

Abstract
Background
Under normoxic conditions, cancer cells use aerobic glycolysis as opposed to glucose oxidation for energy production; this altered metabolism correlates with poor outcomes in neuroblastoma. Hypoxia-inducible factor-1α (HIF-1α) and pyruvate dehydrogenase kinase 4 (PDK4) regulate aerobic glycolysis, while pyruvate dehydrogenase phosphatase 2 (PDP2) promotes glucose oxidation. Here, we sought to determine whether gastrin-releasing peptide receptor (GRP-R) signaling regulates glucose metabolism.
Procedure
Neuroblastoma cell lines, BE(2)-C and SK-N-AS, were used. PCR microararay for glucose metabolism was performed on GRP-R silenced cells. Target protein expression was validated using Western blotting and VEGF ELISA. Cobalt chloride (CoCl2) was used to induce chemical hypoxia. Efficacy of targeting PDK regulation in neuroblastoma was assessed using dichloroacetate (DCA) by conducting cell viability assays and Western blotting for apoptotic markers.
Results
Silencing GRP-R decreased HIF-1α expression and blocked VEGF expression and secretion in both normoxic and CoCl2 induced hypoxia. PCR array analysis identified that GRP-R silencing reduced PDK4 and increased PDP2 mRNA expression. These findings were validated by Western blotting. CoCl2 induced hypoxia increased VEGF secretion, HIF-1α, and PDK4 expression. PDK4 silencing decreased HIF-1α expression and VEGF expression and secretion. DCA treatment decreased BE(2)-C and SK-N-AS proliferation while promoting cell death. GRP-R silencing and DCA treatment synergistically halted BE(2)-C proliferation.
Conclusions
We report that GRP-R regulates glucose metabolism in neuroblastoma by modulating HIF-1α, PDK4 and PDP2. PDK4 regulates glucose metabolism, in part, via regulation of HIF-1α. Synergistic consequences of GRP-R inhibition and DCA treatment may suggest a novel therapeutic strategy for the treatment of aggressive neuroblastoma.
Keywords: Glycolysis, neuroblastoma, GRP-R, PDK4, HIF-1α, VEGF
INTRODUCTION
Neuroblastoma is the most common extracranial malignancy of infants and children, accounting for 6–8% of all childhood cancers and over 15% of pediatric cancer-related deaths (1). Despite recent advances in therapy, the survival rate for advanced stage disease remains dismal at less than 50%. Research advances have ushered in treatment modalities, which successfully induce neuroblastoma cell death. Unfortunately, they also cause damage to normal cells, with subsequent dose-limiting toxicity. In light of this fact, it is critical that we shift our scientific research in neuroblastoma towards identifying molecular pathways that are specific to the biology of cancer cells. Originally described over 80 years ago by Otto Warburg, cancer cells have been known to preferentially use aerobic glycolysis for energy production, despite the presence of oxygen (2). In other words, instead of relying on mitochondrial glucose oxidation for maximal energy production, cancer cells preferably shunt pyruvate, the end product of aerobic glycolysis, away from glucose oxidation and convert it into lactate. Historically known as the Warburg effect, this altered metabolic phenotype has long been correlated with malignant progression, poor clinical outcome (3) and has also been implicated in promoting tumor aggressiveness in neuroblastoma (4).
Several factors are critical for the altered glucose metabolism seen in cancer cells. The relatively nutrient and oxygen – poor conditions that exist within the tumor microenvironment are a driving force for the Warburg effect (5). Under these conditions, hypoxia inducible factor – 1 alpha (HIF-1α) becomes stable and accumulates, which induces the transcription of glucose transporters, glycolytic enzymes, and acts as a mediator to regulate tumor energy metabolism, survival, neovascularization, and invasion (6). Additionally, a regulatory enzyme that plays a key role in glucose metabolism is the pyruvate dehydrogenase complex (PDC). Its key role is the conversion of pyruvate to acetyl CoA and results in the net reduction of glucose (7). PDC is subject to inactivation by pyruvate dehydrogenase kinase (PDK) and activation by pyruvate dehydrogenase phosphatase (PDP), of which there are four isoenzymes (PDK1, PDK2, PDK3 and PDK4) in the PDK family (8) and two isoenzymes (PDP1 and PDP2) in the PDP family (9).
G-protein coupled receptors (GPCR) are part of a large family of cell surface receptors which play a critical role in cancer development, progression, invasion and metastasis (10). We have previously shown that gastrin releasing peptide receptor (GRP-R) is a member of the GPCR family, and its increased expression correlates with neuroblastoma tumor aggressiveness (11). Furthermore, we have reported that GRP-R silencing decreases the expression of key regulators of protein synthesis and cell metabolism, via the PI3K/AKT signaling (12), a pathway known to stimulate aerobic glycolysis in cancer cells (5). GRP-R is critical in the development and progression of neuroblastoma and our previous data illustrates that this receptor signaling helps to sustain cancer cell metabolism. However, whether GRP-R signaling interacts with other pathways critical in altered glucose metabolism has yet to be determined. Therefore, the purpose of this study was to determine whether GRP-R signaling plays a significant role in altering glucose metabolism in neuroblastoma.
METHODS
Materials
Cell lysis buffer was obtained from Cell Signaling Technology (Beverly, MA). Anti β-actin monoclonal antibody, fetal bovine serum (FBS) and dichloroacetate (DCA) were from Sigma (St. Louis, MO). NuPAGE Novex 4–12% Bis–Tris Gel and Lipofectamine 2000 were purchased from Life Technologies (Grand Island, NY). Horseradish Peroxidase (HRP)-conjugated secondary antibodies against mouse IgG, rabbit IgG and human vascular epithelial growth factor (VEGF) antibody were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies against PDK4 and phospho-PDH E1α (ser 293) were from Novus Biological (Littleton, CO). HIF-1α antibody was purchased from BD bioscience (Franklin Lakes, NJ). Antibodies against hexokinase-1 (HK1), Poly (ADP-ribose) polymerase (PARP), and caspase 3 were purchased from AbCam (Cambridge, MA).
Cell Culture
Human neuroblastoma cell lines, BE(2)-C and SK-N-AS, were purchased from American Type Culture Collection (Manassas, VA). Cells were maintained in RPMI 1640 medium with L-glutamine (Cellgro Mediatech, Inc. Herndon, VA) supplemented with 10% FBS. Normoxic cell cultures were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. Chemical hypoxia was induced under similar conditions using cobalt chloride (CoCl2; 100 μM) purchased from Sigma and supplemented into culture media. Stable transfection cell lines were established by selecting out transfected cells with G418 (Cellgro, Mediatech) at 300 μg/ml as previously described (12).
Small Interfering (si) RNA Transfection
SMARTPool siRNA against human PDK4 (siPDK4), GRP-R (siGRP-R), and non-targeting control (siNTC) were purchased from Dharmacon, Inc (Lafayette, CO). Transient transfection was carried out with Lipofectamine 2000 transfection reagent according to the manufacturer’s protocol. Cells were seeded on 6-well plates for RNA or protein preparation and 96-well plates for cell growth assays. After 24 h incubation, media were replaced to serum-free RPMI 1640 containing siRNA (100 pmol) and transfection reagent. Cells were harvested for assays daily for three consecutive days after transfection with the siRNA duplexes. The experiments were repeated on at least three separate occasions.
RNA Isolation and PCR Array
Total cellular RNA extraction was carried out using RNAqueous kit (Ambion, Inc., Austin, TX) according to manufacturer’s instructions. RNAs isolated were quantified by spectrophometer. RNA (1 μg) was used to synthesize cDNA using High Capacity cDNA reverse transcription kit (Life Technologies). Human Glucose Metabolism PCR Array kit profiling expression of key genes involved in the regulation and enzymatic pathways of glucose metabolism was purchased from Qiagen (Frederick, MD). Real time-PCR was performed using the BioRad Thermocycler CFX96. Data analysis was determined by software supplied from the manufacturer.
Western Blot Analysis
Whole-cell lysates were prepared using cell lysis buffer with 1 mM PMSF and incubated on ice for 30 to 60 min. Total protein (20 μg/lane) was resolved on NuPAGE Novex 4–12% Bis–Tris gels and electrophoretically transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA). Nonspecific binding sites were blocked with 5% milk in TBST (120 mM Tris–HCl, pH 7.4, 150 mM NaCl, and 0.05% Tween 20) for 1 h at room temperature. Target proteins were detected by using rabbit or mouse anti-human antibodies (1:500 to1000 dilution) for overnight at 4°C. The membranes were washed three times and incubated with secondary antibodies (1:10,000 dilution) conjugated with HRP. Immune complexes were visualized using the enhanced chemiluminescence system (Amersham Biosciences, Arlington, IL). Equal loading and transfer were confirmed by blotting the same membrane with β-actin antibody (1:5000 dilution). Data are representative of three independent experiments with nearly identical results.
Cell Proliferation Assay
Cells were seeded in 96-well plates at a density of 5 × 103 cells/well in RPMI 1640 culture media with 10% FBS and grown for up to 3 days after treatment. Cell proliferation was assessed by using Cell-Counting Kit-8 (Dojindo Molecular Technologies, Inc., Gaithersburg, MD) daily. Each point was performed in triplicate, and the experiments were repeated three times. The values, corresponding to the number of viable cells, were read at OD450 with FlexStation3 Microplate Reader (Molecular Devices, CA).
VEGF ELISA
The supernatant from cultured cells was collected at the indicated time points and VEGF levels were measured using human VEGF ELISA kit (R&D systems Inc. Minneapolis, MN) according to the manufacturer’s instructions. All experiments were performed on at least two separate occasions.
Statistical Analysis
The statistical analyses were performed using student t-test for comparisons between the treatment groups. A p value of < 0.05 was considered significant. All values are presented as mean ± SEM for the indicated number of independent samples.
RESULTS
GRP-R Silencing Downregulates HIF-1α and VEGF Expression
HIF-1α plays a crucial role in enhancing the survival of cancer cells via its central involvement in the transcription of genes that promote the Warburg effect (13). To determine whether GRP-R regulates HIF-1α, we first examined the effect of GRP-R silencing on HIF-1α protein expression under both normoxic and chemically induced hypoxic conditions. Western blotting demonstrated that silencing GRP-R in BE(2)-C cells (shGRP-R) decreased protein expression of HIF-1α and its downstream target, VEGF, as compared to controls (shCON) in both normoxia and hypoxia (Fig. 1A). Treatment with CoCl2 enhanced protein expression of HIF-1α and VEGF as compared to normoxic BE(2)-C cells. Using a VEGF ELISA and cell culture supernatant, we also found a significant decrease in VEGF secretion after GRP-R silencing in both normoxic and hypoxic conditions. These findings demonstrate that GRP-R signaling plays a role in regulating HIF-1α expression and its downstream target VEGF, suggesting a potential role for GRP-R in regulating tumor glucose metabolism.
Figure 1. GRP-R silencing downregulates HIF-1α and VEGF expression.
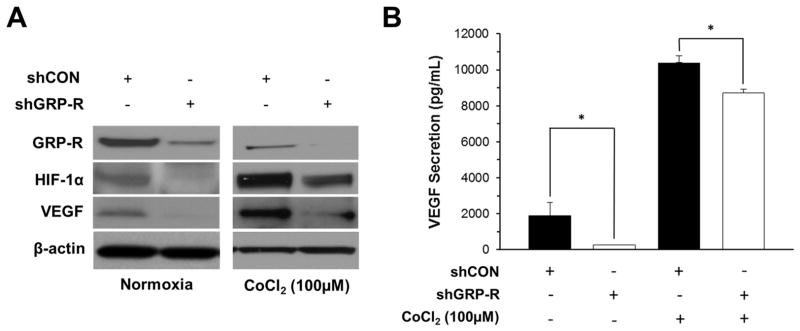
A. Western blotting was used to demonstrate decreased HIF-1α and VEGF protein expression in BE(2)-C/shGRP-R cells under normoxic and chemically induced hypoxia using CoCl2 (100 μM). β-actin demonstrated relatively equal loading. B. BE(2)-C/shCON and shGRP-R cells were seeded onto 6-well plates and cultured for 48 h, then supernatant was collected for VEGF ELISA. ELISA was completed in triplicate (means ± SEM; *=p<0.05 vs. shCON).
GRP-R Regulates Key Enzymes Involved in Glucose Metabolism
Glucose metabolism has several critical points of regulation, including activation and inhibition of the PDC. This enzyme complex is controlled via three major mechanisms, reversible phosphorylation/dephosphorylation (short-term regulation); balancing the redox state and acetyl-CoA/CoA ratio; and transcription activity of regulatory enzymes (long-term regulation) (14). To examine the effect of GRP-R silencing on glucose metabolism on neuroblastoma cells, we collected RNA from both BE(2)-C/shCON and BE(2)-C/shGRP-R cells, and used it in a PCR array specific for regulators of glucose metabolism. As compared to BE(2)-C/shCON cells, PDK4 levels were decreased by 5.61 fold (Fig. 2A) while the levels of PDP2 were increased by 7.50 fold (Fig. 2A) in GRP-R silenced cells. Other isoforms of PDK (PDK 1, 2 and 3) and PDP1 were not significantly changed in our microarray (results not shown). These results were validated in our MYCN amplified BE(2)-C and non-MYCN amplified SK-N-AS cell lines via Western blotting under normoxic conditions (Fig. 2B). Specifically, silencing GRP-R decreased protein expression of PDK4 and increased PDP2 in both cell lines (Fig. 2B). HK1 is another known regulator of glycolytic flux that is activated by HIF1α expression and served as a downstream marker of HIF-1α activity. Silencing GRP-R decreased protein expression of HK1 in SK-N-AS and BE(2)-C cells (Fig. 2B). Inactivation of the PDC by PDK4 has many downstream effects including mitochondrial dysfunction and inhibition of apoptosis, both of which favor tumor proliferation (15). Silencing GRP-R downregulates PDK4 and promotes PDP2 expression in neuroblastoma, providing further insight into how this signaling pathway modulates these key regulators of aerobic glycolysis.
Figure 2. GRP-R regulates key genes involved in glucose metabolism.
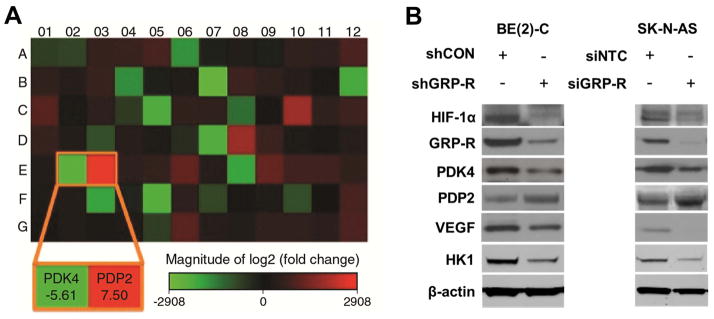
A. Human Glucose Metabolism PCR Array was performed with RNA isolated from BE(2)-C/shCON and BE(2)-C/shGRP-R cells. The altered gene expression was exhibited with heat map. The expression of PDK4 was decreased 5.61-fold and PDP2 was increased 7.5-fold. B. Alteration of HIF-1α, GRP-R, PDK4, PDP2, VEGF, and HK1 protein expression with GRP-R silencing (shGRP-R, siGRP-R) was confirmed by Western blotting in both BE(2)-C and SK-N-AS cells under normoxic conditions. β-actin demonstrated relatively equal loading.
Hypoxia and PDK4 Regulate the Expression of HIF-1α and VEGF
Hypoxic stress is known to upregulate the transcription of genes that promote aerobic glycolysis and subsequently promote neuroblastoma aggressiveness (16). Our studies demonstrate that treating BE(2)-C cells with CoCl2 substantially increased PDK4 and HIF-1α protein expression as compared to untreated cells (Fig. 3A). Furthermore, silencing PDK4 (siPDK4) decreased protein expression of HIF-1α in both normoxic and CoCl2 induced hypoxic conditions (Fig. 3A). We then performed a VEGF ELISA in control vs. siPDK4 neuroblastoma cells, with and without CoCl2. As demonstrated in Fig. 3B, the secretion of VEGF increased under hypoxia, in both siNTC and siPDK4 cells, as compared to untreated cells. Silencing PDK4 significantly decreased VEGF secretion under normoxic conditions. PDK4 silencing also decreased VEGF secretion in CoCl2 induced hypoxia, but this trend failed to reach statistical significance in our trials. Our findings indicate that PDK4 is involved in the regulation of HIF-1α expression and subsequently affects the secretion of its downstream targets, as illustrated here with VEGF.
Figure 3. Hypoxia and PDK4 regulate the expression of HIF-1α and VEGF.
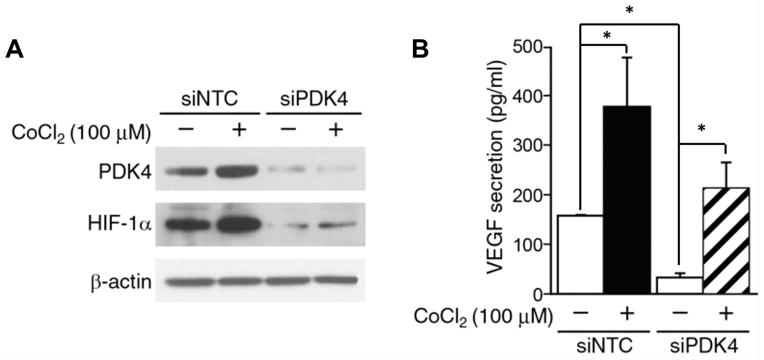
A. Cells were transfected with non-targeting control or PDK4 siRNA for 24 h, and then were treated with CoCl2 (100 μM) for 24 h. PDK4 and HIF-1α expression were detected by Western blotting. PDK4 silencing decreased CoCl2 induced HIF-1α expression. β-actin demonstrated relatively equal loading. B. VEGF secretion was measured using cell culture supernatant by ELISA. ELISA was completed in triplicate (means ± SEM; *=p<0.05 vs. siNTC).
DCA Inhibits Phosphorylation of the Pyruvate Dehydrogenase Complex (PDC) and Decreases Cell Proliferation
DCA, a known inhibitor of the PDK family (17), has been shown to shift cancer cells towards glucose oxidation in NSCLC, breast (18), endometrial (19) and prostate (20) cancers, resulting in the induction of apoptosis. PDK-mediated phosphorylation of the pyruvate dehydrogenase E1α (PDH E1α) inactivates PDC, blocking pyruvate decarboxylation necessary for glucose oxidation. Consistent with this published literature, we were able to block PDH E1α phosphorylation (pPDH E1α) using DCA. This was exhibited in a dose-dependent fashion with a markedly decreased protein expression of pPDC seen at concentrations of 20 and 50 mM (Fig. 4A). DCA significantly decreased cell proliferation in both our BE(2)-C/shCON and SK-N-AS cell lines. Specifically, DCA treatment at 20 mM blocked neuroblastoma proliferation in at 24, 48, and 72 h in both cell lines (Figs. 4B, C). Low-dose treatment with DCA (10 mM) resulted in more modest changes in cell proliferation with statistically significant changes in cell growth observed only at 72 h in BE(2)-C/shCON cells and only at 48 h in SK-N-AS cells (Figs. 4B, C).
Figure 4. Dichloroacetate (DCA) inhibits phosphorylation of the pyruvate dehydrogenase complex (PDC) and decreases cell proliferation.

A. BE(2)-C cells were treated with DCA, a PDK inhibitor at various concentrations (0 – 50 mM) for 48 h. Protein expression of phospho-PDC (pPDH E1α) and total PDC (PDH E1α) were detected by Western blotting. DCA (20 and 50 mM) inhibited the PDC phosphorylation. β-actin demonstrated relatively equal loading. B. Cell proliferation was measured by plating BE(2)-C/shCON (shCON) and BE(2)-C/shGRP-R (shGRP-R) cells in 96-well plates at a density of 5 × 103 cells per well in RPMI 1640 culture medium with 10% fetal bovine serum and treated with DCA at 10 and 20 mM. Cell viability was assessed daily using Cell-Counting Kit-8 (mean± SEM, *=p<0.05 vs. control). C. Cell proliferation was also measured in SK-N-AS cells treated with DCA at 10 mM and 20 mM using Cell-Counting Kit-8 (mean ± SEM, *=p<0.05 vs. control). D. Immunoblotting for HIF-1α, PARP, and caspase 3 was completed for BE(2)-C and SK-N-AS cells treated with DCA (20 - 50 mM). β-actin demonstrated relatively equal loading.
We also completed cell proliferation assays using our stably transfected BE(2)-C/shGRP-R cell line to assess for potential synergistic consequences of GRP-R and PDK inhibition. DCA treatment blocked BE(2)-C/shGRP-R proliferation, as compared to non-treated BE(2)-C/shGRP-R controls (Fig. 4B). Notably, no statistically significant difference in cell viability was noted in BE(2)-C/shGRP-R cells at 48 (p=0.11) and 72 h (p=0.69) following treatment when compared to 24 h after treatment time point, demonstrating that cellular growth was halted with combined GRP-R silencing and PDK inhibition using DCA (20 mM; Fig. 4B). BE(2)-C/shGRP-R cells were also more susceptible to low dose (10 mM) DCA inhibition, as compared to BE(2)-C/shCON, with statistically significant inhibition of cell proliferation noted at 24, 48, and 72 h after treatment. These findings suggest that the DCA treatment has a synergistic effect with silencing GRP-R. Thus, DCA and GRP-R appear to have partially overlapping mechanisms in regulating PDK activity but likely utilize additional mechanisms to elicit their synergistic antiproliferative effects in our neuroblastoma cell lines.
Immunoblotting for caspase 3 and PARP was performed on BE(2)-C and SK-N-AS cells as markers of apoptosis induction with DCA treatment (Fig. 4D). Notably, caspase 3 and PARP cleavage products were increased in BE(2)-C cells, suggesting DCA treatment induced apoptosis (Fig. 4D). Full-length PARP expression was strikingly increased in the SK-N-AS cells with DCA treatment, and activated PARP cleavage products were appreciable via immunoblotting (Fig. 4D). Intriguingly, full-length caspase 3 protein expression also increased with DCA treatment, but no discernable caspase 3 cleavage products were observed in DCA-treated SK-N-AS cells (Fig. 4D). PARP activation has been implicated in multiple cell death mechanisms, including both apoptosis and necrosis via energy depletion. These findings suggest that multiple mechanisms of cell death may be elicited by DCA treatment in neuroblastoma cell lines (21). Here, we highlight that DCA can be used effectively to inhibit PDC phosphorylation, a key regulator of glucose oxidation, and that this small molecule induces cell death and has synergistic antiproliferative effects with GRP-R inhibition in halting neuroblastoma growth.
DISCUSSION
One of the key differences observed in cancer cells is the way in which solid tumors utilize glucose. Known as the Warburg effect, tumor cells have been shown to maintain lactate production regardless of oxygen tension, shunting glucose away from oxidation towards aerobic glycolysis (22). Several known regulators of glucose metabolism include the family of hypoxia inducible factors (HIFs), the c-Myc oncogene, members of the AKT family and the tumor suppressor, p53 (23–25). Given previous work from our laboratory demonstrating a critical role of GRP-R signaling in neuroblastoma development and malignant potential (12, 26), we hypothesized that GRP-R may also be critical in regulating tumor glucose metabolism.
Our studies showed that silencing of GRP-R modulates the protein expression of HIF-1α and its downstream marker VEGF under both normoxia and chemically induced hypoxia. Our PCR array for glucose metabolism and confirmatory Western blots demonstrated that silencing GRP-R down-regulated PDK4 expression, a kinase known to promote aerobic glycolysis in tumor cells. We have also demonstrated that blocking GRP-R increased expression of PDP2, a phosphatase known to activate the PDC responsible for the moving pyruvate into the TCA cycle for glucose oxidation (6). Notably, differential expression of PDK4 and PDP2 with GRP-R silencing was also observed in both our MYCN amplified BE(2)-C and non-MYCN amplified SK-N-AS cell lines, suggesting the regulatory role of GRP-R is not dependent upon MYCN amplification. We also silenced PDK4 in BE(2)-C cells to assess its role in regulating glucose metabolism. PDK4 silencing decreased protein expression of HIF-1α and VEGF protein expression under both normoxic and chemical induced hypoxic conditions. VEGF secretion was diminished with PDK4 silencing but only was only statistically significant under normoxic conditions. Knowing that HIF-1α, PDK4, and PDP2 are key components in mediating the Warburg phenotype in tumor cells, our results suggest that signaling through GRP-R plays a significant role in modulating glucose metabolism in neuroblastoma.
There are multiple triggers that can induce the activity or expression of genes that facilitate aerobic glycolysis/Warburg effect in cancer cells. HIF-1α has been identified as a key regulator of this phenomenon (13, 27). Our study illustrates two potential avenues by which GRP-R modulates the activity of HIF-1α. First, GRP-R could directly regulate HIF-1α expression levels. Alternatively, GRP-R may regulate degradation of HIF-1α. In well-oxygenated cells, HIF-1α is hydroxylated via prolyl hydrolase domain (PHD) proteins for subsequent proteasomal degradation. The activity of PHD proteins is driven by oxygen tension and high levels of α-ketoglutarate, an intermediate of glucose oxidation (27). As such, GRP-R may regulate HIF-1α expression by regulating intermediates of glucose oxidation, such as α-ketoglutarate. Interestingly in this study, we have described that GRP-R reciprocally regulates PDK4 and PDP2 expression. These findings suggest that silencing GRP-R may promote glucose oxidation and potentially favor the creation of α-ketoglutarate, a known promoter of HIF-1α degradation. Our current study does not address how GRP-R regulates HIF-1α protein levels, but our observations invite further inquiry into the mechanism by which GRP-R regulates neuroblastoma glucose metabolism.
Hypoxia has a positive effect on the expression of various PDK isoforms (28, 29). In line with these findings, under chemically induced hypoxia via CoCl2, we were also able to show increased expression of both PDK4 and HIF-1α in neuroblastoma cells. Subsequently, after silencing PDK4, the expression of HIF-1α was decreased, as compared with controls. This is in contrast to other studies that demonstrate HIF-1α, acting as a regulator of PDK4 (30). While inhibition of HIF-1α by other PDK isoforms has been shown (31), this is the first time that PDK4 inhibition has been shown to decrease HIF-1α expression in neuroblastoma. In this study, targeting PDK4 not only has a negative effect on HIF-1α expression but it can also modulate its downstream targets. Given that HIF-1α is known to be associated with tumor aggressiveness (32), regulation of this transcription factor via inhibition of PDK4 could therefore be essential in preventing malignant tumor behavior in neuroblastoma.
Finally, identifying new mechanisms that contribute to neuroblastoma tumor aggressiveness is necessary to developing novel treatment options for this disease. DCA has the ability to inhibit the PDK family and induce cells to switch from aerobic glycolysis to glucose oxidation. We have demonstrated in this study that treating neuroblastoma with DCA blocks cell growth and promotes cell death. DCA promotes apoptosis via caspase 3 and PARP activation in BE(2)-C cells. Interestingly, we failed to demonstrate caspase 3 activation in SK-N-AS cell, while PARP cleavage was induced. PARP activation is known to mediate cell death via both apoptosis and energy-dependent cell necrosis (21). Given its known role in regulating glucose oxidation, our findings suggest that DCA may utilize both apoptosis and cell necrosis via energy depletion to mediate cell death in neuroblastomas. Silencing GRP-R and blocking PDK with DCA halted BE(2)-C cell viability with no discernable growth noted over the appreciable 72 h observation period. These findings suggest that silencing GRP-R and treatment with DCA have synergistic antiproliferative effects. DCA is known to specifically target the mitochondria of malignant cells and have relatively little effect on normal cells (33), but there has been some discrepancy in the literature about the effectiveness in the treatment of neuroblastoma (34). However given its success in other cancers and our preliminary data showing its ability to prevent the phosphorylation of the PDC, decrease cell proliferation, and its synergistic effects with GRP-R silencing, we believe this drug warrants further investigation as a therapeutic adjunct in the treatment of malignant neuroblastoma.
Acknowledgments
This work was supported by a grant R01 DK61470 from the National Institutes of Health.
Footnotes
CONFLICTS OF INTEREST
The authors declare that no competing interests exist.
References
- 1.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369(9579):2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O. Iron, the Oxygen-Carrier of Respiration-Ferment. Science. 1925;61(1588):575–582. doi: 10.1126/science.61.1588.575. [DOI] [PubMed] [Google Scholar]
- 3.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 4.Matsushita K, et al. Glycolysis inhibitors as a potential therapeutic option to treat aggressive neuroblastoma expressing GLUT1. J Pediatr Surg. 2012;47(7):1323–1330. doi: 10.1016/j.jpedsurg.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Y, et al. Emerging metabolic targets in cancer therapy. Front Biosci. 2011;16:1844–1860. doi: 10.2741/3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McFate T, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. 2008;283(33):22700–22708. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel MS, Korotchkina LG. Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation: complexity of multiple phosphorylation sites and kinases. Exp Mol Med. 2001;33(4):191–197. doi: 10.1038/emm.2001.32. [DOI] [PubMed] [Google Scholar]
- 8.Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem. 1995;270(48):28989–28994. doi: 10.1074/jbc.270.48.28989. [DOI] [PubMed] [Google Scholar]
- 9.Huang B, et al. Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J Biol Chem. 1998;273(28):17680–17688. doi: 10.1074/jbc.273.28.17680. [DOI] [PubMed] [Google Scholar]
- 10.Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10(1):47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- 11.Kim S, et al. Gastrin-releasing peptide is a growth factor for human neuroblastomas. Ann Surg. 2002;235(5):621–629. doi: 10.1097/00000658-200205000-00003. discussion 629–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiao J, et al. Gastrin-releasing peptide receptor silencing suppresses the tumorigenesis and metastatic potential of neuroblastoma. Proc Natl Acad Sci U S A. 2008;105(35):12891–12896. doi: 10.1073/pnas.0711861105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr. 2007;39(3):231–234. doi: 10.1007/s10863-007-9081-2. [DOI] [PubMed] [Google Scholar]
- 14.Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans. 2006;34(Pt 2):217–222. doi: 10.1042/BST20060217. [DOI] [PubMed] [Google Scholar]
- 15.Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Targeting glucose metabolism: an emerging concept for anticancer therapy. Am J Clin Oncol. 2011;34(6):628–635. doi: 10.1097/COC.0b013e3181e84dec. [DOI] [PubMed] [Google Scholar]
- 16.Qing G, et al. Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1alpha. Cancer Res. 2010;70(24):10351–10361. doi: 10.1158/0008-5472.CAN-10-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003;43(7):683–691. [PubMed] [Google Scholar]
- 18.Bonnet S, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008;109(3):394–402. doi: 10.1016/j.ygyno.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao W, et al. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 2008;68(11):1223–1231. doi: 10.1002/pros.20788. [DOI] [PubMed] [Google Scholar]
- 21.Koh DD, TM, Dawson VL. Mediation of cell death by poly(ADP-ribose) polymerase 1. Pharmacologic Research. 2005;52:5–14. doi: 10.1016/j.phrs.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 22.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 23.Elstrom RL, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64(11):3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 24.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66(18):8927–8930. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 25.Matoba S, et al. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 26.Lee S, et al. FAK is a critical regulator of neuroblastoma liver metastasis. Oncotarget. 2012 doi: 10.18632/oncotarget.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. The Journal of clinical investigation. 2013;123(9):3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283(42):28106–28114. doi: 10.1074/jbc.M803508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Lee JH, et al. Hypoxia induces PDK4 gene expression through induction of the orphan nuclear receptor ERRgamma. PLoS One. 2012;7(9):e46324. doi: 10.1371/journal.pone.0046324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sutendra G, et al. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene. 2012 doi: 10.1038/onc.2012.198. [DOI] [PubMed] [Google Scholar]
- 32.Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 33.Vella S, Conti M, Tasso R, Cancedda R, Pagano A. Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells. Int J Cancer. 2012;130(7):1484–1493. doi: 10.1002/ijc.26173. [DOI] [PubMed] [Google Scholar]
- 34.Niewisch MR, et al. Influence of dichloroacetate (DCA) on lactate production and oxygen consumption in neuroblastoma cells: is DCA a suitable drug for neuroblastoma therapy? Cell Physiol Biochem. 2012;29(3–4):373–380. doi: 10.1159/000338492. [DOI] [PubMed] [Google Scholar]