

Abstract
Chiral, dimeric natural products containing complex structures and interesting biological properties have inspired chemists and biologists for decades. A seven step total synthesis of the axially chiral, dimeric tetrahydroxanthone natural product rugulotrosin A is described. The synthesis employs a one-pot Suzuki coupling/dimerization to generate the requisite 2,2'-linked biaryl linkage. Highly selective point-to-axial chirality transfer was achieved using palladium catalysis with achiral phosphine ligands. Single X-ray crystal diffraction data was obtained to confirm both the atropisomeric configuration and absolute stereochemistry of rugulotrosin A. Computational studies are described to rationalize the atropselectivity observed in the key dimerization step. Comparison of the crude fungal extract with synthetic rugulotros in A and its atropisomer verified that nature generates a single atropisomer of the natural product.
Due to hindered rotational barriers of highly substituted biaryls and related compounds, a type of stereochemistry in the form of axial chirality is generated, often termed atropisomerism.1 Atropisomerism widely exists in nature and can lead to biological stereoselectivity for molecular targets.2,3,4 Moreover, in contrast to traditional sp3 carbon-centered chirality, control of axial chirality and atropisomer selectivity remains highly challenging especially in the context of complex natural product synthesis.5,6,7,8
Tetrahydroxanthones,9 secondary fungal metabolites, are an emerging family of natural products. Dimeric tetrahydroxanthones10 including the secalonic acids11,12 (Figure 1, a) display intriguing anticancer and antibacterial properties. During the last decade, tetrahydroxanthones containing biaryl axial chirality including the 2,2’-linked natural product13 rugulotrosin A (1) and the 2,4’-linked congener rugulotrosin B (2) (Figure 1, b) have been isolated and characterized. Related hybrid chromone lactone/tetrahydroxanthones including the 2,4'-linked heterodimer gonytolide E (3)14 and the 4,4'-linked chromone lactone homodimer gonytolide A (4)15 have also been reported. Recently, these interesting molecules have attracted significant interest in the synthetic community as evidenced by reports of syntheses of both monomeric16,17,18,19,20,21,22 and dimeric natural products. 23 However, syntheses of tetrahydroxanthones bearing axial chirality including rugulotrosin A (1) and related compounds have not been reported.
Figure 1.

Axially Chiral, Dimeric Tetrahydroxanthone Natural Products and Point-to-Axial Chirality Transfer Strategy. a) Structures of the dimeric tetrahydroxanthone natural products secalonic acids B and D; b) Structures of representative axially chiral tetrahydroxanthone natural products; c) Select literature examples of remote point-to-axial chirality transfer; d) Strategy for the synthesis of rugulotrosin A.
In order to establish axial chirality for rugulotrosin A, we envisioned that a point-to-axial chirality transfer strategy could be implemented. In the last few decades, point-to-axial chirality transfer has been widely used in atropselective synthesis. There are several different approaches for point-to-axial chirality transfer: (a) use of an ortho-auxilary5,6 or chiral leaving group 24 for intermolecular, atropselective coupling; (b) utilization of low-energy conformers derived from existing chirality centers25,26 or chiral auxiliaries5,6 to execute intramolecular couplings; (c) transfer of chiral sp3 carbon to axial chirality;27, 28 (d) and use of remote and existing chirality centers to influence atropselectiviy using intermolecular oxidative/metal-mediated couplings.29,30,31,32,33 For example, Shaw and coworkers reported that the chiral monomer 5 underwent oxidative coupling to generate atropisomer 6 in 76:24 dr which could be improved to higher selectivity using a chiral catalyst system29 (Figure 1, c). Likewise, Lipshutz and coworkers performed an atropselective cross coupling to access the korupensamine B precursor 9 using boronic acid 7 and iodotetrahydroisoquinoline partner 831 (Figure 1, c). In our studies, we considered a remote asymmetric induction strategy for point-to-axial chirality transfer for the rugulotrosin synthesis in the absence of chiral auxiliaries. In the case of dimerization of tetrahydroxanthone substrate 10, it was of interest to determine whether the chirality of the remote stereocenters could be transferred to the axial chirality of ortho, ortho dimer 11 (Figure 1, d), a product bearing a different coupling pattern than previous examples. In this case, we planned to employ transition metal catalysis to bring the two monomers in close proximity to enhance potential for chirality transfer.
Results and Discussion
Our synthesis was initiated with chromone 12 which was prepared in greater than 50 gram batches and purified by recrystallization (Figure 2). Utilizing diisopropylsilyl ditriflate to activate chromone 12 to a siloxybenzopyrylium species,22 vinylogous addition of 2-trimethylsiloxyfuran was achieved which was followed by hydrogenation to afford chromone lactone 13 on adecagram scale (89 % yield, dr > 10:1) after purification by trituration (Supplementary Information S4–S19). Further treatment of 13 with NaH in THF led to the production of epi-blennolide C 14 through Dieckmann cyclization. Due to the propensity of thevinylogous acid moiety of tetrahydroxanthone 14 to both epimerization and tetrahydroxanthone-to-chromone lactone rearrangement, we utilized the neutral methylating reagent trimethylsilyldiazomethane to obtain the desired O-methylated product 16 in 61 % yield along with the separable isomer 15 (20 % yield). After phenol-directed ortho-iodination of 16, the derived aryl iodide 17 was subjected to kinetic acylative resolution using Birman's catalyst 34 on a gram scale (s factor >200).35 Chiral monomer (−)-17 was isolated in 46 % yield (99 % ee) and the O-acylated product (+)-18 was obtained in 46 % yield (99 % ee) after recrystallization.
Figure 2.

Scalable Syntheses of Enantiopure Tetrahydroxanthone Monomers: Key steps involve siloxyfuran addition to a benzopyrylium species (12 to (±)-13) followed by kinetic acylative resolution of (±)-17 using the Birman catalyst.
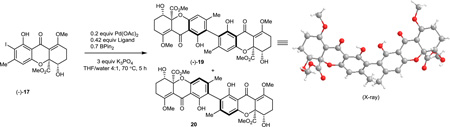
After obtaining the two enantiopure tetrahydroxanthone monomers, our initial plan was to employ our stannane dimerization strategy which successfully led to syntheses of secalonic acids A and D.23 However, after evaluation of various conditions, we observed only trace stannylation of iodides 17 or 18 (Table 1) which may be due to steric hindrance of the aryl iodide substrate. Subsequently, we considered that the less bulky borate may more readily undergo transmetallation. Gratifyingly, iodide (−)-17 was found to be stable under basic conditions and to produce an inseparable mixture of dimeric products (dr = 88: 12) under one-pot Suzuki coupling conditions using Pd(OAc)2/SPhos36 and bis (pinacolato) diboron. We subsequently attempted to establish the stereochemistry of the major atropisomer by NMR analysis and by acid-mediated deprotection of both atropisomers 19 and 20 to 1 and 23 (Figure 4). However, both efforts failed as the axial chirality center is remote from existing stereocenters leading to almost identical 1H NMR spectra for both a tropisomers of rugulotrosin A. Fortunately, a single crystal of the major atropisomer (−)-19 was obtained by recrystallization (CH2Cl2/MeOH) from its mixture with 20. X-ray crystal structure analysis of (−)-19 (CCDC#1022610) was achieved which verified that the relative stereochemistry was identical to that found in natural rugulotrosin A.13
Table 1.
One-pot Suzuki Dimerization of Chiral Tetrahydroxanthone Monomer (−)-17.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ligand | Conversion (yield) | d.r. | Entry | Ligand | Conversion (yield) | d.r. |
| 1 | 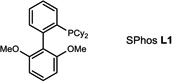 |
68 % (45 %) | 88 : 12 | 7 | 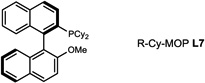 |
44 % (24 %) | 95.5 : 4.5 |
| 2 | 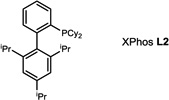 |
<10 % conv | 8 | 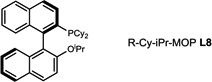 |
40 % conv | 90 : 10 | |
| 3 | 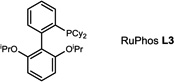 |
50 % (27 %) | 93 : 7 | 9a | 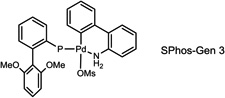 |
72–82 % (61 %) | 89 : 11 |
| 4 | 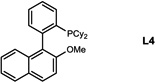 |
65 % (44 %) | 87 : 13 | 10a | 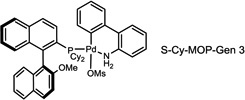 |
45 % (~30 %) | 80 : 20 |
| 5 | 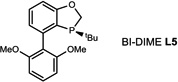 |
no reaction | |||||
| 6 | 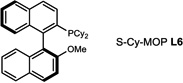 |
36 % conv | 81 : 19 | 11a,b | 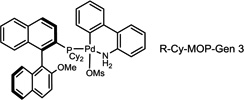 |
62 % (44 %) | 95.5 : 4.5 |
Conditions for Pd precatalyst: 0.2 equiv Pd precatalyst, 0.7 BPin2, 3 equiv K3PO4, THF/water 4:1, 70 °C, 5 h.
0.6 equiv BPin2 was used.
Figure 4.

Syntheses of (−) and (+)-rugulotrosin A and atrop-rugulotrosin A. a) Synthesis of ent-rugulotrosin A; b) Synthesis of rugulotrosin A and comparison rotation data with natural sample; c) Synthesis of atrop-rugulotrosin A; d) Synthesis of ent-atroprugulotrosin A; e) Comparison between the natural extract and synthetic rugulotrosins. HPLC-DAD (210 nm) (Zorbax C18 column, gradient elution H2O/MeCN plus 0.05% HCO2H)analysis of 21 dayPenicillium nov. sp. (MST-F8741) cultures extracted with (a) MeCN, or (b) MeOH, compared against (c) natural (+)-1, (d) synthetic (+)-1, (e) (−)-23, and (f) a mixture of (+)-1, (−)-1, (−)-23 and (+)-23).
During our evaluation of dimerization conditions, we found that water was essential and that the ratio of atropisomers was dependent on ligands for palladium rather than solvent, temperature, or reaction time. Accordingly, we focused our efforts on the influence of ligands on atropisomeric selectivity (Table 1). After extensive ligand screening, we found that monophosphines were crucial for the success of there action, and that bidentate ligands such as XantPhos or DPEPhos (not shown) failed to provide the desired dimeric products. Besides SPhos(L1), the related ligand RuPhos (L2) provided dimeric products in even higher atropselectivity (93:7). Interestingly, we found when the 2' position of the monophophine ligand did not contain an oxygen atom (e.g. carbon atom in XPhos L2 or a nitrogen atom (DavePhos, not shown)), dimeric products were obtained in trace amounts. After experiments with the achiral ligand L4, which provided similar results to SPhos, we anticipated that related chiral ligands may achieve efficient control of atropselectivity.37 We began this investigation with (R) and (S)-BI-DIME ligands L5,38,39 which recently have shown excellent atropselectivities in biaryl couplings. However, neither ligand was able to perform the one-pot dimerization of (−)-12. Gratifyingly, chiral versions of the SPhos ligands, S-Cy-MOP (L6) and R-Cy-MOP (L7),40, 41, 42 both successfully produced dimeric products. The ligand R-Cy-MOP provided a satisfactory dr (95.5:4.5) and the mismatched ligand S-Cy-MOP afforded a lower dr (81:19), both favoring the same major atropisomer. Even with the increased bulk of ligand L8, the dr did not noticeably improve. In order to enhance product yield, we prepared the Buchwald 3rd generation precatalysts43 with SPhos, R-Cy-MOP, and S-Cy-MOP. Indeed, use of the precatalyst increased yields for these cases and maintained at ropselectivity. Use of bis (neopentyl glycolato) diboron ortetrahydroxydiboron44, 45 as boron transfer reagents led only to trace amounts of dimeric products.
The acylated, enantiopure monomer (+)-18 was also investigated in the Suzuki dimerization which generated a separable 3:1 mixture of atropisomersin 36 % yield (Figure 3). Fortunately, an X-ray crystal structure of the minor atropisomer (−)-22 (CCDC#1022611) was obtained to verify relative and absolute stereochemistry. Introduction of a bulk yester onto the secondary alcohol of the monomer (e.g. OCOPh) did not affect the ratio of atropisomers.
Figure 3.

One-pot Suzuki Dimerization of Chiral Tetrahydroxanthone Monomers (+)-18: Optimal conditions shown using Pd(OAc)2 and SPhos as achiral ligand.
After obtaining and establishing stereochemistry for several protected dimers, we anticipated that we could access all atropisomers and enantiomers of rugulotrosin A. Dimer (−)-19 was treated with 3M HCl/MeOH for 10 min which afforded rugulotrosin A ((−)-1) in 89 % yield (Figure 4, a). In a similar manner, (+)-rugulotrosin A was obtained from acidic hydrolysis of (+)-21 (Figure 4, b). With both enantiomers of rugulotrosin A in hand, natural rugulotrosin A was unambiguously assigned as (+)-1 though comparison of rotation data. Similarly, atrop-rugulotrosin A (−)-23 and entatrop rugulotrosin A (+)-23 were synthesized using similar protocols from the protected precursors (−)-22 and (+)-22 (Figure 4, c and d). To verify the stability of the axially chiral biaryl moiety, we heated rugulotrosin A (+)-1 in toluene (1 mg/mL) at 100 °C for 12 h and 150 °C for 3 h. Under both conditions, we did not observe formation of atrop rugulotrosin. This is supported by a high barrier of rotation of rugulotrosin A, calculated as 47.7 kcal/mol using the B3LYP/6-31G(d) level of theory (see Supplementary Information S52–S58).
In order to rationalize the axial chirality selectivity observed in the dimerization, we modeled the geometry of the expected intermediate diaryl palladium (II) complexes46 using SPhos (L1) as ligand (Figure 5). Conformational analysis was carried out by systematic six-fold rotation about the C-Pd bonds, followed by optimization of candidate structures at the B3LYP/LanL2DZ level of theory.47 The five lowest energy structures are represented in Figure 5. From this analysis, the three lowest energy structures 24a – 24c all have dihedral angles (C1, C2, C2’, C1’) for the monomer fragments which would afford atropisomer (−)-19 (precursor to rugulotrosin) after stereospecific reductive elimination (Figure 5,a). The next two higher energy structures 25a and 25b display a reversed dihedral angle which should lead to atropisomer 20 (Table 1). Relative energies correlate with the steric fit of components. Space filling models of structures 24a-c and 25a-b (Supplementary Information S34–S52) show a more crowded arrangement of the latter, in part because of steric interactions between the methyl ester and the cyclohexyl group. The distances between the hydrogen of the methyl ester to the hydrogen on the cyclohexyl of the SPhos ligand are 2.4 and 2.5 Å for conformers 25a and 25b, respectively. Based on literature precedent,47 we assume that these catalyst-reactant complexes are under thermodynamic equilibration; additional computational studies will be required to predict barriers to interconversion. It thus appears that product stereochemistry is determined by reactant and catalyst assembly in the Pd (II) complex; this provides a basis for prediction of stereochemistry in future atropselective couplings.
Figure 5.

Computational Studies for Atropselective Suzuki Dimerization. Conformational analysis was optimized at the B3LYP/LanL2DZ level of theory. Dihedral angles were measured by C1, C2, C2’, C1’and are shown in green. a) Conformers leading to (−)-19; b) Conformers leading to 20.
After assigning the absolute configuration of rugulotrosin A, we employed our library of synthetic rugulotrosins to determine whether Penicillium nov. sp. (MST-F8741) was capable of producing atrop-rugulotrosin A. To this end, separate 21 day fermentations of MST-F8741 were extracted with either MeCN (Figure 4,e(a)) or MeOH (Figure 4, e(b)) and were subsequently analyzed using an HPLC-DAD method optimized for the resolution of rugulotrosin A (Figure 4, e(c,d)) and atrop-rugulotrosin A (Figure 4, e(e)). To further enhance sensitivity, these analyses were also carried out using HPLC-DAD-ESIM Sprotocols augmented by single ion extraction (SIE) (Supplementary Information, S22). As these analytical studies failed to detect any trace of atrop-rugulotrosin in MST-F8741 extracts, we conclude that the rugulotrosin biosynthetic pathway present in Penicillium nov. sp. (MST-F8741) operates with high atropisomer fidelity.
As natural rugulotrosin A had previously been reported to exhibit antibacterial activity,13 we also tested the synthetic rugulotrosins ((+)-1, (−)-1, (−)-23, and (+)-23) against several strains of Gram-positive and Gram-negative bacteria. Consistent with earlier reports,(+)-1 exhibited antibacterial activity against Bacillus subtilis (ATCC 6633) (IC50 = 2.1 µM) and Staphylococcus aureus (ATCC 25923) (IC50 = 6 µM). Of note, the enantiomer (−)-1 was also active against B. subtilis (ATCC 6633) (IC50 = 0.7 µM) and S. aureus (ATCC 25923) (IC50 = 18 µM), whereas the antibacterial activity of the atropisomer (−)-23 was weak and limited to B. subtilis (ATCC 6633) (IC50 = 10 µM) and (+)-23 lacked any appreciable antibacterial activity. None of the rugulotrosins (+)-1, (−)-1, (−)-23, or (+)-23 exhibited activity against the Gram-negative bacteria Escherichia coli (ATCC 25922) and Pseudomonas aeruginosa (ATCC 27853), nor did they exhibit cytotoxicity (IC50>30 µM) against human colon (SW620) and lung (NCI-H460) cancer cells.
Conclusion
In conclusion, we have developed a concise, atropselective approach to rugulotrosin A and stereoisomers through point-to-axial chirality transfer which has facilitated determination of the absolute configuration of rugulotrosin A. Computational studies modelling the geometry of intermediate diaryl palladium (II) complexes have provided a rationale for the atropselectivity observed in the key Suzuki dimerization. These studies highlight the utility of SPhos and related bulky monophosphine ligands48 in palladium couplings both to bring monomers in proximity and enhance remote steric effects leading to atropselectivity. Through HPLC analysis of fungal extracts and synthetic samples, we have determined that Penicillium nov. sp. (MST-F8741) generates rugulotrosin A in an atropselective manner. Moreover, the atropisomers and enantiomers of rugulotrosin A were found to have different activities against Gram-positive bacteria illustrating the importance of stereochemistry on target selectivity. Further studies regarding atropselective syntheses of dimeric natural products are currently under investigation and will be reported in due course.
Supplementary Material
Acknowledgements
Financial support from the NIH (GM-099920) is gratefully acknowledged. The authors gratefully acknowledge Dr. Jeffrey Bacon for crystal structure determination, Drs. Bo Qu and Chris Senanayake for kindly providing both (R) and (S)-BI-DIME ligands, and E. Lacey for supplying extracts of Penicillium nov. sp. (MST-F8741). Research at the BU-CMD was supported by NIH grant GM-067041. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) which is supported by National Science Foundation grant number OCI-1053575.
Footnotes
Author contributions
T.Q. and J.A.P., Jr. conceived of the project, designed and carried out the experiments, analyzed the data and wrote most of the paper. S.L.S.-J. and R.P.J. performed computational studies. Z.G.K. and R.J.C. performed natural extract comparisons and biological studies. All authors discussed the results and commented on the manuscript.
Supplementary information is available in the online version of the paper. Reprints and permission information is available online at www.nature.com/reprints.
Competing financial
The authors declare no competing financial interests.
References
- 1.Moss GP. Basic terminology of stereochemistry. Pure. Appl. Chem. 1996;68:2193–2222. [Google Scholar]
- 2.Zask A, Murphy J, Ellestad GA. Biological stereoselectivity of atropisomeric natural products and drugs. Chirality. 2013;25:265–274. doi: 10.1002/chir.22145. [DOI] [PubMed] [Google Scholar]
- 3.Clayden J, Moran WJ, Edwards PJ, LaPlante SR. The challenge of atropisomerism in drug discovery. Angew. Chem. Int. Ed. 2009;48:6398–6401. doi: 10.1002/anie.200901719. [DOI] [PubMed] [Google Scholar]
- 4.LaPlante SR, Edwards PJ, Fader LD, Jakalian A, Hucke O. Revealing atropisomer axial chirality in drug discovery. Chem. Med. Chem. 2011;6:505–513. doi: 10.1002/cmdc.201000485. [DOI] [PubMed] [Google Scholar]
- 5.Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Atroposelective synthesis of axially chiral biaryl compounds. Angew. Chem. Int. Ed. 2005;44:5387–5427. doi: 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- 6.Bringmann G, Gulder T, Gulder TAM, Breuning M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 2011;111:563–639. doi: 10.1021/cr100155e. [DOI] [PubMed] [Google Scholar]
- 7.Kozlowski MC, Morgan BJ, Linton EC. Total synthesis of chiral biaryl natural products by asymmetric biaryl coupling. Chem. Soc. Rev. 2009;38:3193–3207. doi: 10.1039/b821092f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liau BB, Milgram BC, Shair MD. Total syntheses of HMP-Y1, hibarimicinone, and HMP-P1. J. Am. Chem. Soc. 2012;134:16765–16772. doi: 10.1021/ja307207q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masters K-S, Bräse S. Xanthones from fungi, lichens, and bacteria: The natural products and their synthesis. Chem. Rev. 2012;112:3717–3776. doi: 10.1021/cr100446h. [DOI] [PubMed] [Google Scholar]
- 10.Wezeman T, Masters K-S, Bräse S. Double trouble—the art of synthesis of chiral dimeric natural products. Angew. Chem. Int. Ed. 2014;53:4524–4526. doi: 10.1002/anie.201402384. [DOI] [PubMed] [Google Scholar]
- 11.Franck B, Gottschalk EM, Ohnsorge U, Baumann G. The structure of secalonic acids A and B. Angew. Chem., Int. Ed. Engl. 1964;3:441–442. [Google Scholar]
- 12.Steyn PS. The isolation, structure and absolute configuration of secalonic acid D, the toxic metabolite of Penicillium oxalicum. Tetrahedron. 1970;26:51–57. doi: 10.1016/0040-4020(70)85006-2. [DOI] [PubMed] [Google Scholar]
- 13.Stewart M, et al. Rugulotrosins A and B: two new antibacterial metabolites from an Australian isolate of a Penicillium sp. J. Nat. Prod. 2004;67:728–730. doi: 10.1021/np034038b. [DOI] [PubMed] [Google Scholar]
- 14.Kikuchi H, Isobe M, Kurata S, Katou Y, Oshima Y. New dimeric and monomeric chromanones, gonytolides D-G, isolated from the fungus Gonytrichum sp. Tetrahedron. 2012;68:6218–6223. doi: 10.1021/ol2018449. [DOI] [PubMed] [Google Scholar]
- 15.Kikuchi H, et al. Structures of the dimeric and monomeric chromanones, gonytolides A-C, isolated from the fungus Gonytrichum sp. and their promoting activities of innate immune responses. Org. Lett. 2011;13:4624–4627. doi: 10.1021/ol2018449. [DOI] [PubMed] [Google Scholar]
- 16.Nicolaou KC, Li A. Total syntheses and structural revision of α- and β-diversonolicesters and total syntheses of diversonol and blennolide C. Angew. Chem., Int. Ed. 2008;47:6579–6582. doi: 10.1002/anie.200802632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tietze LF, et al. Enantioselective total synthesis of (−)-diversonol. Chem. Eur. J. 2013;19:4876–4882. doi: 10.1002/chem.201204037. [DOI] [PubMed] [Google Scholar]
- 18.Tietze LF, Ma L, Reiner JR, Jackenkroll S, Heidemann S. Enantioselective total synthesis of (−)-blennolide A. Chem. Eur. J. 2013;19:8610–8614. doi: 10.1002/chem.201300479. [DOI] [PubMed] [Google Scholar]
- 19.Tietze LF, Jackenkroll S, Hierold J, Ma L, Waldecker B. A domino approach to the enantioselective total syntheses of blennolide C and gonytolide C. Chem. Eur. J. 2014;20:8628–8635. doi: 10.1002/chem.201402495. [DOI] [PubMed] [Google Scholar]
- 20.Nising CF, Ohnemüller UK, Bräse S. The total synthesis of the fungal metabolite diversonol. Angew. Chem., Int. Ed. 2006;45:307–309. doi: 10.1002/anie.200502913. [DOI] [PubMed] [Google Scholar]
- 21.Meister AC, et al. Total synthesis of blennolide mycotoxins: design, synthetic routes and completion. Eur. J. Org. Chem. 2014:4861–4875. [Google Scholar]
- 22.Qin T, Johnson RP, Porco JA., Jr Vinylogous addition of siloxyfurans to benzopyryliums: a concise approach to the tetrahydroxanthone natural products. J. Am. Chem. Soc. 2011;133:1714–1717. doi: 10.1021/ja110698n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin T, Porco JA., Jr Total syntheses of secalonic acids A and D. Angew. Chem. Int. Ed. 2014;53:3107–3110. doi: 10.1002/anie.201311260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson JM, Cram DJ. Chiral leaving groups induce asymmetry in syntheses of binaphthyls in nucleophilic aromatic substitution reactions. J. Am. Chem. Soc. 1982;104:881–884. [Google Scholar]
- 25.Evans DA, et al. Nonconventional stereochemical issues in the design of the synthesis of the vancomycin antibiotics: challenges imposed by axial and nonplanar chiral elements in the heptapeptide aglycons. Angew. Chem, Int. Ed. 1998;37:2704–2708. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2704::AID-ANIE2704>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 26.Burns NZ, Krylova ZN, Hanroush RN, Baran PS. Scalable total synthesis and biological evaluation of haouamine A and its atropisomer. J. Am. Chem. Soc. 2009;131:9172–9173. doi: 10.1021/ja903745s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo F, Konkol LC, Thomson RJ. Enantioselective synthesis of biphenols from 1,4-diketones by traceless central-to-axial chirality exchange. J. Am. Chem. Soc. 2011;133:18–20. doi: 10.1021/ja108717r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konkol LC, Guo F, Sarjeant AA, Thomson RJ. Enantioselective total synthesis and studies into the configurational stability of bis murrayaquinone A. Angew. Chem. Int. Ed. 2011;50:9931–9934. doi: 10.1002/anie.201104726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park YS, et al. Synthesis of (−)-viriditoxin: a 6,6’-binaphthopyran-2-one that targets the bacterial cell division protein FtsZ. Angew. Chem., Int. Ed. 2011;50:3730–3733. doi: 10.1002/anie.201007298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipshutz BH, Keith LM. A stereospecific, intermolecular biaryl-coupling approach to korupensamine A en route to the michellamines. Angew. Chem., Int. Ed. 1999;38:3530–3533. [PubMed] [Google Scholar]
- 31.Huang S, Peterson TB, Lipshutz BH. Total synthesis of (+)-korupensamine B via an atropselective intermolecular biaryl coupling. J. Am. Chem. Soc. 2010;132:14021–14023. doi: 10.1021/ja1065202. [DOI] [PubMed] [Google Scholar]
- 32.Coleman RS, Grant EB. Atropdiastereoselective total synthesis of phleichrome and the protein kinase C inhibitor calphostin A. J. Am. Chem. Soc. 1994;116:8795–8796. [Google Scholar]
- 33.Broka CA. Total syntheses of phleichrome, calphostin A, calphostin D. Unusual stereoselective and stereospecific reactions in the synthesis of perylenequinones. Tetrahedron Lett. 1991;32:859–862. [Google Scholar]
- 34.Birman VB, Li X. Homobenzotetramisole: an effective catalyst for kinetic resolution of aryl-cycloalkanols. Org. Lett. 2008;10:1115–1118. doi: 10.1021/ol703119n. [DOI] [PubMed] [Google Scholar]
- 35.Müller CE, Schreiner PR. Organocatalytic enantioselective acyl transfer onto racemic as well as meso alcohols, amines, and thiols. Angew. Chem. Int. Ed. 2010;50:6012–6042. doi: 10.1002/anie.201006128. [DOI] [PubMed] [Google Scholar]
- 36.Barder TE, Walker SD, Martinelli JR, Buchwald SL. Catalysts for Suzuki-Miyaura coupling processes: scope and studies of the effect of ligand structure. J. Am. Chem. Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 37.Masamune S, Choy W, Petersen JS, Sita LR. Double asymmetric synthesis and a new strategy for stereochemical control in organic synthesis. Angew. Chem., Int. Ed. Engl. 1985;24:1–30. [Google Scholar]
- 38.Tang W, et al. A general and special catalyst for Suzuki-Miyaura coupling processes. Angew. Chem. Int. Ed. 2010;49:5879–5883. doi: 10.1002/anie.201002404. [DOI] [PubMed] [Google Scholar]
- 39.Xu G, Fu W, Liu G, Senanayake CH, Tang W. Efficient syntheses of korupensamines A, B and michellamine B by asymmetric Suzuki-Miyaura coupling reactions. J. Am. Chem. Soc. 2014;136:570–573. doi: 10.1021/ja409669r. [DOI] [PubMed] [Google Scholar]
- 40.Hamada T, Chieffi A, Ahman J, Buchwald SL. An improved catalyst for the asymmetric arylation of ketone enolates. J. Am. Chem. Soc. 2002;124:1261–1268. doi: 10.1021/ja011122+. [DOI] [PubMed] [Google Scholar]
- 41.Zhou Y, et al. Enantioselective synthesis of axially chiral multifunctionalized biaryls via asymmetric Suzuki-Miyaura coupling. Org. Lett. 2013;15:5508–5511. doi: 10.1021/ol402666p. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Y, et al. Enantioselective synthesis of axially chiral biaryl monophosphine oxides via direct asymmetric Suzuki coupling and DFT investigations of the enantioselectivity. ACS Catal. 2014;4:1390–1397. [Google Scholar]
- 43.Bruno NC, Tudge MT, Buchwald SL. Design and preparation of new palladium precatalysts for C-C and C-N cross-coupling reactions. Chem. Sci. 2013;4:916–920. doi: 10.1039/C2SC20903A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Little S, Trice J. Tetrahydroxydiboronin: encyclopedia of reagents for organic synthesis. John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 45.Molander GA, Trice SLJ, Kennedy SM, Dreher SD, Tudge MT. Scope of the palladium-catalyzed aryl borylation utilizing bis-boronic acid. J. Am. Chem. Soc. 2012;134:11667–11673. doi: 10.1021/ja303181m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gensch T, et al. Snapshot of the palladium (II)-catalyzed oxidative biaryl bond formation by X-ray analysis of the intermediate diaryl palladium (II) complex. Chem. Eur. J. 2012;18:770–776. doi: 10.1002/chem.201103576. [DOI] [PubMed] [Google Scholar]
- 47.Shen X, Jones GO, Watson DA, Bhayana B, Buchwald SL. Enantioselective synthesis of axially chiral biaryls by the Pd-catalyzed Suzuki-Miyaura reaction: substrate scope and quantum mechanical investigation. J. Am. Chem. Soc. 2010;132:11278–11287. doi: 10.1021/ja104297g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin R, Buchwald SL. Palladium-catalyzed Suzuki-Miyaura cross-coupling reactions employing dialkylbiarylphosphine ligands. Acc. Chem. Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.