

Summary
The helicase RTEL1 promotes t-loop unwinding and suppresses telomere fragility to maintain the integrity of vertebrate telomeres. An interaction between RTEL1 and PCNA is important to prevent telomere fragility, but how RTEL1 engages with the telomere to promote t-loop unwinding is unclear. Here, we establish that the shelterin protein TRF2 recruits RTEL1 to telomeres in S phase, which is required to prevent catastrophic t-loop processing by structure-specific nucleases. We show that the TRF2-RTEL1 interaction is mediated by a metal-coordinating C4C4 motif in RTEL1, which is compromised by the Hoyeraal-Hreidarsson syndrome (HHS) mutation, RTEL1R1264H. Conversely, we define a TRF2I124D substitution mutation within the TRFH domain of TRF2, which eliminates RTEL1 binding and phenocopies the RTEL1R1264H mutation, giving rise to aberrant t-loop excision, telomere length heterogeneity, and loss of the telomere as a circle. These results implicate TRF2 in the recruitment of RTEL1 to facilitate t-loop disassembly at telomeres in S phase.
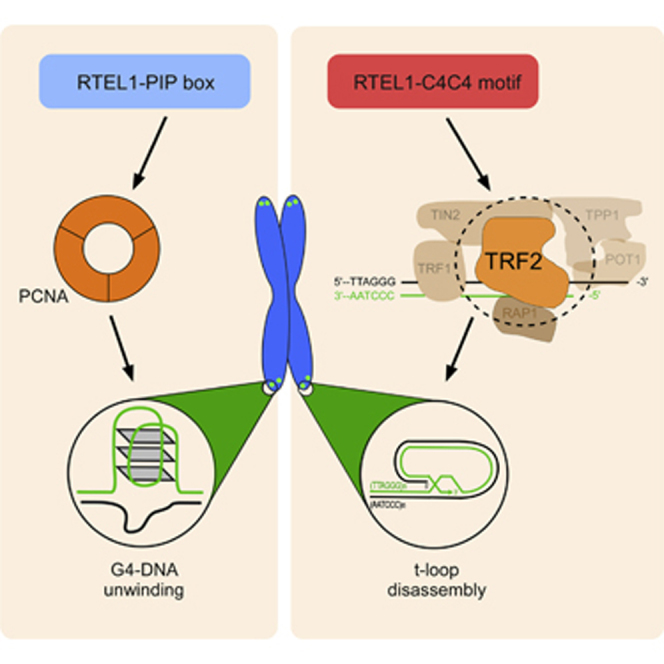
Graphical Abstract
Highlights
-
•
An S phase-specific TRF2-RTEL1 interaction is required for t-loop disassembly
-
•
RTEL1 C4C4 and PIP-box motifs control distinct genome maintenance pathways
-
•
A unique binding site in TRF2 interacts with and recruits RTEL1 to telomeres
-
•
TRF2-RTEL1 interaction is abolished by the disease causing RTEL1R1264H mutation
RTEL1 is an essential DNA helicase that disassembles telomere loops (t-loops) to maintain integrity of chromosome ends. Sarek et al. now establish the mechanism by which RTEL1 is recruited to telomeres to execute t-loop unwinding, which is dependent on an S phase-specific interaction between RTEL1 and the shelterin component TRF2.
Introduction
Vertebrate telomeres are essential nucleoprotein structures that protect chromosome ends from promiscuous DNA repair activities and nucleolytic degradation (for review, see de Lange, 2005). Telomeric DNA is composed of repetitive double-stranded TTAGGG sequences that extend into a 3′ single-stranded overhang on the G-rich strand (Moyzis et al., 1988; Wellinger et al., 1993). Due to the inherent nature of semiconservative DNA replication, telomeres progressively shorten with each cell division. To counter the end replication problem, telomeric repeats can be extended by telomerase, a reverse transcriptase that utilizes an associated RNA moiety (TERC) as a template to add de novo telomeric sequences to the 3′ end of the G-rich strand of the telomere (Greider and Blackburn, 1985; Shippen-Lentz and Blackburn, 1990).
Telomere function is also critically dependent on a complex formed by six telomere-associated proteins, known as shelterin, which comprises TRF1 (telomere repeat binding factor 1), TRF2 (telomere repeat binding factor 2), POT1 (protection of telomeres 1), TIN2 (TRF1-interacting nuclear factor 2), Rap1 (repressor activator protein 1), and TPP1 (TINF2-interacting protein 2) that function to safeguard chromosome ends from the DNA damage response (DDR) and to control telomere maintenance by telomerase (for review, see Palm and de Lange, 2008). Shelterin associates with telomeres by virtue of intrinsic duplex telomeric repeat binding conferred by TRF1 and TRF2 and binding of the single-stranded telomeric 3′ overhang by POT1 (Baumann and Cech, 2001). While TRF1 promotes telomere replication and functions as a negative regulator of telomere length (Sfeir et al., 2009), TRF2 plays a crucial role in telomere end protection (van Steensel et al., 1998). Consequently, loss of TRF1 compromises telomere replication, giving rise to telomere fragility, whereas loss of TRF2 results in loss of the 3′ overhang and extensive chromosome end-to-end fusions (Celli and de Lange, 2005; Sfeir et al., 2009).
Electron microscopy and stochastic optical reconstruction microscopy have revealed that telomeres can adopt lasso-like configurations called t-loops (Doksani et al., 2013; Griffith et al., 1999), which are believed to form as a result of strand invasion of the 3′ single-stranded telomeric DNA into upstream duplex TTAGGG repeats (Griffith et al., 1999). As the 3′ end of the telomere is buried within the t-loop, this structure has been proposed to safeguard the chromosome ends against nucleolytic attack or promiscuous DNA repair activities (for review, see O’Sullivan and Karlseder, 2010). The amino-terminal basic domain of the shelterin component TRF2 is important for stabilizing t-loops and acts to protect the structure from unscheduled nucleolytic processing (Wang et al., 2004). TRF2 is also essential for t-loop formation in vivo (Doksani et al., 2013), which likely contributes to its key role in end protection.
Mechanisms must also exist to disassemble t-loops to allow telomerase access to the 3′ end and/or to avoid collisions with the replisome during S phase. RTEL1 (regulator of telomere length 1) (Ding et al., 2004) is an essential helicase with intrinsic D-loop-disrupting activity that is co-opted to telomeres to dismantle t-loops (Uringa et al., 2012). In the absence of RTEL1, t-loops are inappropriately resolved by the SLX1-SLX4 nuclease complex, leading to catastrophic telomere length changes and loss of the telomere as double-stranded telomere circles (T-circles or TCs) (Vannier et al., 2012). RTEL1 also executes a second function to counteract the formation of telomeric guanine quadruplex (G4) DNA structures during telomere replication, which are a major source of telomere fragility (Vannier et al., 2012). Direct binding of RTEL1 to proliferating cell nuclear antigen (PCNA) is critical for preventing telomere fragility but is dispensable for t-loop disassembly (Vannier et al., 2013). The importance of RTEL1 in human disease has been established with the identification of variants that confer increased risk of human brain tumors (Shete et al., 2009; Wrensch et al., 2009). Mutations in RTEL1 also give rise to Hoyeraal-Hreidarsson syndrome (HHS), a severe form of the telomereopathy dyskeratosis congenita (DKC; for review, see Vannier et al., 2014). HHS is characterized by its similarity to DKC, but patients also present with additional complications including bone marrow failure, microcephaly, and immunodeficiency (Vulliamy et al., 2006). So far, 18 distinct RTEL1 mutations have been identified in HHS (for review, see Vannier et al., 2014), but how these mutations impact on the known functions of RTEL1 is not known.
Here, we define the mechanism by which RTEL1 is recruited to telomeres to promote t-loop disassembly, which we show is dependent on an S phase-specific interaction with the shelterin component TRF2. We demonstrate that the TRF2 interaction site in RTEL1 maps to an uncharacterized C4C4 metal-binding motif, which is commonly mutated in HHS patients. We establish that the p.R1264H mutation within the C4C4 motif, which has a carrier frequency of 1 in 100 in the Ashkenazi Jewish population (Fedick et al., 2014), compromises the RTEL1-TRF2 interaction, leading to catastrophic t-loop resolution accompanied by rapid changes in telomere length, telomere loss, and TC formation. Conversely, we identify a single amino acid substitution (p.I124D) within the TRFH dimerization domain of TRF2 that specifically abolishes RTEL1 binding and prevents its recruitment to telomeres. Remarkably, the TRF2 p.I124D mutation gives rise to rapid changes in telomere length, telomere loss, and TC formation and thereby phenocopies the p.R1264H mutation in RTEL1. Our results reveal that t-loops are highly dynamic and regulated structures whose assembly and disassembly is coordinated during the cell cycle.
Results
RTEL1 Interacts Directly with TRF2
RTEL1 serves a dual function in telomere integrity; it suppresses telomere fragility by unwinding telomeric G4 DNA and also disassembles t-loops to prevent catastrophic loss of the telomere (Vannier et al., 2012). Recently, we have shown that RTEL1’s ability to suppress telomere fragility strictly depends on an interaction with PCNA. Unexpectedly, however, the RTEL1-PCNA interaction was found to be dispensable for t-loop unwinding (Vannier et al., 2013), suggesting the existence of a separate mechanism for recruiting RTEL1 to telomeres. We reasoned that RTEL1 could be recruited to telomeres via an interaction with one of the shelterin components, which are the major constituents of vertebrate telomeres.
To test this hypothesis, we first modified a human bacterial artificial chromosome (BAC) carrying the RTEL1 genomic locus by inserting a FLAP tag (containing GFP and Flag) at the N terminus (NFLAP) of the RTEL1 coding sequence using homologous recombination (Poser et al., 2008). Extracts from HEK293 cells stably expressing NFLAP-tagged RTEL1 at near endogenous levels were subjected to immunoprecipitation for 5 of the 6 endogenous shelterin components and then blotted for RTEL1. Although known partner shelterin components were present in immunoprecipitates of endogenous TRF1, TPP1, or POT1, NFLAP-tagged RTEL1 was not detectable (Figure S1A). However, blotting of immunoprecipitates of endogenous TRF2, and to a lesser extent Rap1, revealed an association with NFLAP-tagged RTEL1 (Figure 1A). Importantly, these interactions were resistant to benzonase treatment, indicating that the interactions between TRF2/Rap1 and RTEL1 are independent of DNA/RNA bridging. Furthermore, TRF2 immunoprecipitated RTEL1 from lysates that were mock depleted with IgG but not from extracts immunodepleted for NFLAP-tagged RTEL1 with a GFP antibody (Figure 1B), thus demonstrating the specificity of the interaction. The interaction between RTEL1 and TRF2/Rap1 was also observed by reciprocal coimmunoprecipitation. TRF2 was coprecipitated from HEK293 cells with NFLAP-tagged RTEL1, but not with irrelevant control protein GFP-ALC1 (Figure 1C). Silencing of Rap1 with lentivirus-mediated RNA interference (shRNA) did not significantly affect binding of RTEL1 to TRF2 (data not shown), which raised the possibility that RTEL1 interacts indirectly with Rap1 and directly with TRF2. To confirm direct binding of RTEL1 to TRF2 in vitro, we performed GST pull-down assays with purified full-length and truncated versions of human TRF2 and affinity purified NFLAP-RTEL1. Western blotting analysis revealed that NFLAP-RTEL1 associates with full-length TRF2 protein (GST-TRF2) and TRF2 lacking the N-terminal basic domain (GST-TRF2ΔB), but not with the basic N-terminal domain (GST-ΔB) or GST alone (Figure 1D). Taken together, these data indicate that RTEL1 directly interacts with TRF2.
Figure 1.

RTEL1 Directly Interacts with TRF2 Predominantly in S Phase
(A) Whole-cell extracts of control (−benz.) and benzonase-treated (+benz.) 293 FLAP-tagged RTEL1 cells were immunoprecipitated using anti-TRF2 or anti-Rap1 antibodies. Protein complexes were analyzed by immunoblotting for GFP, TRF2, and Rap1.
(B) Extracts shown in (A) were immunodepleted with anti-GFP antibody (GFP depl.) or with control IgG (IgG depl.). The GFP- and control-depleted extracts were subjected to immunoprecipitation by anti-TRF2 antibody and analyzed by western blotting as indicated.
(C) Whole-cell extracts of 293 FLAP-tagged RTEL1 cells shown in (A), HEK293 cells, and HEK293 cells transfected with expression vector for ALC1-GFP were immunoprecipitated using GFP-TRAP beads. Immunocomplexes were analyzed by western blotting with indicated antibodies.
(D) Whole-cell extracts from 293 FLAP-tagged RTEL1 cells were incubated with GST control, GST-TRF2, GST-TRF2ΔB, or GST-ΔB. The bound complexes were immunoprecipitated with anti-GFP antibody analyzed by western blotting against GST and GFP.
(E) 293 N-FLAP-RTEL1 cells stably expressing Myc-tagged TRF2 were either cultured asynchronously (A) or released from double-thymidine block (S, G2, and M phase) or from thymidine plus nocodazole block (G1 phase). Cell extracts were analyzed for input (right panels) or were immunoprecipitated with antibodies against Flag (left panel). The immunoblots were probed with anti-Flag and anti-Myc antibodies. Asterisk indicates the nonspecific cross-reactivity detected with Flag antibody.
(F) RTEL1v5 MEFs stably expressing Myc-tagged TRF2 were cultured as indicated in (E). The graph depicts quantification of the interaction spots per cell between RTEL1 and TRF2 as determined by in situ PLA assay. Data are representative of two independent experiments as mean ± SD (∗∗∗∗p < 0.0001, one-way ANOVA). See also Figures S1 and S2.
TRF2 Associates with RTEL1 in a Cell Cycle-Dependent Manner
Since RTEL1 only transiently associates with telomeres (Uringa et al., 2012), we next examined whether the association of RTEL1 with TRF2 varied throughout the cell cycle. To this end we synchronized HEK293 NFLAP-RTEL1 cells and RTEL1v5 mouse embryonic fibroblasts (MEFs), stably expressing Myc-tagged TRF2, by double thymidine block or by incubation with thymidine followed by nocodazole treatment (Figures S1B and S1C). Although a low level of association between RTEL1 and TRF2 was detected throughout the cell cycle, the RTEL1-TRF2 interaction is significantly increased at the G1-to-S transition and peaked in S phase (Figures 1E and S1D). Similarly, we observed that the RTEL1-TRF2 interaction is significantly enhanced in S phase RTEL1v5 mouse cells as determined by proximity ligation assay PLA (Figures 1F and S1E) and is unaffected by inhibiting DNA replication (Figure S2). These results establish that the RTEL1-TRF2 interaction occurs predominantly during S phase and does not require active DNA replication.
RTEL1 C4C4 Motif Is Critical for Its Association with TRF2
To determine the minimal TRF2 interaction site within RTEL1, we conducted a peptide array binding experiment, as previously described (Ward et al., 2010). We reasoned that the TRF2 interaction site was unlikely to map to the N-terminal helicase motif and so focused our attention on the C terminus of RTEL1, which contains three recognizable motifs: a PIP box (PCNA binding), a harmonin N-like domain (Faure et al., 2014), and a C4C4 motif (unknown function). Binding of GST-TRF2 to a 20-mer peptide-scanning array encompassing the C-terminal region of RTEL1 identified a single TRF2-binding peptide that overlapped with the uncharacterized C4C4 motif (Figure 2A).
Figure 2.

RTEL1 Interacts with TRF2 through Cysteine-Rich C4C4 Domain
(A) Immunoblotting of a peptide array for the C-terminal 1147–1369 amino acids of human RTEL1. The arrays were incubated either with GST or with human TRF2 fused with GST, and bound proteins were examined by immunoblotting against GST.
(B) Whole-cell extracts from HEK293 cells were incubated in the absence (−EDTA) or presence (+EDTA) of 50 mM EDTA either with GST or the C4C4 domain (corresponding to human RTEL11234–1292 region) fused to GST. The bound complexes were immobilized to glutathione-linked Sepharose beads followed by western blotting analysis against TRF2 and GST.
(C) Schematic representation of mammalian RTEL1 protein. Full-length RTEL1 is segmented into functional domains shown as colored boxes. Asterisks indicate locations of mutations introduced into human or mouse C4C4 domain.
(D) HEK293 cells were stably transduced with lentiviruses carrying an empty vector (ctrl) or full-length versions of the wild-type Flag-RTEL1 (WT), single-mutant Flag-RTEL1R1264H (R/H), or double-mutant Flag-RTEL1C1279A/C1282A (C/A) as indicated. Whole-cell extracts were immunoprecipitated with anti-TRF2 antibody, and western blotting was carried out to detect the interaction between TRF2 and Flag-tagged RTEL1.
(E) Cell extracts as indicated in (D) were incubated with GST control or human TRF2 fused to GST. The protein complexes were immobilized to glutathione-linked Sepharose beads followed by western blotting analysis against Flag and GST.
(F) Whole-cell extracts of MSK-41 fibroblasts and BJ-hTERT control fibroblasts were immunoprecipitated with anti-RTEL1 antibody. Immunocomplexes were analyzed with antibodies as indicated.
(G) The well-characterized interaction between TRF2 and Rap1 was included as a specificity control for the PLA assay. The graphs depict quantification of the interaction spots per cell as determined by PLA using antibodies as indicated.
(H) Quantification of the interaction between TRF2 and RTEL1 as determined by in situ PLA assay in MSK-41, RPE-1, and BJ-hTERT cells with indicated antibodies. Dashed lines indicate nucleus outlines (as determined by DAPI in blue). Data in (G) and (H) are representative of two independent experiments as mean ± SD (∗∗∗∗p < 0.0001, one-way ANOVA). See also Figure S3.
To confirm that the C4C4 motif of RTEL1 is sufficient to confer binding to TRF2 and to determine if this interaction requires metal coordination, we generated recombinant GST and GST-C4C4 motif fusions and performed pull-downs of endogenous TRF2 from HEK293 cell extracts in the absence (−EDTA) or presence (+EDTA) of EDTA. As shown in Figure 2B (lane 2), the lower isoform of endogenous TRF2 is specifically pulled down from whole-cell extract by GST-C4C4, but not with GST alone (Figure 2B; lane 1). Addition of EDTA to the pull-down, which is predicted to abolish metal coordination of the C4C4 motif, resulted in the complete loss of TRF2 binding (Figure 2B; lane 4). These results reveal that the C4C4 motif of RTEL1 is sufficient for binding to TRF2, and this requires metal coordination.
To assess the biological importance of the C4C4 domain for RTEL1 activities, we introduced C4C4 mutations by site-directed mutagenesis into the human and mouse versions of RTEL1 (see map in Figure 2C). The RTEL1R1264H (human)/RTEL1R1237H (mouse) mutation corresponds to the HHS disease-causing mutation (Ballew et al., 2013) and is located between the second and third metal-coordinating pair of cysteines (Kellenberger et al., 2005). We also generated two cysteine-to-alanine substitutions (RTEL1C1279A/C1282A in human or RTEL1C1252A/C1255A in mouse) that are predicted to disrupt the fourth pair of cysteines coordinating a second metal ion (Kellenberger et al., 2005).
First, we evaluated whether the mutations within the C4C4 motif affect the RTEL1-TRF2 interaction. To this end U2OS cells stably expressing empty vector, wild-type (WT) Myc-C4C4 (an equivalent to the human RTEL11234–1292 region), or two C4C4 mutants, Myc-C4C4R1264H and Myc-C4C4C1279A/C1282A, were analyzed by immunoblotting with anti-Myc antibody. The decreased steady-state abundance of the mutated human C4C4 motif suggests that the mutations might impact on its stability (Figure S3A). To account for the reduced expression, we compensated Myc-C4C4 protein levels of both mutants in immunoprecipitation experiments. Western blotting analysis revealed that endogenous TRF2 was efficiently immunoprecipitated with WT Myc-C4C4 peptide but not with either mutant (Figure S3B). We also introduced the C4C4 motif mutations into the full-length human RTEL1 and found that endogenous TRF2 interacted exclusively with WT Flag-RTEL1, whereas mutations within the C4C4 motif abolished RTEL1’s ability to bind TRF2 (Figure 2D). These results were also confirmed by an in vitro GST pull-down experiment, in which we observed robust interaction of GST-TRF2 with full-length Flag-tagged WT RTEL1, but not with RTEL1R1264H or double-mutant RTEL1C1279A/C1282A (Figure 2E).
Finally, we examined the interaction of TRF2 with RTEL1 in normal BJ human fibroblasts and in patient-derived MSK-41 cells harboring inherited homozygous RTEL1R1264H HHS disease-causing mutations (Ballew et al., 2013). As shown in Figure 2F, the endogenous RTEL1-TRF2 interaction is undetectable in MSK-41 cells (lane 1) but is readily detectable in normal BJ human fibroblasts (Figure 2F, lane 2). An interaction between endogenous TRF2 and Rap1, which served as a positive control, was observed by PLA assay in BJ fibroblasts, retinal-pigmented epithelial RPE1 cells, and in the patient-derived MSK-41 fibroblasts (Figure 2G). In contrast, the RTEL1-TRF2 interaction was only detectable by PLA assay in BJ fibroblasts and RPE1 cells but not in the MSK-41 fibroblasts (Figure 2H). These results establish that the interaction of RTEL1 and TRF2 is compromised by the RTEL1 p.R1264H mutation in HHS.
Mutations in RTEL1 C4C4 Motif Result in Aberrant Telomeres in Metaphase
To investigate the impact of C4C4 motif mutations on the telomere functions of RTEL1, we complemented conditional RTEL1F/F MEFs with retroviruses expressing either empty vector, full-length WT V5-RTEL1, single-mutant V5-RTEL1R1237H, or double-mutant V5-RTEL1C1252A/C1255A. Transient expression of Cre recombinase in the transduced RTEL1F/F MEFs resulted in the expected loss of the floxed RTEL1 alleles and concomitant elimination of the endogenous RTEL1 protein within 96 hr, leaving the V5-RTEL1 WT or mutants as the only source of RTEL1 protein (Figures 3A and 3B). Conditional deletion of RTEL1 in MEFs leads to rapid accumulation of TCs as a consequence of processing of persistent t-loops by the SLX1/4 nuclease (Vannier et al., 2012). Cells expressing empty vector and MEFs complemented either with V5-RTEL1R1237H or V5-RTEL1C1252A/C1255A exhibited very high levels of TCs after eliminating the RTEL1 floxed alleles. In contrast, RTEL1-deficient cells expressing WT V5-RTEL1 exhibited low levels of TCs, comparable to control virus-infected cells (Figure 3C).
Figure 3.

Alterations of RTEL1 in the C4C4 Domain Result in Telomeric Dysfunction
(A) PCR analysis of genomic DNA isolated from RTEL1F/F MEFs stably expressing empty vector (ctrl), wild-type V5-RTEL1 (WT), single-mutant V5-RTEL1R1237H (R/H), or double-mutant V5-RTEL1C1252A/C1255A (C/A) performed 96 hr after infection with control- or Cre-expressing adenovirus.
(B) Western blotting analysis of cells described in (A) to monitor loss of endogenous RTEL1 after Cre introduction and to determine complementation efficiency with ectopic wild-type and RTEL1 mutants.
(C) Phi29-dependent telomere circles (TCs; upper panel), detected in the same cells as detailed in (A). The extent of [32P] incorporation was quantified (bottom panel) from the autoradiographs and the level of [32P] incorporation was normalized to the empty vector control, which was arbitrarily assigned a value of 100%. Data represent the average of three independent experiments ± SD (∗∗p < 0.01, ∗∗∗p < 0.001, Student’s t test).
(D and E) Quantification of telomere fragility (D) and telomere loss (E) per metaphase, determined in cells described in (A). Representative images of the telomere FISH on metaphases are shown on the right panel in (E) with asterisks depicting telomere loss. Data represent the average of three independent experiments as mean ± SD from at least 60 metaphases (∗∗∗∗p < 0.0001; one-way ANOVA). Green, TTAGGG signal; blue, DAPI.
(F) Quantification of the frequency of nuclei with telomere dysfunction-induced foci (TIFs) from the same cells as in (A). Cells with ≥ 2 telomeric 53BP1 foci were scored as TIF positive. Data are representative of three independent experiments as mean ± SD; n > 100 nuclei per single experiment (∗p < 0.05, ∗∗p < 0.01; Student’s t test).
(G) Quantification (left panel) of telomere-sister chromatid exchange (T-SCE) events per metaphase, detected in cells shown in (A). Representative CO-FISH images after labeling leading (green) and lagging (red) strand telomeres are indicated in the right-hand panel. The yellow arrows show chromosomes that display recombination between telomeres. Data are representative of the mean ± SD from at least 40 metaphases (∗∗∗∗p < 0.0001; one-way ANOVA). See also Figure S4.
Next, we examined the levels of telomere fragility and telomere loss in complemented RTEL1F/F MEFs by fluorescent in situ hybridization (FISH) using telomere-specific probes. As expected, inactivation of RTEL1 in RTEL1F/F MEFs, 96 hr after Cre infection, resulted in high levels of fragile telomeres, which manifest as multiple spatially distinct telomere FISH signals (Figure 3D). However, inactivation of RTEL1 in RTEL1F/F MEFs complemented with either WT V5-RTEL1, V5-RTEL1R1237H, or V5-RTEL1C1252A/C1255A did not result in significant induction of telomere fragility when compared to cells complemented with an empty vector control (Figure 3D). Strikingly, inactivation of RTEL1 in RTEL1F/F MEFs expressing V5-RTEL1R1237H or V5-RTEL1C1252A/C1255A resulted in a significant increase in telomere loss, corresponding to 6.2 ± 2.8 and 5.8 ± 3.3 telomeres that lacked a telomere FISH signal on one or both sister chromatid ends, which is comparable to the levels observed in RTEL1 null cells (7.5 ± 3.2) and significantly different from WT V5-RTEL1-complemented cells that present 2.7 ± 1.4 ends lacking detectable telomeric signals in WT V5-RTEL1-complemented cells (Figure 3E). These results establish that a functional C4C4 motif in RTEL1 is essential for suppressing TCs and telomere loss, but is dispensable for preventing telomere fragility.
We next evaluated how loss of the RTEL1-TRF2 interaction might affect the DDR at telomeres. To this end we quantified the number of dysfunctional telomeres by scoring for 53BP1 colocalization with telomeres. Conditional deletion of RTEL1 in RTEL1F/F MEFs expressing V5-RTEL1R1237H or V5-RTEL1C1252A/C1255A resulted in enhanced telomere-dysfunction-induced foci (TIFs), corresponding to 7.2% ± 0.5% and 8.3% ± 1.3% foci per nucleus compared to 2.5% ± 1.6% TIFs per nucleus in the cells expressing WT V5-RTEL1 (Figures 3F and S4). Thus, disrupting the TRF2 binding site on RTEL1 causes a mild DDR that resembles the phenotype of RTEL1 deletion.
To determine whether loss of the RTEL1-TRF2 interaction induces telomere recombination, we employed chromosome orientation (CO)-FISH to monitor the frequency of telomere sister chromatid exchange (T-SCEs). Inactivation of RTEL1 in RTEL1F/F MEFs expressing V5-RTEL1R1237H or V5-RTEL1C1252A/C1255A resulted in a 3-fold increase in the number of T-SCEs when compared to WT V5-RTEL1-complemented cells (Figure 3G). Collectively, these results lead us to propose that recruitment of RTEL1 to telomeres by TRF2 is important for t-loop disassembly and prevents catastrophic loss of the telomere, but is dispensable for efficient telomere replication.
The RTEL1 C4C4 Motif Is Dispensable for Genome-wide Replication and for the RTEL1-PCNA Interaction
Given that a functional C4C4 motif and RTEL1-TRF2 interaction is required for t-loop disassembly but is dispensable for preventing telomere fragility, we wished to determine the impact of C4C4 motif mutations on genome-wide replication dynamics using the molecular combing method (Michalet et al., 1997). As previously described (Vannier et al., 2013), the progression of sister replication forks, 96 hr after inactivation of RTEL1, in RTEL1F/F MEFs is significantly reduced relative to wild-type cells and is associated with increased fork asymmetry and elevated origin usage. In contrast, RTEL1F/F MEFs complemented with V5-RTEL1R1237H or V5-RTEL1C1252A/C1255A exhibited comparable replication fork dynamics to that of V5-RTEL1 WT cells (Figures 4A and 4B). This suggests that the C4C4 motif is dispensable for normal replication throughout the genome.
Figure 4.

C4C4 Domain on RTEL1 Is Dispensable for Genome-wide Replication
(A) Quantification of the replication fork dynamics of RTEL1F/F MEFs complemented with empty vector (ctrl), wild-type V5-RTEL1 (WT), single-mutant V5-RTEL1R1237H (R/H), or double-mutant V5-RTEL1C1252A/C1255A (C/A). Cells were subjected to DNA combing at 96 hr after infection with control- or Cre-expressing adenovirus. The first two bars in the graph depict replication fork dynamics in the non-complemented RTEL1F/F MEFs (∗∗∗∗p < 0.0001, two-tailed Mann-Whitney test). Data are represented as mean ± SD.
(B) Quantification of fork asymmetry from the same cells as indicated in (A) by measuring IdU/CldU right- and left-moving sister forks.
(C) RTEL1F/F MEFs as detailed in (A) were subjected to in situ PLA assay with anti-V5 and anti-PCNA antibodies. To monitor the specificity of the PLA assay, RTEL1+/+v5 MEFs (PIP WT) that retain an intact RTEL1 PIP motif served as a positive control and RTEL1IA/IAv5 PIP box mutant MEFs (PIP IA) that lack RTEL1-PCNA interaction were included as a negative control. Dashed lines indicate nucleus outlines (as determined by DAPI in blue).
(D) Quantification of PLA assay from the same cells as shown in (C). Data are representative of two independent experiments as mean ± SD (∗∗∗∗p < 0.0001, one-way ANOVA).
(E) Whole-cell extracts from RTEL1F/F MEFs as indicated in (A) were subjected to immunoprecipitation with anti-RTEL1 antibody. Protein complexes were analyzed by immunoblotting against PCNA and RTEL1.
These results predict that mutations in the C4C4 motif should not affect the RTEL1-PCNA interaction, which we first examined using in situ PLA. As expected, loss of RTEL1 abolished the RTEL1-PCNA interaction detectable by PLA in RTEL1F/F MEFs complemented with empty vector (Figures 4C and 4D). In contrast, RTEL1-depleted cells complemented with V5-RTEL1R1237H and V5-RTEL1C1252A/C1255A C4C4 mutations retained the RTEL1-PCNA interaction, which corresponded to 13.4 ± 2.9 and 11.6 ± 2.0 PLA foci per nucleus when compared to 11.5 ± 2.1 foci per nucleus in RTEL1F/F MEFs expressing WT V5-RTEL1 (Figures 4C and 4D). Similar results were obtained in coimmunoprecipitation experiments from the same cells (Figure 4E). We suggest that the C4C4 motif is dispensable for the RTEL1-PCNA interaction and does not contribute to the global and telomere replication functions of RTEL1 in cells.
TRF2 Interacts with RTEL1 through Its TRFH Dimerization Domain
To identify the region of TRF2 that interacts with RTEL1, we synthesized peptide arrays of overlapping 20-mer peptides (offset by a single amino acid) that comprised 150 amino acids of TRF2, equivalent to human TRF243–183. We found that GST-tagged full-length human RTEL1 but not GST alone interacted with a peptide corresponding to human TRF2106–125 (Figure S5A), which is located within the TRFH dimerization domain at the helix1-helix2 boundary (Figure 5A). To confirm this result, we purified untagged human TRF2TRFH, a 6xHis-SUMO-C4C4 motif (human RTEL11234–1292), and 6xHis-SUMO control. 6xHis-SUMO-C4C4 but not control bound to TRF2TRFH at both low and high salt concentrations as confirmed by Coomassie staining of the eluted material and resolution on SDS-PAGE gels (Figure 5B). Consistent with our previous result that the binding of TRF2 to the C4C4 motif is dependent on metal coordination (Figure 2B), we found that the interaction of TRF2TRFH with 6xHis-SUMO-C4C4 is greatly reduced by the addition of EDTA (Figure S5B).
Figure 5.

TRF2 Interacts with RTEL1 through TRFH Domain
(A) Schematic view of the domain organization of mammalian TRF2. Horizontal bar represents full-length TRF2 protein with functional domains shown as colored boxes. A close-up view of the RTEL1-binding peptide sequence and its mouse counterpart are also indicated.
(B) Human TRFH recombinant proteins were incubated with nickel-agarose bead-immobilized control 6xHis-SUMO or 6xHis-SUMO-C4C4 (equivalent to human RTEL11234–1292 region). The input controls are depicted in lanes 1 and 2.
(C) Peptide substitution array of the 20-residue RTEL1-binding peptide. The sequence corresponding to the first left column of the array represents the unsubstituted peptide. The array was incubated with a recombinant human C4C4 peptide (RTEL11234–1292) fused with 6xHis-SUMO, and bound proteins were evaluated by immunoblotting against 6xHis.
(D) Whole-cell extracts from 293 FLAP-tagged RTEL1 cells (upper panels) or RTEL1v5 MEFs (bottom panels) stably expressing an empty vector (ctrl), wild-type Myc-TRF2 (WT), or mutant Myc-TRF2I124D (I/D) were immunoprecipitated with anti-Flag or anti-V5 antibodies, respectively. Immunocomplexes were analyzed with antibodies as indicated.
(E) Whole-cell extracts from cells shown in (D) were immunoprecipitated with anti-Myc antibody. Protein complexes were analyzed with antibodies against Rap1, TRF1, and Myc.
(F) ChIP for telomeric DNA associated with shelterin proteins in TRF2F/− MEFs stably expressing lentiviral constructs as in (D) assessed 96 hr after infection with control- or Cre-expressing adenovirus. The input signal consists of 5% of the amount of chromatin per immunoprecipitation.
(G) Quantification of the telomeric DNA signal, relative to each input signal. Data represent the average fold change of telomeric DNA recovered from two independent experiments as mean ± SD. The level of [32P] incorporation detected in MEFs expressing empty vector (ctrl), after 96 hr transduction with control adenovirus (Ad-GFP), was arbitrarily assigned a value of 1. See also Figures S5 and S6.
To identify key residues in TRF2 that are necessary for the TRF2-RTEL1 interaction, we generated a substitution peptide array in which the human TRF2106–125 peptide was systematically substituted (one amino acid substitution at a time) with each of the 20 natural amino acids. Probing this array with 6xHis-SUMO-C4C4 revealed that several amino acid substitutions at isoleucine 121 severely reduced the interaction (Figure 5C). In contrast, a purified 6xHis-SUMO control peptide did not bind to any spot within the TRF2106–125 substitution peptide array, confirming specificity of the interaction (data not shown). To test if the TRF2 p.I121 mutation compromises the RTEL1 interaction in vivo, we introduced the corresponding mutation (p.I124D) into full-length mouse TRF2, which was used to stably transduce HEK293 FLAP-tagged RTEL1 cells and RTELv5 MEFs with retroviruses along with controls (Figure S6). Strikingly, we found that the p.I124D mutation completely abolished binding of TRF2 to Flag-RTEL1 or V5-RTEL1 (Figure 5D). Thus, isoleucine 124 within the TRF2TRFH domain is critical for the TRF2-RTEL1 interaction in vivo.
Mutant TRF2I124D Interacts with Shelterin Proteins and Is Recruited to Telomeres
To examine how the p.I124D mutation affects TRF2 activities at telomeres, we subjected whole-cell extracts of HEK293 FLAP-tagged RTEL1 cells stably expressing an empty vector, WT Myc-TRF2, or Myc-TRF2I124D to immunoprecipitation with anti-Myc or control IgG. While the TRF2I124D mutation specifically abolished the TRF2-RTEL1 interaction (Figure 5D), it did not measurably affect its ability to interact with the shelterin proteins TRF1 and Rap1 (Figure 5E). Since TRF2I124D is not in close proximity to the TIN2 binding site, which is separate from the TRFH domain, and maps to residues 352–365 (Chen et al., 2008, 2011), it is unlikely that this mutation affects interaction with TIN2.
Next, we examined whether the TRF2I124D mutant affects telomere binding of itself or other shelterin components by chromatin immunoprecipitation (ChIP). Complemented TRF2F/− cells were crosslinked with formaldehyde and DNA slot blots were hybridized with a TTAGGG probe to determine relative levels of protein-associated telomeric DNA (Figure 5F). As expected, Cre-mediated deletion of TRF2 in cells expressing empty vector resulted in a severe reduction in telomeric binding of TRF2 accompanied by a 5-fold reduction of TRF1 and Rap1 associated with telomeres (Figure 5G). Importantly, TRF2F/− cells complemented with Myc-TRF2 or Myc-TRF2I124D retained WT or mutant TRF2 as well as endogenous shelterin proteins at telomeres (Figure 5G). These results suggest that the TRF2I124D mutant does not compromise the ability of TRF2 to associate with telomeres and does not measurably impact on telomeric binding by TRF1 and Rap1.
TRF2I124D Phenocopies RTEL1 C4C4 Motif Mutants
To gain further insight into the impact of the TRF2I124D mutant at telomeres, we conducted TC amplification assays and telomere FISH experiments to examine telomere stability and telomere length changes in these cells. We first examined TRF2F/− MEFs complemented with empty vector, Myc-TRF2, or Myc-TRF2I124D for the presence of TCs. Strikingly, inactivation of TRF2 in TRF2F/− MEFs complemented with Myc-TRF2I124D resulted in a 3-fold induction of TCs when compared to cells complemented with WT Myc-TRF2 (Figure 6C). Intriguingly, this result is comparable to the TC ratio observed between WT and C4C4 motif mutants expressed in RTEL1F/F MEFs (Figure 3C). As previously described (Celli and de Lange, 2005), Cre-mediated deletion of TRF2 in TRF2F/− MEFs complemented with empty vector results in chromosome end-to-end fusions with telomeric DNA signals evident at the junction of each fusion (Figure 6D). In contrast, TRF2F/− MEFs complemented with either WT Myc-TRF2 or Myc-TRF2I124D did not present with telomere fusions (Figure 6D). Since loss of TIN2 gives rise to telomere fusions (Takai et al., 2011), these data further suggest that the TRF2I124D mutation does not impact on TRF2-TIN2 binding.
Figure 6.

Disruption of the RTEL1 Binding Site in the TRFH Domain of TRF2 Leads to Abnormal Telomere Phenotypes
(A) PCR analysis of genomic DNA isolated from TRF2F/− MEFs stably expressing empty vector (ctrl), wild-type Myc-TRF2 (WT), or mutant Myc-TRF2I124D (I/D) evaluated 96 hr after infection with control- (left) or Cre-expressing (right) adenovirus.
(B) Western blotting analysis of the cells described in (A) to monitor loss of endogenous TRF2 upon Cre expression and to determine complementation efficiency with ectopic wild-type and mutant TRF2. The asterisk indicates endogenous TRF2.
(C) Phi29-dependent telomere circles (TCs; left panel) detected in the same cells as indicated in (A). The extent of [32P] incorporation was quantified (right panel) from the autoradiographs, and the level of [32P] incorporation by cells expressing Myc-TRF2 (WT) upon Cre was arbitrarily assigned a value of 100%. Data represent the average of two independent experiments as mean ± SD (∗∗∗∗p < 0.001; Student’s t test).
(D–F) Quantification of telomere fusions (D), telomere fragility (E), and telomere loss (F) per metaphase determined from the same cells as in (A). Data represent mean ± SD from at least 60 metaphases (∗∗∗∗p < 0.0001; one-way ANOVA). Representative images of the telomere FISH experiments are shown to the right of (D) and (F). The asterisks indicate loss of telomere signal. Green, TTAGGG signal; blue, DAPI.
(G) Quantitative telomere FISH analysis (Q-FISH), to monitor telomere length heterogeneity, was performed 96 hr after Cre introduction in TRF2F/− MEFs complemented with wild-type Myc-TRF2 (WT) or mutant Myc- TRF2I124D (I/D). Mean fluorescence ± SD was calculated over thousands of events, and intensity was measured in arbitrary units of fluorescence (a.u.f.).
(H) Quantification of the frequency of nuclei with telomere dysfunction-induced foci (TIFs), assessed from the same cells as described in (A). Cells with ≥ 2 telomeric 53BP1 foci were scored as TIF positive. Data are representative of three independent experiments as mean ± SD; n > 100 nuclei per single experiment (∗∗∗∗p < 0.0001; Student’s t test).
(I) Quantification (left panel) of telomere-sister chromatid exchange (T-SCE) events per metaphase, determined from the cells as indicated in (A). Representative CO-FISH images after labeling leading (green) and lagging (red) strand telomeres are shown on the right. The yellow arrows show chromosomes with recombination events. Data represent mean ± SD from at least 40 metaphases (∗∗∗∗p < 0.0001; one-way ANOVA). See also Figure S7.
While we found no evidence that the TRF2I124D mutation is associated with telomere fusions or fragility (Figures 6D and 6E), we did observe a high incidence of telomere loss following inactivation of TRF2 in TRF2F/− MEFs complemented with Myc-TRF2I124D. Telomere FISH analysis revealed that disruption of the RTEL1-TRF2 interaction, associated with the TRF2I124D mutation, resulted in the loss of a telomere signal on 13 ± 7.7 chromosome ends per metaphase. This contrasted with the loss of 5.1 ± 2.8 telomeres per metaphase in cells expressing WT Myc-TRF2 (Figure 6F). To address whether the TRF2I124D mutation gives rise to telomere length heterogeneity, we conducted quantitative fluorescence in situ hybridization (Q-FISH) to measure the intensity of the telomere signal from images of FISH-stained metaphases. Analysis of TRF2F/− MEFs expressing Myc-TRF2I124D, performed 96 hr after Cre introduction, revealed increased telomere length heterogeneity when compared to cells with WT Myc-TRF2 (Figure 6G). Interestingly, these changes in the mean telomere length, which included a statistically significant increase in both short and long telomeres, resembled RTEL1 deficiency, and more specifically the defect associated with mutations in the C4C4 motif (Vannier et al., 2012).
To examine the impact of the TRF2I124D mutation on the DDR, we quantified the number of 53BP1 foci that colocalize with telomeric DNA. TIF analysis revealed that conditional deletion of TRF2 in TRF2F/− MEFs expressing empty vector control resulted in 71% of telomeres with TIFs. In contrast, deletion of TRF2 in TRF2F/− MEFs complemented with either WT Myc-TRF2 or Myc-TRF2I124D mutant diminished the percentage of TIF foci at telomeres by 10.5-fold and 6.1-fold, respectively (Figures 6H and S7). These results suggest that loss of the TRF2-RTEL1 interaction, which compromises the recruitment of RTEL1 to telomeres, elicits a mild telomere-associated DDR, which is comparable in severity to the DDR observed at telomeres in RTEL1-deficient cells or associated with the RTEL1R1264H mutation.
Finally, we performed CO-FISH analysis to examine the frequency of recombination at telomeres induced by the loss of the TRF2-RTEL1 interaction. Inactivation of TRF2 in TRF2F/− MEFs complemented with Myc-TRF2I124D resulted in a greater than 5-fold induction in the frequency of T-SCEs with preferential addition of lagging telomere DNA when compared with cells expressing WT Myc-TRF2 (Figure 6I). These results indicate that the recruitment of RTEL1 to telomeres via its interaction with TRF2 protects against telomeric recombination. Overall, these results establish that TRF2 recruits RTEL1 to telomeres to facilitate t-loop disassembly.
Discussion
Telomeres are dependent on two distinct activities of RTEL1 to maintain the integrity of chromosome ends: RTEL1 disassembles t-loops and also counteracts the formation of G4-DNA secondary structures to prevent telomere loss and telomere fragility, respectively (Vannier et al., 2012). Our previous study established that RTEL1 interacts with the replisome through a PIP-box-dependent interaction with PCNA, which is important for the replication functions of RTEL1 (Vannier et al., 2013). Surprisingly, however, the RTEL1-PCNA interaction is dispensable for t-loop disassembly, which suggested the existence of a distinct mechanism for recruiting RTEL1 to telomeres to promote t-loop unwinding. Here, we establish that RTEL1 is transiently recruited to telomeres to promote t-loop unwinding through an S phase-specific interaction with the shelterin component TRF2 (see Figure 7 for a model). Failure to disassemble t-loops has severe consequences for telomere integrity, which may reflect the need to avoid collisions between t-loops and the replication fork, allow telomerase access to the 3′ end of the telomere, and/or counteract the formation of t-loops in trans between sister telomeres.
Figure 7.

Model of How the S Phase-Specific Interaction of RTEL1 with TRF2 Orchestrates T-Loop Disassembly at Telomeres
RTEL1 disassembles t-loop at telomeres through a process that requires S phase-specific interaction with TRF2 (left). Mutations within C4C4 domain of RTEL1 (p.R1264H) or alterations in the TRF2TRFH dimerization domain (p.I124D) abolish RTEL1-TRF2 complex. Failure to recruit RTEL1 to telomeres leads to persistent t-loops, which are inappropriately resolved by the SLX1/4 nuclease complex, resulting in the formation of TCs, telomere loss, and telomere length heterogeneity.
RTEL1’s ability to interact directly with TRF2 is mediated by a cysteine-rich C4C4 motif, which is predicted to bind two zinc ions to form a cross-braced structure (Kellenberger et al., 2005). The finding that addition of EDTA is sufficient to abolish the TRF2-RTEL1 interaction underscores the importance of C4C4 metal coordination for this association. Of direct clinical relevance is the recent discovery of mutations within the C4C4 motif that are causal for the telomeropathy HHS, including RTEL1R1264H and RTEL1C1244R mutations (Ballew et al., 2013; Le Guen et al., 2013). Strikingly, the carrier frequency of the RTEL1R1264H mutation within the Ashkenazi Orthodox Jewish and general Ashkenazi Jewish populations is 1% and 0.45%, respectively, which has prompted the need for urgent screening to identify heterozygous carriers (Ballew et al., 2013; Fedick et al., 2014). We demonstrate here that the RTEL1-TRF2 interaction is compromised by the p.R1264H mutation in HHS patient-derived cells and in RTEL1R1264H mutant MEFs.
In contrast to the RTEL1 PIP-box mutation, which eliminates binding to PCNA and specifically affects the replication functions of RTEL1, the C4C4 mutations have no measurable impact on normal replication dynamics, telomere fragility, or the interaction with PCNA. Instead, the C4C4 mutation conferred rapid accumulation of TCs concomitant with telomere length changes and telomere loss, which we attribute to the inappropriate resolution of the t-loop by the SLX1/4 nuclease complex (Figure 7 and Vannier et al., 2012). Thus, we propose that the PIP-box and C4C4 motif convey distinct and genetically separable functions, which impact on the targeting of RTEL1 to replication forks and telomeres, respectively.
Our analysis of the RTEL1 binding sites in TRF2 identified a single point mutation, TRF2I124D, located in helix2 of the TRF2TRFH dimerization domain (Fairall et al., 2001), which abolishes the RTEL1-TRF2 interaction. Importantly, TRF2I124D is not in close proximity to the binding sites for TIN2 or Rap1 (Chen et al., 2008, 2011) and does not affect its interaction with TRF1 or Rap1, nor its ability, or the ability of other shelterin components, to bind to telomeres. Furthermore, unlike loss of TRF2, which gives rise to telomere de-protection, telomere end-to-end fusions, and a massive induction of the DDR at telomeres, the TRF2I124D mutation gives rise to a phenotype that is remarkably similar to that observed for the RTEL1R1264H mutation. Both TRF2I124D and RTEL1R1264H mutant cells present with high levels of TCs, telomere length heterogeneity, telomere loss, increased telomere SCEs, and a moderate induction of the DDR at telomeres, which we have previously attributed to the inappropriate resolution of persistent t-loops by the SLX1/4 nuclease complex (Vannier et al., 2012). Thus, we propose that the phenotypes of the TRF2I124D and RTEL1R1264H mutations reflect a failure to recruit RTEL1 to telomeres during S phase, which is necessary to transiently disassemble t-loops. Given the phenotypic similarity of TRF2I124D and RTEL1R1264H mutations, our results also raise the possibility that TRF2 coordinates both the assembly and disassembly of t-loops during the cell cycle.
Previous studies have established that the N-terminal basic domain of TRF2 binds to and protects telomeric Holliday junctions (HJ)/D-loop structures from cleavage by endonucleases, which may play a role in the formation and protection of t-loops (Poulet et al., 2009; Wang et al., 2004). The N-terminal basic domain of TRF2 could also be used to target RTEL1 to the telomere HJ/D-loop during S phase, which must be unwound to release the 3′ telomere end to disassemble the t-loop. It is intriguing to note that the RTEL1 interaction site within the TRFH domain of TRF2 is adjacent to the N-terminal basic domain of TRF2 that confers telomere HJ/D-loop binding (Fouché et al., 2006; Poulet et al., 2009). Given their close proximity, it will be important to determine if the binding of RTEL1 to TRF2 induces an allosteric change in the adjacent basic domain, which could alter HJ/D-loop association to facilitate unwinding by RTEL1 to dismantle the t-loop.
In future studies it will be important to further define how the RTEL1-TRF2 interaction is controlled during the cell cycle and how this is limited to S phase cells. While active replication is not required for the RTEL1-TRF2 interaction, it is intriguing to note that TRF2 is a likely CDK substrate and could be subject to cell cycle-dependent modification. Indeed, TRF2 contains numerous potential CDK phosphorylation sites and can be phosphorylated by a cyclin A-CDK2 complex in vitro (Chi et al., 2008). However, the importance of these putative CDK sites in TRF2 and whether or not they contribute to the RTEL1-TRF2 interaction remains unclear. In addition, TRF2 is detected as two closely migrating bands (Bilaud et al., 1997). How and why RTEL1 preferentially binds to the lower isoform is unclear, but may reflect differences in post-translational modifications (PTMs) that could be determinants of the interaction. Finally, it is also important to consider that RTEL1 may be subject to PTMs during the cell cycle, but this remains ill defined and warrants future investigation. An improved understanding of the cell cycle control of RTEL1 and TRF2 is likely to provide important insights into the assembly and disassembly of t-loops and its contribution to the end protection problem.
Experimental Procedures
Cell Culture Procedures
SV40-LT-immortalized RTEL1F/F (Vannier et al., 2012), TRF2F/− (Celli and de Lange, 2005; a gift from Titia de Lange, The Rockefeller University), RTEL1v5+/+, and RTEL1v5IA/IA (Vannier et al., 2013) MEFs were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 15% fetal bovine serum (Invitrogen), L-glutamine, and penicillin-streptomycin. HEK293, FT 293, Phoenix Ampho 293, BJ-hTERT, RPE-1, patient-derived MSK-41 fibroblasts, and U2OS osteosarcoma cells were kept in DMEM with 10% FBS. Production of retro- and lentiviral supernatants was performed as described earlier (Sarek et al., 2010). RTELF/F MEFs were transduced with pBABE-puromycin retroviruses expressing an empty vector, mouse V5-tagged WT, or mutant RTEL1. TRF2F/− MEFs were infected with pLPC-puromycin retroviruses expressing control vector, mouse WT, or mutant Myc-tagged TRF2. The human U2OS cells were transduced with pLPC-puromycin retroviruses carrying Myc-tagged WT or mutant alleles of the C4C4 domain. HEK293 cells were infected with pHAGE-PGK-puromycin lentiviral constructs to express WT or mutants of human Flag-tagged RTEL1. Transduced cells were selected with puromycin (3 μg/ml) for 2–6 days. Deletion of floxed alleles in RTELF/F and TRF2F/− MEFs was carried out with Ad-GFP-Cre adenovirus (Vector Biolabs), and cells were genotyped by PCR at 96 hr post-infection as described earlier (Celli and de Lange, 2005; Vannier et al., 2012).
In Situ PLA
Cells were plated on coverslips at density 5 × 104 in 24-well plates and left in culture conditions overnight. The next day cells were pre-extracted in CSK buffer (10 mM PIPES [pH 6.8], 100 mM NaCl, 300 mM sucrose, 3 mM magnesium chloride, 1 mM EGTA, and 0.5% Triton X-100) fixed with 5% formaldehyde (Thermo Scientific) for 10 min, permeabilized with PBS containing 0.5% (v/v) NP-40 for 5 min, and blocked for 30 min with goat serum (5%) in PBS. PLA was performed following the manufacturer’s instructions using the Duolink anti-Mouse MINUS and anti-Rabbit PLUS In Situ PLA probes and the Duolink In Situ Detection Reagents Red (Olink Bioscience). Images were acquired with a Zeiss Axio Imager M1 microscope equipped with an ORCA-ER camera (Hamamatsu) and using the Volocity 4.3.2 software (Improvision).
Acknowledgments
We wish to thank Titia de Lange for providing conditional TRF2F/− mouse embryo fibroblasts, plasmids, and antibodies. We also thank Sharon Savage for HHS patient cells and Peter Cherepanov for advice with protein purification of the C4C4 motif. We also thank Nicola O’Reilly and Dhira Joshi (London Research Institute) for peptide array synthesis. In addition, we thank all of the members of our laboratory for helpful discussions. Research in the DNA damage response laboratory of S.J.B. is funded by Cancer Research UK and by a European Research Council (ERC) Advanced Investigator Grant (RecMitMei). S.J.B. is a recipient of a Royal Society Wolfson Research Merit Award. G.S. and S.P. are funded by the EMBO Long-Term Fellowship. J.-B.V. is funded by a long-term fellowship from ERC. This work was also supported by GM56888 and The Starr Cancer Consortium (J.H.J.P.).
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information
References
- Ballew B.J., Joseph V., De S., Sarek G., Vannier J.B., Stracker T., Schrader K.A., Small T.N., O’Reilly R., Manschreck C. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet. 2013;9:e1003695. doi: 10.1371/journal.pgen.1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P., Cech T.R. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–1175. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- Bilaud T., Brun C., Ancelin K., Koering C.E., Laroche T., Gilson E. Telomeric localization of TRF2, a novel human telobox protein. Nat. Genet. 1997;17:236–239. doi: 10.1038/ng1097-236. [DOI] [PubMed] [Google Scholar]
- Celli G.B., de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- Chen Y., Yang Y., van Overbeek M., Donigian J.R., Baciu P., de Lange T., Lei M. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science. 2008;319:1092–1096. doi: 10.1126/science.1151804. [DOI] [PubMed] [Google Scholar]
- Chen Y., Rai R., Zhou Z.R., Kanoh J., Ribeyre C., Yang Y., Zheng H., Damay P., Wang F., Tsujii H. A conserved motif within RAP1 has diversified roles in telomere protection and regulation in different organisms. Nat. Struct. Mol. Biol. 2011;18:213–221. doi: 10.1038/nsmb.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y., Welcker M., Hizli A.A., Posakony J.J., Aebersold R., Clurman B.E. Identification of CDK2 substrates in human cell lysates. Genome Biol. 2008;9:R149. doi: 10.1186/gb-2008-9-10-r149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- Ding H., Schertzer M., Wu X., Gertsenstein M., Selig S., Kammori M., Pourvali R., Poon S., Vulto I., Chavez E. Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell. 2004;117:873–886. doi: 10.1016/j.cell.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Doksani Y., Wu J.Y., de Lange T., Zhuang X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell. 2013;155:345–356. doi: 10.1016/j.cell.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairall L., Chapman L., Moss H., de Lange T., Rhodes D. Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol. Cell. 2001;8:351–361. doi: 10.1016/s1097-2765(01)00321-5. [DOI] [PubMed] [Google Scholar]
- Faure G., Revy P., Schertzer M., Londono-Vallejo A., Callebaut I. The C-terminal extension of human RTEL1, mutated in Hoyeraal-Hreidarsson syndrome, contains harmonin-N-like domains. Proteins. 2014;82:897–903. doi: 10.1002/prot.24438. [DOI] [PubMed] [Google Scholar]
- Fedick A.M., Shi L., Jalas C., Treff N.R., Ekstein J., Kornreich R., Edelmann L., Mehta L., Savage S.A. Carrier screening of RTEL1 mutations in the Ashkenazi Jewish population. Clin. Genet. 2014 doi: 10.1111/cge.12459. [DOI] [PubMed] [Google Scholar]
- Fouché N., Cesare A.J., Willcox S., Ozgür S., Compton S.A., Griffith J.D. The basic domain of TRF2 directs binding to DNA junctions irrespective of the presence of TTAGGG repeats. J. Biol. Chem. 2006;281:37486–37495. doi: 10.1074/jbc.M608778200. [DOI] [PubMed] [Google Scholar]
- Greider C.W., Blackburn E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Griffith J.D., Comeau L., Rosenfield S., Stansel R.M., Bianchi A., Moss H., de Lange T. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- Kellenberger E., Dominguez C., Fribourg S., Wasielewski E., Moras D., Poterszman A., Boelens R., Kieffer B. Solution structure of the C-terminal domain of TFIIH P44 subunit reveals a novel type of C4C4 ring domain involved in protein-protein interactions. J. Biol. Chem. 2005;280:20785–20792. doi: 10.1074/jbc.M412999200. [DOI] [PubMed] [Google Scholar]
- Le Guen T., Jullien L., Touzot F., Schertzer M., Gaillard L., Perderiset M., Carpentier W., Nitschke P., Picard C., Couillault G. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum. Mol. Genet. 2013;22:3239–3249. doi: 10.1093/hmg/ddt178. [DOI] [PubMed] [Google Scholar]
- Michalet X., Ekong R., Fougerousse F., Rousseaux S., Schurra C., Hornigold N., van Slegtenhorst M., Wolfe J., Povey S., Beckmann J.S., Bensimon A. Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science. 1997;277:1518–1523. doi: 10.1126/science.277.5331.1518. [DOI] [PubMed] [Google Scholar]
- Moyzis R.K., Buckingham J.M., Cram L.S., Dani M., Deaven L.L., Jones M.D., Meyne J., Ratliff R.L., Wu J.R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. USA. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan R.J., Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010;11:171–181. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W., de Lange T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Poser I., Sarov M., Hutchins J.R., Hériché J.K., Toyoda Y., Pozniakovsky A., Weigl D., Nitzsche A., Hegemann B., Bird A.W. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat. Methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulet A., Buisson R., Faivre-Moskalenko C., Koelblen M., Amiard S., Montel F., Cuesta-Lopez S., Bornet O., Guerlesquin F., Godet T. TRF2 promotes, remodels and protects telomeric Holliday junctions. EMBO J. 2009;28:641–651. doi: 10.1038/emboj.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarek G., Järviluoma A., Moore H.M., Tojkander S., Vartia S., Biberfeld P., Laiho M., Ojala P.M. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog. 2010;6:e1000818. doi: 10.1371/journal.ppat.1000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A., Kosiyatrakul S.T., Hockemeyer D., MacRae S.L., Karlseder J., Schildkraut C.L., de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shete S., Hosking F.J., Robertson L.B., Dobbins S.E., Sanson M., Malmer B., Simon M., Marie Y., Boisselier B., Delattre J.Y. Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippen-Lentz D., Blackburn E.H. Functional evidence for an RNA template in telomerase. Science. 1990;247:546–552. doi: 10.1126/science.1689074. [DOI] [PubMed] [Google Scholar]
- Takai K.K., Kibe T., Donigian J.R., Frescas D., de Lange T. Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol. Cell. 2011;44:647–659. doi: 10.1016/j.molcel.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uringa E.J., Lisaingo K., Pickett H.A., Brind’Amour J., Rohde J.H., Zelensky A., Essers J., Lansdorp P.M. RTEL1 contributes to DNA replication and repair and telomere maintenance. Mol. Biol. Cell. 2012;23:2782–2792. doi: 10.1091/mbc.E12-03-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B., Smogorzewska A., de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- Vannier J.B., Pavicic-Kaltenbrunner V., Petalcorin M.I., Ding H., Boulton S.J. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149:795–806. doi: 10.1016/j.cell.2012.03.030. [DOI] [PubMed] [Google Scholar]
- Vannier J.B., Sandhu S., Petalcorin M.I., Wu X., Nabi Z., Ding H., Boulton S.J. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science. 2013;342:239–242. doi: 10.1126/science.1241779. [DOI] [PubMed] [Google Scholar]
- Vannier J.B., Sarek G., Boulton S.J. RTEL1: functions of a disease-associated helicase. Trends Cell Biol. 2014;24:416–425. doi: 10.1016/j.tcb.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Vulliamy T.J., Marrone A., Knight S.W., Walne A., Mason P.J., Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107:2680–2685. doi: 10.1182/blood-2005-07-2622. [DOI] [PubMed] [Google Scholar]
- Wang R.C., Smogorzewska A., de Lange T. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 2004;119:355–368. doi: 10.1016/j.cell.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Ward J.D., Muzzini D.M., Petalcorin M.I., Martinez-Perez E., Martin J.S., Plevani P., Cassata G., Marini F., Boulton S.J. Overlapping mechanisms promote postsynaptic RAD-51 filament disassembly during meiotic double-strand break repair. Mol. Cell. 2010;37:259–272. doi: 10.1016/j.molcel.2009.12.026. [DOI] [PubMed] [Google Scholar]
- Wellinger R.J., Wolf A.J., Zakian V.A. Saccharomyces telomeres acquire single-strand TG1-3 tails late in S phase. Cell. 1993;72:51–60. doi: 10.1016/0092-8674(93)90049-v. [DOI] [PubMed] [Google Scholar]
- Wrensch M., Jenkins R.B., Chang J.S., Yeh R.F., Xiao Y., Decker P.A., Ballman K.V., Berger M., Buckner J.C., Chang S. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat. Genet. 2009;41:905–908. doi: 10.1038/ng.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.