

Abstract
Genetic variation underlying hypothalamic pituitary adrenal (HPA) axis over-activity in healthy controls and patients with severe forms of major depression has not been well explored but could explain risk for cortisol dysregulation.
95 participants were studied: 40 patients with psychotic major depression (PMD); 26 patients with nonpsychotic major depression (NPMD); and 29 healthy controls (HC). Collection of genetic material was added one third of the way into a larger study on cortisol, cognition, and psychosis in major depression. Subjects were assessed using the Brief Psychiatric Rating Scale, the Hamilton Depression Rating Scale and the Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders. Blood was collected hourly for determination of cortisol from 6pm to 9am and for the assessment of alleles for 6 genes involved in HPA Axis regulation.
Two of the 6 genes contributed significantly to cortisol levels, psychosis measures or depression severity. After accounting for age, depression, and psychosis, and medication status, only allelic variation for the glucocorticoid receptor gene (GR) accounted for significant variance for mean cortisol levels from 6pm to 1am (r2=.317) and from 1am to 9am (r2=.194). Interestingly, neither depression severity nor psychosis predicted cortisol variance. In addition, GR and corticotropin-releasing hormone receptor 1 (CRH-R1) contributed significantly to psychosis measures and CRH-R1 contributed significantly to depression severity rating.
Keywords: Major depression, psychosis, cortisol, glucocorticoid genes, HPA axis
Introduction
The stress responsive hypothalamic pituitary adrenal (HPA) axis has been implicated in the pathophysiology of mood, anxiety, cognitive, and other disorders. The Axis consists of stimulating forward and feedback inhibition loops involving the brain, pituitary, and adrenal glands to produce glucocorticoid end products. Several reports have found that particularly patients with major depression with psychotic features (PMD) demonstrate elevated activity of the HPA axis as compared with nonpsychotic depressives (NPMD) or healthy controls (HC) (1-10). PMD patients have been reported to demonstrate significantly elevated 24-hour urinary free cortisol levels (1), plasma adrenocorticotropin hormone (ACTH) levels (11), afternoon (1-4 pm) cortisol levels (4), nocturnal serum cortisol levels (6), blunted response to a mineralocorticoid agonist (7), as well as high rates of non-suppression on challenge with dexamethasone and very high post-dexamethasone plasma levels (2, 3, 5, 8, 10). Higher cortisol levels have been associated with impaired cognitive functioning in healthy controls and depressed patients (12, 13).
The HPA axis, which plays an important role in how humans adapt to environmental stressors, consists of the paraventricular nucleus (PVN) of the hypothalamus, the anterior pituitary and the adrenal cortex, and is regulated in part through a finely balanced feedback system, the disruption of which may contribute to depression. The axis can be activated by various neuronal signaling molecules, including serotonin, noradrenaline and neuropeptide Y, depending on the type of stress stimulus (14). Once released the corticotropin-releasing hormone (CRH) stimulates its receptors CRHR1 and CRHR2, which act synergistically to modify the extent and duration of the stress response (15), with arginine vasopressin (AVP) and the urocortins providing additional regulation. The eventual result is the synthesis and release of the glutocorticord cortisol. Cortisol can act through both the mineralocorticoid receptors (NR3C2) and glucocorticoid receptors (NR3C1) and imbalances between these two receptors has been implicated in a maladaptive stress response and depression (16). The glucocorticoid receptor (NR3C1) is widely expressed throughout the body and brain and is involved in a number of physiological processes, many of them linked to the stress response, these include metabolic functions, cellular differentiation, and the nervous and immune system (17), with NR3C2 acting as a counterbalance in a similar to fashion as CRHR2 to promote recovery and a return to homeostasis (16).
Genes for the various hormone components and their receptors have been identified. CRH binds to CRH-R2 with less intensity than it does to CRH-R1. In contrast, urocortin stimulates CRH-R1 and CRH-R2 at equal potency. Urocortin also binds to urocortion 1-3 receptors. CRH-R2 distribution in primate brain overlaps with typical stress circuits (18) and may play a role in HPA stress responses in man. The currently prevailing theory is that CRHR1 and CRHR2 work synergistically, with CRHR2 coming more into play in the recovery phase (14). This theory has evolved from the notion that it was involved primarily in feeding behaviors (18). Individually AVP only has a weak ability to stimulate the release of adrenocorticotropic hormone (ACTH) but working synergistically with CRH, it can enhance ACTH release from the pituitary corticotrophes, via AVPR1b and CRHR1 respectively.
Genetic variation at the corticotrophin releasing hormone (CRH) locus, in the genes encoding its binding protein and type 1 and 2 receptors have been associated with increased risk for developing panic disorder (19), post-traumatic stress disorder and major depression (20) particularly in the face of early abuse (21-23) as well as response to antidepressants in several trials (24). Haplotypes for the glucocorticoid receptors (GR) pointed to a relationship with risk for more aggressive disease progression in multiple sclerosis suggestive of decreased GR sensitivity (25). However, an earlier paper on GR variation had failed to find an association with GR insensitivity (26). Recent studies on specific single nucleotide polymorphisms for GR indicate they are associated with altered GR sensitivity (27) and increased risk for impaired working memory in healthy controls (28); as well as increased risk for attention deficit disorder (29), and depression (30, 31). For the FKBP5 co-chaperone for the glucocorticoid receptor (GR), genetic variants appear to confer risk for rapidly occurring and treatment responsive episodes of depression with altered responses to CRH/dexamethasone challenge (DST) in depression (32), as well as for developing major depression and post-traumatic stress disorder (33, 34) and dexamethasone non-suppression (35).
Variation in HPA axis genes has not been explored in PMD even though the disorder has been reported to have the most marked HPA axis abnormality. Herein, we report on genetic variants for components of the HPA axis and their relationship to depression, psychosis and evening/nocturnal cortisol levels in patients with PMD, NPMD, and healthy controls. The specific aims of this project were to determine: 1) do HPA axis genes predict cortisol levels; 2) do HPA axis genes predict depression or psychosis severity.
Methods
Participants
Psychiatric participants were recruited through inpatient and outpatients facilities at Stanford University or self-referred from online and print study advertisements. Forty-nine patients with psychotic major depression (PMD) and 52 patients with nonpsychotic major depression (NPMD) participated in two waves of a larger study on HPA axis in depression (HPA-2 and HPA-3). Part way through the first wave of study, collection of blood samples for genetic analysis began. The majority of available subjects consented to provide blood for genetic analyses.
Psychotic and nonpsychotic depressed patients were required to have a minimum score of 21 on the Hamilton Depression Rating Scale (HDRS;(36)) and a minimum score of 6 on the Thase Core Endogenomorphic Scale (37), with one exception of a PMD who scored 5. These latter two criteria were designed to ensure inclusion of participants with similar minimum levels of severity of endogenous-type symptoms. PMDs were also required to have a minimum of 5 on the positive symptom subscale of the Brief Psychotic Rating Scale (BPRS; (38), which consists of four items: conceptual disorganization, suspiciousness, hallucinations, and unusual thought content. A score of 4 on the PSS indicates no positive symptoms. NPMD subjects had no history of psychotic symptoms. All patients met the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) criteria for a current major depressive episode, with or without psychotic features.
Healthy comparison subjects were recruited through online and print study advertisements. Overall 56 healthy controls participated in the larger HPA study and 29 provided blood samples. Healthy controls (HC) were assessed for Axis I disorders with theStructured Clinical Interview for DSM-IV (SCID; (39). They were required to have a score less than 6 on the HDRS and have no psychotic symptoms as measured by the BPRS positive symptom subscale. Furthermore, they had no current or history of Axis I psychiatric illness. Participants were allowed to continue their psychiatric medications but were required to maintain a stable medication regimen for at least 1 week prior to the start of the study. Depressed patients were taking a combination of antidepressants, antipsychotics, anxiolytics, and mood stabilizers.
Exclusion criteria for all groups included major medical illnesses, history of seizures or major head trauma, abnormal clinical laboratory tests, pregnant or lactating females, and under the age of 18. Participants were excluded for unstable or untreated hypertension, cardiovascular disease, or endocrine disorders. Subjects on steroids (i.e. prednisone or steroidal inhalers) were not included. Additionally, patients who were actively suicidal, met criteria for primary Obsessive-Compulsive Disorder or Post-traumatic Stress Disorder, or had a history of substance abuse or electroconvulsive therapy in the previous six months were excluded from the study.
A number of subjects recruited were not included in the present analyses because of medications that interfere with cortisol levels. As oral contraceptives and hormone replacement treatments dramatically increase cortisol, all subjects on these medications were removed from the cortisol analyses, as well as several subjects who had missing or incomplete cortisol data. In toto, 27 cases were excluded from the study. Thus, 40 patients with psychotic depression, 26 patients with nonpsychotic depression, and 29 healthy controls participated in this genetics study.
Procedure
The study was approved by the Stanford University Institutional Review Board, and all subjects gave written informed consent before screening. Eligibility screening procedures included the SCID, HAM-D, BPRS, clinical laboratory tests, comprehensive metabolic panel, urine drug screening, and urine screening for pregnancy in female subjects. If participants met inclusion criteria at the eligibility screening, they were returned for baseline procedures.
At baseline, participants were re-administered the HAMD (36) and the BPRS (38). Subjects participated in blood sampling overnight on the Stanford Hospital GCRC. An intravenous line was inserted at 4PM and blood collected hourly for cortisol from 6pm to 9am the next day. Cortisol data on a subset (40 subjects) were previously reported (6) and included 19 PMDs, 10 NPMDs, and 11 HCs.
Cortisol Determination
The blood samples were kept frozen at −80 degrees until analysis. Cortisol assays were conducted by the Brigham Women’s Hospital, General Clinical Research Laboratory in Boston. The analytic sensitivity for cortisol was 0.4 micrograms/dl with a coefficient of variation of less than 7.9%.
Because of the natural diurnal rhythm of the cortisol slopes, the 15-hour blood collection period was divided into two phases based on the apparent nadir: the evening level from 1800 to 0100 hours and the morning level from 0100 to 0900 hours. These epochs correspond to the natural descending and ascending slopes of the cortisol rhythm and are based on the nadirs observed in previous studies (6) as well as in this sample. Mean time of nadir across the groups was 12:32 am (SD=2.26). Figure 1a shows the curve of the cortisol values of the overall group from 6 pm until 9 am.
Figure 1.
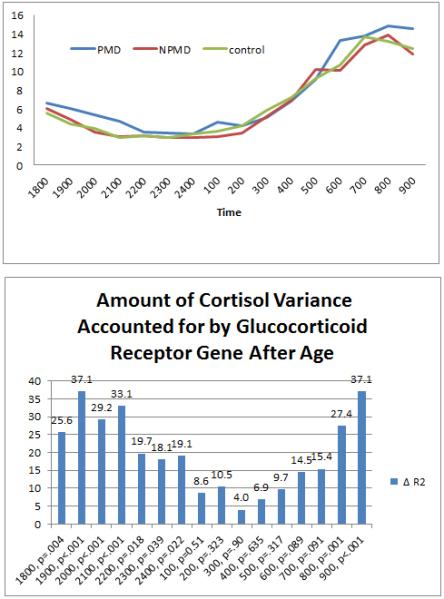
a. Mean Cortisol values across all subjects from 6 pm to 9 am.
b. Amount of cortisol variance accounted for by GR gene at each time point.
Genetics
Blood was collected for assessment of genetic markers at the following HPA axis loci: CRH, CRH-R1, CRH-R2, NR3C1 (glucocorticoid receptor, GR), NR3C2 (mineralocorticoid receptor, MR), and FKBP5. Genetic markers were selected using a standard protocol (40) that utilizes these criteria: 1) The marker has demonstrated (based on literature) or predicted (based on bioinformatic analysis) effects on protein abundance or function; 2) The marker has been associated with depression, psychosis, memory performance or the HPA axis regulation; 3) The marker is located in a region of interest: exons, 3′UTR, intron boundaries (41), or one of the many regulatory elements involved in gene expression; 4) The marker is a good predictor (“tagging SNP”) for the other polymorphisms in the gene due to high linkage disequilibrium (LD); 5) The tagging marker complements other selected markers to provide good LD coverage of the gene.
LD bins and the selection of tagging single-nucleotide polymorphisms (SNPs) were predicted using Haploview and the unrelated individuals from the HapMap CEU (N = 60 individuals of European descent collected in UTAH) (42), which was the closest match to our study sample. De Bakker (43) showed that ‘tagging’ SNPs selected using the CEPH cohort retained a reasonable level of effectiveness across multiple non-African populations. However, in some cases, HapMap either does not contain a particular gene or genomic region, or a particular marker of interest was not present within the HapMap CEU cohort. In these cases, we utilized other SNP databases such as dbSNP, UCSC, Illumina, Seattle SNPs and SNPper. In general only SNPs with a minor allele frequency of 5% or greater were considered.
DNA was extracted from EDTA-treated whole blood using the Gentra Puregene kit (Qiagen, Valencia, CA). Genotyping was performed using Taqman real-time PCR (Applied Biosystems, Foster City, CA). All genotypes were tested for deviation from Hardy Weinberg equilibrium.
Tables 1 and 2 summarize the SNPs and their frequencies that were assayed for each HPA axis gene. 70 SNPs were assessed, distributed in the following manner: CRH (6 SNPs), CRHR1 (21 SNPs), CRH-R2 (18 SNPs), NR3C2 (MR; 15 SNPs), NR3C1 (GR; 10 SNPs), and FKBP5 (2 SNPs). SNPs that had no or minimal variability in our sample were excluded from the analysis. If SNPs had fewer than 5 subjects across all 3 groups homozygous for the rare allele, they were collapsed into the heterozygous group.
Table 1.
HPA axis Genes Assessed
| Gene | Function | Brain Location in humans | SNPs Studied |
|---|---|---|---|
| CRH | Stimulates HPA axis; mediates stress responses in amygdala |
See below for CRH--R1 and CRH-R2 |
N=6 **rs10098823 rs3176921 ^rs5030875 rs5030877 rs6999100 ^rs7350113 |
| CRHR1 | Mediates stress responses and HPA axis stimulation. Binds primarily to CRH. |
Distributed widely throughout brain with particular concentration in the amygdala, paraventricular nucleus of the hypothalamus cortex – prefrontal, cingulate, striate, and insular. Also located in locus coeruleus, cerebellar cortex, solitary tract nucleus, thalamus, and striatum. |
N=21 rs110402 *rs12938031 rs16940674 rs171440 rs17689966 rs242924 rs242940 rs242948 **rs4076452 **rs4792887 **rs4792888 rs7209436 ^rs3785877 ^rs4792825 rs4458044 rs12944712 **rs17763104 **rs2664008 **rs17763658 **rs242942 **rs11657992 |
| CRHR2 | Involved in appetitive behaviors and stress; binds to both CRH and urocortin |
The distribution of CRH R- 2 in nonhuman primate is similar to CRH-R1 and includes cortex, amygdala, and hippocampus. Also reported in choroid plexus nucleus prepostus, and hypothalamus (supraoptic and paraventicular nuclei). Limited distribution in rat brain. |
N=18 **rs2240403 rs2267712 rs2267715 rs2267717_coded rs2270007 rs255100 **rs4723003 **rs7812133 rs255102 rs975537 rs2190242 rs2267716 **rs2284216 rs2284217 **rs4723000 **rs12701020 **rs17159371 rs929377 |
| NR3C1 (GR) |
Feedback inhibition of HPA axis; cognition; immune response |
Cortex widely, hypothalamus, amygdala, hippocampus |
N=10 *rs6195 **rs6198 rs33388 ^rs2918419 rs10052957 rs10482633 **rs12521436 **rs12655166 **rs17209258 rs41423247 |
| NR3C2 (MR) |
Inhibitory control of HPA axis; memory; blood pressure |
Hypothalamus, hippocampus, amygdala |
N=13 **rs5525 *rs5530 rs1879829 rs2070951 **rs2272089 rs3910052 rs4835488 rs6535578 rs7658048 **rs7694064 **rs10213471 *rs17024360 **rs17484245 rs2070950 **rs5522 |
| FKBP5 | Co-chaperone to heat shock protein for GR; stabilizes GR confirmation |
See GR | N=2 rs1360780 rs3800373 |
Little or no variance in the SNPs; SNP not used in analyses
Less than 5 total in the rare homozygous, collapsed into the heterozygous SNP group
Only 2 SNP variations present
Table 2.
Chromosomal position, allelic distribution and role of all genotyped NR3C1 (Glucocorticoid Receptor) SNPs
| Position | Minor allele (major allele) |
MAF1 (Chr. n ) |
HWP | Role | |
|---|---|---|---|---|---|
| rs6198 | 142657621 | C(t) | 14.9% (n=188)* |
1.00 | 3′UTR |
| rs17209258 | 142673397 | G(a) | 15.4% (n=188) |
0.63 | Intron |
| rs33388 | 142697295 | T(a) | 46.3% (n=188) |
0.30 | Intron |
| rs2918419 | 142722353 | G(a) | 10.5% (n=190) |
0.65 | Intron |
| rs10482633 | 142750533 | C(a) | 18.4% (n=190) |
0.77 | Intron |
| rs41423247 | 142778575 | C(g) | 31.1% (n=190) |
1.00 | Intron |
| rs6195 aka rs56149945 | 142779317 | G(a) | 1.1% (n=190) |
1.00 | Asn>Ser at codon 363 (exon 2) |
| rs10052957 | 142786701 | A(g) | 28.9% (n=190) |
0.87 | Promoter or Intron |
| rs12655166 | 142809272 | C(t) | 18.4% (n=190) |
0.69 | Intron |
| rs12521436 | 142817607 | A(g) | 19.5% (n=190) |
0.52 | Promoter |
n=number of chromosomes
Relative positions on chromosome 5 were taken from dbSNP build 131
Abbreviations: Chr., Chromosome; HWP, Hardy-Weinberg P value; MAF, Minor Allele Frequency; UTR, Untranslated region
Data Analyses
We used a gene-centric testing strategy in lieu of a SNP-specific approach. Gene-wide tests have several advantages (44, 45). When the primary scientific interest is in the gene as a whole and its impact on a particular pathway, inconsistencies among individual SNP results can be difficult to interpret. In addition, performing a single global test of a gene first eliminates the need for a multiple testing penalty, which would impose a more stringent significance threshold and reduce power. Individual SNPs can still be evaluated individually once the gene as a whole has been found to have a significant association. A number of novel, gene-centric testing strategies have recently been proposed (46, 47). However, given the small sample sizes, we opted to use a simple strategy employing traditional, well-established statistical F-tests of multiple predictors for multiple linear regression models.
All analyses were conducted using SPSS statistical software. First, differences in cortisol among the groups were examined with ANOVA. Next, the relationship between HPA axis variation and individual genes was examined controlling for the well-known effects of age on serum cortisol levels. In addition, because this sample consists of both patients with depression and healthy comparison subjects, severity of depression and psychosis were also accounted for prior to examining the gene contribution. For each of the six genes, we conducted multiple regressions for cortisol levels with the following predictors: age, psychosis and depression severity, use of psychiatric medications, and all available SNPs for that gene. This model was compared to several more parsimonious models: one with age, psychosis and depression severity, and medication status; one model with age, psychosis and depression severity and another with age only. There is a wide variety of psychiatric medications that patients were taking and the sample is not large enough to break them down into specific medication classes. Thus, if patients were taking daily antidepressants, antipsychotics, anxiolytics, or mood stabilizers, they were deemed positive for the daily psychiatric medication variable. A single gene-centric test for each gene was performed evaluating the contribution of all SNPs simultaneously, using a standard F-tests for multiple predictors to compare it to the model with age, psychosis and depression severity. If the gene-centric tests reached significance for an association of overall genetic variation with cortisol response, individual SNPs within the gene were subsequently evaluated based on coefficients and p-values for that SNP in the presence of age, depression severity, psychosis, and all other SNPs.
We performed linear regression analyses to assess the contributions of each gene to depression and psychosis severity individually. Depression severity was the total score of the 21-item HAMD and psychosis severity was the total score on the Positive Symptom Subscale of the BPRS. There was a strong correlation between depression and psychosis severity in our sample (r=.555, p<.001). Thus the variance associated with the other clinical symptom (depression or psychosis) was accounted for in the first step. Then, all SNPs for a specific gene were again entered as independent predictors and a gene-wide test was performed based on the significance of the entire model. Again, individual SNP contributions were evaluated based on the same multi-SNP model only if the overall gene test reached significance. The reported effect of each SNP was adjusted for all other SNPs in the gene. We performed similar analyses for psychosis, with depression severity variance removed first, using multiple linear regression.
We then examined the relative contributions of haplotypes for NR3C1 using SNPs first reported by Koper et al. (26). Based on these SNPs, we tested 6 haplotypes, previously identified by van Winsen et al. (25) For each reported haplotypes, individuals were coded as carriers if they carried at least one copy of every allele in the haplotype and as non-carriers if they carried no copies for one or more alleles in the haplotype. Each haplotype was then tested separately for association with cortisol, depression and psychosis. The cortisol regression included age at the first step, depression and anxiety at step 2, and the given haplotype. Additional linkage disequilibrium (LD) analysis was carried out using Haploview v4.2 (Broad Institute, Cambridge, MA).
Results
Demographic characteristics are summarized in Table 3. The three groups were well matched on age and gender. Patients with PMD had significantly higher total HDRS scores than did the NPMD’s or HC’s. Differences between the patients primarily reflected psychosis-related symptoms in the PMD group. Mean scores on the Thase Endogenomorphic Subscale were also significantly higher in the PMD patients, although, the differences between clinical groups were relatively small. On the BPRS, PSS scores were understandably higher in the PMD group than in the other two groups.
Table 3.
Demographic and Clinical Measures in Subjects
| PMD (N= 40) |
NPMD (N=26 ) |
HC (N=29 ) |
Test Value | Post-hoc comparison |
|
|---|---|---|---|---|---|
|
| |||||
| Age | 39.33 (14.0) |
40.81 (12.9) |
35.41 (14.2) |
F(2,92)=1.16, ns | |
|
| |||||
| Gender | χ2(2)= 850, ns | ||||
| Female | 20 | 16 | 16 | ||
| Male | 20 | 10 | 13 | ||
|
| |||||
| Ethnicity^ | X2(6)=6.25, ns | ||||
| Caucasian | 30 | 20 | 19 | ||
| African-American | 3 | 2 | 2 | ||
| Asian | 5 | 2 | 8 | ||
| Latino | 2 | 2 | 0 | ||
|
| |||||
| Any Daily Psychiatric Medication Use* |
36/40 | 12/26 | 0/29 | ||
|
| |||||
| Education | 15.10(2.9) | 15.35 (1.9) |
15.64 (2.1) |
F(2,92)=418, ns | |
|
| |||||
| HDRS Total | 28.90 (5.03) |
23.73 (3.3) |
.69 (1.0) | F(2,92)=508.4, p<.001 |
PMD > NPMD > HC |
|
| |||||
| HDRS Endogenormophic Subscale |
9.1 (2.1) | 8.15 (17) |
.14 (.35) | F(2,92)=275.6, p<.001 |
PMD > NPMD > HC |
|
| |||||
| BPRS Total* | 45.10 (7.2) |
33.38 (4.5) |
18.79 (1.4) |
F(2, 92)=207.1, p<.001 |
PMD > NPMD > HC |
|
| |||||
| BPRS PSS* | 10.08 (4.1) |
4.23 (.58) |
4.10 (.41) | F(2, 92)=54.75, p<.001 |
PMD > (NPMD = HC) |
|
| |||||
| Cortisol 6pm – 1 am |
4.93 (2.7) | 3.65 (1.7) |
3.65 (1.5) | F(2,92)=4.19, p=.018 |
PMD > (NPMD = HC) |
|
| |||||
| Cortisol 1 am – 9 am |
9.73 (4.7) | 8.68 (2.6) |
8.93 (2.2) | F(2,92)=.800, ns | |
|
| |||||
| Haplotype Carriers | |||||
| Hap 1 | 33/38 | 20/26 | 23/29 | ||
| Hap 2 | 16/38 | 15/26 | 14/29 | ||
| Hap 3 | 7/38 | 10/26 | 10/29 | ||
| Hap 4 | 10/38 | 6/26 | 7/29 | ||
| Hap5 | 1/38 | 0/26 | 1/29 | ||
| Hap 6 | 2/38 | 2/26 | 0/29 | ||
Ethnicity was assessed because SNP prevalence may differ between ethnic groups.
Any daily psychiatric medication includes use of antidepressants, antipsychotics, anxiolytics, or mood stabilizers
Mean evening cortisol from 6 pm to 1 am was significantly different between the 3 clinical groups, F(2,92)=4.19, p=.018, with PMD higher than the other two groups, who did not differ from one another (see Figure 1a). Mean 1 am – 9 am cortisol was not different between the groups (F(2,92)=.800, ns). These cortisol findings are consistent with our previous findings (6).
Only one of the 6 genes—NR3C1 (GR) -- significantly predicted evening cortisol from 6pm-1am. By itself, age accounted for 5.1% of the variance (F(1,91)=4.93, p=.029. Together depression and psychosis accounted for an additional 5.2% of the variance but did not reach significance (change r2 = F(2,89)=2.58, p=.082). Daily psychiatric medication use accounted for an additional 5.3% of the variance (change r2 = F(1,88)=5.48, p=.022). Genetic variation in NR3C1 considering all SNPs simultaneously accounted for an additional 28.8% of the variance (change r2 = F(10,78) =4.04 p<.001). Five of the 9 SNPs in this gene attained individual statistical significance in this model (rs33888, p=.002; rs2918419 p=001; rs10052957, p<.001; rs10482633, p=.002; and rs41423247, p=.009). The overall model with age, depression, psychosis, medications, and NR3C1 genetic variation together significantly accounted for 44.4% of the variance, F(14,78)=4.45, p<.001. None of the other five genes (CRH, CRH-R1, CRH-R2, NR3C2, or FKBP5) significantly predicted evening cortisol (see supplemental material).
Similarly, only genetic variation in NR3C1 significantly predicted early morning cortisol from 1am to 9am. Age accounted for 15.0% of the variance (F(1,91)=16.00, p<.001. Together depression and psychosis accounted for an additional 0.6% of the variance, but did not reach significance (change r2 = F(2,89)=.297, p=.74). The use of daily psychiatric medications accounted for an additional 4.3% of the variance (change r2 = F(1,88)=4.77, p=.032). Genetic variation in NR3C1 considering all SNPs accounted for an additional 17.1% of the variance (change r2 = F(10,78)=2.38, p=.019). Four of the 9 individual NR3C1 SNPs in this gene attained significance individually (rs2918419, p=.017; rs10052957, p=.001; rs10482633, p=.017; and rs12655166, p=.048). Three of the SNPs, rs2918419, rs10052957, and rs10482633, predicted both evening and early morning cortisol. As indicated in Figure 2, the significant SNPs are generally in low LD with one another, other than an r2 of 0.50 observed between rs10052957 and rs10482633. Together, age, depression and psychosis severity, psychiatric medication use, and NR3C1 account for 37.0% of the variance (F(13,79)=3.56, p<.001). None of the other five genes (CRH, CRH-R1, CRH-R2, NR3C2, or FKBP5) significantly predicted early morning cortisol (see supplemental material).
Figure 2.

Linkage disequilibrium (LD) map of the glucocorticoid receptor (NR3C1) locus showing pairwise r2 values between single nucleotide polymorphisms genotyped in this study. This shows that with the exception of rs6198 and rs10482633 which trend towards the .80 threshold for strong LD the tested SNPs are independent in this population.
Contributions of NR3C1 for each time point between 6 pm to 9 am were then calculated. As indicated in Figure 1b, significant contributions were observed from 6 pm to 1 am and at 8 and 9 am but not from 1 to 7 am. This is consistent with cortisol loadings to NR3C1 primarily at high cortisol concentrations (i.e., low affinity). The mean coefficient of variation (SD / Mean) for hourly cortisol levels was .687, with a range of .435 - .984, indicating that the distribution of significant genetic contribution to cortisol was not just due to lower variance around the nadir.
We then performed linear regression to predict depression severity with all subjects combined. Only one gene, CRHR1, predicted depression. At the first step, psychosis alone accounted for 28.4% of the variance, F(1,88)=34.8, p<.001. Once psychosis was accounted for, the CRHR1 gene accounted for an additional 24.2% of the variance, (F(20,68)=1.73, p=.049. Together psychosis and CRHR1 accounted for 52.5% of the variance F(21, 68)=3.59, p<.001). Only one SNP attained significance-- rs4076452 (p=.019) and two were of trend significance—rs4792825, p=.079 and rs17763658, p=.089. No other gene (CRH, CRHR2, NR3C1, NR3C2, or FKBP5) was significant nor trended to predict depression severity (see supplemental material).
Of the 6 genes examined, both NR3C1 and CRHR1 significantly predicted psychosis severity once depression had been accounted for. After accounting for depression severity, the NR3C1 (GR) gene predicted 12.7% of the variance, F(9,82)=2.35, p=.044. Two SNPs in this gene trended toward individual significance for an association with psychosis—rs6198, p=.093 and rs12521436, p=.056. After accounting for depression severity, CRHR1 gene also predicted psychosis, F(20,68)=2.07, p=.014, accounting for 27.1% of the variance. Three SNPs in this gene attained individual statistical significance, rs110402, p=.023; rs4076452, p=.010; rs17763658, p=.040 and three trended toward significance: rs4792887, p=.10; rs4792888, p=.072; and rs4792825, p=.087. The other four genes (CRH, CRHR2, NR3C2, and FKBP5) did not predict psychosis (see supplemental material).
For all analyses, results were similar for the full sample (N=95) and for the subsample with European Caucasians only (N=69). In the Caucasian sample, the overall prediction of evening cortisol by NR3C1, with age, depression, psychosis, and daily psychiatric medications accounted for, held strong (change r2 = 37.5, p<.001) as did the results for morning cortisol (change r2=26.2, p=.003). Regression results predicting psychosis once depression was accounted for were also similar. For psychosis, results accounted for similar amount of variance but were no longer significant. NR3C1 accounted for 16.5% of the variance, p=.083 and CRHR1 accounted for 27.0% of the variance, p=.26. For depression, results accounted for similar amounts of variance but were no longer trending towards significance: CRHR1 accounted for 23.5% of the variance, p=.42.
We followed our positive findings for NR3C1 with additional haplotype analyses of previously reported GR haplotypes (25, 26). No haplotype was significantly associated, with cortisol, psychosis or depression (see Supplemental materials).
Discussion
The data presented indicate that variation in NR3C1 (GR) gene is associated with elevations in cortisol levels (accounting for 29% of cortisol variance) and with psychosis in depression. NR3C1 mediated feedback has long been thought to play a potential role in the hypercortisolemia of major depression and was in part the basis of the application of the DST to assessing major depression with melancholia. However, two meta-analyses have indicated that the most significant contributor to DEX non-suppression in depression is the presence of psychosis (3, 8). Our data indicate that NR3C1 genetic variation contributes significantly to both psychosis in depression and to hypercortisolemia per se and suggest elevated cortisol levels may reflect deficits in feedback. However, DST’s were not performed in this study and thus we cannot be certain that these particular variants do account for poorer GC feedback.
A number of specific NR3C1 SNPs contributed significantly to hypercortisolemia after accounting for the effects of other SNPs, age, depression and psychosis severity and medication. (48, 49). Importantly, our data suggest that although medication effects contribute significantly for about 5% of the variance of cortisol levels, the majority of the variance (29%) is due to NR3C1 genetic variation. The Bcl1 (rs41423247) variant in NR3C1 appeared to contribute to nocturnal cortisol. This intronic SNP has no known effect on receptor function but has been reported to be associated with: poor negative feedback (50); elevated hormone responses to stress (51) increased risk for depression in premenopausal women (30); hypersensitivity to glucocorticoids and risk for depression (31); decreased activation of the prefrontal cortex on functional magnetic resonance imaging (f-MRI) in healthy controls (28); as well as decreased NR3C1 transcription in the dorsolateral prefrontal cortex in patients with schizophrenia or bipolar disorder (52). It is likely in LD with a functional NR3C1 polymorphism. In the postmortem study, a dose dependent effect of the C allele was observed with C/C homozygotes demonstrating the lowest levels of expression. In our sample, we also observed a dose effect of the C allele on cortisol levels from 6pam-1am across all three groups after age was accounted for. It predicted 7.2% of the variance --F(1,92)=7.58, p=.007. PMD patients demonstrate poorer performance on prefrontal mediated—as well as other--tasks (13, 53), and most recently, we reported that prefrontal activation on f-MRI is significantly decreased in PMD in response to working and verbal memory tasks. These data point to a potential role for this SNP in the development of hypercortisolemia and its cognitive sequelae. Of the other contributing NR3C1 SNP’s, rs33388 has been associated with poorer responses to lithium in bipolar disorder (54) and rs2918419 with insulin resistance in men- a possible sequelae of elevated cortisol levels (55). Both rs33388 and rs2918419 are intronic, and their effects are likely due to LD with one or more functional polymorphisms in NR3C1. Interestingly, rs41423247, rs33388, and rs2918419 are not in strong LD, suggesting that there may be more than one important as yet unknown functional variant that account for clinical prediction.
Regarding psychosis, the NR3C1 SNP rs6198 trended toward significance. This SNP is located in the 3′ UTR of exon 9β, and hence could affect mRNA stability or have other regulatory functions. Although rs6198 was not a significant predictor of hypercortisolemia, it is in moderate LD with the intronic SNP rs10482633 (r2 = 0.79), which was a predictor of cortisol levels. The rs6198 SNP is associated with alteration in mRNA stability of the GR-beta isoform (50) and decreased GR sensitivity (26). It has also recently been associated with attention deficit disorder (29). Attentional deficits have been reported by our group in PMD cohort (53). In addition, the SNP may play a role in smoking behavior (56).
Our results also indicate that variation in the CRHR1 gene confers risk for both depression and psychosis, accounting for over 24% of each. CRH has effects via CRHR1 on both the HPA axis as well as on extra-hypothalamic CRHR1, particularly in the amygdala where it appears not to be under feedback inhibition control and mediates the cognitive and behavioral responses to stress (57). The possible effects in the amygdala may explain the lack of relationship of CRH in this study to cortisol levels. Of interest is that we have reported that PMD patients demonstrate smaller amygdalae in comparison to NPMD’s and HC’s.
Of the 3 SNP’s that were significant contributors to psychosis, rs110402 has been perhaps studied best. A few years ago, Ressler’s group reported an interaction of the SNP with early abuse in increasing risk for developing later depression (17). More recent reports indicate it appears to increase risk for major depression (58) and alcoholism or alcohol use disorder. Ray et al (59) reported an interaction of this allele with early trauma on increasing risk for alcoholism; however, the increased risk for alcohol abuse disorder may be independent of early abuse (60) and this SNP interact with another (rs3811939) to increase risk. Functionally, the SNP is associated with altered f-MRI responses to emotional stimuli in the amygdala, nucleus accumbens and hypothalamus on f-MRI (61) and increased cortisol responses to a Trier Social Stress Test (62). Of the SNP’s that were of trend significance, rs4792887 has been associated with the development of suicidal ideation in the face of mild stress and has also been thought to represent a locus for a gene-environment interaction (63).
We also determined the contribution of CRHR1 SNPs to depression alone by excluding those subjects with a history of psychosis. Genetic variation in CRHR1 overall separated NPMD from HC’s, χ2(20)=36.35, p=.011, R-square=67.4, correct classification=83.0%. Early child abuse has been associated with increased risk for major psychiatric syndromes in adulthood with part of the risk being attributed to CRHR1 SNPs. We are currently assessing our data to determine if we can address this possible contributing factor.
One possible model to explain the relationship of CRH and GR to psychotic depression is to view patients with severe depression (both PMD and NPMD) as demonstrating a different pattern of CRH genetic variation than do HC’s with those depressed patients who develop psychosis and hypercortisolemia also demonstrating alterations in the GR gene. This would argue that PMD reflects alterations in two key components of the HPA axis. Still, the proportion of variance in cortisol levels or psychosis unaccounted for by NR3C1 and CRHR1 suggests that cortisol or NR3C1 or CRH genetic markers alone do not account for the entire risk for PMD. We have hypothesized that the effects of glucocorticoids on other systems—e.g., dopamine-- play a role in the pathophysiology of PMD and as such genetic alterations in these other systems may also confer risk for PMD. We are currently pursuing this hypothesis in other studies in our laboratory.
We did not assess genes for the CRHBP, AVP, or urocortin. These could play a role in cortisol or clinical symptoms but were beyond the scope of this study. There are a number of other limitations to this study, particularly the limited sample size and the number of SNPs assessed, and the lack of DST or another glucocorticoid challenge. The limited sample size is especially relevant for genes with a large numbers of SNPs. Consequently, we made a decision to conduct an overall analysis for each gene, rather than separate analyses for each SNP. While individual SNP results from models with all SNPs in the gene may be underpowered, an approach conducting single-SNP analyses without a prior gene-based test could be biased. Further studies on larger samples are required to replicate these findings and to better understand which specific variants account for severe depression.
Supplementary Material
Acknowledgments
In the last three years, Dr. Alan Schatzberg has served as a consultant for the following companies: BrainCells, CeNeRx, Cervel, Eli Lilly, Forest Labs, GSK, Jazz, Lundbeck, Merck, Neuronetics, Novadel, Novartis, Pfizer, PharmaNeuroBoost, Sanofi-Aventis, Sunovion, Synosia, Takeda, Xhale, Xytis, and Wyeth. He has held equity in the following companies: Amnestix, BrainCells, CeNeRx, Cervel, Corcept (co-founder), Delpor, Forest Labs, Merck, Neurocrine, Novadel, Pfizer, PharmaNeuroBoost, Somaxon, Synosia, and Xhale. Dr. Schatzberg is a named inventor on pharmacogenetic use patents on prediction of antidepressant response and glucocorticoid antagonists in psychiatry.
Dr. Greer Murphy has served as a consultant for Brain Resource, Ltd.
Fredric B. Kraemer has served as a consultant for BASF.
Several authors have been named on a provisional use patent related to the findings in this manuscript, including Alan Schatzberg, Laura Lazzeroni, Greer Murphy, and Jennifer Keller.
Aspects of this study were supported by grants from the Pritzker Foundation, NIH MH50604 to Alan Schatzberg, NIH MH19938 to Alan Schatzberg, and NIH/NCRR CTSA award number UL1 RR025744.
Footnotes
The following authors report no Conflicts of Interest: Anna Lembke, Lakshika Tennakoon, Jane Sarginson, and Gordon Williams.
References
- 1.Anton RF. Urinary free cortisol in psychotic depression. Biol Psychiatry. 1987;22(1):24–34. doi: 10.1016/0006-3223(87)90126-0. [DOI] [PubMed] [Google Scholar]
- 2.Arana GW, Barreira PJ, Cohen BM, Lipinski JF, Fogelson D. The dexamethasone suppression test in psychotic disorders. Am J Psychiatry. 1983;140(11):1521–3. doi: 10.1176/ajp.140.11.1521. [DOI] [PubMed] [Google Scholar]
- 3.Arana GW, Wilens TE, Baldessarini RJ. Plasma corticosterone and cortisol following dexamethasone in psychiatric patients. Psychoneuroendocrinology. 1985;10(1):49–60. doi: 10.1016/0306-4530(85)90038-1. [DOI] [PubMed] [Google Scholar]
- 4.Belanoff JK, Kalehzan M, Sund B, Fleming Ficek SK, Schatzberg AF. Cortisol activity and cognitive changes in psychotic major depression. Am J Psychiatry. 2001;158(10):1612–6. doi: 10.1176/appi.ajp.158.10.1612. [DOI] [PubMed] [Google Scholar]
- 5.Evans DL, Burnett GB, Nemeroff CB. The dexamethasone suppression test in the clinical setting. Am J Psychiatry. 1983;140(5):586–9. doi: 10.1176/ajp.140.5.586. [DOI] [PubMed] [Google Scholar]
- 6.Keller J, Flores B, Gomez RG, Solvason HB, Kenna H, Williams GH, et al. Cortisol circadian rhythm alterations in psychotic major depression. Biol Psychiatry. 2006;60(3):275–81. doi: 10.1016/j.biopsych.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 7.Lembke A, Gomez R, Tenakoon L, Keller J, Cohen G, Williams GH, et al. The mineralocorticoid receptor agonist, fludrocortisone, differentially inhibits pituitary-adrenal activity in humans with psychotic major depression. Psychoneuroendocrinology. 2012 doi: 10.1016/j.psyneuen.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson JC, Davis JM. DST studies in psychotic depression: a meta-analysis. Am J Psychiatry. 1997;154(11):1497–503. doi: 10.1176/ajp.154.11.1497. [DOI] [PubMed] [Google Scholar]
- 9.Posener JA, DeBattista C, Williams GH, Chmura Kraemer H, Kalehzan BM, Schatzberg AF. 24-Hour monitoring of cortisol and corticotropin secretion in psychotic and nonpsychotic major depression. Arch Gen Psychiatry. 2000;57(8):755–60. doi: 10.1001/archpsyc.57.8.755. [DOI] [PubMed] [Google Scholar]
- 10.Schatzberg AF, Rothschild AJ, Stahl JB, Bond TC, Rosenbaum AH, Lofgren SB, et al. The dexamethasone suppression test: identification of subtypes of depression. Am J Psychiatry. 1983;140(1):88–91. doi: 10.1176/ajp.140.1.88. [DOI] [PubMed] [Google Scholar]
- 11.Posener JA, DeBattista C, Williams GH, Kraemer HC, Kalehzan BM, Schatzberg AF. 24-Hour Monitoring of Cortisol and Corticotropin Secretion in Psychotic and Nonpsychotic Major Depression. Arch Gen Psychiatry. 2000;57(8):755–60. doi: 10.1001/archpsyc.57.8.755. [DOI] [PubMed] [Google Scholar]
- 12.Lupien SJ, Gillin CJ, Hauger RL. Working memory is more sensitive than declarative memory to the acute effects of corticosteroids: a dose-response study in humans. Behav Neurosci. 1999;113(3):420–30. doi: 10.1037//0735-7044.113.3.420. [DOI] [PubMed] [Google Scholar]
- 13.Gomez RG, Fleming SH, Keller J, Flores B, Kenna H, DeBattista C, et al. The neuropsychological profile of psychotic major depression and its relation to cortisol. Biol Psychiatry. 2006;60(5):472–8. doi: 10.1016/j.biopsych.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Leonard BE, Myint A. The psychoneuroimmunology of depression. Hum Psychopharmacol. 2009;24(3):165–75. doi: 10.1002/hup.1011. [DOI] [PubMed] [Google Scholar]
- 15.Hsu SY, Hsueh AJ. Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nat Med. 2001;7(5):605–11. doi: 10.1038/87936. [DOI] [PubMed] [Google Scholar]
- 16.De Kloet ER. Hormones and the stressed brain. Ann N Y Acad Sci. 2004;1018:1–15. doi: 10.1196/annals.1296.001. [DOI] [PubMed] [Google Scholar]
- 17.Turner JD, Alt SR, Cao L, Vernocchi S, Trifonova S, Battello N, et al. Transcriptional control of the glucocorticoid receptor: CpG islands, epigenetics and more. Biochem Pharmacol. 2010;80(12):1860–8. doi: 10.1016/j.bcp.2010.06.037. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez MM, Young LJ, Plotsky PM, Insel TR. Autoradiographic and in situ hybridization localization of corticotropin-releasing factor 1 and 2 receptors in nonhuman primate brain. J Comp Neurol. 1999;408(3):365–77. [PubMed] [Google Scholar]
- 19.Keck ME, Kern N, Erhardt A, Unschuld PG, Ising M, Salyakina D, et al. Combined effects of exonic polymorphisms in CRHR1 and AVPR1B genes in a case/control study for panic disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(7):1196–204. doi: 10.1002/ajmg.b.30750. [DOI] [PubMed] [Google Scholar]
- 20.Liu Z, Zhu F, Wang G, Xiao Z, Wang H, Tang J, et al. Association of corticotropin-releasing hormone receptor1 gene SNP and haplotype with major depression. Neurosci Lett. 2006;404(3):358–62. doi: 10.1016/j.neulet.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 21.Bradley RG, Binder EB, Epstein MP, Tang Y, Nair HP, Liu W, et al. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch Gen Psychiatry. 2008;65(2):190–200. doi: 10.1001/archgenpsychiatry.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ressler KJ, Bradley B, Mercer KB, Deveau TC, Smith AK, Gillespie CF, et al. Polymorphisms in CRHR1 and the serotonin transporter loci: gene × gene × environment interactions on depressive symptoms. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(3):812–24. doi: 10.1002/ajmg.b.31052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ressler S, Radhi J, Aigner T, Loo C, Zwerschke W, Sergi C. Insulin-like growth factor-binding protein-3 in osteosarcomas and normal bone tissues. Anticancer Res. 2009;29(7):2579–87. [PubMed] [Google Scholar]
- 24.Binder EB, Owens MJ, Liu W, Deveau TC, Rush AJ, Trivedi MH, et al. Association of polymorphisms in genes regulating the corticotropin-releasing factor system with antidepressant treatment response. Arch Gen Psychiatry. 2010;67(4):369–79. doi: 10.1001/archgenpsychiatry.2010.18. [DOI] [PubMed] [Google Scholar]
- 25.van Winsen LM, Manenschijn L, van Rossum EF, Crusius JB, Koper JW, Polman CH, et al. A glucocorticoid receptor gene haplotype (TthIII1/ER22/23EK/9beta) is associated with a more aggressive disease course in multiple sclerosis. J Clin Endocrinol Metab. 2009;94(6):2110–4. doi: 10.1210/jc.2008-2194. [DOI] [PubMed] [Google Scholar]
- 26.Koper JW, Stolk RP, de Lange P, Huizenga NA, Molijn GJ, Pols HA, et al. Lack of association between five polymorphisms in the human glucocorticoid receptor gene and glucocorticoid resistance. Hum Genet. 1997;99(5):663–8. doi: 10.1007/s004390050425. [DOI] [PubMed] [Google Scholar]
- 27.van Rossum EF, van den Akker EL. Glucocorticoid resistance. Endocr Dev. 2011;20:127–36. doi: 10.1159/000321234. [DOI] [PubMed] [Google Scholar]
- 28.El-Hage W, Phillips ML, Radua J, Gohier B, Zelaya FO, Collier DA, et al. Genetic modulation of neural response during working memory in healthy individuals: interaction of glucocorticoid receptor and dopaminergic genes. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.145. [DOI] [PubMed] [Google Scholar]
- 29.Fortier ME, Sengupta SM, Grizenko N, Choudhry Z, Thakur G, Joober R. Genetic evidence for the association of the hypothalamic-pituitary-adrenal (HPA) axis with ADHD and methylphenidate treatment response. Neuromolecular Med. 2013;15(1):122–32. doi: 10.1007/s12017-012-8202-1. [DOI] [PubMed] [Google Scholar]
- 30.Krishnamurthy P, Romagni P, Torvik S, Gold PW, Charney DS, Detera-Wadleigh S, et al. Glucocorticoid receptor gene polymorphisms in premenopausal women with major depression. Horm Metab Res. 2008;40(3):194–8. doi: 10.1055/s-2007-1004541. [DOI] [PubMed] [Google Scholar]
- 31.van Rossum EF, Binder EB, Majer M, Koper JW, Ising M, Modell S, et al. Polymorphisms of the glucocorticoid receptor gene and major depression. Biol Psychiatry. 2006;59(8):681–8. doi: 10.1016/j.biopsych.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;36(12):1319–25. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- 33.Mehta D, Gonik M, Klengel T, Rex-Haffner M, Menke A, Rubel J, et al. Using polymorphisms in FKBP5 to define biologically distinct subtypes of posttraumatic stress disorder: evidence from endocrine and gene expression studies. Arch Gen Psychiatry. 2011;68(9):901–10. doi: 10.1001/archgenpsychiatry.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimmermann P, Bruckl T, Nocon A, Pfister H, Binder EB, Uhr M, et al. Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: results from a 10-year prospective community study. Am J Psychiatry. 2011;168(10):1107–16. doi: 10.1176/appi.ajp.2011.10111577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299(11):1291–305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery, and Psychiatry. 1980;23:49–65. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thase ME, Hersen M, Bellack AS, Himmelhoch JM, Kupfer DJ. Validation of a Hamilton subscale for endogenomorphic depression. J Affect Disord. 1983;5(3):267–78. doi: 10.1016/0165-0327(83)90050-2. [DOI] [PubMed] [Google Scholar]
- 38.Overall JE, Gorham DE. The brief psychiatric rating scale. Psychological Reports. 1961;10:799–812. [Google Scholar]
- 39.First MB, Spitzer RL, Gibbone M, Williams JBW. Structured Clinical Interview for the DSM-IV-TR Axis I Disorders - Patient Edition. 1997. [Google Scholar]
- 40.Pettersson FH, Anderson CA, Clarke GM, Barrett JC, Cardon LR, Morris AP, et al. Marker selection for genetic case-control association studies. Nat Protoc. 2009;4(5):743–52. doi: 10.1038/nprot.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lomelin D, Jorgenson E, Risch N. Human genetic variation recognizes functional elements in noncoding sequence. Genome Res. 2010;20(3):311–9. doi: 10.1101/gr.094151.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449(7164):851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Bakker PI, Graham RR, Altshuler D, Henderson BE, Haiman CA. Transferability of tag SNPs to capture common genetic variation in DNA repair genes across multiple populations. Pac Symp Biocomput. 2006:478–86. [PubMed] [Google Scholar]
- 44.Ballard DH, Cho J, Zhao H. Comparisons of multi-marker association methods to detect association between a candidate region and disease. Genet Epidemiol. 2010;34(3):201–12. doi: 10.1002/gepi.20448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, Dobie DJ, et al. Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the Consortium on the Genetics of Schizophrenia. Am J Psychiatry. 2011;168(9):930–46. doi: 10.1176/appi.ajp.2011.10050723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gauderman WJ, Murcray C, Gilliland F, Conti DV. Testing association between disease and multiple SNPs in a candidate gene. Genet Epidemiol. 2007;31(5):383–95. doi: 10.1002/gepi.20219. [DOI] [PubMed] [Google Scholar]
- 47.Kwee LC, Liu D, Lin X, Ghosh D, Epstein MP. A powerful and flexible multilocus association test for quantitative traits. Am J Hum Genet. 2008;82(2):386–97. doi: 10.1016/j.ajhg.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garrett A, Kelly R, Gomez R, Keller J, Schatzberg AF, Reiss AL. Aberrant brain activation during a working memory task in psychotic major depression. Am J Psychiatry. 2011;168(2):173–82. doi: 10.1176/appi.ajp.2010.09121718. [DOI] [PubMed] [Google Scholar]
- 49.Kelly R, Garrett A, Cohen J, Gomez RG, Lembke A, Keller J, et al. Altered brain function underlying verbal memory encoding and retrieval in psychotic major depression. Psychiatry Research: Neuroimaging. 2012 doi: 10.1016/j.pscychresns.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Claes S. Glucocorticoid receptor polymorphisms in major depression. Ann N Y Acad Sci. 2009;1179:216–28. doi: 10.1111/j.1749-6632.2009.05012.x. [DOI] [PubMed] [Google Scholar]
- 51.Kumsta R, Entringer S, Koper JW, van Rossum EF, Hellhammer DH, Wust S. Sex specific associations between common glucocorticoid receptor gene variants and hypothalamus-pituitary-adrenal axis responses to psychosocial stress. Biol Psychiatry. 2007;62(8):863–9. doi: 10.1016/j.biopsych.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 52.Sinclair D, Fullerton JM, Webster MJ, Shannon Weickert C. Glucocorticoid receptor 1B and 1C mRNA transcript alterations in schizophrenia and bipolar disorder, and their possible regulation by GR gene variants. PLoS One. 2012;7(3):e31720. doi: 10.1371/journal.pone.0031720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schatzberg AF, Posener JA, DeBattista C, Kalehzan BM, Rothschild AJ, Shear PK. Neuropsychological deficits in psychotic versus nonpsychotic major depression and no mental illness. Am J Psychiatry. 2000;157(7):1095–100. doi: 10.1176/appi.ajp.157.7.1095. [DOI] [PubMed] [Google Scholar]
- 54.Szczepankiewicz A, Rybakowski JK, Suwalska A, Hauser J. Glucocorticoid receptor polymorphism is associated with lithium response in bipolar patients. Neuro Endocrinol Lett. 2011;32(4):545–51. [PubMed] [Google Scholar]
- 55.Syed AA, Halpin CG, Irving JA, Unwin NC, White M, Bhopal RS, et al. A common intron 2 polymorphism of the glucocorticoid receptor gene is associated with insulin resistance in men. Clin Endocrinol (Oxf) 2008;68(6):879–84. doi: 10.1111/j.1365-2265.2008.03175.x. [DOI] [PubMed] [Google Scholar]
- 56.Rovaris DL, Mota NR, de Azeredo LA, Cupertino RB, Bertuzzi GP, Polina ER, et al. MR and GR functional SNPs may modulate tobacco smoking susceptibility. J Neural Transm. 2013 doi: 10.1007/s00702-013-1012-2. [DOI] [PubMed] [Google Scholar]
- 57.Funk CK, O’Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26(44):11324–32. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishitobi Y, Nakayama S, Yamaguchi K, Kanehisa M, Higuma H, Maruyama Y, et al. Association of CRHR1 and CRHR2 with major depressive disorder and panic disorder in a Japanese population. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(4):429–36. doi: 10.1002/ajmg.b.32046. [DOI] [PubMed] [Google Scholar]
- 59.Ray LA, Sehl M, Bujarski S, Hutchison K, Blaine S, Enoch MA. The CRHR1 gene, trauma exposure, and alcoholism risk: a test of G × E effects. Genes Brain Behav. 2013;12(4):361–9. doi: 10.1111/gbb.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ribbe K, Ackermann V, Schwitulla J, Begemann M, Papiol S, Grube S, et al. Prediction of the risk of comorbid alcoholism in schizophrenia by interaction of common genetic variants in the corticotropin-releasing factor system. Arch Gen Psychiatry. 2011;68(12):1247–56. doi: 10.1001/archgenpsychiatry.2011.100. [DOI] [PubMed] [Google Scholar]
- 61.Hsu DT, Mickey BJ, Langenecker SA, Heitzeg MM, Love TM, Wang H, et al. Variation in the corticotropin-releasing hormone receptor 1 (CRHR1) gene influences fMRI signal responses during emotional stimulus processing. J Neurosci. 2012;32(9):3253–60. doi: 10.1523/JNEUROSCI.5533-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahon PB, Zandi PP, Potash JB, Nestadt G, Wand GS. Genetic association of FKBP5 and CRHR1 with cortisol response to acute psychosocial stress in healthy adults. Psychopharmacology (Berl) 2013;227(2):231–41. doi: 10.1007/s00213-012-2956-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wasserman D, Sokolowski M, Rozanov V, Wasserman J. The CRHR1 gene: a marker for suicidality in depressed males exposed to low stress. Genes Brain Behav. 2008;7(1):14–9. doi: 10.1111/j.1601-183X.2007.00310.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.