay or prevent many chronic diseases in model organisms (Kennedy et al, 2014). The new study by Dai et al highlights the cross talk between multiple cellular stress responses and its role in malignant growth. Understanding the effect of this cross talk on the aging metabolome will be fundamental to develop new therapeutic interventions for treatment of age-onset diseases, including cancer.
Acknowledgments
We are grateful to Gordon R. Gray for helpful comments on the manuscript. Our laboratory is financially supported by the Academy of Finland, the Sigrid Jusélius Foundation, the Finnish Cancer Organizations and Åbo Akademi University.
References
- Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Tang Z, Cao J, Zhou W, Li H, Sampson S, Dai C. Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. EMBO J. 2015;34:275–293. doi: 10.15252/embj.201489062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- Hwang AB, Ryu EA, Artan M, Chang HW, Kabir MH, Nam HJ, Lee D, Yang JS, Kim S, Mair WB, Lee C, Lee SS, Lee SJ. Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2014;111:E4458–E4467. doi: 10.1073/pnas.1411199111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Moskophidis D, Mivechi NF. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011;14:91–103. doi: 10.1016/j.cmet.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss-Coray T, Sierra F. Geroscience: linking aging to chronic disease. Cell. 2014;159:709–713. doi: 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron. 2013;78:94–108. doi: 10.1016/j.neuron.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A, Sergelius C, Vasara J, Blom MAH, Elsing AN, Roos-Mattjus P, Sistonen L. Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc Natl Acad Sci USA. 2013;110:E3388–E3397. doi: 10.1073/pnas.1305275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A, Sistonen L. HSF1 at a glance. J Cell Sci. 2014;127:261–266. doi: 10.1242/jcs.132605. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XN, Chen LP, Bai Q, Ma L, Li DC, Zhang JM, Gao C, Lei ZN, Zhang ZB, Xing XM, Liu CX, He ZN, Li J, Xiao YM, Zhang AH, Zeng XW, Chen W. PP2A-AMPKα-HSF1 axis regulates the metal-inducible expression of HSPs and ROS clearance. Cell Signal. 2014;26:825–832. doi: 10.1016/j.cellsig.2014.01.002. [DOI] [PubMed] [Google Scholar]