

Abstract
In mammals, glucocorticoids (GCs) and their intracellular receptor, the glucocorticoid receptor (GR), represent critical checkpoints in the endocrine control of energy homeostasis. Indeed, aberrant GC action is linked to severe metabolic stress conditions as seen in Cushing's syndrome, GC therapy and certain components of the Metabolic Syndrome, including obesity and insulin resistance. Here, we identify the hepatic induction of the mammalian conserved microRNA (miR)-379/410 genomic cluster as a key component of GC/GR-driven metabolic dysfunction. Particularly, miR-379 was up-regulated in mouse models of hyperglucocorticoidemia and obesity as well as human liver in a GC/GR-dependent manner. Hepatocyte-specific silencing of miR-379 substantially reduced circulating very-low-density lipoprotein (VLDL)-associated triglyceride (TG) levels in healthy mice and normalized aberrant lipid profiles in metabolically challenged animals, mediated through miR-379 effects on key receptors in hepatic TG re-uptake. As hepatic miR-379 levels were also correlated with GC and TG levels in human obese patients, the identification of a GC/GR-controlled miRNA cluster not only defines a novel layer of hormone-dependent metabolic control but also paves the way to alternative miRNA-based therapeutic approaches in metabolic dysfunction.
Keywords: glucocorticoid signalling, LDLR, LSR, miRNA-379, VLDL triglyceride
Introduction
The hypothalamic–pituitary–adrenal (HPA) endocrine axis is a critical physiological stress circuit to maintain body homeostasis during diverse situations such as trauma, exercise, or nutrient deprivation, mediated in major parts through adrenal glucocorticoid hormone (GC) action (Rose & Herzig, 2013). In metabolic control, GC signaling acts as a major counter-regulatory system against insulin action, and states of either endogenous or exogenous GC deficiency or excess, for example, Addison's disease, Cushing's syndrome, or GC therapy, respectively, are characterized by severe perturbations in systemic energy metabolism that closely mimic aspects of the Metabolic Syndrome (Vegiopoulos & Herzig, 2007). Indeed, aberrantly elevated GC activity and/or levels are discussed to be linked to the major components of the syndrome, including obesity, insulin resistance, hyperglycemia, and systemic dyslipidemia (Anagnostis et al, 2009). GC levels have been found to be elevated in insulin-resistant patients and are strongly associated with a hyperglycemic and fatty liver phenotype (Phillips et al, 1998). In congruence to this, recent clinical trials using inhibitors of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), the key enzyme in pre-receptor GC generation, have been proven safe and effective in reducing hepatic liver fat content in obese humans (Stefan et al, 2014), suggesting that GC can—at least in part—drive major aspects of metabolic dysfunction as associated with human obesity.
At the molecular level, GC action is mediated through the glucocorticoid receptor (GR), a member of the nuclear receptor transcription factor family (Opherk et al, 2004; Lemke et al, 2008). In liver, numerous direct protein-encoding target genes of the GR have been identified by both classical and high-throughput chromatin recruitment studies (Wang et al, 2004), mediating the GC/GR impact on hepatic gluconeogenesis, triglyceride (TG), and bile acid metabolism (Herzig et al, 2001; Lemke et al, 2008: Rose et al, 2011). In contrast to the well-described protein mediators of GC/GR metabolic action, additional layers of the GC/GR-dependent molecular network and mechanisms of metabolic fine-tuning still remain elusive.
Recently, a class of small non-coding RNAs (microRNAs, miRs) has emerged as a critical but as-yet largely unexplored layer of metabolic control. Indeed, individual miRNAs have been found to regulate diverse aspects of energy homeostasis, including pancreatic beta-cell insulin secretion, adipose tissue lipid storage, and hepatic cholesterol and lipid handling (Rottiers & Naar, 2012). However, no miRNA targets of the endocrine GC/GR axis in metabolic dysfunction have been identified to date, prompting us to test the hypothesis that distinct miRNAs serve as molecular output effectors of the HPA-GC/GR metabolic control circuit.
Results
The miR-379/410 cluster represents a direct GR target in liver
To define GC/GR-dependent miRNA networks with immediate relevance in metabolic dysfunction, we initially performed large-scale miRNA expression profiling of livers from wild-type and db/db diabetic mice as a model for diabesity-related hyperglucocorticoidemia (Lemke et al, 2008). A total of 36 miRNAs were found to be up- or down-regulated more than twofold between these animals (Fig1A). Next, we aligned this data set with a second miRNA signature, resulting from differential expression profiling between mice lacking the GR specifically in hepatocytes and controls (Rose et al, 2011). Data cross-comparison revealed a set of 10 miRNAs that showed significant down-regulation in response to GR deficiency. Some of these miRNAs were simultaneously induced in obesity-related diabetes (Fig1A), indicating that these miRNAs represent bona fide mediators of GC/GR-driven metabolic dysfunction. Intriguingly, 6 out of 10 GC/GR-targeted miRNAs are located in the miR-379/410 genomic cluster that is conserved between mammalian species (Glazov et al, 2008), which resides on mouse and human chromosomes 12 and 14, respectively. Indeed, selective expression analysis verified the induction of five cluster members in db/db as well as in mice chronically treated with the GC analogue dexamethasone (DEX) by real-time PCR (Supplementary Fig S1A). The up to fourfold, GR-dependent induction of miR-379 in diabetic mice (Fig1B) and its relatively high abundance in liver (Supplementary Fig S1B) next prompted us to investigate the general importance of this regulation in other models of elevated GC action. To this end, we analyzed liver extracts from healthy New Zealand black (NZB) mice compared to New Zealand obese (NZO) mice, the latter representing a multigenic obesity model (Leiter & Reifsnyder, 2004). In congruence with the hyperglucocorticoidemia in this model (Rose et al, 2010), miR-379 expression was found to be elevated significantly when compared to corresponding controls (Fig1B), thereby correlating with circulating corticosterone levels in these mice (Supplementary Fig S1C), supporting the hypothesis that particularly miR-379 is a key output of the GC/GR endocrine pathway under conditions of metabolic dysfunction. In support of this possibility, levels of miR-379 were consistently found to be diminished in models of GR deficiency, including both miRNA-induced (Rose et al, 2011) and genetic (Opherk et al, 2004) hepatocyte-specific GR deficiency (Fig1C and D, Supplementary Fig S1D). Indeed, chromatin immunoprecipitation using GR-specific antibody experimentally verified the recruitment of the receptor to a binding site within the miR-379 promoter (Fig1E). A similar result had been previously reported in GR Chip-sequencing data deposited in human genome database (Supplementary Fig S1F), suggesting that the miR-379/410 cluster represents a direct transcriptional target of the GR. To verify the functional significance of GR DNA binding to the miR-379 locus in vivo, we examined mice harboring a dimerization-defective GR mutant (GRdim) that impairs receptor DNA binding to full GREs (Opherk et al, 2004). Wild-type mice chronically treated with DEX exhibited higher hepatic levels of miR-379 as compared with controls, while the genetic impairment of GR DNA-binding capacity in GRdim mice completely abrogated this effect (Supplementary Fig S1A), thereby validating a DNA binding-dependent regulatory function of the GC/GR pathway for miR-379 expression in the liver. Of note, DEX treatment of isolated primary mouse hepatocytes (Supplementary Fig S1E) and human liver slices (Fig1F) led to the induction of miR-379 irrespective of culture glucose levels (data not shown), demonstrating the cell autonomy of the observed effects. Co-treatment with the GR-antagonist RU-486 completely abolished the DEX effects, thereby confirming GR specificity (Fig1F).
Figure 1.
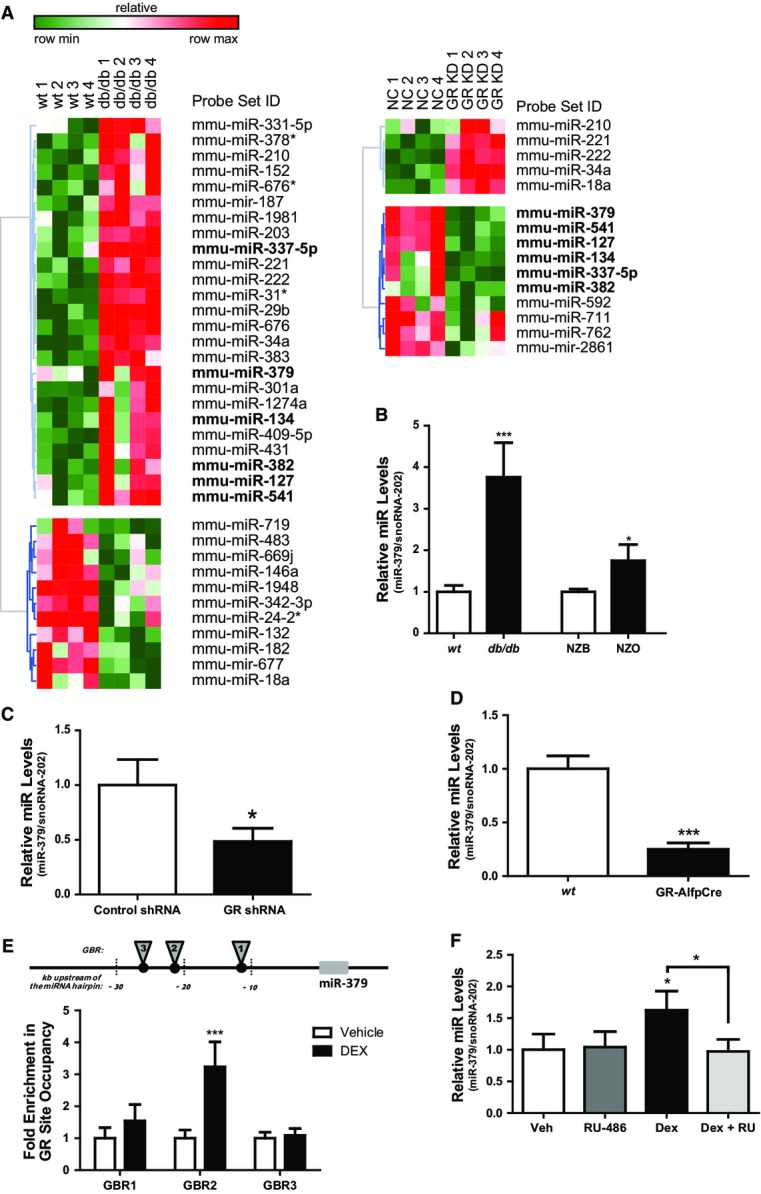
- Heatmap showing relative miRNA expression between wild-type (wt) and db/db mice (n = 4), and between mice treated with negative control (NC) and GR-specific miRNA recombinant adeno-associated virus (rAAV) (n = 4). Higher and lower expression is displayed in red and green, respectively. Commonly regulated miRNAs in the miR-379/410 cluster are shown in bold. Differentially regulated miRNAs are ≥ twofold, P < 0.05.
- Quantitative miR-379 PCR levels in livers of db/db, New Zealand obese (NZO) mice, and corresponding controls—wt and New Zealand black (NZB) (n ≥ 4). Bar graphs show mean ± SEM; t-test: ***P < 0.001 or *P < 0.05.
- Quantitative miR-379 levels in livers of db/db mice treated with control or GR-specific shRNA adenovirus (n = 5). Bar graphs show mean ± SEM; t-test: *P < 0.05.
- Hepatic miR-379 levels as determined by RT–qPCR analysis in wt and hepatocyte-specific GR knockout mice (GR-AlfpCre) (n = 4). Bar graphs show mean ± SEM; t-test: ***P < 0.001.
- Chromatin immunoprecipitation (ChIP) qPCR for validation of GR-binding regions (GBR) upstream miR-379 hairpin: 1 (−11,197 to −111,268), 2 (−21,021 to −21,135), and 3 (−26,761 to −26,793). Fold enrichment of GR-binding site occupancy relative to negative control, anti-HA. (n = 4). Bar graphs show mean ± SEM; t-test: ***P < 0.001.
- miR-379 levels in human liver treated ex vivo with or without 1 μM RU486 and 0.1 μM DEX (n = 4). snoRNA-202 was used for normalization of miRNA levels. Bar graphs show mean ± SEM; ANOVA (with post hoc test): *P < 0.05.
miR-379 silencing in liver affects circulating VLDL-TG levels
The identification of miR-379 as a direct target of GC/GR signaling with aberrant hepatic expression in diabesity prompted us to next explore the functional importance of miR-379 for hepatic and systemic energy homeostasis. To this end, we efficiently and specifically silenced miR-379 in the liver by locked nucleic acid (LNA) antisense technology in wild-type animals (Fig2A, Supplementary Fig S2A and C). While miR-379-specific LNA delivery had no influence on serum alanine aminotransferase (ALT) (Supplementary Fig S2B), hepatic glycogen (Supplementary Fig S2D), TG (Supplementary Fig S2E), and serum cholesterol (Supplementary Fig S2F) levels as well as glucose tolerance (Supplementary Fig S2G), miR-379 deficiency significantly lowered total serum TG levels (Fig2B) and tended to decrease total serum cholesterol (Fig2C), indicating that hepatic miR-379 principally controls metabolism of circulating TG. Indeed, serum fast-protein liquid chromatography (FPLC) revealed that liver silencing of miR-379 robustly reduced levels of VLDL-associated TG, while only mildly lowering cholesterol lipoprotein loading (Fig2D, Supplementary Fig S2H). Consistent with the role of elevated VLDL-TG as a major independent risk factor for cardiovascular complications in the Metabolic Syndrome and its tight association with insulin resistance (Nordestgaard & Varbo, 2014), diminished VLDL-TG in miR-379 LNA-treated mice correlated with improved insulin sensitivity (Supplementary Fig S2I), inhibition of FOXO1 and phosphoenolpyruvate carboxykinase (PEPCK) (Supplementary Fig S2J and K), as well as consistent improvement in pyruvate tolerance (Supplementary Fig S2L), while blood glucose levels remained overall unaltered (Supplementary Fig S2M).
Figure 2.
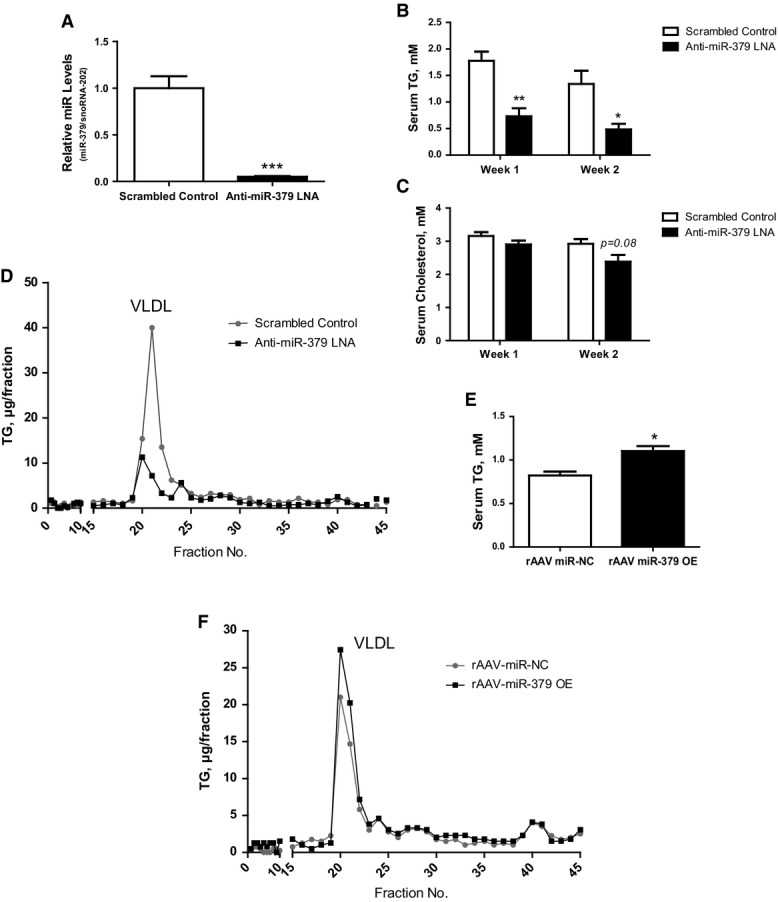
- RT–qPCR analysis of miR-379 expression in C57Bl/6J (wt) mice treated with anti-miR-379 or scrambled control locked nucleic acid (LNA) (n = 5). Bar graphs show mean ± SEM; t-test: ***P < 0.001.
- Serum triglyceride (B) and cholesterol (C) levels after 1 and 2 weeks of LNA treatment of same animals as in (A) under fed conditions. Bar graphs show mean ± SEM; ANOVA (with post hoc test): **P < 0.01 or *P < 0.05.
- Triglyceride profiles of fast-protein liquid chromatography (FPLC)-fractionated serum of same animals as in (A). Very-low-density lipoprotein (VLDL) peak is indicated.
- Serum triglyceride levels of wt mice overexpressing miR-379 or scrambled control miRNA via hepatocyte-specific rAAV vectors (n = 4). Bar graphs show mean ± SEM; t-test: *P < 0.05.
- Triglyceride profiles of FPLC-fractionated serum of same animals as in (E). VLDL peak is indicated.
To next determine whether elevation of hepatic miR-379 expression was sufficient to cause systemic dyslipidemia in a more chronic setting, we employed a recombinant adeno-associated virus (rAAV) delivery system allowing the expression of the miRNA specifically in liver parenchymal cells but not in other liver cell types for a period of several months (Rose et al, 2011). Eight weeks after rAAV delivery, mice with hepatocyte-specific miR-379 overexpression (Supplementary Fig S2N) that did not affect serum levels of liver damage markers (Supplementary Fig S2O) displayed significantly higher levels of serum TG associated with the VLDL fraction (Fig2E and F) but maintained normal cholesterol homeostasis (Supplementary Fig S2P and Q).
miR-379 acts through LSR- and LDLR-dependent hepatic lipid re-uptake
The observed regulation of serum VLDL-TG by miR-379 next prompted us to explore the mechanistic basis of this effect. Systemic TG metabolism is determined by the relative balance of hepatic and peripheral lipid uptake and release, correlating with de novo TG formation (lipogenesis) and fatty acid (FA) β-oxidation in the liver (Wang et al, 2012). In this respect, miR-379-specific LNA treatment of mouse hepatocytes had no influence on FA oxidation as determined by metabolic flux analysis (Supplementary Fig S3A). Also, adipose tissue-associated lipoprotein lipase activity (Supplementary Fig S3B) and hepatic VLDL release were unaffected by miR-379 deficiency as compared to control littermates (Supplementary Fig S3C). In contrast, hepatic uptake of radiolabeled VLDL (Supplementary Fig S3D) and orally administered labeled TG (Fig3A and B) was significantly and specifically elevated upon miR-379 knockdown in both db/db and wild-type mice, respectively, suggesting that the control of circulating VLDL-TG by hepatic miR-379 is not predominantly conferred by alterations in hepatic or peripheral lipid synthesis or utilization pathways but rather relies on enhanced hepatic VLDL-TG clearance.
Figure 3.

- Serum 3H-triolein in a lipid tolerance test in db/db mice treated with an anti-miR-379 or scrambled control Tough Decoy (TuD) construct delivered by rAAV (n = 5–6). Mice were fasted for 16 h and given a 100-μl oral fat load of olive oil spiked-in with 3H-triolein. Inset: area under the curve. Line graphs show mean ± SEM; ANOVA (with post hoc test): ***P < 0.001 or **P < 0.01.
- Organ distribution of 3H-triolein radioactivity from animals in (A). BAT, brown adipose tissue; pgWAT, perigonadal white adipose tissue; GCM, gastrocnemius skeletal muscle. Bar graphs show mean ± SEM; t-test: *P < 0.05.
- Protein levels of LSR and LDLR from livers of animals treated with anti-miR-379 or scrambled control LNA (n = 5), same animals as in Fig2A. Shown are the LSR-α (68 kDa) and LSR-β (56 kDa) subunits and the glycosylated LDLR (between 100 and 130 kDa) protein.
- Protein levels of LSR and LDLR from livers of wild-type (wt) mice treated with dexamethasone (1 mg/kg BW) or isotonic saline for 28 days. (n = 7–8).
- Western blot of liver extracts from wt or LDLRKO mice treated with control or LSR shRNA-containing adenovirus and with anti-miR-379 or scrambled control LNA (n = 7).
- Serum triglyceride levels of same animals as in (E) under fed conditions. Bar graphs show mean ± SEM; ANOVA (with post hoc test): **P < 0.01 or *P < 0.05.
- Flag-LSR protein levels in HEK293 cells treated with miR-379 mimics. 100 nM cel-miR-293b was used as the control mimic. [-]: untransfected.
- miR-379 target validation of LSR (H) and LDLR (I) using a dual-luciferase reporter gene assay. miRNA binding site (MBS) predicted by RNA22 and MiRTiF and the corresponding mutated (mut) sequence were cloned into psiCHECK™-2 vector. Bar graphs show mean ± SEM; ANOVA (with post hoc test): ***P < 0.001.
In congruence with this hypothesis, bioinformatic miRNA target gene screening (Supplementary Fig S4A) revealed that miR-379 was predicted to bind genes involved in hepatic lipid (re-) uptake, most notably including the major hepatic lipid re-uptake transporters, lipolysis-stimulated lipoprotein receptor (LSR, Supplementary Fig S3I), and low-density lipoprotein receptor (LDLR, Supplementary Fig S3J) (Ishibashi et al, 1993; Narvekar et al, 2009). Both, LSR and LDLR expression, have previously been found to be severely compromised in hypertriglyceridemic db/db mice, and liver-specific inactivation of both LSR and LDLR triggered the elevation of serum VLDL-TG in healthy wild-type animals by specifically preventing hepatic re-uptake of apolipoprotein B-associated TG from the circulation (Narvekar et al, 2009; Foley et al, 2013).
In line with the in silico predictions, LNA-mediated miR-379 silencing led to the up-regulation of hepatic LSR and LDLR protein expression in wild-type animals (Fig3C, Supplementary Fig S3K), and DEX treatment inhibited LSR and LDLR protein levels as compared with controls (Fig3D, Supplementary Fig S3L), overall suggesting that miR-379 may regulate systemic VLDL-TG through its inhibitory impact on LSR and LDLR function. To address this hypothesis functionally, we performed genetic rescue experiments using LNA/shRNA adenovirus co-administration in wild-type mice, preventing the miR-379 LNA-dependent induction of LSR protein expression through simultaneous shRNA-mediated inhibition of LSR (Supplementary Fig S4B–D). Ten days after LNA/adenovirus co-delivery into the tail vein of mice, miR-379 silencing triggered hypotriglyceridemia (Supplementary Fig S4E) while LSR inhibition alone caused an elevation of circulating VLDL-TG as described when compared with control littermates (Narvekar et al, 2009). Of note, the treatments left cholesterol parameters mostly unaffected (Supplementary Fig S4F). In the absence of functional hepatic LSR, miR-379 silencing still exerted an effect on total and VLDL-associated serum TG levels (Supplementary Fig S4E and data not shown), suggesting that LSR alone does not explain the impact of miR-379 on systemic TG metabolism. Consistent with this assumption, LDLR protein levels were increased upon double miR-379/LSR knockdown (Supplementary Fig S4B and C). To test for a potential compensatory role of LDLR in the absence of LSR, we next employed wild-type and LDLR knockout (KO) animals (Ishibashi et al, 1993) in LNA/LSR shRNA adenovirus co-injection experiments to obtain a functional LSR/LDLR double deficiency in the liver (Fig3E, Supplementary Fig S4G–I). While miR-379 silencing reduced serum cholesterol and TG in wild-type and/or LDLRKO mice (Fig3F, Supplementary Fig S4J), the effect of miR-379 LNA delivery was completely abrogated in LDLR/LSR double-deficient mice (Fig3F), overall demonstrating that miR-379 determines circulating TG levels through a double impact on two components of hepatic TG clearance, that is, LSR and LDLR. Consistent with this notion, treatment of cultured cells with miR-379 mimics inhibited both protein expression (Fig3G) and luciferase reporter genes carrying wild-type LSR and LDLR miR-379 target sequences, while leaving mutated target sites unaffected (Fig3H and I), thereby validating the specificity of the observed effects.
Hepatic silencing of miR-379 counteracts obesity-related hypertriglyceridemia
Antisense strategies targeting specific miRNAs have emerged as attractive therapeutic options for several human pathologies, including metabolic complications and viral diseases, thus recently prompting first clinical trials in this direction (Stenvang et al, 2012). Given the current search for efficient and specific therapeutic modalities to overcome hypertriglyceridemia as the major cardiovascular risk factor in patients with the Metabolic Syndrome (Watts et al, 2013), we were tempted to address the therapeutic potential of miR-379 in obesity-associated hyperlipidemia.
To this end, we silenced hepatic expression of miR-379 by LNA delivery into 12-week-old db/db mice with fully established hyperglucocorticoidemia, hyperlipidemia, and hyperglycemia. Seven days after LNA delivery, hepatic miR-379 deficiency (Supplementary Fig S5A) led to a marked reduction in serum VLDL-TG to wild-type levels (Fig4A and C) and improved blood glucose levels along with an ameliorated insulin resistance index (Supplementary Fig S5B, Fig4B). In line with results from wild-type animals, miR-379 LNA treatment had no major effect on circulating cholesterol and corticosterone levels (Supplementary Fig S5C–E). Indeed, in congruence with the ApoB TG clearance properties of hepatic LSR and LDLR (Narvekar et al, 2009) and a concomitant induction of lipogenic gene expression (Supplementary Fig S5F), miR-379 deficiency promoted accumulation of TG in the liver (Supplementary Fig S5G) and caused a significant induction of both LSR and LDLR protein expression as compared with control LNA-treated animals (Fig4D, Supplementary Fig S5H). To corroborate these findings in an independent multigenic obesity model, we treated New Zealand obese (NZO) mice with miR-379-specific or control LNA (Supplementary Fig S5I). In line with the results from the db/db studies, silencing of hepatic miR-379 significantly reduced serum VLDL-TG levels with a minor impact on serum cholesterol (Supplementary Fig S5J and K, Fig4E) and elevated hepatic protein expression of both LDLR and LSR (Fig4F, Supplementary Fig S5L).
Figure 4.
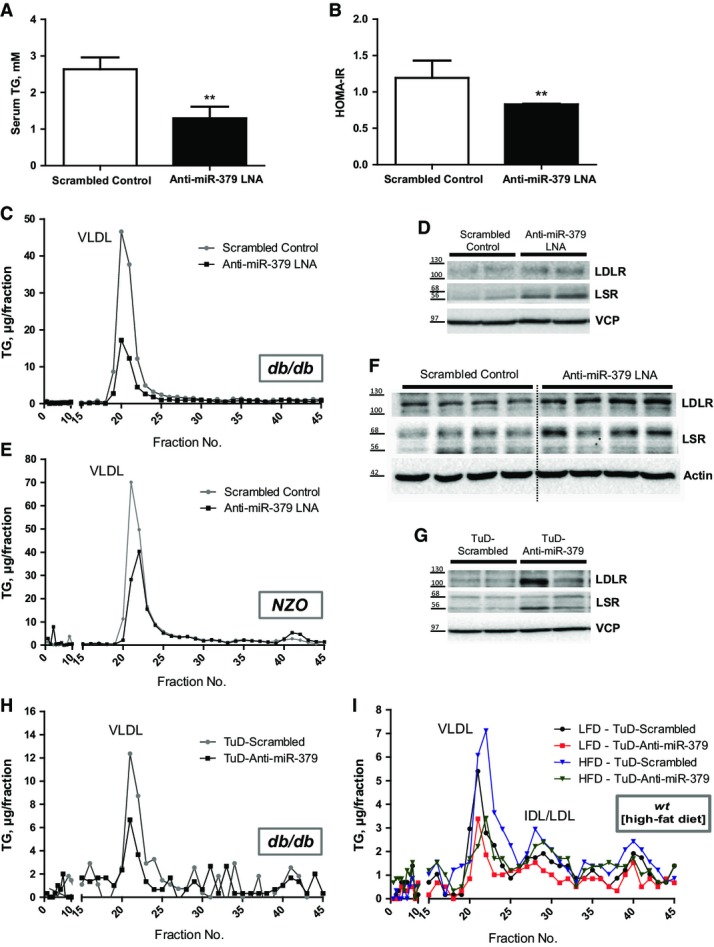
- Serum triglyceride levels in db/db mice treated with anti-miR-379 or scrambled control LNA (n = 7). Bar graphs show mean ± SEM; t-test: **P < 0.01.
- Homeostatic model assessment for insulin resistance (HOMA-IR) of same animals as in (A). Bar graphs show mean ± SEM; t-test: **P < 0.01.
- Triglyceride profiles of FPLC-fractionated serum of same animals as in (A) under fed conditions. VLDL peak is indicated.
- Protein levels of LSR and LDLR from livers of same animals as in (A). Shown are the LSR-α (68 kDa) and LSR-β (56 kDa) subunits and the glycosylated LDLR (between 100 and 130 kDa) protein.
- Triglyceride profiles of FPLC-fractionated serum of NZO mice treated with anti-miR-379 or scrambled control LNA (n = 7). VLDL peak is indicated.
- Protein levels of LSR and LDLR from livers of same animals as in (E).
- Protein levels of LSR and LDLR from livers of db/db mice treated with an anti-miR-379 or scrambled control Tough Decoy (TuD) construct delivered by rAAV 28 days post-injection (n = 8).
- Triglyceride profiles of FPLC-fractionated serum of same animals as in (G) under fed conditions. VLDL peak is indicated.
- Triglyceride profiles of FPLC-fractionated serum of C57Bl/6J mice fed with low-fat (10%) or high-fat (60%) diet for 12 weeks and treated with either anti-miR-379 or scrambled control Tough Decoy (TuD) construct delivered by rAAV. Mice were sacrificed 4 weeks after virus injection at 18 weeks of age. VLDL peak is indicated.
To validate our findings by an independent experimental system, we designed and employed a novel miR-379-specific Tough Decoy (TuD-Anti-miR-379) (Haraguchi et al, 2009) approach in db/db animals (Supplementary Fig S6A). Hepatic AAV-mediated delivery of TuD-Anti-miR-379 under control of a hepatocyte-specific promoter (Kulozik et al, 2011) induced LSR and LDLR protein expression in liver (Fig4G) and substantially decreased serum VLDL-TG levels in 14-week-old db/db mice (Fig4H, Supplementary Fig S6B), while only weakly impairing serum cholesterol levels (Supplementary Fig S6C and D). Importantly, TuD-Anti-miR-379 delivery into wild-type animals fed a high-fat diet for 12 weeks (Supplementary Fig S6E–H) also lowered circulating VLDL-TG and cholesterol levels as demonstrated by FPLC (Fig4I, Supplementary Fig S6I and J) even in the absence of overt hyperglucocorticoidemia under these conditions (Supplementary Fig S6K), indicating that miR-379 also controls systemic TG homeostasis under conditions of dietary energy excess.
Hepatic miR-379 levels correlate with serum cortisol and TG in human obese patients
Together, these studies demonstrate that the GC/GR-inducible miR-379 pathway represents a critical checkpoint in systemic dyslipidemia in diabesity and that selective targeting of miR-379 in the liver can be used as a therapeutic measure against GC-dependent hypertriglyceridemia. To finally test the validity of this assumption in humans, we investigated hepatic miR-379 expression in a clinically well-defined cohort of more than 70 individuals (Supplementary Table S1). In line with our experimental findings in animals, hepatic miR-379 levels were lower in lean individuals as compared with obese subjects (Fig5A) and positively correlated with serum cortisol and TG levels in this patient cohort as well as in the obese-only sub-cohorts, respectively (Fig5B–E), indicating that the GC/GR-dependent control of the miR-379 pathway represents a conserved (patho)physiological mechanism in obesity-dependent lipid homeostasis.
Figure 5.

- Hepatic miR-379 levels in obese patients (n = 64) and healthy controls (n = 10). Bar graphs show mean ± SEM; t-test: ***P < 0.001.
- Serum triglyceride (B) and cortisol (C) levels in obese and healthy control individuals. Bar graphs show mean ± SEM; t-test: **P < 0.01 or *P < 0.05.
- Correlation analyses of human hepatic miR-379 and serum cortisol (D) or triglycerides (E) (n = 74). Shown are the Spearman's rank coefficients and P-values. Sample population was stratified into diabetic obese, non-diabetic obese, and controls based on statistically significant interaction effects of the groups and serum parameters in a multiple linear regression model.
Discussion
Our study identifies the first functional miRNA pathway downstream of the mammalian HPA-GC/GR signaling axis by demonstrating that miRNAs in the 379/410 cluster are key outputs of the GC/GR endocrine pathway under conditions of metabolic dysfunction (Fig6).
Figure 6.
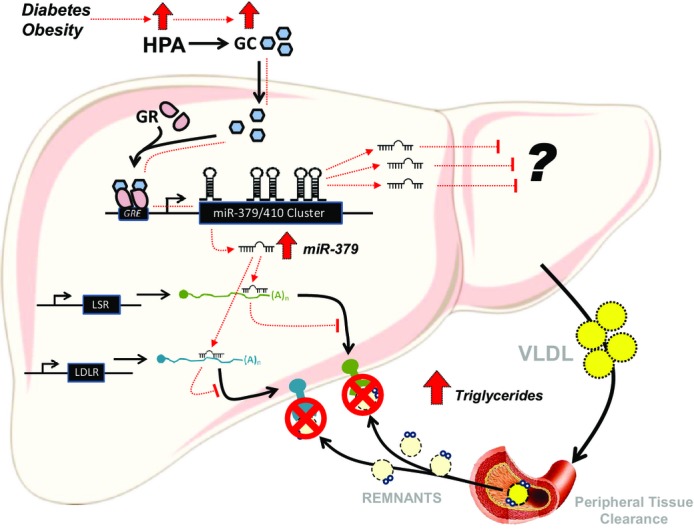
GR-dependent control of miR-379 in lipid metabolism
HPA, hypothalamic–pituitary–adrenal axis; GC, glucocorticoids; GR, glucocorticoid receptor; GRE, glucocorticoid response elements; miR, microRNA; LSR, lipolysis-stimulated lipoprotein receptor; LDLR, low-density lipoprotein receptor; VLDL, very-low-density lipoprotein receptor.
Glucocorticoids regulate the expression of its target via binding with the GR and subsequent interaction of the complex to a specialized region of the gene. In case of miRNAs in the cluster, receptor dimerization might be necessary for the GC action (Supplementary Fig S1A) together with other transcription factors and co-regulators (Supplementary Fig S1F). By using the ChIP-configured UCSC Human Genome Browser, we were able to locate possible GR-binding sites in the vicinity of the hsa-miR-379 locus (Supplementary Fig S1F). Previous GR ChIP-Seq results in C2C12 myotubes (Kuo et al, 2012) and 3T3-L1 adipocytes (Yu et al, 2010) likewise pinpointed GR-binding sites around the mmu-miR-379 gene. One of these sites proved to be a direct binding site in the expression of the miRNA in the liver (Fig1E). Interestingly, the data we have mined from the human genome browser displayed features reminiscence of GC/GR regulation. First, upstream miR-379 gene are several DNase-hypersensitive regions. It has been reported that these DNase-prone sites are observed prior to GR binding in the liver (Grange et al, 1989). Second, in these DNase-hypersensitive sites, the GR-binding regions occur with the ChIP-Seq hits for transcriptional activators known to work with the GR like p300, the liver hepatocyte nuclear factor 4A, and CEBPB. The latter has been recently demonstrated to keep chromatin accessibility in the liver and allows GR recruitment to regulatory elements (Grontved et al, 2013).
Recently, miRNAs in the miR-379/410 cluster have been demonstrated to be important in postnatal weaning of mouse newborns allowing the liver to adapt to changing glucose and lipid requirements of the organism (Labialle et al, 2014). In rodents, the postnatal weaning period is accompanied by adrenocortical quiescence known as the ‘stress non-responsive period’ (SNRP) where GC levels are low allowing the animals to withstand stress and favor anabolic reactions for the growth of the organism. This SNRP has been shown to be counterintuitive in any other life stages (Sapolsky & Meaney, 1986). In humans, several studies have reported that prenatal maternal and postnatal stressors increase the risk for type 2 diabetes, obesity, and elevated VLDLs in later life (Maniam et al, 2014). These reports, together with the mechanism we proposed of how a GC-responsive miRNA in the cluster can regulate systemic TG and VLDL levels, suggest a very interesting unifying model of how all these are interrelated. Follow-up studies are necessary to explore other miRNAs induced by GCs in the cluster as they may also unravel another layer of metabolic control by miR-379/410 cluster members.
Of note, the miR-379/410 cluster region has been genetically linked to growth deficiency, respiratory insufficiency, precocious puberty, obesity, and type 1 diabetes in both mice and humans (Moon et al, 2002; Kagami et al, 2008; Wallace et al, 2010). As consequences of aberrant GC/GR action significantly overlap with these phenotypes, it is tempting to speculate that the HPA/GC/GR pathway—at least partly—serves as a master control also of non-metabolic targets of the miR-379/410 cluster, thereby establishing the GR–miR-379/410 axis as a critical molecular hub for hormone-dependent metabolic and developmental control throughout mammalian evolution.
Given the importance of GC function for human (patho-)physiology and the correlation of miR-379 with metabolic parameters in a human patient cohort, it is likely that modulation of miR-379 activity will significantly impact distinct features of systemic energy homeostasis also in humans (Fig5). In this respect, the levels of serum TG are contributed by the TG-rich lipoproteins (TRLs), primarily chylomicrons and VLDL. The levels of VLDL-TGs in turn depend on peripheral clearance via lipoprotein lipase (LPL) action, rate of secretion by the liver, and clearance of lipoprotein remnants. The knockdown of miR-379 in the liver of wt and db/db mice did not affect LPL activity and the rate of VLDL secretion (Supplementary Fig S3B and C), supporting the hypothesis that TRL clearance represents the dominant mechanism of miR-379 action.
Consistent with previous reports, our data indicate that (i) the liver is the major site of TRL clearance under normal room temperature conditions (Fig3B, Supplementary Fig S3D) and (ii) TRL clearance occurs remarkably fast (Redgrave, 1973; Spady, 1992; Pirillo et al, 2014). Indeed, chylomicron and VLDL remnants, which are similar in size and composition, have previously been found to be rapidly cleared by the liver (Mahley & Huang, 2007; Williams & Chen, 2010), which is consistent with our TRL/VLDL uptake studies. Also, recent kinetic studies in obese, diabetic men suffering from hypertriglyceridemia demonstrated that basal VLDL-TG clearance was significantly lower compared to healthy controls (Sondergaard et al, 2012), which is in congruence with our data in human patients, showing that hepatic miR-379 levels correlate with both serum cortisol and TG levels in obese individuals (Fig5D and E). While hepatic VLDL hypersecretion clearly represents a critical feature in obesity-related hypertriglyceridemia (Ginsberg, 2002), these data allow the hypothesis that also defects in hepatic VLDL clearance may significantly contribute to lipid metabolic dysfunction in the Metabolic Syndrome.
Mechanistically, TRL clearance works in several steps involving the action of LDLR, LRP (LDLR-related protein), LSR, VLDLR, and HSPGs (heparan sulfate proteoglycans) (MacArthur et al, 2007). The TRL remnant particles are sequestered into the hepatic Space of Disse from endothelial fenestrae which are absent in adipose tissue capillaries (Yen et al, 2008). Our in silico miRNA target gene screening showed that miR-379 targets most of these receptors. However, we were only able to identify LSR and LDLR as valid and functional targets of miR-379. This is consistent with reports stating that both LSR and LDLR are severely compromised in hyperglucocorticoidemic db/db mice (Narvekar et al, 2009) as well as our functional characterization of miR-379 deficiency upon which these receptors are up-regulated, an effect that can be rescued by simultaneous LSR/LDLR double-knockdown (Fig3E and F; Supplementary Fig S4G–I). In line with these findings, also the LDLR has been reported to effectively clear TG as seen in the mild but significant elevation of serum TG levels in LDLR KO mice, and demonstrated by recent TG-rich lipoprotein uptake studies (Ishibashi et al, 1996; Foley et al, 2013). Of note, the LDLR was found to represent the predominant TG clearance pathway under conditions of chronic metabolic dysfunction (i.e. diet-induced obesity (DIO)) (Foley et al, 2013), acting independently from the HSPG system, which is consistent with our findings in db/db, NZO, and DIO mouse models (R.M. de Guia, A.J. Rose, A. Sommerfeld, A. Krones-Herzig, M. Berriel Diaz, S. Herzig, unpublished data).
In both mice and humans, high levels of GC in the blood are correlated with dysfunctional metabolic phenotypes as associated with the Metabolic Syndrome and Cushing's syndrome. Whether obesity is associated with increased cortisol levels is still a matter of debate. However, multiple studies actually show increased cortisol in obese humans (Anagnostis et al, 2009) which we have also seen in our study (Fig5). In addition, recent clinical data argue for a contribution of GC signaling to fatty liver phenotypes in human obese patients (Stefan et al, 2014). In the present study, LNA- and TuD-mediated knockdown of miR-379 resulted in decreased total serum and VLDL triglycerides in db/db, NZO, and high-fat diet-fed mice (Fig4) with tendencies to likewise affect cholesterol levels (Supplementary Fig S5D and K). Interestingly, our rather short-term HFD feeding experiment did not lead to elevations in circulating GC levels, thus likely representing a non-GC-associated obesity phenotype. Importantly, knockdown of miR-379 was still able to lower circulating TG levels under these conditions (Fig4I), indicating (i) that miR-379 has additional as yet not identified targets under HFD conditions and (ii) that miR-379 inactivation could still have therapeutic benefits in obese patients without elevation in circulating GC levels.
These results, therefore, present potentially new way to manage hypertriglyceridemia in obesity and other GC-related pathologies. To date, high statin dosage, fibrates, niacin, and omega-3 fatty acids are being used to treat hypertriglyceridemia. Most of these pharmacotherapies, alone or in combination, can reduce human serum TG levels from 20 to 50% (Kramer, 2013), thereby reflecting the same range of reduction we observed in mouse models upon miR-379 knockdown. Although additional studies are needed, we did not observe any harmful effects of LNA- and AAV-mediated TuD in mice. Our LNA-based miR-379 inhibition supports the growing interest in the use of LNA-based therapeutics in the management and treatment of diseases (Esau et al, 2006; Rayner et al, 2011). The use of vector-encoded TuD, on the other hand, is a relatively new mode inhibiting miRNAs which could have potential application in the future (Bak et al, 2013). Since anti-hyperlipidemic antisense drugs have been approved by the FDA recently (Kramer, 2013), the identification of a GC/GR-controlled miRNA may thus not only pave the way to alternative strategies in minimization of side effects in GC therapy but also as-yet non-explored miRNA-based approaches in targeting hypertriglyceridemia and other components of the Metabolic Syndrome.
Materials and Methods
Recombinant viruses
For long-term inactivation of the GR, adeno-associated viruses (AAV) encoding GR-specific miRNA (miR) were established as described previously (Rose et al, 2011). Recombinant viruses were produced in 293T cells through triple co-transfection of the recombinant viral backbone vector with the helper plasmids. Viruses were purified using the iodixanol method, dialyzed, and titrated by qPCR.
For liver-specific overexpression of miR-379, the same recombinant viral backbone vector was used as above. miR-379 genomic sequence ± 100 bp was PCR amplified with primers harboring BglII and SalI restriction sites. PCR product was then digested with the appropriate restriction enzymes and cloned into pcDNA6.2-GW/EmGFP-miR (BLOCK-iTTM PolII miR RNAi Expression Vector Kit; Invitrogen, Darmstadt, Germany). The construct was then tested in Hepa1–6 cell line and primary hepatocytes together with the corresponding control prior to sub-cloning into the AAV vector. Adenoviruses expressing LSR-specific or non-specific control oligonucleotides were cloned as described previously (Berriel Diaz et al, 2008; Narvekar et al, 2009). Viruses were purified using the CsCl method and dialyzed against phosphate-buffered saline containing 10% glycerol prior to animal injection.
Design and synthesis of LNA oligonucleotides
The LNA oligonucleotides were synthesized as unconjugated and fully phosphorothioated oligonucleotides (Exiqon A/S, Vedbaek, Denmark). The perfectly matching 15-mer LNA-anti-miR oligonucleotide (5′-GTTCCATAGTCTACC-3′) was complementary to nucleotides 2–16 in the mature miR-379 sequence. The scrambled control LNA oligonucleotide was synthesized with the following sequence: 5′-ACGTCTATACGCCCA-3′.
Construction of anti-miR-379 Tough Decoy
The Tough Decoy (TuD) construct and corresponding scrambled control were designed as previously described (Haraguchi et al, 2009). Briefly, TuD-Anti-miR-379 was designed to contain two miR-379 binding sites in a stem1-miR-binding-site-stem2 structure (Supplementary Table S2). The scrambled control was designed using siRNA Wizard 3.1 (http://www.sirnawizard.com/scrambled.php). The oligonucleotide pairs were annealed and cloned into pcDNA™6.2-GW/EmGFP (Invitrogen, Darmstadt, Germany). The construct was tested in vitro and the efficiency of knockdown assessed by RT–qPCR and Northern blot (data available upon request). The decoy sequences were then sub-cloned into the AAV vector.
Animal experiments
Male, 8- to 12-week-old C57BL/6J, db/db, New Zealand black (NZB), and New Zealand obese (NZO) were obtained from Charles River Laboratories (Brussels, Belgium). GRdim/dim, GRAlfpCre, and corresponding wild-type littermates were obtained and bred as previously described (Rose et al, 2011). All mice were maintained on a 12-h light/dark cycle with regular unrestricted diet and had free access to drinking water. In all cases, mice were acclimated to the housing facility and control diet food (% kcal: 70 carbohydrate, 20 protein, 10 fat; Research Diets Inc, New Brunswick, NJ, USA) for at least 1 week prior to experimentation. For high-fat diet studies, C57BL/6J mice were fed for 12 weeks with 60% kcal fat-containing diet (Research Diet D12492) or 10% kcal fat (Research Diet D12450B) as control.
To examine the effects of long-term liver GR knockdown on the expression of miRs, mice were administered with 5 × 1011 viral genomes of non-specific negative control or GR-specific miR bearing AAV. Mice were kept on normal diet for 6 weeks after which fasted overnight for 14–16 h and then euthanized.
In additional experiments, 7- to 10-week-old male homozygous GR dimerization deficient (GRdim/dim) (Reichardt et al, 1998) and wild-type littermates were treated daily i.p. with 9α-fluoro-16α-methylprednisolone (DEX, 1 mg/kg; Sigma-Aldrich, Munich, Germany) or vehicle (Veh, 2% ethanol in isotonic saline) for 28 days after which they were sacrificed in the overnight fasted state. To examine the effects of genetic hepatocyte-specific knockout of GR in mice, liver samples from GRAlfpCre (Opherk et al, 2004) and wild-type littermates were taken in the fasted state as previously reported (Lemke et al, 2008).
To investigate the metabolic effects of the loss of miR-379 in the liver, C57BL/6J, NZO, and db/db mice (The Jackson Laboratory, ME, USA) were injected i.v. with 15 mg/kg BW of anti-379-specific LNA or scrambled control (both reconstituted in water and diluted with isotonic saline) for two consecutive days. Mice were then euthanized 7 days or 14 days post-injection in the random fed state. An independent study was done using liver-specific rAAV Tough Decoy constructs. Mice (db/db or high-fat diet-fed) were administered i.v. with 5 × 1011 viral genomes of TuD-Anti-miR-379 or TuD-scrambled miR-binding-site-containing AAV. Mice were kept on normal diet until sacrificed at week 4.
To evaluate whether the metabolic effects of miR-379 inhibition are still observable upon knockdown of LSR, C57BL/6J mice were injected i.v. with adenoviruses expressing LSR-specific or non-specific shRNAs as described previously (Narvekar et al, 2009). Three days after virus injection, mice were injected with anti-miR-379-specific LNA or scrambled control as indicated above. Mice were then euthanized after 7 days post-LNA injection.
To investigate the LSR and LDLR regulation by miR-379, LDLR knockout mice (LDLRKO) (The Jackson Laboratory) were utilized. LDLRKO animals were injected with adenoviruses expressing LSR-specific or non-specific shRNAs as described above. Three days after virus injection, mice were injected with anti-miR-379-specific LNA or scrambled control as indicated above. Mice were then euthanized after 7 days post-LNA injection. The experiment was done alongside C57BL/6J mice injected with the non-specific control adenovirus and anti-miR-379-specific LNA or scrambled control.
Organs including liver, brown adipose tissues, perigonadal fat pads, heart, intestine, and gastrocnemius muscles were collected after the corresponding time periods, weighed, snap-frozen, and used for further analysis. Whole blood was also collected for serum isolation which was then kept at −80°C until needed for analyses. Total body fat content was determined by an EchoMRI body composition analyzer (Echo Medical Systems, Houston, TX, USA).
In each animal experiment, mice were randomly assigned to each group. Number of animals per group to detect biologically significant effect sizes was calculated using appropriate statistical sample size formula and indicated in the biometrical planning section of the animal license submitted to the governing authority. Blinding was not done during animal group allocation but in some measurements made in the study (i.e. Hepatic VLDL Release Assay). Animal handling and experimentation was performed in accordance with the European Union directives and the German animal welfare act (Tierschutzgesetz) and approved by local authorities (Regierungspräsidium Karlsruhe).
Tissue and serological metabolite and hormone analyses
To ensure an analysis representative of the whole liver, frozen liver samples were pulverized (Bessman Tissue Pulveriser, 189–476; VWR International GmbH, Darmstadt, Germany) and subsequent tissue powder aliquots were weighed and prepared for analyte extractions. Hepatic lipids were extracted as described previously (Herzig et al, 2003).
Serum levels of glucose were measured using an automated analyzer (One Touch; Lifescan, Neckargemünd, Germany). Commercially available kits were used to quantify serum, triglycerides (Sigma-Aldrich), cholesterol (RANDOX, Crumlin, NIR), non-esterified fatty acids (WAKO, Neuss, Germany), ketone bodies (WAKO), alanine amino transferase (Fischer Scientific, Schwerte, Germany), corticosterone (Assay Designs, Ann Arbor, MI, USA), and insulin (Mercodia, Uppsala, Sweden). Liver TG, cholesterol, free fatty acids, and ketone bodies were analyzed from the extracts using the same commercial kits as for serum analysis. Values were calculated as millimol per liter (serum) or per gram wet tissue (liver).
Fast-protein liquid chromatography
Serum from specified number of mice per experimental group was pooled and subjected to fast-protein liquid chromatography as previously described (Lichtenstein et al, 2007). Cholesterol and TG were measured in the eluted fractions using commercial kits as above. Values were calculated as μg of triglyceride or cholesterol per fraction.
Hepatic VLDL release
Very-low-density lipoprotein production was determined after tyloxapol (Sigma, Munich, Germany) injection as described (Mandard et al, 2006). Briefly, seven db/db mice per group were fasted for 4 h. Forty (40) μl of blood was drawn from each mouse by cutting the tip of the tail. The volume of tyloxapol in μl required per mouse was approximately three times that of the weight of the mouse in grams, that is, a 25-g mouse required 75 μl of a 20% solution. Specified amounts were administered via the tail vein, and 40 μl blood samples were taken every 50 min for 2.5 h. The mice were eventually sacrificed after 300 min. The serum TG values were determined (see above) and plotted versus time.
Lipid and lipoprotein clearance studies
Oral lipid tolerance test with 3H-triolein
Oral fat tolerance test was performed essentially as described previously (Bartelt et al, 2011). In particular, overnight (∽16 h, 15–09 h) fasted mice were orally gavaged with 100 μl olive oil (O1514; Sigma-Aldrich, DEU) containing 0.37 MBq of [9,10-3H(N)]-triolein (Hartmann Analytic GmbH, DEU) per mouse. 50 μl of tail vein blood samples was collected for serum isolation before and at 1, 2, and 4 h after oral fat administration. Mice were sacrificed after collection of blood at the last time point. The liver, brown adipose tissue, perigonadal white adipose tissue, gastrocnemius skeletal muscle, heart, and small intestine were collected, weighed, and snap-frozen in LN2. VLDL was isolated from 10 μl serum using precipitation buffer (K613-100; BioVision, CA, USA) and re-suspended in PBS. The VLDL precipitation/isolation method was validated by measuring the ApoB levels and absence of ApoA1 (data not shown). Serum and VLDL-TG were quantified as indicated above. Aliquots of tissues were extracted using Solvable™ (Perkin Elmer Inc, Rodgau, Germany, DEU). The serum, VLDL isolates, and tissues extracts were counted for 3H activity using standard scintillation counting (Packard 2200CA Tri-Carb Liquid Scintillation analyzer; Packard Instruments, Meriden, CT, USA). Tissue clearance rates were calculated based upon the specific activity of the administered 3H-olive oil (cpm/mol), assuming that 100% of the 3H-triolein was incorporated into the olive oil, along with tissue counts (cpm) and mass (g), and time (h) of collection after administration.
VLDL isolation
Very-low-density lipoprotein was isolated from serum samples obtained from 6 to 20 wt mice by ultracentrifugation as described (Redgrave et al, 1975). Briefly, 3.5 ml serum was put in a polyallomer tube SW40Ti and mixed with 1.39 g KBr. The mixture was over-layered with 332.8 ml of a KBr solution (D = 1.063, 1.019, and 1.006 g/ml) and run for 18 h at 217,000 g in an ultracentrifuge (Beckman Coulter GmbH, Krefeld, DEU). VLDL was carefully removed from the topmost gradient and subsequently measured for TG content.
VLDL-3H TG clearance
Ex vivo VLDL labeling and clearance study in mice were performed as previously described (Bartelt et al, 2011) with some modifications. The turnover study was conducted by intravenous injection of 2 mg VLDL-TG with 0.37 MBq of [9,10-3H(N)]-triolein (Hartmann Analytic GmbH, DEU) per mouse. The isolated VLDL-TG was labeled by adding the calculated amount into a tube containing dried 3H-triolein. After very gentle agitation at 4°C overnight, the mixture was re-suspended in 0.9% saline. Mice were fasted for 4 h. 25 μl of blood samples was collected for serum isolation at different times after i.v. tail vein administration of VLDL: 0.5, 1, 2, and 4 min. Mice were sacrificed after collection of blood at the last time point. The liver, brown adipose tissue, perigonadal white adipose tissue, and gastrocnemius skeletal muscle were collected, weighed, and snap-frozen in LN2. The organs were processed as described above. The serum and tissues extracts were counted for 3H activity using standard scintillation counting (Packard 2200CA Tri-Carb Liquid Scintillation analyzer; Packard Instruments). Serum 3H-VLDL levels were calculated based upon the specific activity of the administered 3H-TG-VLDL (cpm/mol), assuming that 100% of the 3H-triolein was incorporated into the VLDL. Tissue clearance rates were calculated based upon the specific activity of the administered 3H-TG-VLDL (cpm/mol), along with tissue counts (cpm) and mass (g), and time (min) of collection after administration.
LPL activity
LPL activity measurements were performed as described (Jones et al, 2013) using frozen adipose tissue samples.
Glucose tolerance test
Glucose tolerance tests were performed as described (Kulozik et al, 2011).
Pyruvate tolerance test
Mice were fasted for approximately 16–18 h before the experiment. Prior to pyruvate injection, the body weight of the mouse was measured and the basal glucose level was determined by bleeding the mouse from the tail and measuring with the use of glucometer. The mice were then injected intraperitoneally with 10 μl/g body weight of 20% (w/v) pyruvate solution (in 0.9% NaCl diluent) giving a dose of 2 g/kg BW. The measurement of blood glucose was taken by sampling blood from the tail at 20, 60, 120, and 240 min after injection.
RNA isolation and quantitative taqman RT–PCR
Total RNA was extracted from homogenized mouse liver or cell lysates using Qiazol reagent and miRNeasy® Mini Kit (Qiagen, Hilden, Germany). cDNA was prepared by reverse transcription using the M-MuLV enzyme and Oligo dT primer (Fermentas, St Leon-Rot, Germany). cDNAs were amplified using assay-on-demand kits and an ABI PRISM 7700 Sequence detector (Applied Biosystems, Darmstadt, Germany). RNA expression data were normalized to levels of TATA-box binding protein (TBP) RNA. For miRNA RT–PCR, Applied Biosystems’ TaqMan® MiRNA Reverse Transcription Kit (Life Technologies GmbH, Darmstadt, Germany) was used together with TaqMan® MiRNA Assay Primer. snoRNA-202 was used as internal control for miRNA quantifications.
Protein analysis
Protein was extracted from frozen organ samples, cultured cell lines or primary cells in cell lysis buffer (Rohm et al, 2013), and 10 mg of protein was loaded onto 5–12% SDS–polyacrylamide gels and blotted onto nitrocellulose membranes. Western blot assays were performed as described (Herzig et al, 2001) using antibodies specific for LSR (DKFZ, Heidelberg, Germany), LDLR (ab30532; Abcam), FoxO1 (9454; Cell Signalling), p-FoxO1 (9461; Cell Signalling), actin (A5441; Sigma), or VCP (ab11433; Abcam). Band optical densities were quantified using ImageJ software (NIH).
Histological examinations
For hematoxylin and eosin (H&E) and oil red O lipid staining, 5-mm cryosections of liver embedded in Tissue Tek OCT compound (Sakura, Netherlands) were fixed in Baker's formol. Nuclei were counterstained with hematoxylin.
Cell culture and transfections
Cells were cultured with 10% FBS in DMEM (HEK293, Hepa1–6, and FAO hepatoma; ATCC, VA, USA) supplemented with glutamine in the presence or absence of antibiotics depending on the mode of transfection application. For in vitro dexamethasone treatments, cells were conditioned in culture for 2 weeks in charcoal-stripped FBS prior to steroid treatment. Plasmid constructs, miR mimics (Fisher Scientific, Schwerte, Germany), and LNAs (Exiqon A/S) were transfected into cells using PEI (Polysciences Europe GmbH, Eppelheim, Germany), Promofectin (for primary hepatocytes) (Promocell GmbH, Heidelberg, Germany), Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany), or CaCl2 method according to the manufacturer's or using the standard protocol. Plasmid constructs were transfected from 50 to 2,000 ng amounts, while miR mimics and LNAs were transfected from 50 to 100 nM concentrations. In all cell culture studies, cells were transfected at 50% confluency. The effect of overexpression/knockdown constructs, mimics, or LNAs were evaluated 24–48 h post-transfection.
Primary mouse hepatocytes were isolated and cultured as described (Klingmuller et al, 2006).
Reporter constructs and dual-luciferase assay
Oligonucleotides with integrated XhoI and NotI sites corresponding to conserved LSR and LDLR miR-379 binding sites (Supplementary Table S2) predicted by RNA22 were annealed and cloned into psiCHECK™-2 vector. Matching oligos containing the mutated binding site sequence were also prepared. For the dual-luciferase assay, HEK293A cells at 8,000 cells/well density were seeded in 96-well plate. The cells were transfected with 75 ng of the psiCHECK™-2 vector and 5 nmol of miR mimics (Fisher Scientific) using Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany). Cel-miR-239b was utilized as a negative control. Firefly and Renilla luciferase activities were measured 48 h after transfection using a dual-luciferase reporter assay kit (Promega, Mannheim, Germany). Renilla luciferase activity was normalized using firefly luciferase activity.
Seahorse β-oxidation metabolic flux assay
Mitochondrial β-oxidation activity was determined using an XF96 Extracellular Flux Analyser (Seahorse, Copenhagen, Denmark). Prior to the experiment, 1,000 Hepa1–6 hepatoma cells were seeded per well in a 96-well format and transfected with miR-379 overexpression and knockdown constructs using Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany). The assay was performed according to the manufacturer's protocol and using 200 μM BSA-palmitate conjugate, 0.5 mM carnitine (Sigma-Aldrich), and 100 μM etomoxir (Sigma-Aldrich). Oxygen consumption rate was calculated and normalized to protein content as determined by sulforhodamine B staining.
Chromatin immunoprecipitation
Liver tissue powder samples from DEX or vehicle-treated wild-type mice were fixed with formaldehyde in PBS, and ChIP assays were performed as described (Canettieri et al, 2003) using antibodies specific for GR (Cat. No. sc-1004: Santa Cruz Biotechnology Inc, Heidelberg, Germany), or non-specific anti-HA antibody (Cat. No. sc-805: Santa Cruz Biotechnology Inc, Germany). Precipitated DNA fragments were analyzed by PCR amplification using primers directed against three distinct promoter regions within the miR-379 locus. Identical results were obtained by using a second, isotype-specific control antibody.
Microarray expression profiling
miR profiling was performed on hepatic total RNA extracts from [1] wild-type and db/db mice and from [2] control mice and rAAV-treated mice for liver-specific GR knockdown. RNA isolation, cDNA and cRNA synthesis, and hybridization to arrays of type GeneChip® miRNA 2.0 Array (Affymetrix, Freiburg, Germany) were performed according to the manufacturer's recommendations. Four arrays per group were hybridized. Microarray data were analyzed based on ANOVA using a commercial software package (Micro Array Solution, version 1.0; SAS Institute, Cary, NC, USA). Standard settings were used, except for the following specifications: Log-linear mixed models were fitted for values of perfect matches, with genotype considered to be constant and the array identification, random. Custom CDF with Unigene-based gene/transcript definitions was used to annotate the arrays. Affected biological pathways reflected by the differential gene expression were determined by ORA based on Fisher's exact test. Microarray data were deposited in the GEO data repository.
Northern blotting
Twenty to forty microgram of total RNA was separated on 15% TBE–urea polyacrylamide gels and semi-dry transferred to Hybond N+ membrane (Amersham, GE Healthcare, Germany). Complete LNA probe complementary to miR-379, miR-122, and let-7a was designed as described in the miRNA registry (http://www.sanger.ac.uk/). 20 pmol of the probe was labeled with T4 polynucleotide kinase (New England Biolabs) and 20 μCi of [γ-32P] ATP (250 μCi; Perkin Elmer Inc). Hybridizations were performed at 50°C for 16 h in a 100 ml of the small RNA hybridization buffer (formamide, 1 M NaCl, 0.2 M sodium phosphate, 0.04 M EDTA, Denhardt's solution, SDS). The membrane was washed several times and exposed to an erased phosphor screen which was then scanned and analyzed after at least 4 h. For stripping and re-probing, the membrane was incubated at RT with boiling stripping solution (0.02× SSC · 0.01% SDS) and hybridized with another probe after 30 min of pre-hybridization. U6RNA probe (5′-CACGAATTTGCGTGTCATCCTT-3′) was used as loading control.
Ex vivo human liver study
To examine the effects of glucocorticoid treatment to human hepatic miRs, a healthy piece of freshly re-sected liver from an individual (age: 51 years, sex: female) undergoing liver surgery to remove a tumor was utilized. From this sample, small pieces (10–20 mg) were carefully dissected and incubated (37°C, 5% CO2) in quadruplicate in non-adhesive tissue culture plates (Greiner Bio-One, Frickenhausen, Germany) in Williams’ medium E (+ 2 mM l-glutamine and 0.1% BSA; Biochrom AG, Berlin, Germany) containing 0.11% EtOH (Vehicle), 1 μM RU486 (Sigma-Aldrich), 0.1 μM DEX (Sigma-Aldrich), and 1 μM RU486 + 0.1 μM DEX. Samples were incubated for 30 min prior to treatment with or without RU486. Incubations were ceased at 3 h after treatment upon which the tissue pieces were washed with PBS, rapidly frozen in liquid nitrogen, and stored at −80°C until needed. Each volunteer gave written informed consent before participation, and the study was approved by the local ethics committee.
Human liver samples
Subjects
miR-379 expression was investigated in liver tissue samples obtained from 10 Caucasian healthy donors and 64 Caucasian obese men and women with (n = 27) or without (n = 37) type 2 diabetes who underwent open abdominal surgery for Roux-en-Y bypass, sleeve gastrectomy or elective cholecystectomy. A small liver biopsy was taken during the surgery, immediately frozen in liquid nitrogen, and stored at −80°C until further preparations. The phenotypic characterization of the cohort has been performed as described previously (Kloting et al, 2010). All baseline blood samples were collected between 8 and 10 am after an overnight fast. All study protocols have been approved by the ethics committee of the University of Leipzig (363-10-13122010 and 017-12-230112) in accordance with the principles of the WMA Declaration of Helsinki. All participants gave written informed consent before taking part in the study.
hsa-miR-379 studies
Human miR-379 mRNA expression was measured by quantitative real-time RT–PCR in a fluorescent temperature cycler using the TaqMan assay, and fluorescence was detected on an ABI PRISM 7000 sequence detector (Applied Biosystems). Total RNA was isolated as described above. Applied Biosystems’ TaqMan® MiRNA Reverse Transcription Kit (Life Technologies GmbH) was used together with TaqMan® MiRNA Assay Primer for hsa-miR-379 to quantify the miRNA level. RNU58B was used as internal control for miRNA quantifications.
Bioinformatics and miRNA target site analysis
UCSC Genome Browser (http://genome.ucsc.edu/index.html) was utilized to identify putative promoter region of the miRNA and scan the region for results of experiments from transcription factor ChIP-Seq (Meyer et al, 2013). For miR target prediction, RNA22 (https://cm.jefferson.edu/rna22v1.0/) and MiRTiF (http://mirtif.bii.a-star.edu.sg/) were utilized. DAVID Bioinformatics Resources 6.7 (http://david.abcc.ncifcrf.gov/) was used for functional annotations of the predicted targets which include filtering for liver targets and cluster annotation.
Statistical analyses
All analyses were carried out with SigmaPlot v.12 software (Systat Software GmbH, Erkrath, Germany). For each experiment, means and SEM of continuous variables were determined. Statistical analyses with two groups were performed using Student's or paired t-test in one-factorial designs. For multifactorial study designs, two-way ANOVA was used when appropriate. Holm–Sidak post hoc was applied when significant differences were found with an overall significance level < 0.05. In cases of non-conformity to normality, nonparametric tests were utilized (i.e. Mann–Whitney U-test, Kruskal–Wallis test). Multiple linear regression modeling was utilized in human data which included the major variables (serum cortisol, TG, and hepatic miR-379 levels) and respective interaction with stratifying categorical variable (controls, diabetic obese, non-diabetic obese) to detect multicollinearity. Correlation of variables was determined using Pearson's or Spearman's rank correlation analysis.
Acknowledgments
We thank Carolyn Algire, Peter Angel, Johannes Backs, Alexander Ernst, Katharina Genreith, Adriano Maida, Maria Muciek, Anja Reimann, Michaela Schäfer, Sigrid Stöhr, Christian Stoy, Diana Tichy, Xiaoyue Wang, and Julia Winter for technical support as well as Marc Montminy and Tobias Schafmeier for advice on the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft (He3260/7-2) and the Helmholtz Association (ICEMED, Metabolic Dysfunction) to SH.
Author contributions
RMG, AJR, AS, OS, DS, AZ, YF, AK-H, TS, MK, CS, NG, GD-T, SD, NK, and MBD performed experiments. MB supervised and conducted patient sample collection and analyses. SH designed and directed research and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Review Process File
References
- Anagnostis P, Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP. Clinical review: the pathogenetic role of cortisol in the metabolic syndrome: a hypothesis. J Clin Endocrinol Metab. 2009;94:2692–2701. doi: 10.1210/jc.2009-0370. [DOI] [PubMed] [Google Scholar]
- Bak RO, Hollensen AK, Primo MN, Sorensen CD, Mikkelsen JG. Potent microRNA suppression by RNA Pol II-transcribed ‘Tough Decoy’ inhibitors. RNA. 2013;19:280–293. doi: 10.1261/rna.034850.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, Eychmuller A, Gordts PL, Rinninger F, Bruegelmann K, Freund B, Nielsen P, Merkel M, Heeren J. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17:200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- Berriel Diaz M, Krones-Herzig A, Metzger D, Ziegler A, Vegiopoulos A, Klingenspor M, Muller-Decker K, Herzig S. Nuclear receptor cofactor receptor interacting protein 140 controls hepatic triglyceride metabolism during wasting in mice. Hepatology. 2008;48:782–791. doi: 10.1002/hep.22383. [DOI] [PubMed] [Google Scholar]
- Canettieri G, Morantte I, Guzman E, Asahara H, Herzig S, Anderson SD, Yates JR, 3rd, Montminy M. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat Struct Biol. 2003;10:175–181. doi: 10.1038/nsb895. [DOI] [PubMed] [Google Scholar]
- Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Foley EM, Gordts PL, Stanford KI, Gonzales JC, Lawrence R, Stoddard N, Esko JD. Hepatic remnant lipoprotein clearance by heparan sulfate proteoglycans and low-density lipoprotein receptors depend on dietary conditions in mice. Arterioscler Thromb Vasc Biol. 2013;33:2065–2074. doi: 10.1161/ATVBAHA.113.301637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg HN. New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation. 2002;106:2137–2142. doi: 10.1161/01.cir.0000035280.64322.31. [DOI] [PubMed] [Google Scholar]
- Glazov EA, McWilliam S, Barris WC, Dalrymple BP. Origin, evolution, and biological role of miRNA cluster in DLK-DIO3 genomic region in placental mammals. Mol Biol Evol. 2008;25:939–948. doi: 10.1093/molbev/msn045. [DOI] [PubMed] [Google Scholar]
- Grange T, Roux J, Rigaud G, Pictet R. Two remote glucocorticoid responsive units interact cooperatively to promote glucocorticoid induction of rat tyrosine aminotransferase gene expression. Nucleic Acids Res. 1989;17:8695–8709. doi: 10.1093/nar/17.21.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grontved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, Aguilera G, Hager GL. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. 2013;32:1568–1583. doi: 10.1038/emboj.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi T, Ozaki Y, Iba H. Vectors expressing efficient RNA decoys achieve the long-term suppression of specific microRNA activity in mammalian cells. Nucleic Acids Res. 2009;37:e43. doi: 10.1093/nar/gkp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92:883–893. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Herzig S, Hedrick S, Morantte I, Koo SH, Galimi F, Montminy M. CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature. 2003;426:190–193. doi: 10.1038/nature02110. [DOI] [PubMed] [Google Scholar]
- Ishibashi S, Perrey S, Chen Z, Osuga J, Shimada M, Ohashi K, Harada K, Yazaki Y, Yamada N. Role of the low density lipoprotein (LDL) receptor pathway in the metabolism of chylomicron remnants. A quantitative study in knockout mice lacking the LDL receptor, apolipoprotein E, or both. J Biol Chem. 1996;271:22422–22427. doi: 10.1074/jbc.271.37.22422. [DOI] [PubMed] [Google Scholar]
- Jones A, Friedrich K, Rohm M, Schafer M, Algire C, Kulozik P, Seibert O, Muller-Decker K, Sijmonsma T, Strzoda D, Sticht C, Gretz N, Dallinga-Thie GM, Leuchs B, Kogl M, Stremmel W, Diaz MB, Herzig S. TSC22D4 is a molecular output of hepatic wasting metabolism. EMBO Mol Med. 2013;5:294–308. doi: 10.1002/emmm.201201869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagami M, Sekita Y, Nishimura G, Irie M, Kato F, Okada M, Yamamori S, Kishimoto H, Nakayama M, Tanaka Y, Matsuoka K, Takahashi T, Noguchi M, Masumoto K, Utsunomiya T, Kouzan H, Komatsu Y, Ohashi H, Kurosawa K, Kosaki K, et al. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet. 2008;40:237–242. doi: 10.1038/ng.2007.56. [DOI] [PubMed] [Google Scholar]
- Klingmuller U, Bauer A, Bohl S, Nickel PJ, Breitkopf K, Dooley S, Zellmer S, Kern C, Merfort I, Sparna T, Donauer J, Walz G, Geyer M, Kreutz C, Hermes M, Gotschel F, Hecht A, Walter D, Egger L, Neubert K, et al. Primary mouse hepatocytes for systems biology approaches: a standardized in vitro system for modelling of signal transduction pathways. Syst Biol (Stevenage) 2006;153:433–447. doi: 10.1049/ip-syb:20050067. [DOI] [PubMed] [Google Scholar]
- Kloting N, Fasshauer M, Dietrich A, Kovacs P, Schon MR, Kern M, Stumvoll M, Bluher M. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. 2010;299:E506–E515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- Kramer W. Novel drug approaches in development for the treatment of lipid disorders. Exp Clin Endocrinol Diabetes. 2013;121:567–580. doi: 10.1055/s-0033-1351258. [DOI] [PubMed] [Google Scholar]
- Kulozik P, Jones A, Mattijssen F, Rose AJ, Reimann A, Strzoda D, Kleinsorg S, Raupp C, Kleinschmidt J, Muller-Decker K, Wahli W, Sticht C, Gretz N, von Loeffelholz C, Stockmann M, Pfeiffer A, Stohr S, Dallinga-Thie GM, Nawroth PP, Berriel Diaz M, et al. Hepatic deficiency in transcriptional cofactor TBL1 promotes liver steatosis and hypertriglyceridemia. Cell Metab. 2011;13:389–400. doi: 10.1016/j.cmet.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc Natl Acad Sci USA. 2012;109:11160–11165. doi: 10.1073/pnas.1111334109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labialle S, Marty V, Bortolin-Cavaille ML, Hoareau-Osman M, Pradere JP, Valet P, Martin PG, Cavaille J. The miR-379/miR-410 cluster at the imprinted Dlk1-Dio3 domain controls neonatal metabolic adaptation. EMBO J. 2014;33:2216–2230. doi: 10.15252/embj.201387038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiter EH, Reifsnyder PC. Differential levels of diabetogenic stress in two new mouse models of obesity and type 2 diabetes. Diabetes. 2004;53(Suppl 1):S4–S11. doi: 10.2337/diabetes.53.2007.s4. [DOI] [PubMed] [Google Scholar]
- Lemke U, Krones-Herzig A, Berriel Diaz M, Narvekar P, Ziegler A, Vegiopoulos A, Cato AC, Bohl S, Klingmuller U, Screaton RA, Muller-Decker K, Kersten S, Herzig S. The glucocorticoid receptor controls hepatic dyslipidemia through Hes1. Cell Metab. 2008;8:212–223. doi: 10.1016/j.cmet.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Lichtenstein L, Berbee JF, van Dijk SJ, van Dijk KW, Bensadoun A, Kema IP, Voshol PJ, Muller M, Rensen PC, Kersten S. Angptl4 upregulates cholesterol synthesis in liver via inhibition of LPL- and HL-dependent hepatic cholesterol uptake. Arterioscler Thromb Vasc Biol. 2007;27:2420–2427. doi: 10.1161/ATVBAHA.107.151894. [DOI] [PubMed] [Google Scholar]
- MacArthur JM, Bishop JR, Stanford KI, Wang L, Bensadoun A, Witztum JL, Esko JD. Liver heparan sulfate proteoglycans mediate clearance of triglyceride-rich lipoproteins independently of LDL receptor family members. J Clin Invest. 2007;117:153–164. doi: 10.1172/JCI29154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Huang Y. Atherogenic remnant lipoproteins: role for proteoglycans in trapping, transferring, and internalizing. J Clin Invest. 2007;117:94–98. doi: 10.1172/JCI30889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandard S, Zandbergen F, van Straten E, Wahli W, Kuipers F, Muller M, Kersten S. The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J Biol Chem. 2006;281:934–944. doi: 10.1074/jbc.M506519200. [DOI] [PubMed] [Google Scholar]
- Maniam J, Antoniadis C, Morris MJ. Early-Life Stress, HPA Axis Adaptation, and Mechanisms Contributing to Later Health Outcomes. Front Endocrinol (Lausanne) 2014;5:73. doi: 10.3389/fendo.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, Raney BJ, Pohl A, Malladi VS, Li CH, Lee BT, Learned K, Kirkup V, Hsu F, Heitner S, Harte RA, et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon YS, Smas CM, Lee K, Villena JA, Kim KH, Yun EJ, Sul HS. Mice lacking paternally expressed Pref-1/Dlk1 display growth retardation and accelerated adiposity. Mol Cell Biol. 2002;22:5585–5592. doi: 10.1128/MCB.22.15.5585-5592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvekar P, Berriel Diaz M, Krones-Herzig A, Hardeland U, Strzoda D, Stohr S, Frohme M, Herzig S. Liver-specific loss of lipolysis-stimulated lipoprotein receptor triggers systemic hyperlipidemia in mice. Diabetes. 2009;58:1040–1049. doi: 10.2337/db08-1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626–635. doi: 10.1016/S0140-6736(14)61177-6. [DOI] [PubMed] [Google Scholar]
- Opherk C, Tronche F, Kellendonk C, Kohlmuller D, Schulze A, Schmid W, Schutz G. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol. 2004;18:1346–1353. doi: 10.1210/me.2003-0283. [DOI] [PubMed] [Google Scholar]
- Phillips DI, Barker DJ, Fall CH, Seckl JR, Whorwood CB, Wood PJ, Walker BR. Elevated plasma cortisol concentrations: a link between low birth weight and the insulin resistance syndrome? J Clin Endocrinol Metab. 1998;83:757–760. doi: 10.1210/jcem.83.3.4634. [DOI] [PubMed] [Google Scholar]
- Pirillo A, Norata GD, Catapano AL. Postprandial lipemia as a cardiometabolic risk factor. Curr Med Res Opin. 2014;30:1489–1503. doi: 10.1185/03007995.2014.909394. [DOI] [PubMed] [Google Scholar]
- Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011;478:404–407. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redgrave TG. Cholesterol feeding alters the metabolism of thoracic-duct lymph lipoprotein cholesterol in rabbits but not in rats. Biochem J. 1973;136:109–113. doi: 10.1042/bj1360109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redgrave TG, Roberts DC, West CE. Separation of plasma lipoproteins by density-gradient ultracentrifugation. Anal Biochem. 1975;65:42–49. doi: 10.1016/0003-2697(75)90488-1. [DOI] [PubMed] [Google Scholar]
- Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schutz G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- Rohm M, Sommerfeld A, Strzoda D, Jones A, Sijmonsma TP, Rudofsky G, Wolfrum C, Sticht C, Gretz N, Zeyda M, Leitner L, Nawroth PP, Stulnig TM, Diaz MB, Vegiopoulos A, Herzig S. Transcriptional cofactor TBLR1 controls lipid mobilization in white adipose tissue. Cell Metab. 2013;17:575–585. doi: 10.1016/j.cmet.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Vegiopoulos A, Herzig S. Role of glucocorticoids and the glucocorticoid receptor in metabolism: insights from genetic manipulations. J Steroid Biochem Mol Biol. 2010;122:10–20. doi: 10.1016/j.jsbmb.2010.02.010. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Diaz MB, Reimann A, Klement J, Walcher T, Krones-Herzig A, Strobel O, Werner J, Peters A, Kleyman A, Tuckermann JP, Vegiopoulos A, Herzig S. Molecular control of systemic bile acid homeostasis by the liver glucocorticoid receptor. Cell Metab. 2011;14:123–130. doi: 10.1016/j.cmet.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Herzig S. Metabolic control through glucocorticoid hormones: an update. Mol Cell Endocrinol. 2013;380:65–78. doi: 10.1016/j.mce.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2012;13:239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Meaney MJ. Maturation of the adrenocortical stress response: neuroendocrine control mechanisms and the stress hyporesponsive period. Brain Res. 1986;396:64–76. doi: 10.1016/s0006-8993(86)80190-1. [DOI] [PubMed] [Google Scholar]
- Sondergaard E, Sorensen LP, Rahbek I, Gormsen LC, Christiansen JS, Nielsen S. Postprandial VLDL-triacylglycerol secretion is not suppressed in obese type 2 diabetic men. Diabetologia. 2012;55:2733–2740. doi: 10.1007/s00125-012-2624-z. [DOI] [PubMed] [Google Scholar]
- Spady DK. Hepatic clearance of plasma low density lipoproteins. Semin Liver Dis. 1992;12:373–385. doi: 10.1055/s-2008-1040407. [DOI] [PubMed] [Google Scholar]
- Stefan N, Ramsauer M, Jordan P, Nowotny B, Kantartzis K, Machann J, Hwang JH, Nowotny P, Kahl S, Harreiter J, Hornemann S, Sanyal AJ, Stewart PM, Pfeiffer AF, Kautzky-Willer A, Roden M, Haring HU, Furst-Recktenwald S. Inhibition of 11beta-HSD1 with RO5093151 for non-alcoholic fatty liver disease: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2:406–416. doi: 10.1016/S2213-8587(13)70170-0. [DOI] [PubMed] [Google Scholar]
- Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3:1. doi: 10.1186/1758-907X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275:43–61. doi: 10.1016/j.mce.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet. 2010;42:68–71. doi: 10.1038/ng.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci USA. 2004;101:15603–15608. doi: 10.1073/pnas.0407008101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Gray NE, Kuo T, Harris CA. Regulation of triglyceride metabolism by glucocorticoid receptor. Cell Biosci. 2012;2:19. doi: 10.1186/2045-3701-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GF, Ooi EM, Chan DC. Demystifying the management of hypertriglyceridaemia. Nat Rev Cardiol. 2013;10:648–661. doi: 10.1038/nrcardio.2013.140. [DOI] [PubMed] [Google Scholar]
- Williams KJ, Chen K. Recent insights into factors affecting remnant lipoprotein uptake. Curr Opin Lipidol. 2010;21:218–228. doi: 10.1097/MOL.0b013e328338cabc. [DOI] [PubMed] [Google Scholar]
- Yen FT, Roitel O, Bonnard L, Notet V, Pratte D, Stenger C, Magueur E, Bihain BE. Lipolysis stimulated lipoprotein receptor: a novel molecular link between hyperlipidemia, weight gain, and atherosclerosis in mice. J Biol Chem. 2008;283:25650–25659. doi: 10.1074/jbc.M801027200. [DOI] [PubMed] [Google Scholar]
- Yu CY, Mayba O, Lee JV, Tran J, Harris C, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS One. 2010;5:e15188. doi: 10.1371/journal.pone.0015188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Review Process File