

Abstract
Mer is a receptor tyrosine kinase implicated in acute lymphoblastic leukemia (ALL), the most common malignancy in children. The currently available data provide a rationale for development of Mer kinase inhibitors as cancer therapeutics that can target both cell autologous and immune-modulatory anti-tumor effects. We have previously reported several series of potent Mer inhibitors and the objective of the current report is to identify a chemically dissimilar back-up series that might circumvent potential, but currently unknown, flaws inherent to the lead series. To this end, we virtually screened a database of ∼3.8 million commercially available compounds using high-throughput docking followed by a filter involving Structural Protein-Ligand Interaction Fingerprints (SPLIF). SPLIF permits a quantitative assessment of whether a docking pose interacts with the protein target similarly to an endogenous or known synthetic ligand, and therefore helps to improve both sensitivity and specificity with respect to the docking score alone. Of the total of 62 experimentally tested compounds, 15 demonstrated reliable dose-dependent responses in the Mer in vitro kinase activity assay with inhibitory potencies ranging from 0.46 μM to 9.9 μM.
Introduction
Acute lymphoblastic leukemia (ALL) is the most frequent type of cancer in children and accounts for nearly 30% of all pediatric cancers[1]. Particularly, the T-cell ALL subtype has a poorer prognosis, with a 5-year relapse-free survival rate of 60–75% even with effective treatment[2]. Extensive conventional chemotherapeutic treatment often results in toxic side effects, such as organ damage, secondary malignancy or emergent chemoresistance[3]. Mer receptor tyrosine kinase, ectopically expressed in at least 50% of pediatric T-cell ALL samples, has been shown to play a role in ALL genesis[1, 3]. Moreover, Mer is not expressed in normal T- and B-lymphocytes. Overall, the currently available data support a hypothesis that Mer kinase inhibitors might be developed into selective therapeutics for ALL. We have previously reported several series of potent Mer inhibitors, including compound 2 (see Figure 1) [4], resulting from structure-based design[4-9]. While our Mer project is progressing through IND enabling studies with an initial clinical candidate from this series, we are also working on identifying a chemically dissimilar back-up series that might circumvent potential flaws inherent to the current lead series. In such an endeavor, often referred to as lead- or scaffold-hopping, virtual screening, either structure- or pharmacophore-based, is often a tool of choice.
Figure 1.

Reference ligand structures for SPLIF scoring.
In Structure-based Virtual Screening (SB-VS), each small-molecule ligand is docked into the putative binding pocket of the protein in a number of energetically acceptable binding modes called poses [10], for each of which binding affinity is assessed using a scoring function [11]. While it is now generally accepted that most of the popular docking algorithms perform fairly well in generating sound poses, the scoring functions most often fail to adequately evaluate the binding affinity[12-18]. Hence, even the optimistic success rates that are generally reported in SB-VS benchmark studies[17, 18] might often be insufficient when screening large chemical libraries against a novel target with an objective to experimentally test 50 to 100 virtual hits. Therefore, all possible means must be deployed to improve the odds of getting a sizable number of confirmed actives out of very small sets of virtual hits. Of special interest are scoring approaches that can take advantage of known ligand-bound protein structures (e.g., enzyme-bound substrates) as these are likely to capture molecular interactions that are most important for high affinity binding. Here we made use of an approach termed Structural Protein-Ligand Interaction Fingerprints (SPLIF) that exploits this general idea of quantifying and comparing ligand-protein interactions[19]. In particular, in SPLIF, 3D-structures of interacting ligand and protein fragments are explicitly encoded in the fingerprint. Consequently, all possible interaction types that may occur between the fragments (e.g., π–π, CH–π, etc) are implicitly encoded into SPLIF. The reported fingerprints are used for calculation a normalized quantitative score that expresses the similarity between the interaction profile of a docking pose and that of a reference protein-ligand complex.
The study involved screening a collection of 3.8 million commercially available compounds using a popular docking tool Glide[20] followed by a SPLIF-rescoring step and a cluster-based triage. Eventually, the 62 selected virtual hits were purchased and their inhibitory potency was assessed in the Mer Microfluidic Capillary Electrophoresis assay.
Materials and Methods
Small-molecule Dataset
The virtual collection of commercially available compounds was created from 5 large catalogs: Asinex, ChemDiv, Enamine, IBS and Life Chemicals. These vendors have been selected because they have their own, up-to-date stocks, offer affordable prices and high availability rates and are able to satisfy our shipping requirements. The resulting collection features ∼3.8 million compounds and is updated on a semi-annual basis via SD files provided by the vendors. The files used in this study have been uploaded between July and December of 2011. Virtually all compounds satisfied our usual pre-VS filters, i.e., a softened version of the Lipinski rules[21] (2+ violations of Number of H-bond donors < 6, Number of H-bond acceptors < 12, Molecular Weight between 200 and 600, ALogP < 5.5) and REOS[22]. Chemical structures of all screened compounds were cleaned using PipelinePilot software[23]. The cleaning protocol included salt stripping, mixture splitting, functional group standardization and charge neutralization. Ionizable compounds were converted to their most probable charged species at pH 7.4. Pipeline Pilot was then used for 3D conversion.
Docking
Small-molecule structures were docked into the active site of the target proteins using the Glide program [20] in standard docking precision (Glide SP). The binding region was defined by a 20Å × 20Å × 20Å box centered on a reference ligand. A scaling factor of 0.8 was applied to the van der Waals radii. Default settings were used for all the remaining parameters. The top 3 poses were generated for each ligand and subjected to SPLIF scoring.
Structural Protein-Ligand Interaction Fingerprints (SPLIF)
SPLIF scoring consists of two steps: 1) generating SPLIF for the current docking pose and 2) calculating similarity between the current and reference SPLIFs. The details of the technique have been described in our earlier work[19]. In this study, Functional Connectivity Fingerprints up to the second closest neighbor (FCFP4) from the Pipeline Pilot software[23] were used as SPLIF bits. The SPLIF-based similarity score was calculated as follows:
| (1) |
where NUMLA is the number of Unique Matching Ligand Atoms, i.e., atoms constituting the matching circular fragments of the docking pose compared to the reference (on the ligand side); NULA is the number of Unique Ligand Atoms, i.e., atoms constituting all interaction fingerprints of the docking pose (on the ligand side); NUMPA is the number of Unique Matching Protein Atoms, i.e., atoms constituting the matching circular fragments of the docking pose compared to the reference (on the protein side); NUPA is the number of Unique Protein Atoms, i.e., atoms constituting all interaction fingerprints of the docking pose (on the protein side). The whole workflow was implemented in Pipeline Pilot[23]. The constituent algorithms were developed in Pipeline Pilot Script. The current implementation allows processing of ∼10 poses per second in screening mode.
Reference structures
Three high-resolution crystal structures of the Mer protein kinase domain were used in this study: i) in complex with adenosine diphosphate (ADP) (resolution 1.90 Å; PDB code: 3BRB)[24]; (ii) in complex with a weakly potent inhibitor C-52 (1) (resolution 2.80 Å; PDB code: 3BPR)[24]; and in complex with a highly potent inhibitor UNC569 (2), previously reported by us (resolution 2.69 Å; PDB code: 3TCP)[4]. Because we did not intend to mimic the phosphate groups of ATP, as ligands including these interactions are highly unlikely to be cell penetrant, we stripped them to yield the reference ligand 3. The reference ligand structures are shown in Figure 1.
The corresponding PDB files were processed as follows. Hydrogen atoms were added to the protein, the active site was visually inspected and appropriate corrections were made for tautomeric states of histidine residues, orientations of hydroxyl groups, and protonation states of basic and acidic residues. The hydrogen atoms were energy minimized in the MMFF force field[25] using the Macromodel software with the Maestro graphics interface[26] with all the non-hydrogen atoms constrained to their original positions.
Hit analysis and selection
After virtual hits were selected based on a combination of Glide and SPLIF scores, they were subjected to a hit triage process. The triage was based upon a number of objective and subjective criteria. The objective criteria included (i) redundancy reduction, by dropping some ligands belonging to large clusters, i.e., groups of chemically similar compounds; and (ii) elimination of compounds that are highly dissimilar from other virtual hits (also called singletons). Both redundancy reduction and singleton elimination were performed by means of the Pipeline Pilot software[23]. Redundancy reduction consisted of two steps. First, the virtual hits were grouped into clusters with members similar at ≥45% (Tanimoto; ECFP4 fingerprints). The clustering method used at this step was Maximum Dissimilarity clustering without limitation on the maximum number of clusters and with the number of re-center steps set to zero[23]. In the next step, 20% to 50% of compounds were then selected from each cluster in such a way that larger clusters contributed smaller percentages. The output ligands were aligned to their respective Maximum Common Substructures to facilitate the subsequent visual ad hoc selection. To facilitate an ad hoc hit selection / elimination we have created a hit list, in which each cluster was represented by a single (central) compound.
Mer Microfluidic Capillary Electrophoresis assay
Inhibition of Mer kinase activity by analogues was tested using a microfluidic capillary electrophoresis (MCE) assay, in which phosphorylated and unphosphorylated substrate peptides were separated and analyzed through a LabChip EZ Reader[27, 28].
Compound testing was performed in a 384 well, polypropylene microplate in a final volume of 50 μL in 50 mM Hepes, Ph 7.4 containing 0.1% Bovine Serum Albumin (BSA), 0.1% Triton X-100, 10 mM MgCl2 and ATP at 5 μM. All reactions were terminated by addition of 50 μL of 70 mM EDTA. Phosphorylated and unphosphorylated substrate peptides were separated following a 180 minute incubation on a LabChip EZ Reader equipped with a 12-sipper chip in separation buffer supplemented with CR-8 and analyzed using EZ Reader software. The reaction was run at 2 nM enzyme concentration. More details can be found in our previous work[4, 6].
Quality control of compound samples
Quality control of the purchased and screened compounds was performed by diluting 1 μL of DMSO stock solution (10mM concentration) with 49 μL of MeOH. The sealed plate was then directly used to inject 5 μL from each well onto an Agilent 6110 Series LC/MS system with the UV detector set to 220 nm. Samples were injected onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 μM, C18 column at room temperature. A mobile phase of A being H2O + 0.1% acetic acid and B being MeOH + 0.1% acetic acid was used. A linear gradient from 10% to 100% B in 5.0 min was followed by pumping 100% B for another 2 minutes with a flow rate of 1.0 mL/min. Mass spectra (MS) data were acquired in positive ion mode using an Agilent 6110 single quadrupole mass spectrometer with an electrospray ionization (ESI) source. The purity of all compounds was found to be 95% or higher by UV absorption at 220 nm, 254 nm and 280 nm and the MS+1 peak was consistent for the purchased structure.
Results and Discussion
Mer[1] is protein kinase belonging to the receptor tyrosine kinase subfamily that might be considered – after a long record of success stories[29] – as a “low-hanging-fruit” target. However, in our biochemical assay, Mer was sensitive to only a few known kinase inhibitors. Moreover, the nanomolar potency Mer inhibitor UNC569 (compound 2) has shown significant selectivity when screened against a broad protein kinase panel[4] and the outcome of our random diversity screen suggests a very low rate of potent Mer inhibitors in diverse sets of commercially available compounds (see detail below). All the above suggests that a post-docking filtration of docking poses based on prior knowledge of ligand-protein interactions would be of particular interest in order to reduce the false positive and false negative rates that characterizes unfiltered structure-based screening[17, 30].
In this study, the generic VS workflow included the following steps: (i) Glide-based docking and scoring; (ii) SPLIF-based scoring; (iii) hit selection based on the analysis of the Glide and SPLIF scores; and (iv) hit triage. The latter step involved three components: (i) diversity-based selection as described in Materials and Methods; (ii) a subjective triage, such as dropping clusters that the chemists would not like to follow-up on even if they contain some true actives; and (iii) unexpected reasons that result in elimination of some virtual hits from the list, such as compound price or stock depletion. The initial pre-filtering of docking poses by means of a conventional scoring function (G-score in this study) has been validated in our earlier benchmark study [31].
SPLIF Reference System
When characterizing the lead series in our previous study[4], we obtained an X-ray structure of the Mer kinase domain in complex with compound 2. In addition to the latter ligand-protein complex with a highly potent inhibitor, there are two more ligand-bound Mer structures: with a weak inhibitor (ca. 10 μM), C-52 (1), and the co-factor ATP (3). We merged all three reference ligands into a single superligand, so that any docking pose matching one fragment in one reference fingerprint and another fragment in a different reference fingerprint would get a higher SPLIF-score than if it is compared to one reference ligand at a time. Overall, 1,330 SPLIF bits have been generated for this reference Mer-bound superligand. The above SPLIF-bits result from 138 FCFP-bits on the protein side (constituted by 53 unique atoms) and 139 FCFP-bits on the ligand side (36 unique atoms).
Screening workflow and statistics
Glide docking of 3.8 million commercial compounds yielded 1.56 million compounds with poses showing G-scores better than 0 kcal/mol (see Figure 2a for the G-score probability density distribution). The G-score distribution is quasi-normal with a mean at ∼-5 kcal/mol and a standard deviation of ∼1 kcal/mol. To start the selection process, we had to set a G-score threshold that would eliminate the least likely true Mer inhibitors. To this end, we made use of probability density distributions of known Mer actives and inactives. By this time, our chemical optimization program had generated 385 Mer actives (IC50 < 1 μM) and 409 inactives (IC50 > 30 μM). Both actives and inactives were docked using the same protocol as described in Materials and Methods. Their G-score distributions (see Figure 2b) indicate that a G-score threshold of -6 kcal/mol adequately separates actives from inactives with optimal false positives vs false negatives rates. Therefore, assuming similar distributions for the 1.5 million scored compounds, the G-score threshold for the virtual screening campaign was set to -6 kcal/mol, which resulted in a selection of 403,581 compounds.
Figure 2. a) The distribution of Gscores of 1.5 million acceptable compounds for virtual screening after docking to the Mer active site; b) the distributions of Gscores of in-house identified Mer actives and inactives; c) the distribution of SPLIF-scores of remaining 400K compounds after the Gscore filter being applied; d) the distribution of SPLIF-scores of in-house identified Mer actives and inactives.
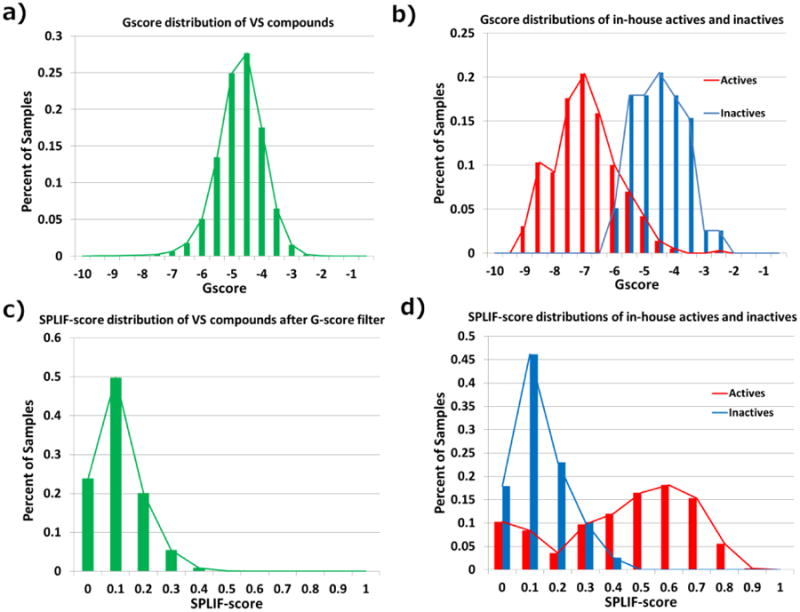
All poses resulting from the G-score selection were submitted to SPLIF-score calculation with the ultimate goal of obtaining a manageable list of hit-candidates that can be subjected to hit triage and, ultimately, experimental testing. Again, we made use of known Mer actives and inactives (as described in the previous paragraph) to determine an optimal SPLIF-score threshold. As can be seen in Figure 2d, an optimal actives/inactives separation occurs at a threshold of ∼0.35. Consistently, the value of 0.35 is where the SPLIF-score distribution for 403,581 preselected compounds approaches zero (see Figure 2c) and hence hits having higher SPLIF-scores may be considered as outliers to the baseline distribution (i.e., distribution of inactives). The retained SPLIF-score threshold of 0.35 resulted in a selection of 10,862 SPLIF-based hit-candidates. These candidates were subjected to a diversity-based selection as described in Materials and Methods. In addition, 544 cluster centers have been visually inspected and a few clusters have been dropped as inappropriate lead-candidates (e.g., nucleotides, steroids, etc.). A few more selected virtual hits were out of stock. Eventually, 62 compounds have been purchased and tested in the Mer microfluidic capillary electrophoresis assay.
It is noteworthy that the SPLIF score alone was not enough to efficiently rank docking poses. Indeed, more than 40,000 compounds satisfy the SPLIF-score threshold of 0.35. This result is consistent with an intuitive anticipation that even if a fraction of a ligand mimics an existing x-ray pose, the rest of it may strongly diminish its binding affinity. Therefore, SPLIF-scoring should only be applied to likely binders, where “likely” means that they need to have a satisfactory preliminary docking score.
Hit analysis
Of the total of 62 experimentally tested compounds, 15 demonstrated reliable dose-dependent responses in the Mer microfluidic capillary electrophoresis assay with inhibitory potencies ranging from 0.46 μM to 9.9 µM (see Figure 3 and Table 1). Remarkably, 2 hits, 4 and 5, demonstrated mid-nanomolar potencies of respectively 0.46 μM and 0.60 μM. Despite relatively low potencies of the other hits, they still may be worth consideration as potential leads due to fairly high ligand efficiencies (LE)[32]. The LE values were calculated as pIC50 normalized by the number of heavy atoms and are given in Table 1. The LE values for the 15 confirmed actives range between 0.17 and 0.34 and are comparable to LE of the current lead (compound 2; LE=0.11).
Figure 3. Chemical structures of 15 confirmed Mer actives.

Table 1. Virtual screening scores and experimental activities for 15 confirmed Mer actives and the reference compound UNC569 (2).
| Cmpd | Catalog ID | Mer IC50 [μM] | Ligand efficiency | G-score [kcal/mol] | G-score rank | SPLIF score | SPLIF rank |
|---|---|---|---|---|---|---|---|
| 2 | 0.0029 | 0.12 | -7.20 | 14,418 | 0.90 | 1 | |
| 4 | F477-3112 | 0.46 | 0.22 | -6.08 | 96,049 | 0.61 | 26 |
| 5 | F477-1435 | 0.60 | 0.21 | -6.92 | 36,837 | 0.47 | 907 |
| 6 | AEM 15393841 | 1.6 | 0.29 | -6.07 | 349,812 | 0.45 | 1,198 |
| 7 | AEM 14526301 | 2.3 | 0.20 | -6.06 | 292,468 | 0.57 | 59 |
| 8 | SYN 15421752 | 2.3 | 0.18 | -6.22 | 123,565 | 0.49 | 510 |
| 9 | T5907809 | 2.5 | 0.23 | -6.11 | 323,271 | 0.56 | 80 |
| 10 | LEG 15385818 | 3.5 | 0.17 | -6.69 | 39,335 | 0.46 | 1,029 |
| 11 | SYN 15385796 | 4.1 | 0.19 | -6.63 | 89,261 | 0.46 | 997 |
| 12 | T0515-4771 | 4.3 | 0.27 | -6.85 | 46,829 | 0.58 | 49 |
| 13 | C301-9568 | 6.2 | 0.22 | -6.24 | 223,491 | 0.50 | 387 |
| 14 | AOP 15805701 | 6.2 | 0.22 | -6.71 | 40,529 | 0.54 | 123 |
| 15 | ART 14509905 | 8.6 | 0.24 | -6.22 | 255,851 | 0.58 | 47 |
| 16 | BAS 03754884 | 8.9 | 0.34 | -6.56 | 111,098 | 0.49 | 575 |
| 17 | LMK 13266598 | 9.0 | 0.19 | -6.29 | 221,409 | 0.57 | 66 |
| 18 | T6375787 | 9.9 | 0.17 | -6.51 | 125,677 | 0.45 | 1,180 |
These 15 hits represent distinguishable scaffolds, such as thieno[3,2-c]quinolones (4 and 5), pyrimidin-2-amines (6, 7, 8, 10, 11, 15 and 17), pyrimidine-fused heterocycles (9, 13, 16 and 18), as well as singletons 12 and 14. Their inhibitory potencies are weaker than the potency of the reference ligand 2, but most of them demonstrate lead-like[33] profiles and may serve as valuable starting points for further chemical optimization. In our previous study we showed that on a broad panel of targets SPLIF-scores were not redundant with respect to other ranking schemas that could have been used in a similar setting (e.g., G-score alone, 2D- or SIFt-similarity to known actives [34]). As can be seen in Table 1, in the current study, G-score ranks for the 15 confirmed actives range from 39,335 to 349,812 (#96,049 for the most potent compound 4), which means that none of them would have been shortlisted for purchase using that method alone.
All confirmed hits provide strong evidence that using SPLIF-score as a post-docking filter does not undermine the scaffold-hopping capacity of SBVS. Indeed, none of the 15 actives feature a chemical scaffold identical to any reference compound (1-3). However, despite the clear structural differences between, for example, the most potent hit 4 and the reference compound 2, their respective docking poses look intuitively similar (see Figure 4) and make the same key interactions.
Figure 4. A docking pose of the most potent hit 4 (green) overlaid with the reference ligand 2 (magenta).

Finally, we also assessed the overall efficiency of our virtual screening run by comparing its hit rate (i.e., 15 hits / 62 tested = 24%) and to that of a random diversity screen. To this end, we have screened 10 randomly selected 384-well (320-ligand) plates in a single-dose run (at 10 μM; in duplicate) in the Mer microfluidic capillary electrophoresis assay. Only six compounds of the 3,200 screened have shown activity beyond a threshold of 30% (that is a consensus of both statistical and potency significance) resulting in 0.12% hit rate (4 hits / 3,200 tested). Therefore, our SPLIF-based SBVS has demonstrated a ∼200-fold (24% / 0.12%) improvement over a random screen.
Conclusions
In this paper, we report new template starting points for inhibitors of Mer, a receptor tyrosine kinase and a potential therapeutic target for the treatment of ALL and other cancers[35]. We performed SBVS against a database of ∼3.8 million commercially available compounds. In order to improve the odds of success, a recently introduced SPLIF-score [31] was used as a post-docking filter that quantitatively assesses whether a docking pose interacts with the protein target similarly to reference ligand. A total of 62 SPLIF-based virtual hits have been purchased and tested in the Mer MCE assay. Fifteen tested compounds demonstrated reliable dose-dependent inhibitory potencies with IC50's ranging from 0.46 μM to 9.9 μM. The hits identified have high ligand efficiencies, show lead-like property profiles and represent new chemical motifs that might be used as starting points for further chemical optimization. Additionally, this study confirms our previous findings [31] that SPLIF can significantly improve the success rate of SBVS while conserving its inherent scaffold-hopping ability.
Supplementary Material
Acknowledgments
This work was supported by the University Cancer Research Fund and Federal Funds from the National Cancer institute, National Institute of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- Mer
Mer Tyrosine Kinase
- ALL
Acute Lymphoblastic Leukemia
- VS
Virtual Screening
- SPLIF
Structural Protein-Ligand Interaction Fingerprints
- ECFP
Extended Connectivity Fingerprint
Footnotes
Supporting Information Available: Chemical structures and biological activities of experimentally tested compounds are available in SD formats. This material is available free of charge via the Internet at http://www.journals.elsevier.com/bioorganic-and-medicinal-chemistry.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Graham DK, Salzberg DB, Kurtzberg J, Sather S, Matsushima GK, Keating AK, Liang X, Lovell MA, Williams SA, Dawson TL, Schell MJ, Anwar AA, Snodgrass HR, Earp HS. Ectopic Expression of the Proto-oncogene Mer in Pediatric T-Cell Acute Lymphoblastic Leukemia. Clin Cancer Res. 2006;12:2662–2669. doi: 10.1158/1078-0432.CCR-05-2208. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Relling MV, Downing JR. Acute Lymphoblastic Leukemia. New Engl J Med. 2004;350:1535–1548. doi: 10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 3.Linger RMA, DeRyckere D, Brandão L, Sawczyn KK, Jacobsen KM, Liang X, Keating AK, Graham DK. Mer receptor tyrosine kinase is a novel therapeutic target in pediatric B-cell acute lymphoblastic leukemia. Blood. 2009;114:2678–2687. doi: 10.1182/blood-2009-03-209247. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Liu J, Yang C, Simpson C, Deryckere D, Van Deusen A, Miley MJ, Kireev D, Norris-Drouin J, Sather S, Hunter D, Korboukh VK, Patel HS, Janzen WP, Machius M, Johnson GL, Earp HS, Graham DK, Frye SV, Wang X. Discovery of Novel Small Molecule Mer Kinase Inhibitors for the Treatment of Pediatric Acute Lymphoblastic Leukemia. ACS Med Chem Lett. 2012;3:129–134. doi: 10.1021/ml200239k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christoph S, DeRyckere D, Sather S, Wang XD, Kireev D, Janzen W, Liu J, Yang C, van Deusen A, Simpson C, Norris-Drouin J, Frye S, Earp HS, Johnson GL, Graham DK. UNC569 As Novel Small Molecule Mer Receptor Tyrosine Kinase Inhibitor for Treatment of ALL. Blood. 2011;118:1111–1112. [Google Scholar]
- 6.Liu J, Zhang W, Stashko MA, Deryckere D, Cummings CT, Hunter D, Yang C, Jayakody CN, Cheng N, Simpson C, Norris-Drouin J, Sather S, Kireev D, Janzen WP, Earp HS, Graham DK, Frye SV, Wang X. UNC1062, a new and potent Mer inhibitor. Eur J Med Chem. 2013;65:83–93. doi: 10.1016/j.ejmech.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang W, McIver AL, Stashko MA, DeRyckere D, Branchford BR, Hunter D, Kireev D, Miley MJ, Norris-Drouin J, Stewart WM, Lee M, Sather S, Zhou Y, Di Paola JA, Machius M, Janzen WP, Earp HS, Graham DK, Frye SV, Wang X. Discovery of Mer specific tyrosine kinase inhibitors for the treatment and prevention of thrombosis. J Med Chem. 2013;56:9693–9700. doi: 10.1021/jm4013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang WH, Zhang DH, Stashko MA, DeRyckere D, Hunter D, Kireev D, Miley MJ, Cummings C, Lee M, Norris-Drouin J, Stewart WM, Sather S, Zhou YQ, Kirkpatrick G, Machius M, Janzen WP, Earp HS, Graham DK, Frye SV, Wang XD. Pseudo-Cyclization through Intramolecular Hydrogen Bond Enables Discovery of Pyridine Substituted Pyrimidines as New Mer Kinase Inhibitors. Journal of Medicinal Chemistry. 2013;56:9683–9692. doi: 10.1021/jm401387j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang W, DeRyckere D, Hunter D, Liu J, Stashko MA, Minson KA, Cummings CT, Lee M, Glaros TG, Newton DL, Sather S, Zhang D, Kireev D, Janzen WP, Earp HS, Graham DK, Frye SV, Wang X. UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J Med Chem. 2014;57:7031–7041. doi: 10.1021/jm500749d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Current computer-aided drug design. 2011;7:146. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang SY, Grinter SZ, Zou X. Scoring functions and their evaluation methods for protein–ligand docking: recent advances and future directions. PCCP. 2010;12:12899–12908. doi: 10.1039/c0cp00151a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirchmair J, Markt P, Distinto S, Wolber G, Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J CAMD. 2008;22:213–228. doi: 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]
- 13.Scior T, Bender A, Tresadern G, Medina-Franco JL, Martínez-Mayorga K, Langer T, Cuanalo-Contreras K, Agrafiotis DK. Recognizing Pitfalls in Virtual Screening: A Critical Review. J Chem Inf Model. 2012;52:867–881. doi: 10.1021/ci200528d. [DOI] [PubMed] [Google Scholar]
- 14.Warren G, Andrews C, Capelli A, Clarke B, LaLonde J. A critical assessment of docking programs and scoring functions. J Med Chem. 2005;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 15.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 16.Ferrara P, Gohlke H, Price D, Klebe G, Brooks CI. Assessing scoring functions for protein-ligand interactions. J Med Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 17.Cross JB, Thompson DC, Rai BK, Baber JC, Fan KY, Hu Y, Humblet C. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J Chem Inf Model. 2009;49:1455–1474. doi: 10.1021/ci900056c. [DOI] [PubMed] [Google Scholar]
- 18.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins: Structure, Function, and Bioinformatics. 2004;57:225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 19.Da C, Kireev DB. Structural Protein-Ligand Interaction Fingerprints (SPLIF) for Structure-Based Virtual Screening: Method and Benchmark Study. Journal of Chemical Information and Modeling. 2014 doi: 10.1021/ci500319f. Article ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 21.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 22.Walters WP, Murcko MA. Prediction of ‘drug-likeness’. Advanced Drug Delivery Reviews. 2002;54:255–271. doi: 10.1016/s0169-409x(02)00003-0. [DOI] [PubMed] [Google Scholar]
- 23.Pipeline Pilot, ver 8.5. Accelrys Software Inc.; 2009. [Google Scholar]
- 24.Huang X, Finerty P, Jr, Walker JR, Butler-Cole C, Vedadi M, Schapira M, Parker SA, Turk BE, Thompson DA, Dhe-Paganon S. Structural insights into the inhibited states of the Mer receptor tyrosine kinase. J Struct Biol. 2009;165:88–96. doi: 10.1016/j.jsb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. Journal of Computational Chemistry. 1996;17:490–519. [Google Scholar]
- 26.Maestro Suite. Schrodinger LLC; 2009. [Google Scholar]
- 27.Bernasconi P, Min Chen, Galasinski S, Popa-Burke I, Bobasheva A, Coudurier L, Birkos S, Hallam R, Janzen WP. A Chemogenomic Analysis of the Human Proteome: Application to Enzyme Families. Journal of Biomolecular Screening. 2007;12:972–982. doi: 10.1177/1087057107306759. [DOI] [PubMed] [Google Scholar]
- 28.Pommereau A, Pap E, Kannt A. Two Simple and Generic Antibody-Independent Kinase Assays: Comparison of a Bioluminescent and a Microfluidic Assay Format. Journal of Biomolecular Screening. 2004;9:409–416. doi: 10.1177/1087057104264175. [DOI] [PubMed] [Google Scholar]
- 29.Pytel D, Sliwinski T, Poplawski T, Ferriola D, Majsterek I. Tyrosine kinase blockers: new hope for successful cancer therapy. Anti-cancer agents in medicinal chemistry. 2009;9:66–76. doi: 10.2174/187152009787047752. [DOI] [PubMed] [Google Scholar]
- 30.McGaughey GB, Sheridan RP, Bayly CI, Culberson JC, Kreatsoulas C, Lindsley S, Maiorov V, Truchon JF, Cornell WD. Comparison of topological, shape, and docking methods in virtual screening. J Chem Inf Model. 2007;47:1504–1519. doi: 10.1021/ci700052x. [DOI] [PubMed] [Google Scholar]
- 31.Da C, Kireev D. Structural Protein–Ligand Interaction Fingerprints (SPLIF) for Structure-Based Virtual Screening: Method and Benchmark Study. Journal of Chemical Information and Modeling. 2014;54:2555–2561. doi: 10.1021/ci500319f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reynolds CH, Tounge BA, Bembenek SD. Ligand Binding Efficiency: Trends, Physical Basis, and Implications. J Med Chem. 2008;51:2432–2438. doi: 10.1021/jm701255b. [DOI] [PubMed] [Google Scholar]
- 33.Teague SJ, Davis AM, Leeson PD, Oprea T. The Design of Leadlike Combinatorial Libraries. Angew Chem Int Ed. 1999;38:3743–3748. doi: 10.1002/(SICI)1521-3773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 34.Deng Z, Chuaqui C, Singh J. Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein-Ligand Binding Interactions. Journal of Medicinal Chemistry. 2003;47:337–344. doi: 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- 35.Earp HS, Graham DK, et al. Role of TAM kinases in cancer. Nature. 2014 in press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.