

Abstract
Reversible protein ubiquitylation plays important roles in various processes including DNA repair. Here, we identify the deubiquitylase USP45 as a critical DNA repair regulator. USP45 associates with ERCC1, a subunit of the DNA repair endonuclease XPF–ERCC1, via a short acidic motif outside of the USP45 catalytic domain. Wild-type USP45, but not a USP45 mutant defective in ERCC1 binding, efficiently deubiquitylates ERCC1 in vitro, and the levels of ubiquitylated ERCC1 are markedly enhanced in USP45 knockout cells. Cells lacking USP45 are hypersensitive specifically to UV irradiation and DNA interstrand cross-links, similar to cells lacking ERCC1. Furthermore, the repair of UV-induced DNA damage is markedly reduced in USP45-deficient cells. ERCC1 translocation to DNA damage-induced subnuclear foci is markedly impaired in USP45 knockout cells, possibly accounting for defective DNA repair. Finally, USP45 localises to sites of DNA damage in a manner dependent on its deubiquitylase activity, but independent of its ability to bind ERCC1–XPF. Together, these results establish USP45 as a new regulator of XPF–ERCC1 crucial for efficient DNA repair.
Keywords: interstrand cross-link, nucleotide excision repair, ubiquitin, UV irradiation
Introduction
Protein ubiquitylation is a post-translational modification that plays a critical role in regulating biological processes (Hershko & Ciechanover, 1998; Kerscher et al, 2006). The first step of the ubiquitylation process involves the covalent attachment via an isopeptide linkage of the C-terminal carboxyl residue of ubiquitin to the epsilon amine group of a Lys residue side chain on the target protein. Extra ubiquitin moieties can then be conjugated to any one of the ubiquitin Lys residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48 and Lys63) or the amino terminal Met1 residue to form polyubiquitin chains (Komander & Rape, 2012). The most extensively studied role for ubiquitylation is Lys48-linked targeting of proteins for 26S proteasome-mediated degradation (Glickman & Maytal, 2002). More recent work has revealed crucial roles for other types of ubiquitylation including monoubiquitylation and other types of polyubiquitin chain, in particular the K63 and K11 linkages (Ikeda & Dikic, 2008). Hybrid polyubiquitin chains made up of diverse ubiquitin linkages have also been detected in cells (Guzzo et al, 2012; Emmerich et al, 2013). The diverse polyubiquitin chains regulate cell functions such as gene transcription, DNA repair and replication, immune cell signalling, intracellular trafficking and many more (Hershko & Ciechanover, 1998; Kulathu & Komander, 2012).
Key features of ubiquitylation that are likely to account for why it has been selected as a major regulatory mechanism is that it is inducible, reversible and can serve as a scaffolding mechanism to recruit diverse sets of proteins that recognise mono- and specific polyubiquitin chains. The enzymes that assemble and disassemble ubiquitin chains underpin the versatility of the ubiquitin system. Three types of ubiquitin enzymes (E1, E2 and E3) control the conjugation of ubiquitin to targets (Pickart, 2001) and approaching 100 deubiquitylases (DUBs) act to reverse ubiquitylation (Nijman et al, 2005).
The catalytic domains of DUBs have been subdivided into five families based on the nature of their catalytic domains, namely ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), Machado–Joseph disease protein domain proteases (MJDs), ovarian tumour proteases (OTUs), monocyte chemotactic protein-induced protein proteases (MCPIP) and JAB/MPN/Mov34 metalloenzyme proteases (JAMM) (Nijman et al, 2005; Komander et al, 2009). JAMM DUBs are metalloproteases, whereas the rest of the families are cysteine proteases (Nijman et al, 2005; Komander et al, 2009). Although several OTU members display marked selectivity towards certain ubiquitin chain linkages, the majority of DUBs, especially those belonging to the USP group, display no significant intrinsic selectivity towards ubiquitin chain topologies (Rago et al, 2007; Faesen et al, 2011; Ritorto et al, 2014).
We were interested in studying the role of ubiquitin-specific-processing protease 45 (USP45), as cancer genome analysis has revealed that ∽12% of prostate cancers and ∽5% of diffuse large B-cell lymphoma display deletions in the USP45 gene (http://www.cbioportal.org). USP45 is also one of the least well-characterised DUBs and is highly conserved with clear homologues in mammals, fish, frog, fly (Drosophila CG4165) and worm (Caenorhabditis elegans CYK-3). USP45 belongs to a group of twelve related USP isoforms (USP3, USP5, USP13, USP16, USP20, USP22, USP33, USP39, USP44, USP49 and USP51) that possess a short N-terminal region specific to each isoform bearing no obvious functional motif (residues 1–61 USP45), followed by an N-terminal zinc finger–ubiquitin-binding protein (Znf-UBP, residues 62–138 USP45) and a conventional catalytic USP peptidase domain (residue 190–814-end of protein in USP45) (Ye et al, 2009; Fig1A).
Figure 1.
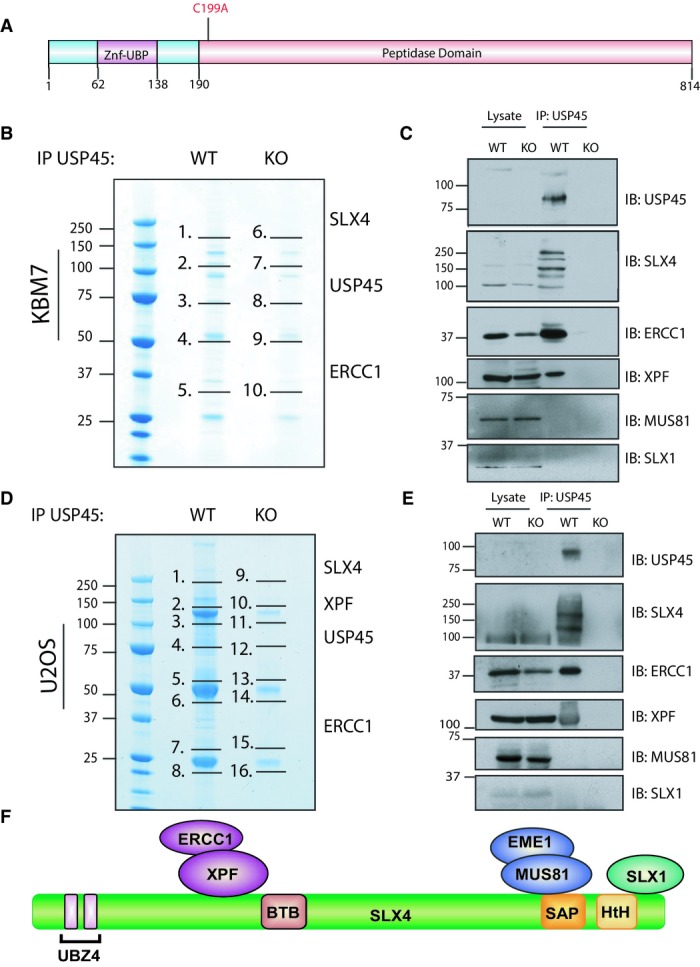
- Schematic representation of the domain architecture of human USP45 protein.
- Endogenous USP45 was immunoprecipitated (IP) from wild-type (WT) and USP45 knockout (KO) KBM7 cells lines. The immunoprecipitates were resolved on a polyacrylamide gel and stained with Instant Blue. The gel was divided into the indicated sections, and proteins identified by mass spectrometry are detailed in Supplementary Table S1. The key proteins that we focus on in this study are indicated.
- Endogenous USP45 was immunoprecipitated from the WT and KO KBM7 cell lines and subjected to immunoblotting with the indicated antibodies.
- Endogenous USP45 was immunoprecipitated from WT and USP45 KO U2OS cells lines and analysed as in (B). Mascot scores of the interactors are detailed in Supplementary Table S2.
- Endogenous USP45 was immunoprecipitated from the WT or KO U2OS cell lines and subjected to immunoblotting with the indicated antibodies. Similar results were obtained in at least two separate experiments.
- Schematic representation of the SLX4 complex.
Source data are available online for this figure
Here, we demonstrate that USP45 acts to regulate the ubiquitylation of the ERCC1 subunit of the XPF–ERCC1 DNA repair nuclease that plays a critical role in the repair of DNA interstrand cross-links (ICL) and in nucleotide excision repair (NER) of UV light-induced DNA damage (McNeil & Melton, 2012). Consistent with USP45 playing a major role in controlling ERCC1–XPF biology, our data reveal that knockout of USP45 significantly sensitises cells to ICLs and UV light, DNA damaging agents that trigger responses controlled by ERCC1–XPF. We also provide evidence that the ability of ERCC1 to translocate to foci of DNA damage is controlled by USP45 and show that ability of cells to repair cyclobutane pyrimidine dimers (CPD), DNA lesions resulting from UV-C treatment, is greatly impaired in USP45 knockout cells. Lastly, we observe that endogenous USP45 rapidly and transiently localises to sites of DNA damage induced by laser irradiation of cells exposed to agents that induce DNA damage in a manner dependent upon the deubiquitylase activity of USP45, but independent of its ability to bind ERCC1–XPF. These data highlight a novel important role for USP45 in controlling ERCC1–XPF and DNA damage responses.
Results
USP45 associates with the ERCC1–XPF endonuclease
To investigate USP45 function in vivo, we raised antibodies that detected endogenous USP45 (Supplementary Fig S1). We also disrupted the USP45 gene in both chronic myelogenous leukaemia KBM-7 cells (Rago et al, 2007; Supplementary Fig S2A) and in U2OS osteosarcoma cell line (Supplementary Fig S2B) to knockout USP45 protein expression. To gain clues to USP45 function, we set out to identify interacting proteins. We used mass spectrometry to characterise proteins that co-immunoprecipitated with endogenous USP45 from the wild-type but not from the knockout KBM7 (Fig1B, Supplementary Table S1 and Source Data to Fig 1) or U2OS cells (Fig1D, Supplementary Table S2 and Source Data to Fig 1). We observed there were a number of proteins that co-precipitated with USP45 from wild-type cells but not from the USP45 knockout cell lines. These included MYH9, MYH10, MYL12B, ZFR and RBMX that have previously been reported to interact with USP45 in large scale DUB interaction screens (Sowa et al, 2009).
We decided to focus on proteins that had not been previously reported to interact with USP45, namely ERCC1, XPF and SLX4. ERCC1–XPF is a heterodimeric DNA repair endonuclease involved in the repair of bulky DNA lesions such as UV-induced DNA damage, and DNA interstrand cross-links (Ciccia et al, 2008; Zhang & Walter, 2014). Around 50% of ERCC1–XPF in cells binds to SLX4, a scaffold protein that binds two other DNA repair endonucleases besides XPF–ERCC1: MUS81-EME1 and SLX1 (Fig1F) (Fekairi et al, 2009; Munoz et al, 2009; Svendsen et al, 2009; Stoepker et al, 2011a; Saito et al, 2013). The SLX4 complex is not required for repair of UV-induced DNA damage but is essential for repair of ICLs (Fekairi et al, 2009; Munoz et al, 2009; Svendsen et al, 2009). Although ERCC1 forms a heterodimer with XPF (Sijbers et al, 1996; Ciccia et al, 2008; McNeil & Melton, 2012), XPF was not detected in KBM7 mass spectrometry analysis of the original USP45 immunoprecipitates (Fig1B). However, immunoblotting analysis confirmed that XPF as well as ERCC1 and SLX4 co-immunoprecipitated with endogenous USP45 from wild-type, but not USP45 knockout KBM7, as well as U2OS, cell lines (Fig1C and E).
USP45 interacts with ERCC1
Previous gel filtration studies have established that the ERCC1–XPF endonucleases exists in two distinct complexes: one that co-elutes with SLX4 in high molecular weight fractions and the other which elutes in lower molecular weight fractions devoid of SLX4 (Munoz et al, 2009). We next undertook gel filtration of HEK293 cells extracts, having confirmed that endogenous ERCC1, XPF and SLX4 co-immunoprecipitated with endogenous USP45 in these cells (Fig2B). Immunoblot analysis revealed that, like XPF–ERCC1, USP45 eluted in two pools. One pool co-eluted in high molecular weight fractions with SLX4 complex components (containing XPF, ERCC1, MUS81 and SLX1) and the other pool co-eluted with XPF and ERCC1 at lower molecular weight fractions (Fig2A). These data suggested that USP45 interacts directly with XPF–ERCC1 and not with SLX4.
Figure 2.

- Extracts of HEK293 cells were analysed by gel filtration on a Superdex 200 10/300 GL preparative grade column (GE Healthcare) in buffer containing 0.15 M NaCl, and every second fraction denatured and analysed by immunoblotting with the indicated antibodies. The white dotted lines indicate that the samples were run on different polyacrylamide gels.
- Endogenous USP45 or ERCC1 was immunoprecipitated from HEK293 cell lysates and subjected to immunoblotting with the indicated antibodies.
- Yeast two-hybrid (Y2H) assays were performed with a GAL4 DNA-binding domain fusion and/or activation domain for each protein indicated in the table to detect interaction between these proteins. Cells grown on media lacking LEU, TRP and HIS (to select for bait and prey plasmids) or tested for lacZ reporter gene activity.
- Endogenous SLX4 and ERCC1 were immunoprecipitated from extracts of WT or ERCC1 KO MEFs (D), or WT or SLX4 KO MEFs (E) and subjected to immunoblotting with the indicated antibodies.
- Extracts of WT or SLX4 KO MEFs were analysed by gel filtration as described in (A). Similar results were obtained in at least two separate experiments.
To define the subunit of XPF–ERCC1 with which USP45 interacts, we undertook a yeast two-hybrid analysis following the approach that we previously employed to characterise the interactions of SLX4 with its nuclease partners (Munoz et al, 2009). This revealed that USP45 interacted with ERCC1, but not with SLX4 or XPF (Fig2C, Supplementary Fig S3). The association of ERCC1 and USP45 was observed to be markedly more robust than interaction with other SLX4 components (Fig2C, Supplementary Fig S3), further suggesting that USP45 associates with ERCC1.
In agreement with USP45 associating with SLX4 indirectly through its ability to bind ERCC1, we found that USP45 did not co-immunoprecipitate with SLX4 from ERCC1 knockout mouse embryonic fibroblasts (MEFs) (Fig2D; McWhir et al, 1993; Melton et al, 1998). Moreover, we observed that in SLX4 knockout MEFs (Castor et al, 2013), co-immunoprecipitation of USP45 and ERCC1 was unaffected (Fig2E) consistent with USP45 binding to ERCC1 and not to SLX4. Previous work has revealed that in a fibroblast cell line derived from a Fanconi anaemia patient deficient in SLX4, the high molecular weight complex of ERCC1–XPF is absent (Stoepker et al, 2011b). To verify how loss of SLX4 impacted on USP45 distribution, we undertook gel filtration analysis of wild-type and SLX4 knockout MEFs. This revealed that the fraction of USP45 (as well as ERCC1 and XPF) that co-elutes with SLX4 in the high molecular weight fractions in extracts derived from wild-type MEFs is lost in SLX4 knockout MEFs although lower molecular weight is unaffected (Fig2F).
USP45 interacts specifically with ERCC1–XPF via its N-terminal 61 residues
Co-immunoprecipitation studies employing fragments of USP45 suggested that the non-catalytic 61 amino acids that lie prior to the Znf-UBP domain are necessary for binding to ERCC1, as truncation of these residues abolishes co-immunoprecipitation with endogenous ERCC1 (Fig3A). Consistent with this notion, a fragment of USP45 encompassing the N-terminal 1–62 residues interacted with ERCC1 with similar affinity to full-length USP45. The N-terminal 62 residues are highly conserved in all orthologues of USP45 we have analysed (Fig3B), but not in other DUBs. We undertook an alanine scan analysis in which fifteen of the most conserved residues within the USP45 [1–62] fragment were individually mutated to Ala, and tested the effect on ERCC1 binding. This revealed that mutation of three acidic adjacent residues (Asp25, Glu26 or Asp27) markedly inhibited interaction with ERCC1 without affecting expression of the USP45 [1–62] fragment (Fig3C, Supplementary Fig S4). A full-length USP45 [Asp25Ala, Glu26Ala] mutant failed to co-immunoprecipitate with ERCC1 (Fig3D). To verify that the interaction with ERCC1–XPF is specific to USP45, we tested whether seven different N-terminal Znf-UBP domain-containing DUBs (USP5, USP16, USP20, USP39, USP44, USP49 and USP51) associated with endogenous ERCC1–XPF after immunoprecipitation. This revealed that USP45, but not any of the other USP isoforms tested, associated with ERCC1–XPF (Fig3E).
Figure 3.
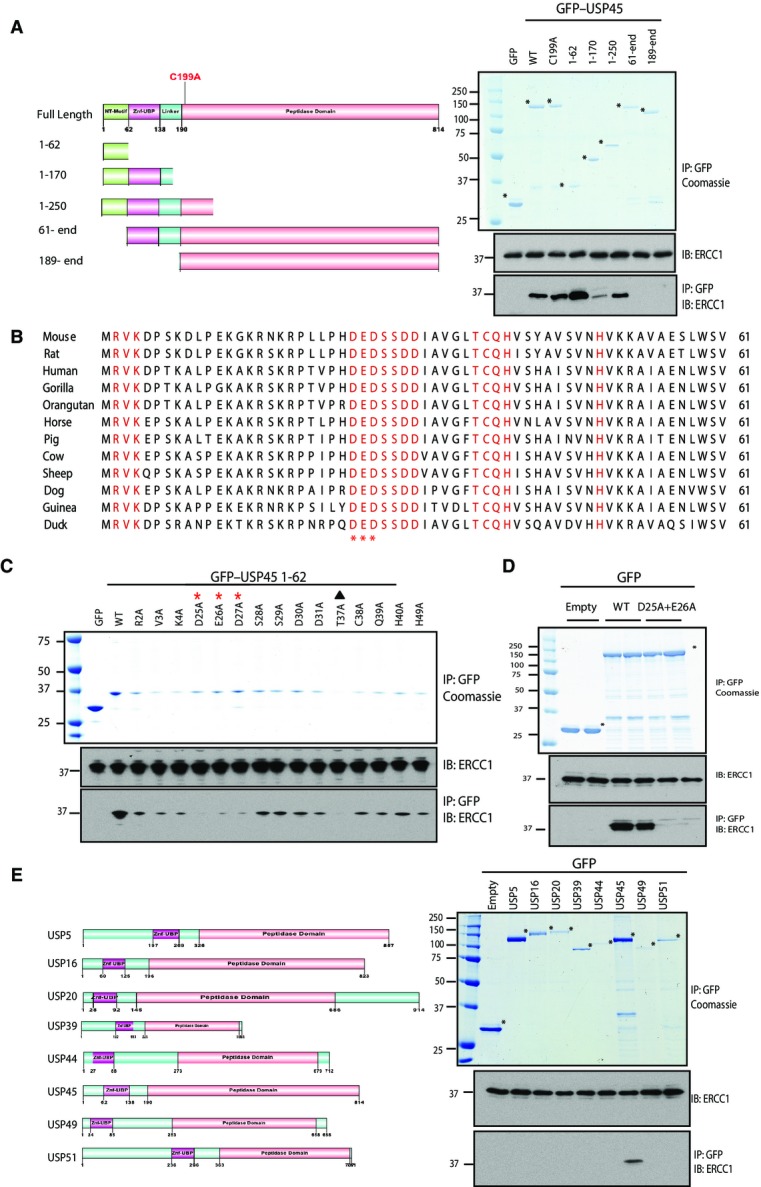
- HEK293 cells were transiently transfected with the indicated GFP fusion constructs encoding full-length or indicated fragments of USP45. Thirty-six hours post-transfection cells were lysed and GFP immunoprecipitated. The immunoprecipitates were resolved on a polyacrylamide gel and stained with Instant Blue or subjected to immunoblotting with the indicated antibodies. GFP empty vector was used as a negative control.
- Sequence alignment of the first 62 residues of USP45 from the indicated species. Conserved residues that were mutated in part (C) are indicated in red. The residues whose mutation in (C) is shown to inhibit binding to ERCC1 are marked with an asterisk. Residue Thr37 (marked with a solid triangle) was found to destabilise expression when mutated to Ala.
- HEK293 cells were transiently transfected with constructs expressing the indicated wild-type or mutant GFP–USP45 [1–62] fragment (C) or GFP full-length USP45 (D) or empty GFP (C and D) as a control and processed as described in (A). LICOR–Odyssey quantitation of results shown in (C) is shown in Supplementary Fig S4.
- As in (C) except HEK293 cells were transfected with the indicated N-terminal Znf-UBP domain-containing DUBs. Similar results were obtained in at least two separate experiments.
USP45 controls ubiquitylation of ERCC1
We reasoned that USP45 might control ubiquitylation of ERCC1, XPF or SLX4. To address this question, HEK293 cells were treated with the proteasome inhibitors lactacystin (Fig4A, left panel) or bortezomib (Fig4A, right panel) for different times before lysis in a buffer containing N-acetylmaleimide to inhibit DUBs dependent on catalytic cysteine residues, and EDTA to inhibit metalloenzyme dependent DUBs. As expected, treatment of cells with lactacystin or bortezomib induced a time-dependent general increase in protein ubiquitylation (lower panel, Fig4A). Furthermore, both inhibitors induced a time-dependent increase in the levels of ERCC1 and provoked the concomitant appearance of a diffuse higher molecular weight species of ERCC1, suggestive of ubiquitylation. In contrast, levels of XPF or SLX4 were not markedly altered, nor was there any evidence for ubiquitylation of these proteins following inhibition of the proteasome (Fig4A).
Figure 4.

- HEK293 cells were treated with the proteasome inhibitor lactacystin (5 μM) or bortezomib (10 μM) for the times indicated. Cells were lysed in the presence of 0.1 M N-ethylmaleimide to inhibit deubiquitylation and lysates were subjected to immunoblotting with indicated antibodies.
- Wild-type (WT) and USP45 knockout (KO) U2OS cells were lysed and subjected to immunoblotting as in (A).
- USP45 KO U2OS cells were transiently transfected with a construct encoding FLAG-ERCC1. Thirty-six hours post-transfection cells were treated with lactacystin (5 μM) for 6 h and lysed with a buffer containing 0.1 M N-ethylmaleimide. Flag-ERCC1 was immunoprecipitated and treated in the absence or presence of 0.1 μg of recombinant USP45 wild-type (WT), non-ERCC1-binding USP45 [Asp25Ala, Glu26Ala] (AA) or catalytically inactive USP45 [Cys199Ala] (CA) for 1 h. The reactions were terminated by addition of sample buffer and analysed by immunoblot and Coomassie staining. Similar results were obtained in at least two separate experiments.
To further investigate the role of USP45, we compared the levels of ERCC1, XPF and SLX4 in wild-type and USP45 knockout U2OS cells (Fig4B). This revealed that disruption of USP45 in U2OS cells induced a marked increase in levels of diffuse high molecular weight species of ERCC1, presumed to be ubiquitylated forms of the protein, that was accompanied by a decrease in levels of the non-ubiquitylated species of ERCC1 migrating at 37 kDa. In contrast, the levels and migration pattern of XPF or SLX4 were unaltered in USP45 knockout cells (Fig4B). Restoring expression of wild-type USP45 in knockout U2OS cells markedly lowered the high molecular weight species of ERCC1 (Fig4B).
To explore whether USP45 can directly catalyse deubiquitylation of ERCC1, we isolated ubiquitylated Flag–ERCC1 (Fig4C) or GFP–ERCC1 (Supplementary Fig S5B), expressed in USP45 knockout U2OS cells treated with lactacystin proteasomal inhibitor. Immunoblot analysis with ERCC1 antibody indicated that ERCC1 was significantly ubiquitylated under these conditions (Fig4C, Supplementary Fig S5B). Treatment of ubiquitylated ERCC1 with wild-type recombinant USP45 induced almost complete deubiquitylation of ERCC1, judged by a loss in the higher molecular weight species of ERCC1 which is accompanied by an increase in the level of non-ubiquitylated ERCC1 (Fig4C). Importantly, the USP45 mutant [Asp25Ala, Glu26Ala] that displays the same intrinsic catalytic activity as wild-type USP45 when assayed using a generic ubiquitin–rhodamine substrate (Supplementary Fig S5A), but that cannot bind ERCC1, failed to deubiquitylate ERCC1 in parallel experiments (Fig4C, Supplementary Fig S5B). As expected, the catalytically inactive USP45 [Cys199Ala] mutant also failed to deubiquitylate ERCC1 (Fig4C, Supplementary Fig S5B). Taken together, these data indicate that disruption of USP45 leads to the accumulation of ubiquitylated forms of ERCC1, but it appears that the overall levels of ERCC1 are not affected. Furthermore, USP45 can deubiquitylate ERCC1 in a manner that requires association of USP45 and ERCC1. In this light, we observed that 5 other DUBs tested (AMSH, Cezanne, OTUB1, UCHL1 and USP27X) failed to deubiquitylate ERCC1 under conditions that wild-type USP45 deubiquitylated ERCC1 (Supplementary Fig S5C). Immunoblot analysis of immunoprecipitated GFP–ERCC1 with chain topology-specific ubiquitin antibodies revealed the presence of both Lys48- and Lys63-specific chain linkages (Supplementary Fig S5C). Both the Lys48 and Lys 63 linkages were sensitive to treatment with wild-type USP45 (Supplementary Fig S5C).
USP45 promotes survival of cells exposed to agents that induce DNA damage responses controlled by ERCC1–XPF endonuclease
Work from many laboratories has shown that ERCC1 is involved in the repair of DNA ICLs and for nucleotide excision repair (NER) of UV-induced DNA damage (Kaye et al, 1980; Westerveld et al, 1984; Hoy et al, 1985; Kondo et al, 1989; Busch et al, 1997). The influence of USP45 on ERCC1 ubiquitylation prompted us to test whether USP45 is required for ICL repair and NER, using our USP45 knockout cells. Before these experiments, we first monitored the growth rate of wild-type and USP45 knockout U2OS cells and found that the knockout cells proliferated more slowly, an effect that was rescued by overexpression of wild-type but not catalytically inactive mutant (Supplementary Fig S6A). This is reminiscent of cells lacking ERCC1 (Weeda et al, 1997). Flow cytometry analysis indicated, however, that USP45 knockout did not influence the proportion of cells in G1, S and G2-M phase in asynchronous cell populations (Supplementary Fig S6B).
We next performed clonogenic survival assays to compare the sensitivity of wild-type and USP45 knockout U2OS cells to mitomycin C (MMC), which induces ICLs, or to UV-C irradiation. This revealed that compared to the wild-type cells, the USP45 knockout U2OS cells were hypersensitive to both MMC (Fig5A and C) and UV-C (Fig5E). Furthermore, expression of wild-type USP45, but not the USP45 [Asp25Ala, Glu26Ala] mutant or the catalytically inactive USP45 [Cys199Ala] mutant, rescued the genotoxin hypersensitivity of USP45 knockout U2OS cells (Fig5A and C). Expression of similar levels of ectopic wild-type and mutant USP45 in USP45 knockout cells was confirmed by immunoblot analysis (Fig5B and D). The slow growth of USP45 knockout cells (Supplementary Fig S6) raised the possibility that sensitivity to UV and ICLs was caused by general sickness, but USP45 knockout U2OS cells were not more sensitive than parental cells to the effects of hydroxyurea, a ribonucleotide reductase inhibitor that causes stalling of replication forks (Yarbro, 1992) (Fig5F). Taken together, the data in this section reveal that the catalytic activity of USP45 is required for repair of ICLs and DNA damage induced by UV light.
Figure 5.
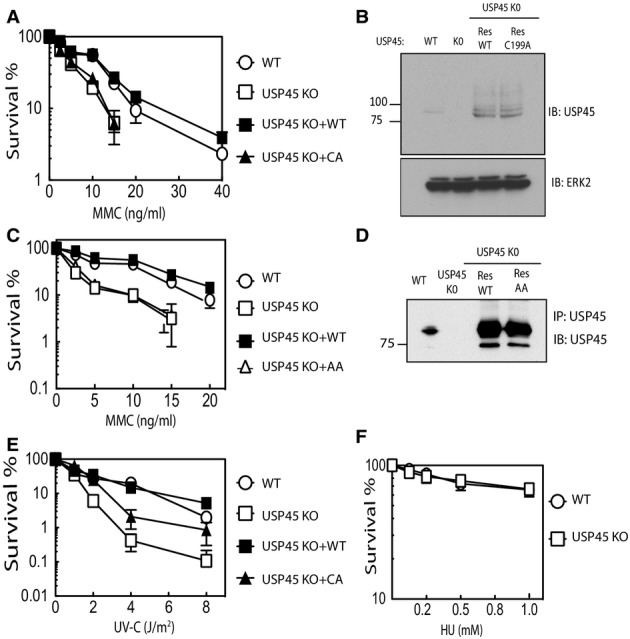
- Clonogenic survival assays were carried out in wild-type (WT), USP45 knockout (KO) or USP45 KO cells re-expressing wild-type USP45 (RESWT) or catalytically inactive USP45 [Cys199Ala] (RESCA). The cells were treated with the indicated doses of mitomycin C (MMC) 16 h. Medium was changed and number of colonies quantified after 10 days. Each data point is the average of 3 experiments undertaken with 6 replicates ± SD.
- Control immunoblot analysis demonstrating that in the USP45 KO U2OS rescue cell lines, similar levels of wild-type and catalytically inactive USP45 [Cys199Ala] were expressed.
- As above, except rescue experiments are undertaken using the non-ERCC1-binding USP45 [Asp25Ala, Glu26Ala] mutant.
- As in (A), except that DNA damage is induced using UV-C irradiation (E) or 16 h hydroxyurea treatment (F). Similar results were obtained in at two separate experiments.
Previous work has suggested that ERCC1 deficiency leads to chromosomal aberrations, including gaps, breaks and radials in metaphase spreads of MMC-treated Ercc1 null cells (Niedernhofer et al, 2004b). We therefore explored the levels of chromosome abnormalities in wild-type and USP45 knockout U2OS cells. This revealed a significant increase of abnormalities in USP45 knockout cells compared to wild-type cells (Supplementary Fig S7).
To investigate whether USP45 exerts its effects through stabilising ERCC1 expression, we monitored ERCC1 levels in wild-type and USP45 knockout cells treated with the protein synthesis inhibitor cycloheximide. This revealed that exposure of cells to cycloheximide for 8 h did not markedly affect ERCC1 levels in either the wild-type or USP45 knockout cells treated in the presence or absence of MMC (Supplementary Fig S8). Moreover, we observed that overexpression of ERCC1 failed to rescue the hypersensitivity of USP45 knockout cells to MMC or UV-C (Supplementary Fig S9). These data therefore indicate that likely the ubiquitylation of ERCC1, which is reversed by USP45, controls an aspect of ERCC1 function other than regulating its stability.
USP45 regulates the recruitment of ERCC1 to the damage
XPF–ERCC1 forms spontaneous foci in cells that probably correspond to telomeres, and we next analysed the impact of USP45 on spontaneous ERCC1 in U2OS cells by quantifying the proportion of cell nuclei with 10 or more ERCC1 foci (Stoepker et al, 2011b). This analysis revealed that USP45 knockout U2OS cells displayed a three to fourfold reduction in nuclei with 10 or more ERCC1 foci compared to wild-type cells (Fig6A and B). Moreover, expression of wild-type USP45, but not catalytically deficient USP45 [Cys199Ala] mutant in these cells, restored the number of spontaneous ERCC1 foci almost to the level seen in wild-type U2OS cells (Fig6A and B).
Figure 6.

- Wild-type (WT), USP45 knockout (KO) or USP45 KO cells re-expressing wild-type USP45 (RESWT) or catalytically inactive USP45 [Cys199Ala] (RESCA) were used to analyse ERCC1 foci formation. Staining and analysis of endogenous ERCC1 foci formation was undertaken before (left panel), or after DNA damage induction by mitomycin C (40 ng/ml, 16 h, middle panel) or UV (20 J/m2 followed by 3-h recovery, right panel). Three independent experiments were performed in which 500 cells per experiment were analysed. Scale bar, 10 μm.
- Proportion of cells displaying more than 10 endogenous ERCC1 foci were quantified. Three independent experiments were performed in which 500 cells per experiment were analysed. Results are the mean of 3 experiments ± SD.
- The wild-type and USP45 knockout U2OS cells were treated with mitomycin C (40 ng/ml, 16 h) and total number of ERCC1 staining foci per cell was quantified in at least 50 independent cells at the indicated times. Similar results were obtained in two separate experiments. The data are presented as the average number of ERCC1 staining foci per cell.
- As in (C), except that γ-H2AX-staining foci were analysed. The data are presented as the average number of γ-H2AX staining foci per cell.
Treatment of cells with genotoxins stimulates the formation of DNA damage-induced XPF–ERCC1 foci. We observed that exposure of wild-type U2OS cells to MMC (24 h) or UV-C (4 h post-UV) caused an approximately sevenfold increase in the number of nuclei with 10 or more ERCC1 foci (Fig6A and B). In contrast, no such increases in ERCC1 foci were observed in USP45 knockout U2OS cells (Fig6A and B). Expression of wild-type USP45, but not the catalytically deficient USP45 [Cys199Ala] mutant in USP45 knockout U2OS cells, restored the number of DNA damage-induced ERCC1 foci to levels observed in wild-type cells (Fig6A and B). Normal DNA damage responses were initiated in wild-type and USP45 knockout cells as judged by formation of γ-H2AX foci after MMC treatment and XPA foci after UV-C treatment (Supplementary Figs S10 and S11). Treatment with the lactacystin proteasomal inhibitor increases ubiquitylation of ERCC1 (Fig4A), but had no significant impact on ERCC1 foci formation in wild-type or USP45 knockout cells (Supplementary Fig S12).
Time-course analysis revealed that in wild-type cells, the number of ERCC1 containing foci was markedly elevated at 24 and 48 h post-MMC treatment and declined to near basal levels within 96 h (Fig6C, Supplementary Fig S13). In contrast, in USP45 knockout cells, the number of ERCC1 containing foci remained low at all time points analysed (Fig6C, Supplementary Fig S13).
We also explored how USP45 knockout impacts on γ-H2AX foci induced by MMC. After 24 h MMC treatment, the number of γ-H2AX containing foci was high in both wild-type and USP45 knockout cells, indicating that normal DNA damage responses were initiated (Fig6D, Supplementary Fig S14). Strikingly, however, the number of γ-H2AX foci remained high in USP45 knockout cells even after 96 h post-MMC treatment under conditions in which the number of foci had returned to near basal levels with 72 h in wild-type cells (Fig6D, Supplementary Fig S14). Similar effects on γ-H2AX foci were reported in Ercc1 knockout MEFs (Niedernhofer et al, 2004b), consistent with the defects in USP45 knockout cells caused by defects in ERCC1 function.
USP45 is recruited to DNA damage sites and controls repair of UV-induced DNA damage
We next explored whether USP45 localises at sites of DNA damage. To this end, we used a monoclonal antibody that recognised endogenous USP45 in immunofluorescence experiments. USP45 showed a diffuse staining pattern in both cytoplasmic and nuclear compartments of wild-type U2OS cells. This pattern was specific for USP45 because no signal was observed in USP45 knockout U2OS cells (Supplementary Fig S1B). Furthermore, the localisation of a GFP-tagged form of USP45 overexpressed in U2OS cells was similar to the endogenous protein (Supplementary Fig S1B). We induced DNA damage in three ways: firstly, cells were incubated with 5-bromo-2′-deoxyuridine and nuclei micro-irradiated along a track in the nucleus with a 355-nm laser; this treatment induces heterogeneous range of DNA lesions. In wild-type cells, USP45 formed a stripe along the track of DNA damage induced in this manner, which also contained the γ-H2AX signal, a well-defined marker of DNA damage (Fig7). Secondly, cells were incubated with the intercalating agent psoralen and nuclei were micro-irradiated along a track using a 355-nm UV-A laser to induce predominantly ICLs (Lachaud et al, 2014). In control experiments, psoralen was replaced by angelicin, which in the presence of UV-A causes formation of intrastrand cross-links (a substrate for NER) instead of ICLs. As shown in Fig7, in wild-type U2OS cells, USP45 localised to tracks of DNA damage induced by psoralen/UV-A and by angelicin/UV-A. In each case, the USP45 stripes contained γ-H2AX. In USP45 knockout U2OS cells, laser stripes contained γ-H2AX signal but not USP45 signal (Fig7). To investigate whether USP45 is also recruited to DNA damage induced by UV-C treatment, which has profound disabling effects in NER-defective cells, U2OS cells were overlaid with microscale pore-containing filters and exposed to a UV-C energy source (254 nm). This revealed recruitment of GFP–USP45, but not GFP alone, to the regions of localised UV irradiation. These same areas staining for USP45 co-localised with sites of staining with an antibody specific for CPD generated by UV-C irradiation (Fig8A) (Gerard et al, 2006; Pathania et al, 2011).
Figure 7.
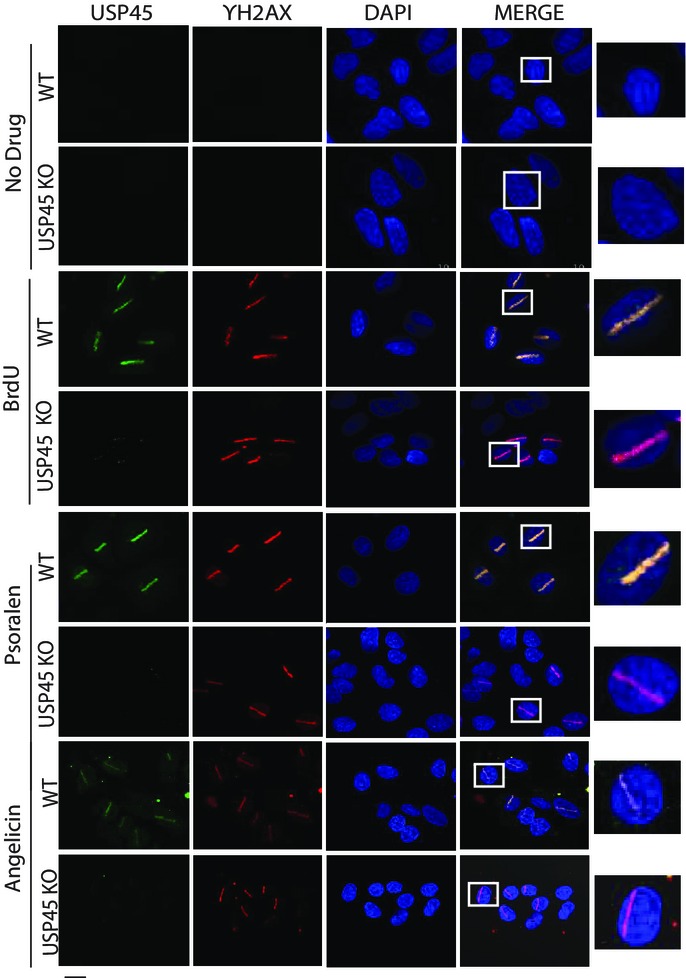
USP45 localises at sites of DNA damage
Representative immunofluorescence images of endogenous wild-type (WT) and USP45 knockout (KO) U2OS cells treated with no drug, BrdU (10 μM, 24 h), psoralen or angelicin (25 μM, 3 h). Cells were fixed 10 min after laser micro-irradiation and stained for localisation of endogenous USP45, phosphorylated γ-H2AX and DAPI. The right-hand panel shows magnification of one micro-irradiated cell. Similar results were obtained in at least three independent experiments. Scale bar, 10 μm.
Figure 8.
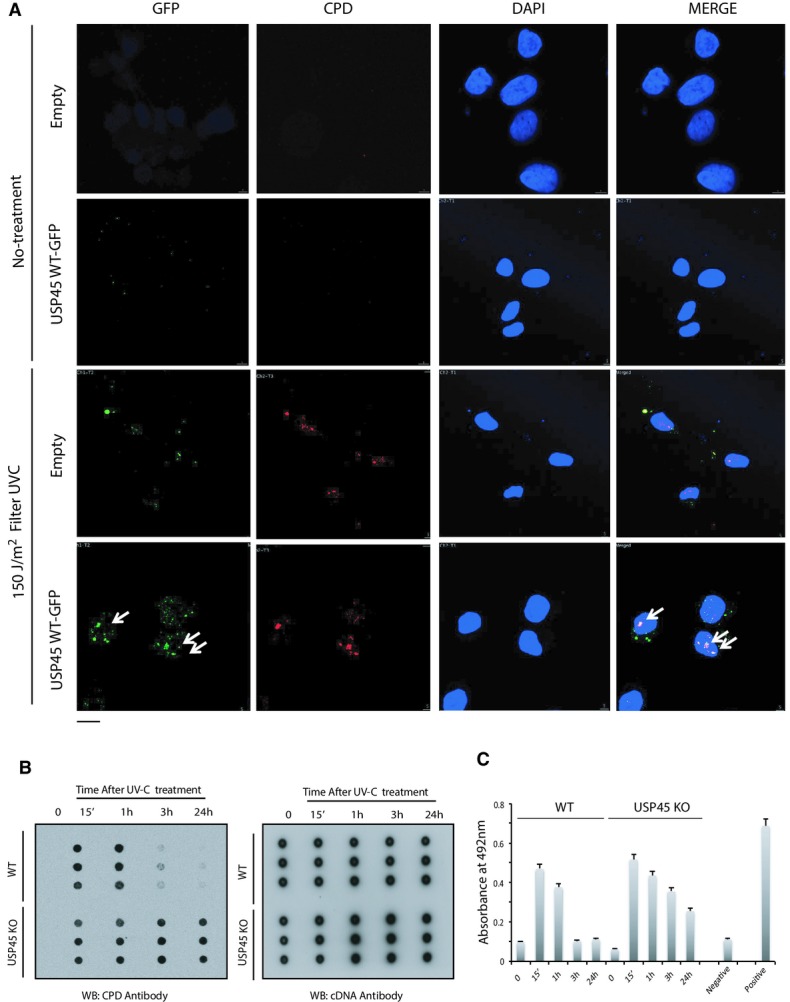
- U2OS cells were transiently transfected with the GFP–USP45 or GFP alone. Thirty-six hours post-transfection cells were irradiated with UV-C (150 J/m2) through a micropore filter (lower panels) or left untreated (upper panels). Five minutes post-irradiation, cells were fixed and co-immunostained with antibodies recognising cyclobutane pyrimidine dimer (CPD) DNA lesions and GFP. The white arrows indicate co-localisation between GFP–USP45 and CPD. Scale bar, 10 μm.
- U2OS wild-type (WT) and USP45 knockout (KO) cells were irradiated with UV-C (20 J/m2). At the indicated post-irradiation times, genomic DNA was extracted and subjected to Southern dot blot analysis using antibodies recognising CPD and dsDNA total antibody. Data are shown from a triplicate experiment in which each blot is derived from independent cells.
- Same as (B) but using a High Sensitivity CPD/Cyclobutane Pyrimidine Dimer Elisa kit (NM-MA-K001) from Cosmo Bio. For the DNA damage detection, the manufacturer protocol was followed. Absorbance at 492 was measured that represent amount of CPDs in each sample at indicated times. Calf thymus DNA, UV-C irradiated (10 J/m2) and not irradiated was used as positive and negative samples, respectively.
Given the importance of XPF–ERCC1 in the repair of UV-induced DNA damage, we next assessed the repair of CPDs in wild-type and USP45 knockout U2OS cells exposed to UV-C irradiation. To this end, we used the dot blot method where genomic DNA is probed with an antibody that recognises CPDs (Patrick, 1977; Schwarz et al, 2002). DNA repair was judged by disappearance of the CPD signal in dot blot analyses of genomic DNA during recovery from UV exposure. As shown in Fig8B, repair of UV-C-induced CPDs in wild-type cells was observed after 3 h and complete by 24 h. In contrast in USP45 knockout cells, significant levels of CPD damage were still observed 24 h after UV-C irradiation (Fig8B, Supplementary Fig S15). Similar results were obtained when we carried out ELISA analysis, instead of dot blot, using a different anti-CPD antibody (Fig8C). Taken together, these data indicate that USP45 localises at sites of UV-induced DNA damage, and at intra- and interstrand cross-links, and that USP45 is required, like ERCC1 for efficient NER.
USP45 is recruited transiently to sites of damage independently of ERCC1–XPF
To study the time course of recruitment of USP45 to sites of laser-induced DNA damage, we stably expressed GFPNLS–USP45 in U2OS cells with a nuclear localisation signal (NLS) on the GFP moiety to ensure nuclear localisation. We induced tracks of DNA damage as described above and monitored GFP fluorescence at sites of laser-induced micro-irradiation after BrdU treatment. This experiment confirmed that GFPNLS–USP45, but not isolated GFPNLS, formed sub-nuclear stripes along the laser tracks under all conditions employed. The recruitment of USP45 to these stripes was rapid, occurring within 30 s and declining thereafter to near basal levels within 5 min (Fig9). We also observed that in contrast to wild-type USP45, the catalytically inactive mutant GFPNLS–USP45 [Cys199Ala] when stably expressed in U2OS cells, was not recruited to laser stripes (Fig9). Moreover, GFPNLS–[USP45 residues 62-end] that lacks the ERCC1-binding motif, was still transiently recruited to laser-induced sites of DNA damage, whereas the ERCC1-binding GFPNLS–[USP45 residues 1–62] fragment was not recruited to sites of DNA damage (Supplementary Fig S16A). To confirm that ERCC1 was not required for recruitment of USP45 to sites of DNA damage, we show that in ERCC1 knockout MEFs, endogenous USP45 is normally recruited to sites of damage (Supplementary Fig S16B and C).
Figure 9.
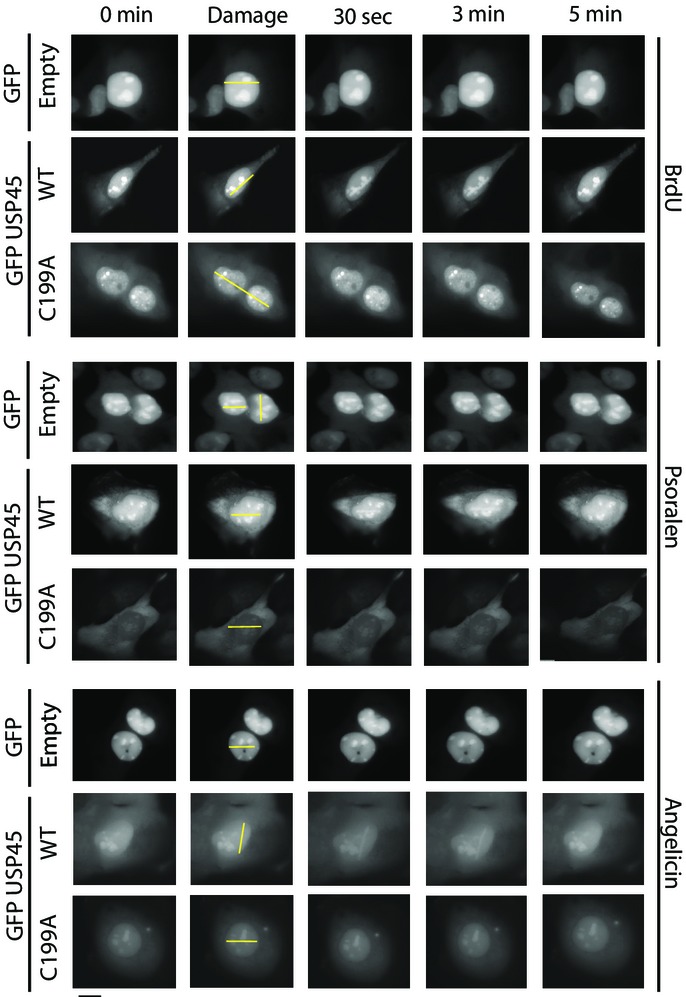
USP45 recruitment to sites of DNA damage is rapid and transient
Live-cell fluorescence analysis of U2OS cells stably expressing either GFPNLS empty, wild-type (WT) USP45 or catalytically inactive USP45 [Cys199Ala]. Cells were treated with BrdU (10 μM, 24 h), psoralen or angelicin (25 μM, 3 h) and images captured at the indicated times after laser micro-irradiation. The location of the micro-irradiation stripe in each cell is indicated with a yellow line. Similar results were obtained in at least two independent experiments. Scale bar, 10 μm.
Discussion
Little is known about how the activity of DNA repair nucleases is controlled in cells. Insufficient activity would compromise DNA repair, whereas excessive activity might result in pathological genome cleavage events. In this study, we identify USP45 as a regulator of ERCC1 function in cells. Mass spectrometry analysis identified ERCC1–XPF and the SLX4 scaffold as potentials targets of USP45 together with a number of other proteins that co-precipitated with USP45. These included MYH9, MYH10, MYL12B, ZFR and RBMX that have previously been reported to interact with USP45 (Sowa et al, 2009). There are two pools of ERCC1–XPF in cells—one is bound to the SLX4 scaffold protein and the other is not (Munoz et al, 2009; Stoepker et al, 2011b). Gel filtration experiments showed that USP45 was associated with both pools of XPF–ERCC1. This observation, together with the findings that USP45 co-precipitates with XPF–ERCC1 in Slx4 null cells and that ERCC1 interacts with USP45 in a yeast two-hybrid assay, indicate that USP45 interacts directly with ERCC1.
Our data reveal that the interaction of ERCC1 with USP45 is specific to this particular DUB as the interaction is mediated by the N-terminal 62 amino acids that are highly conserved in USP45 orthologues, but are not found in other DUBs. We identify a cluster of acidic residues (Asp25, Glu26 and Asp27), which lie within a stretch of seven invariant residues (DEDSSDD, residues 25–31) that are conserved in all USP45 species we have analysed. This acidic motif is essential for the interaction as the USP45 [Asp25Ala, Glu26Ala] mutant fails to bind ERCC1. In future work, it would also be interesting to explore whether this motif enables USP45 to bind specifically to ERCC1 or whether it also enables USP45 to interact with other substrates. Interestingly, all DUBs related to USP45 that have an N-terminal Znf-UBP domain (USP3, USP5, USP13, USP16, USP20, USP22, USP33, USP39, USP44, USP49 and USP51) also possess distinct N-terminal residues that are highly conserved between species but not between USP isoforms. By analogy with USP45, it would be interesting to explore whether these N-terminal motifs also operate to direct interactions of Znf-UBP domain-containing DUBs with their target(s).
Previous work has revealed that several components of the DNA damage repair pathways are controlled by deubiquitylation. These include the deubiquitylation of PCNA and FANCD2 (Garcia-Higuera et al, 2001) catalysed by USP1 (Nijman et al, 2005), deubiquitylation and stabilisation of 53BP1 or claspin by USP28 (Zhang et al, 2006) and more recently is has been reported that USP3 catalyses the deubiquitylation of H2AX and γ-H2AX (Sharma et al, 2014). Thus far, there have not been any reports regarding the regulation of ERCC1–XPF by ubiquitylation, but our results indicate that USP45 counteracts ubiquitylation of ERCC1. Supporting this conclusion is the finding that ubiquitylated forms of ERCC1 accumulate in cells lacking USP45, accompanied by a concomitant decrease in the levels of the non-ubiquitylated form of ERCC1. We also observe that in vitro wild-type USP45 can catalyse the deubiquitylation of ERCC1 that has been immunoprecipitated from cells treated with the proteasome inhibitor lactacystin (Fig4C, Supplementary Fig S5B). Importantly, we find that the USP45 mutant [Asp25Ala, Glu26Ala] that is catalytically active but that cannot bind to ERCC1 (Supplementary Fig S5A) failed to deubiquitylate ERCC1 in vitro, indicating that specific interaction between ERCC1 and USP45 is essential to trigger deubiquitylation. Therefore, a function of USP45 appears to control ubiquitylation of ERCC1.
Cells lacking USP45 are hypersensitive to the same spectrum of genotoxic agents—UV-C and ICL-induced DNA damage—as cells lacking XPF or ERCC1. Like cells defective in XPF or ERCC1, USP45 knockout cells show defects in the repair of UV-induced CPD photoproducts (Fig8B and C, Supplementary Fig S15) and defects in the resolution of H2AX foci during recovery from exposure to ICLs (Fig6D, Supplementary Fig S14). Furthermore, XPF–ERCC1 is the only known protein common to both NER and ICL repair (Niedernhofer et al, 2004a; Muniandy et al, 2009; Orelli et al, 2010; Wang et al, 2011). On this basis, we speculate that is likely that XPF–ERCC1 is the major target of USP45 in controlling DNA repair. Consistent with this notion, the DNA damage sensitivity of USP45 knockout cells is only rescued by wild-type USP45 and not by an active USP45 [Asp25Ala, Glu26Ala] mutant that is unable to bind ERCC1 (Fig5C) or catalytically inactive USP45 [Cys199Ala] (Fig5A). This implies that the rescue of the sensitivity phenotype depends on the interaction between ERCC1 and USP45 as well as its catalytic activity. We cannot exclude the possibility that there may be other substrates of USP45 relevant to genome stability, but our data strongly suggest therefore that ERCC1 is the major substrate of USP45 in promoting DNA repair.
How does ERCC1 deubiquitylation promote DNA repair? Our findings that overexpression of ERCC1 did not rescue the sensitivity of USP45 knockout cells to MMC and UV-C (Supplementary Fig S9) and that USP45 knockout does not affect ERCC1 half-life (Supplementary Fig S8), suggest that the role of USP45 in controlling ERCC1 ubiquitylation goes beyond control of ERCC1 stability and expression levels. Instead ubiquitylation/deubiquitylation appears to control ERCC1 function in a more direct way. In this light, USP45-defective cells also show a striking reduction in the DNA damage-induced formation of ERCC1 foci, which was not rescued by proteasome inhibition (Supplementary Fig S12). This defect in ERCC1 recruitment could in principle account for the DNA repair defects of USP45-defective cells. Thus, the major role of ERCC1 deubiquitylation by USP45 may be to enable ERCC1–XPF to gain access to DNA damage sites. How removal of ubiquitin from ERCC1 impacts on its recruitment to sites of DNA damage at the molecular level will be an interesting avenue for future investigation. It will also be a challenging endeavour because the mechanism of recruitment of ERCC1–XPF to ICLs differs from recruitment to UV-induced damage (JR, unpublished data; Volker et al, 2001). It will also be fascinating to identify the E3 ligase responsible for ERCC1 ubiquitylation and work out how it is regulated.
We showed that USP45 is also recruited to sites of DNA damage lesions, in a manner that is dependent on functional catalytic activity of the deubiquitylase domain, but independent of its ability to bind ERCC1 (Fig9, Supplementary Fig S16). These results suggest that there are additional component(s) to be identified that recruit USP45 to sites of DNA damage. In future work, it would be important to elucidate the mechanism by which USP45 is recruited to sites and define whether there are new USP45 substrates located in the vicinity of DNA lesions. It is interesting to note that a recent paper describing a complementary set of functional screens to identify DUBs involved in DNA damage responses did not identify USP45 (Nishi et al, 2014). One of the screening approaches employed by these authors involved analysing the ability of transiently expressed GFP-tagged DUBs to form stripes at sites of micro-irradiation. However, the authors found that GFP–USP45 is localised mostly in the cytoplasm after transient transfection. This is also consistent with our transient transfection data (not shown). However, we have found that endogenous USP45 is localised in cytoplasm and nucleus (Supplementary Fig S1B) and is robustly recruited to DNA damage stripes (Fig7). In addition, the GFP–USP45 construct we have employed, that is recruited to sites of DNA damage, possesses a nuclear localisation signal in the GFP motif to promote nuclear expression of overexpressed GFP–USP45. We also generated cells that stably express much lower levels of GFP–USP45 than observed under transient transfection conditions, and this might also facilitate the visualisation of USP45 binding to DNA damage sites (Fig9).
Several studies have found ERCC1 to be overexpressed in cisplatin-resistant tumours, and the levels of ERCC1 mRNA and protein have been used to profile tumours to predict responses to platinum compounds (Ciccia et al, 2008; Kirschner & Melton, 2010; McNeil & Melton, 2012). It would be interesting to explore whether there were any mutations that disrupted USP45 in these tumours or other tumours. Indeed, as mentioned in the introduction, cancer genome analysis has revealed that ∽12% of prostate cancers, ∽5% of diffuse large B-cell lymphoma and a number of other tumours display deletions in the USP45 gene (http://www.cbioportal.org). Whilst cisplatin may not be widely used in the therapy of these tumours, as personalisation of medicine moves forward, it would be worth exploring whether patients displaying loss of function mutations in USP45 would be sensitised to platinum therapy. Moreover, it would be interesting to screen unclassified Fanconi anaemia patients, where no gene has been assigned, for mutations in USP45. It would also be of interest to explore whether inhibiting USP45 could be employed as a therapeutic strategy to induce sensitisation of cancer cells to platinum-based therapies.
Materials and Methods
Reagents
Ubiquitin monomer, BSA, Tris, Triton X-100 and DTT, trimethyl-psoralen (TMP), hydroxyurea, angelicin and BrdU were purchased from Sigma-Aldrich. Di-ubiquitin topoisomers (linear, K6-, K11-, K27-, K29-, K33-, K48- and K63-linked ubiquitin–ubiquitin) were purchased from Boston Biochem. Complete protease inhibitors cocktail tablets were obtained from Roche. Tween-20, Colloidal blue staining kit and precast SDS–polyacrylamide Bis-Tris gels (Invitrogen). MMC was from Duchefa.
All recombinant proteins, plasmids and antibodies generated for the present study are available upon request and described in further detail on our reagents website (https://mrcppureagents.dundee.ac.uk/).
Antibodies
The following antibodies were raised by the Division of Signal Transduction Therapy (DSTT) at the University of Dundee in sheep and affinity-purified against the indicated antigens: anti-USP45 (1st, 2nd and 3rd bleed of S052D, residues 756–780 of human USP45) and (1st, 2nd and 3rd bleed of S109D, residues 29–80 of human USP45), ERCC1 (3rd bleed of S185D, residues 1–200 of human ERCC1), mouse ERCC1 (3rd bleed of S161D, residues 1–200 of mouse ERCC1), SLX4 (3rd bleed of S714C, residues 1,535–1,834 of human SLX4), mouse SLX4 (3rd bleed of S819C, residues 166–end of mouse SLX4), mouse XPF (3rd bleed of S164D, residues 1–200 of mouse XPF), SLX1 (3rd bleed of S701B, residues 100–275 human SLX1). Human Mus81 was from Immunoquest. Total FK2 ubiquitin antibody and K48-linkage-specific ubiquitin antibodies (clone Apu2) was from Millipore. GAPDH, rabbit anti-γ-H2AX and K63-linkage-specific ubiquitin antibodies (D7A11) were from Cell Signalling. Secondary antibodies coupled to horseradish peroxidase and human XPF (clone219) were from Thermo Scientific. USP45 for immunofluorescence was from Sigma-Aldrich (H1A029602). Anti-Thymine Dimer mAb (CPD), clone KTM53, was from Kamiya Biomedical Company.
General methods
Restriction enzyme digestions, DNA ligations and other recombinant DNA procedures were performed using standard protocols. All mutagenesis was performed using the QuikChange site-directed mutagenesis method (Stratagene) with KOD polymerase (Novagen). All DNA constructs were verified by DNA sequencing, which was performed by The Sequencing Service, School of Life Sciences, University of Dundee, using DYEnamic ET terminator chemistry (Amersham Biosciences) on Applied Biosystems automated DNA sequencers. DNA for mammalian cell transfection was amplified in E. coli DH5α strain, and plasmid preparation was done using Qiagen Maxi prep Kit according to manufacturer's protocol.
Buffers
Lysis buffer for mammalian cell lysis contained 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% (w/v) Triton, 1 mM sodium orthovanadate, 10 mM sodium glycerophosphate, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 0.27 M sucrose, 0.1% (v/v) 2-mercaptoethanol, 1 mM benzamidine and 0.1 mM PMSF. Buffer A contained 50 mM Tris–HCl (pH 7.5) and 0.1 mM EGTA. TBS-Tween (TTBS) was Tris–HCl (pH 7.5), 0.15 M NaCl and 0.2% (v/v) Tween-20.
Cell culture and transfections
HEK293 and U2OS cells were cultured on 10-cm dishes in DMEM supplemented with 10% (v/v) foetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin. For transfection, each dish of adherent cells was transfected with 5–10 μg of plasmid DNA and 20 μl of 1 mg/ml polyethylenimine (Polysciences) as described previously (Durocher et al, 2002). The cells were cultured for a further 36 h and lysed in 0.3 ml ice-cold lysis buffer per dish, lysates were clarified by centrifugation at 4°C for 15 min at 16,200 g, and the supernatants were frozen in aliquots in liquid nitrogen.
Immunoprecipitation
For immunoprecipitation of GFP, TRAP-GFP beads were used. 0.5–5 mg of lysates was incubated with 10–20 μl of antibody–resin conjugate for 2 h at 4°C under gentle agitation, and the immunoprecipitates were washed three times with lysis buffer containing 0.15 M NaCl and then twice with buffer A. Proteins were eluted by resuspending washed immunoprecipitates in 30 μl of 1× SDS sample buffer.
Immunoblotting
Cell lysates (20 μg), purified proteins or immunoprecipitates in SDS sample buffer were subjected to electrophoresis on a polyacrylamide gel and transferred to nitrocellulose membranes. The membranes were incubated for 1 h with TTBS containing 5% (w/v) skimmed dried milk powder. The membranes were immunoblotted in the same buffer 16 h at 4°C with the indicated primary antibodies. Sheep antibodies were used at a concentration of 1 μg/ml, whereas commercial antibodies were diluted 1,000- to 5,000-fold. The blots were then washed with TTBS and incubated for 1 h at room temperature with secondary HRP-conjugated antibodies diluted 2,500-fold in 5% (w/v) skimmed milk in TTBS. After repeated washes, the signal was detected with the enhanced chemiluminescence reagent and the X-ray films were processed in a Konica Minolta Medical SRX-101 film processor.
Gel filtration
For gel filtration experiments, cell extracts (2 ml containing 5 mg protein) were filtered through a 0.22-μm filter and loaded on a Superdex 200 10/300 GL preparative grade column (GE Healthcare) in 50 mM Tris–HCl 7.4 buffer containing 1 mM EDTA, 0.15 M sodium chloride and 0.1% (v/v) 2-mercaptoethanol. The flow rate was 1.0 ml/min, and fractions of 1 ml (200) were collected. Molecular weight markers (Bio-Rad) were as follows: Dextran blue (2,000 kDa), thyroglobulin (670 kDa) and bovine gamma globulin (158 kDa). Every second fraction was denatured and subjected to Western blot analysis.
Expression of DUB enzymes
For bacterial protein expression, cultures were initially grow at 37°C until OD600 was between 0.4 and 0.6, then the temperature was reduced to 15°C and cultures were induced with 400 μM isopropyl β-d-1-thiogalactopyranoside and grown for further 16–18 h. Cultures were harvested, lysed and affinity purified on glutathione–Sepharose. The purified GST-USP45 was then further purified by ion exchange chromatography on a HiTrap SP-Sepharose column at pH 7.5 with a linear gradient between 0 and 500 mM NaCl. The fractions containing USP45 were pooled and dialysed into 50 mM Tris–HCl pH 7.5, 270 mM sucrose, 0.1 mM EGTA, 0.03% (V/V) Brij-35 and 0.1% (v/v) 2-mercaptoethanol.
Rhodamine DUB assay
Experiments were performed using rhodamine 110-glycine and 20 ng/μl of each DUB at 30°C during time indicated in each figure in 40 mM Tris–HCl buffer at pH 7.6, with 5 mM DTT and 0.05 mg/ml BSA. Samples were prepared in triplicates and 215 analysed in 96-well plates using a Perkin Elmer Envision 2104 multi-label reader at Excitation/Emission 485/535 nm.
In vitro DUB assays
DUB in vitro activity was performed using 100 ng of each DUB. Both enzymes and substrates were freshly mixed in reaction buffer (40 mM Tris–HCl, pH 7.6, 5 mM DTT, 0.005% (w/v) BSA for each run. The reaction mixture was incubated at 30°C for 60 min. The reaction was stopped by adding the SDS loading buffer if the sample was used to run a 8–10% (w/v) bis-acrylamide gel.
Identification of USP45 interacting protein by mass spectrometry
Fifty milligram of cell lysates was pre-cleared by incubation with 50 μl of protein G-Sepharose 1 h at 4°C, filtration and by incubation with 100 μl of pre-immune IgG covalently coupled to protein G-Sepharose for 1 h at 4°C on a rolling shaker. The supernatants were then incubated with 50 μg of anti-USP45-specific antibody covalently coupled to protein G-Sepharose for 1 h at 4°C on a rolling shaker. The immunoprecipitates were washed three times with 10 ml of lysis buffer containing 0.2 M NaCl and twice with 10 ml of buffer A. The beads were resuspended in a total volume of 30 μl of LDS sample buffer (Invitrogen). The samples were then filtered with a 0.44-μm Spin-X filter (Corning), reduced with 10 mM dithiothreitol and alkylated with 50 mM iodoacetamide in 0.1 M NH4HCO3 (30 min at room temperature), boiled and subjected to electrophoresis on a NuPAGE Bis-Tris 4–12% (w/v) polyacrylamide gel. Colloidal Coomassie stained gel was divided into pieces, each of which were washed with 0.1 M NH4HCO3 and 50% (v/v) acetonitrile/50 mM NH4HCO3, dried and incubated with 25 mM triethylammonium bicarbonate with 5 μg/ml trypsin 16 h at 30°C on shaker. The resultant peptides were submitted to LC-MS on a Proxeon EASY-nLC nano-liquid chromatography system coupled to a Thermo-LTQ-Orbitrap mass spectrometer. Data files were searched against the SwissProt human database using Mascot (http://www.matrixscience.com) run on an in-house system, with 10 p.p.m. mass accuracy for precursor ions, a 0.8-Da tolerance for fragment ions, and allowing for carbamidomethyl (C) as fixed modification and for oxidation (M) as variable modification. Peptide mass fingerprinting data analysis was performed using OLMAT (http://www.proteinguru.com/MassSpec/OLMAT).
Laser irradiation and confocal microscopy
U2OS cells were seeded in 35-mm glass bottom dishes at 1 × 106 cells per dish and incubated with psoralen TMP, angelicin (both at 25 μM) for at least 2 h or with BrdU at 10 μM for 24 h (Sigma-Aldrich). Cells were kept in the dark from this point on. A PALM MicroBeam system (Zeiss Microimaging) was employed in this study. We used the 355-nm UV laser through a Plan Fluor 40X/1.25d n.a. oil at 20 and 25% energy for TMP/angelicin and BrdU, respectively, to irradiate a 3 × 20 pixel region internal to the nuclei of the cells. Cells were then subjected to indirect immunofluorescence as describe previously (Davis et al, 2012). Each experiment was done a minimum of three times, and a minimum of 100 cells was PUV-A laser-irradiated per replicate.
Yeast two-hybrid assay
Yeast strain AH109 (Invitrogen) has a HIS3 gene and a lacZ gene regulated by Gal4-binding sites. This strain was transformed with a pAS2.6 plasmid (Invitrogen) encoding USP45 fused to the DNA-binding domain of Gal4 (the plasmid also has the TRP1 selectable marker) and with pACT2.6 plasmids (Invitrogen) encoding XPF and ERCC1 to the Gal4 activation domain. The reverse experiment using USP45 fused to the Gal4 activation domain (these plasmids had the LEU2 selectable marker) and ERCC1 and SLX4 fused to the DNA-binding domain of Gal4. Empty vectors were used as control as well as USP27X (a DUB another member of the USP family). Cells were plated onto minimal medium lacking leucine (LEU) and tryptophan (TRP) and onto synthetic complete media laking LEU and TRP and histidine (HIS) to assay reporter gene activation. Cells from the −LEU −TRP plate were overlaid with a nitrocellulose filter that was frozen in liquid nitrogen. The filter was then placed on a piece of filter paper impregnated with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) to test for lacZ expression that resulted in a blue colour.
Induction of DNA damage and clonogenic cell survival assays
For the cytotoxicity assays, 500 cells were plated in six replicates onto 6-well plates in complete growth medium. After cells attached, they were treated with indicated concentration of MMC for 24 h before medium was replaced with fresh growth medium or with the indicated dose of UV using a Spectrolinker XL-1500 UV cross-linker. The number of colonies with > 100 cells was counted. For each genotype, cell viability of untreated cells was defined as 100%. Data are represented as mean ± SEM from three independent experiments. In FOCI analysis, at least 100 cells of three independent experiments were analysed using ImageJ with plugin PzFOCI.
Analysis of chromosomal aberrations
U2OS cells were initially plated in DMEM containing 10% FBS until cells reached 60–70% confluence. Cell cultures were treated with MMC at a final concentration of 100 ng/ml and diepoxybutane (DEB) at a final concentration of 0.1 μg/ml for 48 h. All cultures were harvested following conventional cytogenetic protocol (Brown & Lawce, 1997). Briefly, the cell cultures were treated with 0.1 μg/ml Colcemid (Irvine Scientific) approximately 30 min before the initiation of harvest. For chromosome preparations, the cells were harvested following conventional cytogenetic protocol of hypotonic treatment (75 mM KCl) and freshly prepared chilled 3:1 acetone:methanol fixation. This was followed by four additional fixation cycles and air-dried slide preparation. The slides were ‘aged’ in a hot oven at 60°C over 16 h, followed by Giemsa staining (Invitrogen). A total of 25 metaphases were scored for each culture.
Analysis of cell cycle by flow cytometry
U2OS and KBM7 cells were analysed for their respective cell cycle phase distribution using flow cytometry. Cells were trypsinised in the case of U2OS, washed with PBS + 0.2% (w/v) BSA and resuspended in flow cytometry tubes. Cells were then fixed by 70% (v/v) ice-cold ethanol and stored at −20°C until analysis. After washing fixed cells once with PBS, RNase A (50 μg/ml) and PI (50 μg/ml) were added to the cells and incubated in the dark at room temperature (25°C) for 20 min. The live cell populations were then subjected to quantitative measurement of DNA content by flow cytometry using a FACSCanto™ (BD Biosciences) and cell cycle distribution and the percentage of G2–S–G1 cells determined by the Watson (pragmatic) modelling algorithm using FlowJo software (Treestar).
UV-C CPD analysis
Cells grown on glass plates (VWR) were either fixed directly with 4% (v/v) paraformaldehyde and permeabilised with 0.2% Triton X-100 in PBS, or pre-extracted before fixation with 0.5% (v/v) Triton X-100 in CSK buffer (cytoskeletal buffer: 10 mM PIPES pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2). For CPD staining, DNA was denatured with 0.5 M NaOH for 5 min. Samples were blocked in 5% (w/v) BSA (bovine serum albumin; Sigma-Aldrich) in PBS supplemented with 0.1% (v/v) Tween before incubation with primary and secondary antibodies conjugated to Alexa-Fluor 488 or 594 (Invitrogen) (Adam et al, 2013). The samples were analysed in a confocal Leica Microscope 510.
Southern dot blot analysis
Cells were treated with UV (20 J/m2), and genomic DNA was extracted at indicated time points using the Qiagen DNeasy blood and tissue extraction kit, as per manufacturer's instructions. Five nanogram of genomic DNA was diluted to 100 μl with 0.5 M of NaOH and 1.5 M of NaCl, and samples were boiled at 95°C to denature the DNA and blotted on a nylon membrane followed by UV cross-linking (DNA containing side facing downwards) to fix DNA onto the membrane. The blot was blocked with 5% (w/v) milk TBST for 1 h at room temperature and incubated with anti-thymidine mouse monoclonal antibody (against CPDs) (Kamiya, USA) overnight and detected with anti-mouse IgG horseradish peroxidase-labelled secondary antibody, similar to Western blotting procedures. Control of total cDNA was measured with Abcam dsDNA antibody (ab27156).
CPD ELISA kit
High Sensitivity CPD/Cyclobutane Pyrimidine Dimer Elisa kit (NM-MA-K001) was from Cosmo Bio. Cells were treated with UV (20 J/m2), and genomic DNA was extracted at indicated time points using the Qiagen DNeasy blood and tissue extraction kit, as per manufacturer's instructions. 0.4 μg/ml of genomic DNA was diluted with 1× assay diluent, heated at 100°C for 10 min. After samples were chill rapidly in an ice for 15 min, 50 μl of each samples was added to each well of the ELISA plate wells pre-coated with protamine sulphate and dry completely during 18 h at 37°C. For the DNA damage detection, the manufacturer protocol was followed. Calf thymus DNA, UV-C irradiated (10 J/m2) and not irradiated was used as positive and negative samples, respectively.
Cell growth assays
Cells (2,000–3,000 per well) WT/K0 USP45 or rescue experiment with WT or Cys199Ala mutant were seeded into the inner of 96-well plates in eight replicates and allowed to attach for 16 h in the case of U2OS cells. Cell viability was determined using CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) according to the manufacturer's instructions. Results were plotted with a best-fit sigmoidal variable slope dose–response curve, and GI50 (growth inhibition by 50%) values were calculated using GraphPad Prism 5.0. The MTS assay was then performed 24, 48, 72, 96 and 120 h post-seeding. Results are presented as the fold change in absorbance over the 5-day period relative to the assay start point (24 h post-seeding or day 0). The cells were assayed in eight replicates.
Acknowledgments
We thank Dennis Castor and Nidhi Nair for providing the Slx4 WT/KO MEF, David Melton for the Ercc1 WT/KO MEF cell lines and Rosemary Clarke for help with flow cytometry. We thank Luis Garcia for his advice on the yeast two-hybrid analysis. We are also grateful to David Campbell for processing mass spectrometry samples. We also thank Richard Ewan for advice on the rhodamine assay. We also express gratitude for the excellent technical support of the MRC-Protein Phosphorylation and Ubiquitylation Unit (PPU) DNA Sequencing Service (coordinated by Nicholas Helps), the MRC-PPU tissue culture team (coordinated by Kirsten Airey and Janis Stark), the Division of Signal Transduction Therapy (DSTT) protein and antibody purification teams (coordinated by Hilary McLauchlan and James Hastie). The Medical Research Council as well as Janssen Pharmaceutica supported this study. We also acknowledge Wellcome Trust support for the microscopy (097945/B/11/Z) and flow cytometry (097945/B/11/Z).
Author contributions
ABP-O, CL and IM planned, undertook and analysed all of the experiments. PS generated the USP45 knockout KBM7 and U2OS cell lines. TM generated the cDNA constructs utilised in this project. ABP-O, IH, JR and DRA conceived of the project, planned the experiments, analysed the results and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12
Supplementary Figure S13
Supplementary Figure S14
Supplementary Figure S15
Supplementary Figure S16
Supplementary Tables S1–S2
Supplementary Materials and Methods and Figure Legends
Review Process File
Source Data for Figure 1
References
- Adam S, Polo SE, Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Brown MG, Lawce HJ. Peripheral blood cytogenetic methods. In: Barch MJ, Knutsen T, Spurbeck J, editors. The AGT Laboratory Manual. Philadelphia: Lippincott-Raven; 1997. pp. 77–89. (eds) [Google Scholar]
- Busch DB, van Vuuren H, de Wit J, Collins A, Zdzienicka MZ, Mitchell DL, Brookman KW, Stefanini M, Riboni R, Thompson LH, Albert RB, van Gool AJ, Hoeijmakers J. Phenotypic heterogeneity in nucleotide excision repair mutants of rodent complementation groups 1 and 4. Mutat Res. 1997;383:91–106. doi: 10.1016/s0921-8777(96)00048-1. [DOI] [PubMed] [Google Scholar]
- Castor D, Nair N, Declais AC, Lachaud C, Toth R, Macartney TJ, Lilley DM, Arthur JS, Rouse J. Cooperative control of holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol Cell. 2013;52:221–233. doi: 10.1016/j.molcel.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, McDonald N, West SC. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem. 2008;77:259–287. doi: 10.1146/annurev.biochem.77.070306.102408. [DOI] [PubMed] [Google Scholar]
- Davis EJ, Lachaud C, Appleton P, Macartney TJ, Nathke I, Rouse J. DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat Struct Mol Biol. 2012;19:1093–1100. doi: 10.1038/nsmb.2394. [DOI] [PubMed] [Google Scholar]
- Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, Cohen P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci USA. 2013;110:15247–15252. doi: 10.1073/pnas.1314715110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faesen AC, Luna-Vargas MP, Geurink PP, Clerici M, Merkx R, van Dijk WJ, Hameed DS, El Oualid F, Ovaa H, Sixma TK. The differential modulation of USP activity by internal regulatory domains, interactors and eight ubiquitin chain types. Chem Biol. 2011;18:1550–1561. doi: 10.1016/j.chembiol.2011.10.017. [DOI] [PubMed] [Google Scholar]
- Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, Coulon S, Dong MQ, Ruse C, Yates JR, 3rd, Russell P, Fuchs RP, McGowan CH, Gaillard PH. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009;138:78–89. doi: 10.1016/j.cell.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gerard A, Polo SE, Roche D, Almouzni G. Methods for studying chromatin assembly coupled to DNA repair. Methods Enzymol. 2006;409:358–374. doi: 10.1016/S0076-6879(05)09021-X. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Maytal V. Regulating the 26S proteasome. Curr Top Microbiol Immunol. 2002;268:43–72. doi: 10.1007/978-3-642-59414-4_3. [DOI] [PubMed] [Google Scholar]
- Guzzo CM, Berndsen CE, Zhu J, Gupta V, Datta A, Greenberg RA, Wolberger C, Matunis MJ. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci Signal. 2012;5:ra88. doi: 10.1126/scisignal.2003485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hoy CA, Thompson LH, Mooney CL, Salazar EP. Defective DNA cross-link removal in Chinese hamster cell mutants hypersensitive to bifunctional alkylating agents. Cancer Res. 1985;45:1737–1743. [PubMed] [Google Scholar]
- Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye J, Smith CA, Hanawalt PC. DNA repair in human cells containing photoadducts of 8-methoxypsoralen or angelicin. Cancer Res. 1980;40:696–702. [PubMed] [Google Scholar]
- Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- Kirschner K, Melton DW. Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res. 2010;30:3223–3232. [PubMed] [Google Scholar]
- Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Kondo S, Mamada A, Miyamoto C, Keong CH, Satoh Y, Fujiwara Y. Late onset of skin cancers in 2 xeroderma pigmentosum group F siblings and a review of 30 Japanese xeroderma pigmentosum patients in groups D, E and F. Photo-dermatology. 1989;6:89–95. [PubMed] [Google Scholar]
- Kulathu Y, Komander D. Atypical ubiquitylation—the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- Lachaud C, Castor D, Hain K, Munoz I, Wilson J, Macartney TJ, Schindler D, Rouse J. Distinct functional roles for the SLX4 ubiquitin-binding UBZ domains mutated in Fanconi anemia. J Cell Sci. 2014;127:2811–2817. doi: 10.1242/jcs.146167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil EM, Melton DW. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 2012;40:9990–10004. doi: 10.1093/nar/gks818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhir J, Selfridge J, Harrison DJ, Squires S, Melton DW. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat Genet. 1993;5:217–224. doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- Melton DW, Ketchen AM, Nunez F, Bonatti-Abbondandolo S, Abbondandolo A, Squires S, Johnson RT. Cells from ERCC1-deficient mice show increased genome instability and a reduced frequency of S-phase-dependent illegitimate chromosome exchange but a normal frequency of homologous recombination. J Cell Sci. 1998;111(Pt 3):395–404. doi: 10.1242/jcs.111.3.395. [DOI] [PubMed] [Google Scholar]
- Muniandy PA, Thapa D, Thazhathveetil AK, Liu ST, Seidman MM. Repair of laser-localized DNA interstrand cross-links in G1 phase mammalian cells. J Biol Chem. 2009;284:27908–27917. doi: 10.1074/jbc.M109.029025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz IM, Hain K, Declais AC, Gardiner M, Toh GW, Sanchez-Pulido L, Heuckmann JM, Toth R, Macartney T, Eppink B, Kanaar R, Ponting CP, Lilley DM, Rouse J. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell. 2009;35:116–127. doi: 10.1016/j.molcel.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004a;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004b;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17:331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Nishi R, Wijnhoven P, le SC, Tjeertes J, Galanty Y, Forment JV, Clague MJ, Urbe S, Jackson SP. Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat Cell Biol. 2014;16:1016–1026. doi: 10.1038/ncb3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelli B, McClendon TB, Tsodikov OV, Ellenberger T, Niedernhofer LJ, Scharer OD. The XPA-binding domain of ERCC1 is required for nucleotide excision repair but not other DNA repair pathways. J Biol Chem. 2010;285:3705–3712. doi: 10.1074/jbc.M109.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Nguyen J, Hill SJ, Scully R, Adelmant GO, Marto JA, Feunteun J, Livingston DM. BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol Cell. 2011;44:235–251. doi: 10.1016/j.molcel.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick MH. Studies on thymine-derived UV photoproducts in DNA–I. Formation and biological role of pyrimidine adducts in DNA. Photochem Photobiol. 1977;25:357–372. doi: 10.1111/j.1751-1097.1977.tb07355.x. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Rago C, Vogelstein B, Bunz F. Genetic knockouts and knockins in human somatic cells. Nat Protoc. 2007;2:2734–2746. doi: 10.1038/nprot.2007.408. [DOI] [PubMed] [Google Scholar]
- Ritorto MS, Ewan R, Perez-Oliva AB, Knebel A, Buhrlage SJ, Wightman M, Kelly SM, Wood NT, Virdee S, Gray NS, Morrice NA, Alessi DR, Trost M. High throughput screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat Commun. 2014;5:4763. doi: 10.1038/ncomms5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito TT, Lui DY, Kim HM, Meyer K, Colaiacovo MP. Interplay between structure-specific endonucleases for crossover control during Caenorhabditis elegans meiosis. PLoS Genet. 2013;9:e1003586. doi: 10.1371/journal.pgen.1003586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz A, Stander S, Berneburg M, Bohm M, Kulms D, van Steeg H, Grosse-Heitmeyer K, Krutmann J, Schwarz T. Interleukin-12 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Nat Cell Biol. 2002;4:26–31. doi: 10.1038/ncb717. [DOI] [PubMed] [Google Scholar]
- Sharma N, Zhu Q, Wani G, He J, Wang QE, Wani AA. USP3 counteracts RNF168 via deubiquitinating H2A and gammaH2AX at lysine 13 and 15. Cell Cycle. 2014;13:106–114. doi: 10.4161/cc.26814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, Carter KC, Shell BK, Evans E, de Jong MC, Rademakers S, de Rooij J, Jaspers NG, Hoeijmakers JH, Wood RD. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86:811–822. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AW, Jaspers NG, Bettecken T, Joenje H, Schindler D, Rouse J, de Winter JP. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet. 2011a;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AW, Jaspers NG, Bettecken T, Joenje H, Schindler D, Rouse J, de Winter JP. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet. 2011b;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- Svendsen JM, Smogorzewska A, Sowa ME, O'Connell BC, Gygi SP, Elledge SJ, Harper JW. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138:63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, van Zeeland AA, Mullenders LH. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–224. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- Wang AT, Sengerova B, Cattell E, Inagawa T, Hartley JM, Kiakos K, Burgess-Brown NA, Swift LP, Enzlin JH, Schofield CJ, Gileadi O, Hartley JA, McHugh PJ. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011;25:1859–1870. doi: 10.1101/gad.15699211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997;7:427–439. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- Westerveld A, Hoeijmakers JH, van Duin M, de Wit J, Odijk H, Pastink A, Wood RD, Bootsma D. Molecular cloning of a human DNA repair gene. Nature. 1984;310:425–429. doi: 10.1038/310425a0. [DOI] [PubMed] [Google Scholar]
- Yarbro JW. Mechanism of action of hydroxyurea. Semin Oncol. 1992;19:1–10. [PubMed] [Google Scholar]
- Ye Y, Scheel H, Hofmann K, Komander D. Dissection of USP catalytic domains reveals five common insertion points. Mol BioSyst. 2009;5:1797–1808. doi: 10.1039/b907669g. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zaugg K, Mak TW, Elledge SJ. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126:529–542. doi: 10.1016/j.cell.2006.06.039. [DOI] [PubMed] [Google Scholar]
- Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair. 2014;19:135–142. doi: 10.1016/j.dnarep.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12
Supplementary Figure S13
Supplementary Figure S14
Supplementary Figure S15
Supplementary Figure S16
Supplementary Tables S1–S2
Supplementary Materials and Methods and Figure Legends
Review Process File
Source Data for Figure 1