

Summary
Our understanding of the host-pathogen relationship in tuberculosis can help guide tuberculosis (TB) drug discovery in at least two ways. First, the recognition that host immunopathology affects lesional TB drug distribution means that pharmacokinetic evaluation of drug candidates needs to move beyond measurements of drug levels in blood, whole lungs or alveolar epithelial lining fluid to include measurements in specific types of lesions. Second, by restricting the replication of M. tuberculosis (Mtb) subpopulations in latent TB infection and in active disease, the host immune response puts Mtb into a state associated with phenotypic tolerance to TB drugs selected for their activity against replicating Mtb. This has spurred a major effort to conduct high throughput screens in vitro for compounds that can kill Mtb when it is replicating slowly if at all. Each condition used in vitro to slow Mtb’s replication and thereby model the phenotypically drug-tolerant state has advantages and disadvantages. Lead candidates emerging from such in vitro studies face daunting challenges in the design of proof-of-concept studies in animal models. Moreover, some non-replicating subpopulations of Mtb fail to resume replication when plated on agar, although their viability is demonstrable by other means. There is as yet no widely replicated assay in which to screen compounds for their ability to kill this ‘viable but non-culturable’ subpopulation. Despite these hurdles, drugs that can kill slowly replicating or non-replicating Mtb may offer our best hope for treatment-shortening combination chemotherapy of TB.
Keywords: drug resistance, immunopathology, non-replication, persistence, phenotypic tolerance, viable but non-culturable
Introduction
The introduction to this volume explained why immunologists belong at the table of TB drug development, alongside microbiologists, medicinal chemists and pharmacologists. This chapter reflects how two scientists at the table view the impact of the host-pathogen interaction on early stage TB drug discovery. One of us is an immunologist struggling to learn chemistry. The other is a chemist, whose own mental immune system has developed a degree of tolerance for immunology. We bring different disciplinary backgrounds to a shared goal, to understand the fundamentals of drug resistance in TB, how the host contributes to them, and how we can turn obstacle into opportunity.
Types of drug resistance in TB
In the following discussion, resistance to a drug is understood in an operational sense as non-susceptibility of the bacterium to the drug when susceptibility was expected in the context of an infected human host. There are three major determinants of drug resistance in TB: host factors, heritable bacterial factors and non-heritable (phenotypic) bacterial factors. The first and the third types, which include prominent influences of the host immune system, typically receive the least attention, but they play an enormous role.
Host-based drug resistance
TB drugs are used because they produced favorable outcomes in most of the people who took them during experimental trials. However, once the drugs enter clinical practice, host factors can contribute to poor outcomes in a substantial proportion of those who use them. Such host factors range from the behavioral—skipping doses or prematurely terminating treatment because the drugs have already made the patient feel better or because they make the patient feel worse—to the metabolic, such as catabolizing the drugs at different rates.
Host immunopathology can confer functional drug resistance by under-dosing certain anatomic regions in people who take their drugs as prescribed and metabolize them as expected. For example, some drugs fail to accumulate in caseous lesions (1). Others may accumulate to different degrees in pneumonitic areas or in the fibrous capsules surrounding caseous lesions. The ability of drugs to access the epithelial lining fluid and the pharmacokinetics of drugs in this site may also be important if understudied factors in determining their activity (2). These localized effects, which can operate at the macroscopic or microscopic scale within an organ, have been detected through the application of liquid chromatography-mass spectrometry to extracts of dissected lesions and the application of positional mass spectrometry imaging to frozen sections (1). The resultant heterogeneity of treatment effects is consistent with but not necessarily the same as what has been revealed by computerized tomography with and without positron emission tomography in patients and non-human primates (3). These and other studies have revealed markedly different responses from one TB lesion to another in the same individual receiving TB drugs systemically (3, 4). In short, the same host can have drug-responsive and drug-non-responsive TB in the same organ at the same time, and in some cases, this may be due to the local nature of the host’s immunopathology.
Immunology has nothing to contribute to the behavioral factors that contribute to the failure of TB drugs and little to contribute to the metabolic ones. However, immunopathology is a major determinant of the intra-organ distribution and hence the efficacy of TB drugs. Reciprocally, drugs may affect immunopathology, given that release of immunostimulatory molecules from dying bacteria may activate tissue-damaging host pathways and release of diffusible antigens may limit effective mycobacterial control. This basis of functional drug resistance provides a potential target for host-directed therapies. For example, modulation of immunopathology may result from agents that affect the amount of cyclic nucleotides (5), the profile of eicosanoids produced by host phagocytes and lymphocytes (6–8), or the action of matrix metalloproteinases (9).
Drug resistance attributable to heritable changes in mycobacteria
Although Mtb does not appear to exchange mobile genetic elements and has a relatively low rate of spontaneous genetic drift, the numbers of bacilli that can accumulate in a host with TB can include individual cells with spontaneous mutations at any locus that does not impose a major fitness cost. Each of the frontline TB drugs now in use is likely to be ineffective against at least one bacillus in a population of 108, whereas it is estimated that a tuberculous lung may contain 1013 Mtb. The mechanisms by which mutations confer resistance include diminished drug uptake or increased efflux, increased drug catabolism, decreased prodrug conversion to an active form, alteration or over-expression of a critical target, or expression of a compensatory pathway (10).
Heritable drug resistance in Mtb is not a problem to which immunology can offer much assistance, except in the sense that host-directed therapy is unlikely to fail on this basis and thereby offers a way around it. Drugs that act on the host rather than on the pathogen cannot encounter resistance based on the pathogen’s failure to accumulate the compound, activate it, over-express its target, and so on. Nonetheless, it remains possible that host-directed therapy could select for a pathogen in which mutation has either relieved dependence on the host pathway that the intervention inhibits or decreased susceptibility to a host pathway that the intervention augments.
Heritable drug resistance has an Achilles’ heel. It is highly unlikely that mutations conferring resistance to more than one drug will arise spontaneously in a single bacillus, unless one mutation upregulates efflux pump(s) that affect more than one drug. Combination chemotherapy with agents directed at different targets and subject to different efflux mechanisms should eradicate all the Mtb in given host, provided that host-based mechanisms of resistance do not stand in the way. Combination chemotherapy was introduced to the practice of medicine in the setting of TB treatment (11), from which it spread to the chemotherapy of cancer and HIV/AIDS. Without combination chemotherapy, the cure rate of TB would be near zero. With it, cure rates of 95% can be obtained.
However, combination chemotherapy has an Achilles’ heel as well. If a person is infected with a strain of Mtb that is already heritably resistant to all but one of the drugs in the combination and this is not appreciated when treatment begins, the resultant disease may be effectively subjected to monotherapy, encouraging the propagation of a strain that is heritably resistant to the last agent as well. Given that treatment is often imperfectly executed in resource-poor settings, it is hardly surprising that strains are now widespread that are heritably resistant to some, most or all of the existing drugs. Resistant isolates were already detected during a pre-approval clinical trial of an agent that was then approved by the FDA on the last day of 2012 (12). Without fundamental changes to treatment regimes as they are actually executed in the field, it is predictable that heritable resistance will limit the effectiveness of any new combination of TB drugs within a few years of its introduction.
What needs to change? To answer this, we need to consider two phenomena collectively called phenotypic tolerance.
Non-heritable drug resistance (phenotypic tolerance)
What needs to change is the duration of treatment. To cure 95% of people with drug-sensitive TB takes 6–8 months of directly observed therapy with multiple drugs that have significant side effects. Economics and human nature conspire to interrupt treatment, which allows spread of strains with heritable resistance. Shortening the duration of treatment should delay the spread of heritable resistance.
Why does treatment take so long? The same drug combinations that take 6 months to cure TB in people can eradicate Mtb in vitro in a few days. What happens in a host to allow Mtb to persist in the face of what should be cidal levels of anti-infectives? As already discussed, host-based factors like drug distribution may play a role. However, another hypothesis is that we are not using the right combination of drugs, based on the biology of the disease. At the root of this concern is the observation that culture conditions designed to mimic the host environment impose phenotypic tolerance in vitro to the present arsenal of TB drugs. Such conditions generate a state of phenotypic tolerance that was called ‘class 2’, because another form of non-heritable drug resistance had been described earlier (13). To set the stage for a detailed discussion of class 2 phenotypic tolerance, we need to review the origins of the concept of phenotypic tolerance in experiments that can, in retrospect, be understood to have identified phenotypic tolerance of class 1.
Class I phenotypic tolerance
Phenotypic tolerance to antibiotics, also called bacterial persistence, was first articulated by Bigger (14) when he exposed penicillin-sensitive Staphyloccus to penicillin and observed that a small proportion of the inoculum, about 1 in 106, survived. When the survivors were rescued by addition of penicillinase, expanded without penicillin, and then exposed to penicillin again, they succumbed in the same proportion as before (14). He concluded that survival of the few during the first exposure to penicillin experiment had not been due to their acquisition of heritable resistance.
Various species of bacteria have shown this behavior in response to different antibiotics, and diverse mechanisms appear to account for it. We now know that the size of the tolerant population can be genetically controlled, for example by toxin-antitoxin pairs (15), even though the basis of resistance to a given anti-infective is not based on mutation.
An influential study of bacterial persistence introduced the use of time-lapse photomicroscopy of bacteria in a microfluidic device, which allowed the investigators to follow the fate of individual bacteria. The few cells in a population of replicating Escherichia coli that survived perfusion with ampicillin were those that had been non-replicating before the ampicillin was applied (16). For some years it was assumed that non-replication accounted for all instances of the phenotypic tolerance displayed by a tiny subpopulation of bacteria in a largely replicating population. However, another time-lapse photomicroscopy study of bacteria in a microfluidic chamber demonstrated that phenotypic tolerance to the prodrug isoniazid on the part of a few cells in a largely replicating population of Mtb was displayed by those cells in which expression of the activating enzyme catalase was stochastically extinguished before the isoniazid was applied (17). There was no difference in the replication rates of the drug-tolerant subpopulation and the drug-susceptible majority (17).
Subsequent studies have demonstrated that the association is variable between phenotypic tolerance of a subpopulation of bacteria in a largely replicating population, and the replication rate of the tolerant subpopulation (18, 19). There may be as many mechanisms for class I phenotypic tolerance as there are epigenetic, transcriptional, post-transcriptional, translational, and post-translational ways to mimic the changes afforded by heritable mutations (13).
The obvious therapeutic response to class I phenotypic tolerance is the same as the therapeutic response to heritable drug resistance: combination chemotherapy with drugs each of which can kill most of the population. To the degree that different subpopulations of bacteria manifest class I phenotypic tolerance to different drugs by different mechanisms, combinations of those drugs should kill all subpopulations.
If combination chemotherapy with currently approved drugs killed all Mtb subpopulations in patients as fast as seen in vitro with the drug concentrations achieved in vivo, then cure should take a few days, not half a year. By this reasoning, class I phenotypic tolerance to drugs active against replicating Mtb does not appear to explain completely the need for prolonged treatment of TB with combination chemotherapy using the drugs that were approved on the basis of their efficacy against replicating Mtb. Class II phenotypic tolerance, discussed next, may offer a better explanation.
Class II phenotypic tolerance of ‘culturable’ and ‘viable but non-culturable’ (VBNC) Mtb While class I phenotypic tolerance is an intrinsic property manifest by a small proportion of bacteria in a population that is largely replicating under standard conditions, class II phenotypic tolerance affects almost all the bacteria in a population and is extrinsic, in that it arises under externally imposed conditions that limit replication (13). The first scientist to demonstrate this was none other than Bigger (14). By lowering the temperature, incubating the bacteria in distilled water, or treating them with boric acid, three interventions that imposed bacteriostasis, Bigger markedly increased the proportion of persisters among staphylococci exposed to penicillin. He correlated this with the cessation of replication and attributed it to an insensitivity of non-replicating organisms to a drug that is active against replicating organisms (14).
Because almost all the Mtb in a non-replicating population display phenotypic tolerance to multiple antibiotics, the individual bacteria in the population must each resist multiple drugs. Thus, resistance of this type cannot be overcome simply by combining those drugs. Instead, new kinds of drugs are needed that can kill non-replicating Mtb (13).
Little is known about the mechanism of class II phenotypic tolerance. One hypothesis is that non-replicating bacteria may be far less reliant than replicating bacteria on the synthesis of cell walls, protein, RNA and DNA, the processes inhibited by most antibiotics. However, Mtb adapt to the stresses that impose non-replication with a robust but distinct transcriptional response (20) and perhaps with cell wall remodeling (21, 22). Thus, non-reliance on macromolecular synthesis is an unsatisfactory explanation for class II phenotypic tolerance. The stresses that impose non-replication may also lead to diminished intrabacterial accumulation of certain drugs, but it is unclear to what extent this contributes to phenotypic tolerance. Table 1 contrasts class I and class II phenotypic tolerance and introduces what may be an important subpopulation of bacteria that display class II phenotypic tolerance: so-called ‘viable but non-culturable’ (VBNC) Mtb.
Table 1.
Classes of phenotypic tolerance
| Class | Population growth state | Persistence phenotype | VBNC Mtb | Inducers of persistence | Speculative mechanisms | Approach to treatment |
|---|---|---|---|---|---|---|
| I | most cells replicating | small minority; different cells tolerate different abx | none | unknown; stochastic | epigenetic, transcriptional, translational or post-translational expression or suppression of any process for which a genetic change can give heritable resistance | combine different drugs |
| II | most cells not replicating | large majority; same cells tolerate many abx | subset of class II persisters |
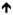 H+, RNI, ROI; H+, RNI, ROI;
 ;O2, C, N, P, Fe ;O2, C, N, P, Fe |
metabolic stress promotes oxidative stress, leading to adaptation | include new kinds of drugs active on non-replicating cells |
An important goal for TB drug discovery is to identify, characterize, and prioritize compounds that can eradicate Mtb populations that contribute to relapse in human disease, so that inclusion of such drugs in a therapy regimen will allow earlier discontinuation of the treatment of TB than is now recommended. The bacteria that contribute to relapse when treatment with current agents is shortened are probably not the organisms sampled in sputum by conventional techniques, since achieving sputum culture-negative status is not well correlated with relapse-free cure (23).
The conventional techniques for monitoring mycobacterial load in TB patients by analysis of their sputum are enumeration of colony-forming units (CFUs) on agar or detection of an increase in biomass proportional to their initial number when Mtb grown from the sputum in liquid culture deplete the oxygen in mycobacterial growth indicator tubes (MGIT). Some investigators contend that persistent, apparently drug-tolerant populations of Mtb may not be those that are detected by the CFU or MGIT techniques. Instead, they may be the VBNC Mtb that have been enumerated in limiting dilution assays as the majority of Mtb in sputum from a substantial subset of patients (24).
VBNC populations have been reported in over 50 species of bacteria (25) by the operational definition that they do not form CFU at the time of study, yet can be detected later as CFU or at the time of study by other means, such as inoculation into a host, where they produce disease. The term is paradoxical in that the alternate means of detection can include forms of culture other than on agar, such as limiting dilution in liquid medium supplemented with medium conditioned by Mtb (24).
Mtb that ceases to replicate can enter states that differ in the requirements for returning to replication and causing disease. In some stressed states, the ability to form CFU is lost unless Mtb first undergoes at least the first two of the following three treatments: (i) prolonged incubation in liquid (ii) after limiting dilution (iii) in the presence of labile factors from mid-log or early stationary phase culture. Other approaches to detecting VBNC Mtb include the demonstration that some organ homogenates from Mtb-infected hosts produce no CFU but cause TB after transfer to another host, and the demonstration that immunosuppression can cause appearance of CFU in Mtb-infected mice that had been drug-treated and apparently cured, because organ homogenates from other members of the same cohort were cultured before immunosuppression and produced no CFU (26).
Conventional in vitro assays for mycobactericidal activity do not take the possibility of VBNC Mtb into account. Unpublished evidence suggests that some compounds with mycobactericidal activity are inactive in vitro against VBNC Mtb, while others can kill VBNC Mtb. The characterization of TB drug candidates for their activity against VBNC is a major area for further study.
Implications for drug discovery
A consideration of the host-pathogen relationships invites three hypotheses. (i) Host conditions in most infected people impose enough stress on Mtb to kill some but not all of the Mtb and to slow or halt the replication of most of the rest for prolonged periods. (ii) In people who go on to develop clinically active TB, host conditions continue to generate some slowly replicating or non-replicating subpopulations of Mtb, and these display class II phenotypic tolerance to drugs that were selected primarily for their ability to kill replicating Mtb. (iii) Combination chemotherapy of TB could produce faster cures than at present, if some of the agents were active against replicating Mtb and some of them were active against non-replicating Mtb, including VBNC Mtb. This last hypothesis was already supported by the treatment time reduction afforded by inclusion of pyrazinamide (discussed below) in combination chemotherapy, but the degree of treatment shortening afforded by pyrazinamide is insufficient and resistance to it is spreading.
The fact that slowly replicating or non-replicating subpopulations in vitro display substantial resistance to most TB drugs selected primarily for their ability to kill replicating Mtb argues that new drugs are needed that are selected on the basis of their ability to kill slowly replicating or non-replicating Mtb—drugs that can not only replace pyrazinamide, but out-do it.
The problem then becomes how to model relevant host conditions in vitro in a form compatible with high throughput screening. Additionally or alternatively, the problem becomes to identify pathways on which Mtb depends for its survival in the metabolic states that confer class II phenotypic tolerance to existing drugs and to find inhibitors of those pathways that can kill intact Mtb while it is replicating slowly or not at all.
Modeling host conditions that induce class II phenotypic tolerance in Mtb
Until 2008, there were, to our knowledge, no drugs or compounds reported that selectively killed non-replicating bacteria in vitro without also killing replicating bacteria of the same species. Very few of the drugs that could kill replicating bacteria could kill the same bacteria when they were non-replicating, and those that did, did so to a much lesser extent. In 2008, thioxothiazolidines were discovered that inhibited dihydrolipoamide acyltransferase, an enzyme involved in Mtb’s intermediary metabolism and antioxidant defense (27). These compounds were markedly and selectively bactericidal to Mtb when its replication was halted by hypoxia, acidification, starvation, exposure to nitrosative stress, or incubation in tissue culture medium designed to mimic human extracellular fluid. The same compounds had no impact on the viability of replicating Mtb (27). This finding encouraged us to conduct high-throughput screens to find more such compounds.
It is unknown if a slow replication rate is a causal factor or only a biomarker for class II phenotypic tolerance. Be that as it may, the putatively physiologic conditions found so far to make an Mtb population relatively resistant to most current TB drugs in vitro are associated with a marked reduction in Mtb’s replication rate. Thus, the authors’ laboratories have designed high throughput screens under conditions resulting in slow replication or non-replication (28), recognizing that in the spectrum of lesion types found in humans with TB, we can only speculate about replication rates.
We operationally define non-replicating conditions as conditions where the CFU count does not significantly change over the period of observation. This definition does not exclude some degree of balanced replication and death. We operationally define conditions for slow replication as those in which the CFU count increases by about one-fifth of that which would occur during exponential growth in standard medium over the same period. This target rate of replication was chosen on the basis of observations in mice that have contained Mtb infection through immune activation so that CFU are stable for a prolonged period. There, evidence suggested that Mtb undergoes balanced death and growth, with the generation time of the growing organisms being about fivefold longer than that of Mtb undergoing exponential growth in standard medium in vitro (29).
When conditions that impose non-replication are rendered more stringent or extended in time, they can lead to loss of CFU. As noted above, loss of CFU is not always accompanied by loss of viability as documented by other means. As yet there is no high throughput assay to screen for compounds that kill VBNC Mtb, that is, Mtb that do not form CFU at the time of study but retain replicative potential. Work to develop such an assay is underway.
In the ensuing years, many experimental compounds have been identified that can kill non-replicating Mtb in vitro extensively enough to reduce CFU by 5–7 log10 over 3–7 days, in some cases to below the limit of detection. Less commonly, compounds have been identified that kill both non-replicating Mtb and replicating Mtb, without toxicity to host cells in vitro, such as nitazoxanide (30) and certain nitrofuranylcalanolides (31). To our knowledge, there has been only one compound whose activity in vitro is selective for non-replicating Mtb that has been shown to be clinically active against TB and to allow a shorter course of curative treatment. That compound, pyrazinamide, was identified over 60 years ago by its activity in mice. Pyrazinamide’s mycobactericidal activity in vitro is so weak that it would probably not have been detected in vitro under typical screening conditions. Of the compounds with extensive, bactericidal activity in vitro that are selectively active against non-replicating Mtb, none, to our knowledge, has yet been tested in clinical trials. Thus, uncertainty remains about how best to identify such compounds and how to test them in preclinical animal models. This mirrors our uncertainty about conditions in the human host-pathogen relationship and our ability to recapitulate those conditions in vitro and in animals.
Complicating matters, slowly replicating or non-replicating states are heterogeneous both as a result of the conditions that impose them and the duration of exposure to those conditions. There are probably overlapping sets of genes on which Mtb is conditionally dependent for survival in different slowly replicating or non-replicating states. Some genes will be essential in common, for example, to maintain energy, reducing power, membrane potential, proton motive force, cell wall integrity, and DNA repair. Others will be essential to survive particular stresses.
These stresses can be exerted in any combination from a list that includes oxidative, nitrosative, acidic, nutritional, micronutritional (such as iron deficiency), hypoxic, and membrane-perturbing (such as by antimicrobial peptides). Various oxidative and nitrosative stresses are chemically and functionally distinct. Exposure of bacteria to one antibiotic at a sublethal concentration can slow their replication and induce class II phenotypic tolerance to the same and other antibiotics (32–34). For example, exposure of Mtb to sublethal concentrations of isoniazid, rifampin, or streptomycin produces oxidative stress and creates dependency on the glyoxylate shunt as a previously unappreciated form of anti-oxidant defense (35).
There is no generally accepted model for non-replicating bacteria. Each condition that can generate a slowly replicating or non-replicating state associated with class II persistence in vitro has a different degree of evidence for its physiologic relevance, and each may create its own set of experimental complications. In humans with TB, it is likely that a combination of the conditions described below occurs in specific lesion types (36). Below are some examples of such conditions.
Starvation
The Loebel model (starvation in phosphate-buffered saline) is unlikely to directly mimic the in vivo environment, because Mtb will probably never completely lack a carbon or nitrogen source in the human host. Nonetheless, starvation may provide a useful model of complex nutritional deficiency that may have relevance to bacilli in vivo (37). Experimentally, the duration of starvation is likely to have a profound effect on the outcome, as starved Mtb draws down its metabolic stores, such as trehalose, glycogen, fatty acids (such as from triacylglycerols) and glutamate. Adaptation to starvation requires the stringent response and the hyper-phosphorylated guanosine transcriptional signaling molecules ppGpp and pppGpp, which are important for TB survival in mice (38), and which have been implicated in clinical isolates by mutations that compensate for rifampicin-resistant alterations in RNA polymerase (39).
One of the early impacts of starvation is reduction of protein synthetic capacity. Protein synthesis can be switched off artificially by withdrawing streptomycin from an unusual mutant of Mtb that has a ribosomal mutation making Mtb dependent on streptomycin to grow (40). An advantage of the method is that it can be applied both in vitro and in mice. A disadvantage is that isolated interference with protein synthesis may not closely model natural states that impose non-replication by impacting multiple pathways.
Alterations in metabolism due to use of a specific carbon source
Based on two key observations, it has long been thought that Mtb in mice is restricted to fatty acids as a carbon source. In 1956, Bloch and Segal observed that Mtb taken directly from mouse lungs respired more oxygen if provided with fatty acids than if provided with carbohydrates (41). This suggested that Mtb had been metabolizing fatty acids in vivo. However, other lipids, such as cholesterol, were not tested, and it was not considered that Mtb in vivo need not necessarily be metabolizing at a maximal rate. That is, the substrate Mtb can metabolize fastest is not necessarily the substrate to which it has access in vivo or the substrate on which it depends in vivo.
The second observation supporting the importance of fatty acids as a carbon source in vivo was the severe attenuation of isocitrate lyase (ICL)-deficient Mtb (42). At the time, what was known about ICL was that it sustained the glyoxylate shunt for anaplerotic incorporation of carbon from fatty acids. The phenotype of ICL deficiency therefore implied that Mtb relies on fatty acids to survive in mice and that providing a fatty acid as sole carbon source, at a level inadequate to support rapid replication, would mimic a physiologic slowly replicating or non-replicating state. However, it later became clear that the icl-null phenotype involves the methylcitrate cycle, the secretion of succinate and the antioxidant role of the glyoxylate shunt in addition to or instead of reliance on fatty acids (35, 43, 44).
The attenuated phenotype of strains of Mtb deficient in glucose phosphorylation (45) and cholesterol metabolism (46) in the same mouse model suggested that Mtb accesses and depends on carbon sources other than fatty acids. Virulence of Mtb in the mouse also requires triosephosphate isomerase (47) and fructose-1,6-bisphosphate aldoase (48), implicating glycolysis and gluconeogenesis in pathogenesis. While lipids could serve as carbon donors for gluconeogenesis, so might amino acids and CO2 (49). Thus, imposition of a non-replicating state solely by restriction of carbon source to a fatty acid may not model a physiologic, host-imposed stress. The evidence is stronger that slow growth of Mtb on cholesterol may do so.
Prolonged stationary phase
It is unknown to what degree prolonged stationary phase in vitro directly mimics an in vivo environment. Attaining stationary phase in different ways and remaining in it for different lengths of time may lead to varying combinations of hypoxia, nutritional deficiency and acidification. In late stationary phase, a large proportion of the Mtb are dead and clumping is a serious issue, making this an impractical model for high throughput screening.
Prior drug treatment
Sublethal treatment with an antimicrobial agent can impair replication and render bacteria phenotypically tolerant to other drugs besides the one applied (34). Bigger (14) demonstrated this when his sublethal treatment of staphylococci with boric acid induced class II phenotypic tolerance to penicillin. Treating a replicating population with one drug and then testing other compounds for their ability to kill the survivors may have the disadvantage of testing populations with an undefined proportion of class I and class II persisters.
Mild acidification
Mild acidification is thought to characterize Mtb-infected sites in part because pyrazinamide is effective in vivo and it is mostly effective in vitro at low pH. However, hypoxia also gives pyrazinamide some efficacy in vitro.
Many Mtb reside in macrophages in vivo. Macrophage phagosomes containing Mtb are mildly acidic (pH <6.2). When macrophages are immunologically activated, their Mtb-containing phagosomes become more acidic (pH 4.5) (50). Moreover, the extracellular fluid at inflammatory sites is likely to become acidic as macrophages, monocytes, dendritic cells, lymphocytes, and neutrophils invade the site and crowd the stromal and parenchymal cells, overburdening the local vascular supply and enforcing increased reliance on glycolysis, with accompanying lactate secretion and diminished lactate removal. TB itself secretes succinic acid to maintain an energized membrane under hypoxic conditions and may contribute to a local acidification of its microenvironment, whether intracellular or extracellular (43, 51).
An experimental complication of using mild acid to impose non-replication is that some compounds that inhibit Mtb in vitro can undergo structural modifications in mild acid. This is sometimes accelerated or capacitated by the presence of nitrite, as discussed below (28). If this is a risk that compounds face in vivo, it is important to set up a screen or counter-screen in which such compounds can be identified and their propensity for such transformation can be considered along with their other pharmacologic properties.
Hypoxia
Some lesional environments are hypoxic, as shown by direct measurement (52). When Mtb is hypoxic, its major alternative electron acceptor in vivo is likely to be nitrate. Mtb readily respires nitrate, and nitrate is a physiologic constituent of human body fluids whose amount is likely to increase in inflammatory sites, for reasons discussed below. Unfortunately, standard Mtb growth media omit nitrate. This omission is a mistake arising from inadequate knowledge of mammalian physiology at the time that standard bacteriologic media were formulated. Thus, much of what we think we know about Mtb’s physiology in hypoxia is based on experiments conducted in non-physiologic culture media (53). For example, the Wayne model of hypoxia has emerged as an in vitro gold standard to mimic the host environment. However, most experiments using the Wayne model omit nitrate and thereby impose a greater degree of respiratory compromise on Mtb than is likely to be physiologic. Nonetheless, the tendency of this model to give dramatically different results for drugs than under aerobic conditions has made the model a mainstay for evaluating new compounds for their activity against non-replicating Mtb.
Nitrite and other reactive nitrogen intermediates (RNIs)
Nitrite, like nitrate, is a physiologic component of human blood and interstitial fluid. Nitrite arises partly from dietary nitrate. Ingested nitrate is absorbed from the gastrointestinal tract, circulates in the blood, and is taken up and concentrated in the salivary glands. It is then secreted into the oral cavity, where it is reduced to nitrite by the anaerobic microbiota in the crevices on the back of the tongue (54). Nitrite also arises from auto-oxidation of nitric oxide produced by nitric oxide synthases, both constitutive and inducible, including the constitutive isoform in vascular smooth muscle and the inducible isoform in bronchial epithelium and activated macrophages (55).
Human macrophages in inflammatory sites, including those in lungs of people with TB, express the inducible isoform of nitric oxide synthase (iNOS or NOS2) (56–62). In contrast, blood monocytes collected from healthy donors rarely express iNOS when differentiated into macrophages in vitro. This is an artifact associated with current methods of using normal blood monocytes in vitro to model inflammatory tissue macrophages (63). In fact, current evidence suggests that monocytes circulating in healthy people are not precursors of the macrophages in many tissues (64).
Mtb itself produces nitrite when it respires nitrate. The degree of hypoxia necessary to ramp up Mtb’s nitrate respiration (by upregulating its nitrate transporter) can be achieved in vitro when Mtb resides within human macrophages, even in cultures carried out in room air (53).
Nitrite affects Mtb in some ways that are distinct from the impact of nitric oxide and in some ways that are the same (53). Moreover, at moderately low pH, nitrite is partially protonated. The nitrous acid so formed can dismutate, generating NO, as well as the stronger nitrosating agents NO2 (the radical, not the anion), N2O3 and N2O5. The NO autoxidizes, generating nitrite again and also nitrate. As noted above, nitrate serves as an alternate electron acceptor of physiologic relevance in hypoxia.
Thus, the physiological relevance of in vitro models for Mtb is improved by including nitrate and nitrite, or alternatively, including nitrite under conditions (mild acidity) that allow for generation of nitrate. Underscoring this, the transcriptional response of Mtb to most of the stresses discussed in this chapter is encompassed in its transcriptional response to RNI arising from NO produced by iNOS (20).
An experimental complication of using mildly acidified nitrite as a physiologic stress to induce a non-replicating state is that some chemical compounds with anti-mycobacterial activity are unstable under these conditions (28). While this is inconvenient, it is likely to reflect events that can occur in the body. Improved pharmacotherapy of TB may require identifying and avoiding these transformations.
Summary: an immunologically inspired approach to TB drug discovery
We do not know enough about Mtb’s biology in the human host to specify an optimal way to screen in vitro for compounds that can kill Mtb whose resistance to standard drugs is largely attributable to class II phenotypic tolerance. However, with the foregoing considerations in mind, we recommend the following approaches:
Screen compounds in a setting that combines multiple stresses that can induce a non-replicating state and for which evidence of physiologic relevance is strong, such as hypoxia, mild acidity, and the presence of nitrite. As noted above, nitrite at acidic pH is an indirect source of nitrate, and nitrate becomes important when modeling in vivo conditions that include hypoxia. Alternatively, screen compounds in several settings that include individual stresses plausibly related to phenotypic tolerance, such as slow growth on cholesterol.
Proceed through the usual subsequent steps, which are not detailed here, such as to inspect for structural alerts, confirm with quality-controlled, resupplied powders, determine the minimum bactericidal concentration, cytotoxicity, and microbicidal spectrum, evaluate the extent and speed of killing, and so on.
Test compounds emerging from the foregoing studies for stability in acidified nitrite, whether or not they were identified in a screen that included acid or nitrite. Consider further those compounds that are stable. Consider also those that are quantitatively converted to a defined product with the desirable properties that had been attributed to the original molecule and that have no major new liabilities. Discard those that are degraded to diverse, undefined, ineffective, or toxic products.
Cross-test compounds emerging from the above studies in replicating models and other non-replicating or slowly replicating models.
For compounds passing the previous filters, test their ability to kill VBNC Mtb in vitro.
Test leading compounds in animal models. There is no consensus on how best to do this, given that this begs the general question whether any animal models adequately predict efficacy of TB drugs in humans, and superimposes on it the specific question of how to use experimental animals to model non-replicating conditions pertinent to human TB.
Proceeding on the premise that animal models, despite their limitations, are indispensable, we suggest that the least costly approaches of highest probable relevance are to treat mice during the chronic phase of infection in the Grosset and/or Kramnik models, that is, beginning at least 4 weeks following low-dose aerosol infection of C57BL/6 or C3HFeB/HeJ mice (65, 66), and to do so with the compound alone and in combination with an agent that kills replicating Mtb, such as isoniazid. Tests in mice would be considered positive if the test agent produces a statistically significant reduction in CFU over the vehicle control when tested alone, or over isoniazid when tested in combination with isoniazid, and does so in independent experiments, without causing toxicity that requires termination of treatment. Positive results would normally lead to tests in combination with other agents.
If CFU assays indicate apparent sterilization, additional experiments should be done in which the apparently sterilizing treatment is followed by administration of glucocorticoids or iNOS inhibitors (67, 68) as immunosuppressive agents, so as to distinguish true sterilization from imposition of latency. Such experiments should motivate additional work in non-human primates to achieve full pre-clinical validation. Models in marmosets and macaques have greater relevance to human TB than mouse models, but are very costly. Moreover, the large volume of organs in non-human primates makes it very difficult to follow total organ CFU as a read-out. However, animal-sparing radiologic read-outs of disease burden are being applied in such models and may ultimately prove sufficiently robust to allow for meaningful assessment of sterilizing activity (3). As in mice, the existence of substantial populations of replicating Mtb along with some non-replicating Mtb complicates using non-human primates for proof-of-concept studies for compounds that are selectively active in vitro against non-replicating Mtb. That complication might be solved by conducting studies in non-human primates in combination with drugs that are selectively active against replicating Mtb, as noted above for mice.
Ultimately, however, we must accept that TB is a human disease, and we can only acquire definitive understanding about it by studying people. We need to conduct larger numbers of more informative clinical experiments, whether they are observational or hypothesis-driven and whether they analyze the impact of genetics, environment, iatrogenic interventions, or the interactions of all three.
Acknowledgments
Preparation of this review was supported by grants from the Bill and Melinda Gates Foundation and NIH (RO1 AI64768, RO1 AI105807 and U19 AI111143) and by the Milstein Program in Translational Medicine. The Department of Microbiology and Immunology at Weil Cornell Medical College is supported by the William Randolph Hearst Foundation. We thank K. Burns-Huang for review of the manuscript.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Dartois V. The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nature reviews Microbiology. 2014;12(3):159–167. doi: 10.1038/nrmicro3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodvold KA, Yoo L, George JM. Penetration of anti-infective agents into pulmonary epithelial lining fluid: focus on antifungal, antitubercular and miscellaneous anti-infective agents. Clinical pharmacokinetics. 2011;50(11):689–704. doi: 10.2165/11592900-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Via LE, et al. Differential virulence and disease progression following Mycobacterium tuberculosis complex infection of the common marmoset (Callithrix jacchus) Infection and immunity. 2013;81(8):2909–2919. doi: 10.1128/IAI.00632-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin PL, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nature medicine. 2014;20(1):75–79. doi: 10.1038/nm.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subbian S, et al. Phosphodiesterase-4 inhibition alters gene expression and improves isoniazid-mediated clearance of Mycobacterium tuberculosis in rabbit lungs. PLoS pathogens. 2011;7(9):e1002262. doi: 10.1371/journal.ppat.1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayer-Barber KD, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511(7507):99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tobin DM, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148(3):434–446. doi: 10.1016/j.cell.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tobin DM, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 2010;140(5):717–730. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elkington PT, D’Armiento JM, Friedland JS. Tuberculosis immunopathology: the neglected role of extracellular matrix destruction. Science translational medicine. 2011;3(71):71ps76. doi: 10.1126/scitranslmed.3001847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsh C. Where will new antibiotics come from? Nature reviews Microbiology. 2003;1(1):65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 11.Devadatta S, et al. Response of patients infected with isoniazid-resistant tubercle bacilli to treatment with isoniazid plus PAS or isoniazid alone. Bulletin of the World Health Organization. 1961;25:807–829. [PMC free article] [PubMed] [Google Scholar]
- 12.Andries K, et al. Acquired Resistance of Mycobacterium tuberculosis to Bedaquiline. PloS one. 2014;9(7):e102135. doi: 10.1371/journal.pone.0102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nathan C. Fresh approaches to anti-infective therapies. Science translational medicine. 2012;4(140):140sr142. doi: 10.1126/scitranslmed.3003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bigger JW. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet. 1944:497–500. [Google Scholar]
- 15.Maisonneuve E, Gerdes K. Molecular mechanisms underlying bacterial persisters. Cell. 2014;157(3):539–548. doi: 10.1016/j.cell.2014.02.050. [DOI] [PubMed] [Google Scholar]
- 16.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305(5690):1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 17.Wakamoto Y, et al. Dynamic persistence of antibiotic-stressed mycobacteria. Science. 2013;339(6115):91–95. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- 18.Amato SM, Orman MA, Brynildsen MP. Metabolic control of persister formation in Escherichia coli. Molecular cell. 2013;50(4):475–487. doi: 10.1016/j.molcel.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Orman MA, Brynildsen MP. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrobial agents and chemotherapy. 2013;57(7):3230–3239. doi: 10.1128/AAC.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schnappinger D, et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. The Journal of experimental medicine. 2003;198(5):693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, 3rd, Blanchard JS. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science. 2009;323(5918):1215–1218. doi: 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vilcheze C, et al. Novel inhibitors of InhA efficiently kill Mycobacterium tuberculosis under aerobic and anaerobic conditions. Antimicrobial agents and chemotherapy. 2011;55(8):3889–3898. doi: 10.1128/AAC.00266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips PP, Nunn AJ, Paton NI. Is a 4-month regimen adequate to cure patients with non-cavitary tuberculosis and negative cultures at 2 months? The international journal of tuberculosis and lung disease: the official journal of the International Union against Tuberculosis and Lung Disease. 2013;17(6):807–809. doi: 10.5588/ijtld.12.0725. [DOI] [PubMed] [Google Scholar]
- 24.Mukamolova GV, Turapov O, Malkin J, Woltmann G, Barer MR. Resuscitation-promoting factors reveal an occult population of tubercle Bacilli in Sputum. American journal of respiratory and critical care medicine. 2010;181(2):174–180. doi: 10.1164/rccm.200905-0661OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oliver JD. Recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS microbiology reviews. 2010;34(4):415–425. doi: 10.1111/j.1574-6976.2009.00200.x. [DOI] [PubMed] [Google Scholar]
- 26.Scanga CA, et al. Reactivation of latent tuberculosis: variations on the Cornell murine model. Infection and immunity. 1999;67(9):4531–4538. doi: 10.1128/iai.67.9.4531-4538.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bryk R, et al. Selective killing of nonreplicating mycobacteria. Cell host & microbe. 2008;3(3):137–145. doi: 10.1016/j.chom.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gold B, et al. Nonsteroidal anti-inflammatory drug sensitizes Mycobacterium tuberculosis to endogenous and exogenous antimicrobials. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(40):16004–16011. doi: 10.1073/pnas.1214188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gill WP, et al. A replication clock for Mycobacterium tuberculosis. Nature medicine. 2009;15(2):211–214. doi: 10.1038/nm.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Carvalho LP, Lin G, Jiang X, Nathan C. Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. Journal of medicinal chemistry. 2009;52(19):5789–5792. doi: 10.1021/jm9010719. [DOI] [PubMed] [Google Scholar]
- 31.Zheng P, et al. Synthetic calanolides with bactericidal activity against replicating and nonreplicating Mycobacterium tuberculosis. Journal of medicinal chemistry. 2014;57(9):3755–3772. doi: 10.1021/jm4019228. [DOI] [PubMed] [Google Scholar]
- 32.Johnson PJ, Levin BR. Pharmacodynamics, population dynamics, and the evolution of persistence in Staphylococcus aureus. PLoS genetics. 2013;9(1):e1003123. doi: 10.1371/journal.pgen.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorr T, Lewis K, Vulic M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS genetics. 2009;5(12):e1000760. doi: 10.1371/journal.pgen.1000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deris JB, et al. The innate growth bistability and fitness landscapes of antibiotic-resistant bacteria. Science. 2013;342(6162):1237435. doi: 10.1126/science.1237435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nandakumar M, Nathan C, Rhee KY. Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nature communications. 2014;5:4306. doi: 10.1038/ncomms5306. [DOI] [PubMed] [Google Scholar]
- 36.Dartois V, Barry CE., 3rd A medicinal chemists’ guide to the unique difficulties of lead optimization for tuberculosis. Bioorganic & medicinal chemistry letters. 2013;23(17):4741–4750. doi: 10.1016/j.bmcl.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Molecular microbiology. 2002;43(3):717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 38.Dahl JL, et al. The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(17):10026–10031. doi: 10.1073/pnas.1631248100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song T, et al. Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the beta’ subunit of RNA polymerase. Molecular microbiology. 2014;91(6):1106–1119. doi: 10.1111/mmi.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sala C, et al. Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrobial agents and chemotherapy. 2010;54(10):4150–4158. doi: 10.1128/AAC.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bloch H, Segal W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. Journal of bacteriology. 1956;72(2):132–141. doi: 10.1128/jb.72.2.132-141.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKinney JD, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406(6797):735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 43.Eoh H, Rhee KY. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(16):6554–6559. doi: 10.1073/pnas.1219375110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eoh H, Rhee KY. Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of Mycobacterium tuberculosis on fatty acids. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(13):4976–4981. doi: 10.1073/pnas.1400390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marrero J, Trujillo C, Rhee KY, Ehrt S. Glucose phosphorylation is required for Mycobacterium tuberculosis persistence in mice. PLoS pathogens. 2013;9(1):e1003116. doi: 10.1371/journal.ppat.1003116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(11):4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trujillo C, et al. Triosephosphate isomerase is dispensable in vitro yet essential for Mycobacterium tuberculosis to establish infection. mBio. 2014;5(2):e00085. doi: 10.1128/mBio.00085-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puckett S, et al. Inactivation of fructose-1,6-bisphosphate aldolase prevents optimal co-catabolism of glycolytic and gluconeogenic carbon substrates in Mycobacterium tuberculosis. PLoS pathogens. 2014;10(5):e1004144. doi: 10.1371/journal.ppat.1004144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beste DJ, et al. (1)(3)C metabolic flux analysis identifies an unusual route for pyruvate dissimilation in mycobacteria which requires isocitrate lyase and carbon dioxide fixation. PLoS pathogens. 2011;7(7):e1002091. doi: 10.1371/journal.ppat.1002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science. 2003;302(5645):654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 51.Watanabe S, et al. Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS pathogens. 2011;7(10):e1002287. doi: 10.1371/journal.ppat.1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Via LE, et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infection and immunity. 2008;76(6):2333–2340. doi: 10.1128/IAI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cunningham-Bussel A, Zhang T, Nathan CF. Nitrite produced by Mycobacterium tuberculosis in human macrophages in physiologic oxygen impacts bacterial ATP consumption and gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(45):E4256–4265. doi: 10.1073/pnas.1316894110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lundberg JO, Weitzberg E, Cole JA, Benjamin N. Nitrate, bacteria and human health. Nature reviews Microbiology. 2004;2(7):593–602. doi: 10.1038/nrmicro929. [DOI] [PubMed] [Google Scholar]
- 55.Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1992;6(12):3051–3064. [PubMed] [Google Scholar]
- 56.Andersson J, Samarina A, Fink J, Rahman S, Grundstrom S. Impaired expression of perforin and granulysin in CD8+ T cells at the site of infection in human chronic pulmonary tuberculosis. Infection and immunity. 2007;75(11):5210–5222. doi: 10.1128/IAI.00624-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi HS, Rai PR, Chu HW, Cool C, Chan ED. Analysis of nitric oxide synthase and nitrotyrosine expression in human pulmonary tuberculosis. American journal of respiratory and critical care medicine. 2002;166(2):178–186. doi: 10.1164/rccm.2201023. [DOI] [PubMed] [Google Scholar]
- 58.Facchetti F, et al. Expression of inducible nitric oxide synthase in human granulomas and histiocytic reactions. The American journal of pathology. 1999;154(1):145–152. doi: 10.1016/S0002-9440(10)65261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nathan C. Inducible nitric oxide synthase in the tuberculous human lung. American journal of respiratory and critical care medicine. 2002;166(2):130–131. doi: 10.1164/rccm.2205016. [DOI] [PubMed] [Google Scholar]
- 60.Nicholson S, et al. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. The Journal of experimental medicine. 1996;183(5):2293–2302. doi: 10.1084/jem.183.5.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rahman S, et al. Compartmentalization of immune responses in human tuberculosis: few CD8+ effector T cells but elevated levels of FoxP3+ regulatory t cells in the granulomatous lesions. The American journal of pathology. 2009;174(6):2211–2224. doi: 10.2353/ajpath.2009.080941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schon T, et al. Local production of nitric oxide in patients with tuberculosis. The international journal of tuberculosis and lung disease: the official journal of the International Union against Tuberculosis and Lung Disease. 2004;8(9):1134–1137. [PubMed] [Google Scholar]
- 63.Vogt G, Nathan C. In vitro differentiation of human macrophages with enhanced antimycobacterial activity. The Journal of clinical investigation. 2011;121(10):3889–3901. doi: 10.1172/JCI57235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Epelman S, Lavine KJ, Randolph GJ. Origin and Functions of Tissue Macrophages. Immunity. 2014;41(1):21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Franzblau SG, et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis. 2012;92(6):453–488. doi: 10.1016/j.tube.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 66.Pan H, et al. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 2005;434(7034):767–772. doi: 10.1038/nature03419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infection and immunity. 1995;63(2):736–740. doi: 10.1128/iai.63.2.736-740.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.MacMicking JD, et al. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(10):5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]