

Abstract
Congenital heart disease is the most common human developmental disorder, affecting ~1:100 newborns, and is the primary cause of birth-defect related deaths worldwide. As a major regulator of receptor tyrosine kinase (RTK), cytokine and G-protein coupled receptor signaling, the non-receptor protein tyrosine phosphatase SHP2 plays a critical role in normal cardiac development and function. Indeed, SHP2 participates in a wide variety of cellular functions, including proliferation, survival, differentiation, migration, and cell-cell communication. Moreover, human activating and inactivating mutations of SHP2 are responsible for two related developmental disorders called Noonan and LEOPARD Syndromes, respectively, which are both characterized, in part, by congenital heart defects. Structural, enzymologic, biochemical, and SHP2 mouse model studies have together greatly enriched our knowledge of SHP2 and, as such, have also uncovered the diverse roles for SHP2 in cardiac development, including its contribution to progenitor cell specification, cardiac morphogenesis, and maturation of cardiac valves and myocardial chambers. By delineating the precise mechanisms by which SHP2 is involved in regulating these processes, we can begin to better understand the pathogenesis of cardiac disease and find more strategic and effective therapies for treatment of patients with congenital heart disorders.
Keywords: PTPN11, SHP2, SHP-2, SHPTP2, phosphatase, signaling, cardiac development, congenital heart disease, valve, myocardium
Introduction
A consequence of post-translational modification, phosphorylation influences the activity, complex association, and/or localization of proteins, and therefore plays a key role in mediating signaling events that control multiple important cellular processes, including proliferation, survival, differentiation, migration, and cell-cell communication. Therefore, it is not surprising that alterations in phosphorylation of proteins, as orchestrated by the balanced actions of protein kinases and protein phosphatases, lead to onset of multiple diseases, including cancers, metabolic diseases, and developmental anomalies.
Congenital heart disease (CHD), the most common human developmental disorder, affects ~1:100 newborns and is the primary cause of birth-defect related deaths worldwide [1]. Though numerous CHD phenotypes of various etiologies exist, the majority of patients present with abnormal valves, membranous septa and/or myocardial defects [2]. Abnormalities in signaling molecules and/or pathways involved in cardiac development are implicated; however, the underlying mechanisms remain poorly understood. In this regard, it was recently discovered that both gain-of-function (GOF) as well as loss-of-function (LOF) mutations in PTPN11, the gene encoding the protein tyrosine phosphatase (PTP) SHP2, induce a variety of cardiac developmental defects, highlighting the importance of fine-tuning phosphorylation events during heart embryogenesis. In this review, we will focus on the newly discovered key functional roles for SHP2 in cardiac development and discuss how the mutations therein might contribute to the onset of CHD.
SHP2 structure and mechanism of action
SHP2, a ubiquitously expressed non-receptor protein-tyrosine phosphatase, contains two SH2 domains, a central PTP catalytic domain and a C-terminal tail with two tyrosine phosphorylation sites and a proline-rich motif (Figure 1). Resolution of the crystal structure and biochemical analyses have elucidated its mechanism of regulation, whereby in the inactive state, the backside loop of the N-SH2 domain folds into the PTP catalytic pocket rendering SHP2 physically and biochemically inactive [3]. Binding of a phosphotyrosyl (pY) peptide to the SH2 domain induces a conformational change, unfolding the structure and allowing substrates to access the catalytic site of the enzyme [4, 5] (Figure 1). The interaction of SHP2 with pY proteins, such as receptor tyrosine kinases (RTK), cytokine receptors and scaffolding adaptors (e.g. FRS, IRS, Gab), mediates downstream signaling events that control proliferation, differentiation, migration, cell cycle progression, and apoptosis (Figure 2).
Figure 1.

SHP2 structure and mechanism of activation upon phosphotyrosyl-peptide binding.
SHP2 contains two SH2 domains (N-SH2, C-SH2), a central PTP catalytic domain and a C-terminal tail with two tyrosine phosphorylation sites (Y542, Y588) and a proline-rich motif. In the absence of receptor stimulation, SHP2 is found in a closed conformation with the N-SH2 backside loop bound to the PTP domain (left). Upon stimulation, SHP2 is recruited to the membrane by phosphotyrosyl-peptide binding, unfolded, and activated, allowing for substrate to bind into the catalytic pocket (right).
Figure 2.
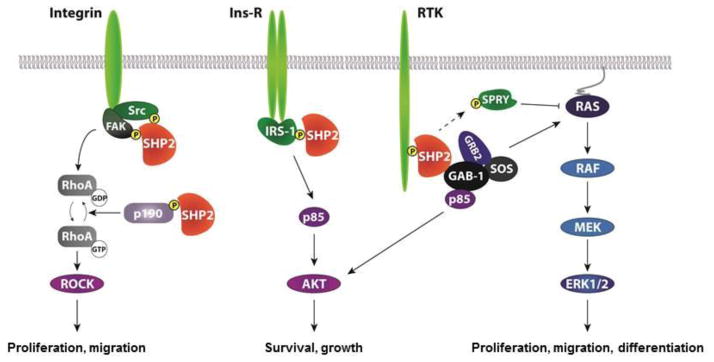
Signaling pathways regulated by SHP2.
SHP2 binds to receptors and adaptor proteins and facilitates positive signaling to multiple signaling pathways involved in important cellular pathways that control proliferation, migration, differentiation, growth, and survival. SHP2 dephosphorylates Src, FAK, and p190RhoGAP, leading to activation of RhoA/ROCK (left panel). SHP2 also dephosphorylates IRS-1 to mediate PI3K/AKT signaling pathway (middle panel). In addition, several mechanisms have been proposed for activation of the MAPK/ERK pathway, including dephosphorylation of the SPROUTY inhibitor and recruitment of GRB2/SOS activators (Right panel), but additional substrates are thought to be involved but have yet to be identified.
SHP2 is required for Ras/ERK pathway activation by most, if not all, RTKs as well as by cytokine receptors and integrins [5–8] (Figure 2). Although there is general agreement that activated SHP2 acts upstream and/or in parallel to Ras, the precise substrate target(s) that promote Ras/ERK activation remain controversial. Recent work implicates SHP2 in RTK-evoked SFK activation, with SFKs, in turn, required for sustained Ras activation [9]. Other targets include inactivation of RasGap [10] and Sprouty inhibitors [11]. Besides this positive signaling function, SHP2 negatively regulates PI3K/AKT pathway in EGFR signaling, but is required for PDGF and IGF-evoked AKT signaling [12]. Additional actions of SHP2 on JAK/STAT [13], NFkB [14], NFAT [15], and RhoGAP [16] have been proposed.
SHP2 mutations and human disease
SHP2 is required for normal development. Heterozygous missense mutations of PTPN11 cause human disease, namely Noonan (NS) and LEOPARD (LS) Syndromes, two related congenital disorders involving cardiac defects (Table 1). Both NS and LS are autosomal dominant disorders characterized by multiple, variably penetrant defects, including proportional short stature, facial dimorphism, and CHDs. The most common cardiac manifestation in NS is pulmonic stenosis (PS) resulting from dysplastic valve leaflets, but stenosis of other valves (mitral valve), atrial septal defects (ASD), ventricular septal defects (VSD), atrioventricular septal defects (AVSD), or more rarely, double outlet right ventricle (DORV) also are seen [17, 18]. Hypertrophic cardiomyopathy (HCM) has also been reported in a few NS patients without SHP2 mutations [19], but genotype-phenotype correlation studies show that only 8% of SHP2 associated-NS patients present with this cardiac disorder [20, 21]. In contrast to NS, the majority of LS patients (~85–90%) develop HCM [22]. However, they have also been demonstrated to have similar valve anomalies to NS as well [22]. In addition, LS patients have a higher prevalence of conduction abnormalities, including left anterior and posterior hemi-blocks, right bundle branch block or complete atrioventricular block, atrial tachycardia or fibrillation, and prolonged PR intervals [23].
Table 1.
Cardiac defects observed in PTPN11-associated Noonan and LEOPARD Syndromes.
| Noonan Syndrome 1/1000 to 1/2000 birth |
LEOPARD Syndrome No epidemiologic data |
|---|---|
| Pulmonary stenosis | |
| Septal defects (ASD, VSD, AVSD, DORV) | Hypertrophic cardiomyopathy |
| Hypertrophic cardiomyopathy | ECG anomalies |
| Aortic coarctation | Valve anomalies (Mitral, Aortic) |
| Mitral valve defects | Pulmonary stenosis |
| Tetralogy of Fallot | Coronary abnormalities |
| Patent ductus arteriosus | Septal defects |
Cardiac defects are listed by order of frequency
Interestingly, the specific PTPN11 mutations that cause either NS or LS are distinct. Most NS mutations reside within the N-SH2 domain interface that allows for the intramolecular interaction with the PTP domain. Therefore, NS mutations disrupt the ability of SHP2 to retain the closed, inactive conformation without affecting the PTP domain activity. Consequently, NS-associated SHP2 mutants display increased basal activity (since they are found in an open conformation) and behave as GOF alleles (since the PTP domain is more accessible to substrate) [24]. In contrast, LS mutations affect conserved residues important for PTP catalysis, leading to LOF of the phosphatase activity [25, 26]; however, as in NS, they also perturb N-SH2/PTP domain interactions, suggesting that both NS and LS mutants can out-compete WT SHP2 for binding to RTKs/scaffolds [25, 27–29]. Therefore, it is also likely that both NS and LS mutants have additional perturbations in non-phosphatase domains of SHP2, functions that possibly play significant and important roles in cardiac development.
SHP2 and early development
Studies investigating the role of SHP2 during early cardiac development have been impeded by the fact the SHP2 homozygous deletion in mice is early embryonic lethal. SHP2 null blastocytes die at the time of implantation (between E3.5–6.5) due to massive apoptosis of both embryonic inner cell mass and trophoblastic cells. Indeed, in the trophoblast, SHP2 promotes cell survival by inducing FGF4-mediated activation of the SFK/RAS/ERK cascade, leading to destabilization of the pro-apoptotic BIM protein [30]. However, the pro-survival role of SHP2 in embryonic cells still remains unknown, particularly since inhibition of the same FGF4/SFK/ERK pathway in embryonic stem (ES) cells impairs proliferation but does not affect survival [31]. In addition, inhibition of SHP2, either by homozygous deletion or by siRNA knock-down in ES cells does not induce apoptosis; rather this inhibition blocks differentiation of cells into each of the three germ layers [32], suggesting that SHP2 is likely also involved in embryonic cell differentiation (Figure 3). Additional studies will be required to assess the anti-apoptotic function of SHP2 during embryonic development prior to gastrulation. In particular, comparison of SHP2 deletion versus SHP2 LOF of the phosphatase activity, using LS mouse models for example, may help elucidate whether SHP2 in early cardiac development relies primarily on phosphatase-dependent and/or phosphatase-independent functions of the enzyme.
Figure 3.

SHP2 is involved in early embryonic and cardiac development through multiple mechansims.
Studies have linked SHP2 to survival mechanisms through signaling pathways in inner cell mass, trophoblastic, and cardiac progenitor cells. In addition, SHP2 is also involved in proliferation, differentiation, and gastrulation convergence and extension movements through activation of distinct signaling pathways.
Cardiac progenitor specification and survival
Studies in Zebrafish and Xenopus embryos using morpholino-mediated knock-down of SHP2, as well as NS and LS SHP2 mutant injections, have bypassed the need for SHP2 in early embryonic development and have allowed for the elucidation of the role for SHP2 during cardiac specification and early cardiac morphogenesis (Table 2). Explantation assays in amphibian and avian embryos have shown that cells of the cardiac lineage are specified and committed at the onset of gastrulation, as cells are ingressing through the primitive streak [33, 34]. Following gastrulation and the formation of the three embryonic germ layers, myocardial and endocardial precursor cells, located in the anterior lateral plate mesoderm, are arranged into bilateral fields, which then merge at their anterior margins to form the cardiac crescent [35]. Next, convergence and fusion of the bilateral heart primordia along the midline initiates the formation of the linear heart tube, which is composed of an external myocardial layer and an internal endocardial layer separated by a myocardial-secreted extracellular matrix called cardiac jelly [35]. Importantly, mesodermal induction and cardiac cell specification and differentiation are governed by multiple signaling pathways. Key players are Nodal, Bone Morphogenetic Protein (BMP), Wnt, and FGF signals, which cooperate to induce cardiac differentiation, as assessed by induced expression of key regulators of the lineage: NKX2.5, GATA4, and TBX5. Myocyte lineage commitment is further marked by the expression of contractile proteins, including myosin light chain 2a (MLC2a) and sarcomeric myosin heavy chain (MHC) [36].
Table 2.
Cardiac phenotypes observed in different animal models of SHP2 mutations.
| Animal | Experimental design | SHP2 mutation | Cardiac phenotype | References |
|---|---|---|---|---|
| Xenopus | Injection of SHP2NS mRNA at the one-cell stage | NS (N308D, D61G, Q79R) | Smaller hearts, Impaired cardiac looping, Incomplete integration of cardiac precursors, Delay in cell cycle, Enrichment in ECM, Disruption of myofibril formation and polarity. | Colon et al. 2012 |
|
| ||||
| Zebrafish | MO-mediated knock-down of SHP2 vs. injection of SHP2NS and SHP2LS mRNA at the one-cell stage | Deletion NS (D61G, T73I) LS (A462T, G465A) |
Defective convergence-extension cell movements during gastrulation | Jopling et al. 2007 |
|
| ||||
| Injection of SHP2NS and SHP2LS mRNA at the one-cell stage | NS (D61G, T73I) LS (A462T, G465A) |
Reduced heart rate, Impaired cardiac looping, Slower directional migration of cardiomyocytes, Impaired ciliogenesis and cilia function in Kupffer’s vesicle (reduced cilia number and length, loss of coordinated flow) | Jopling et al. 2012 | |
|
| ||||
| Mouse | Knock-in, Ubiquitous null mutation, exon 2 replaced by βgal-expressing cassette and spliced acceptor site | Deletion | Inner cell mass and trophoblastic cell death | Yang et al. 2006 |
|
| ||||
| Wnt1-Cre driven excision of floxed exon 3–4 | Deletion | Failure of OFT septation, Anomalies of great arteries | Nakamura et al. 2009 | |
|
| ||||
| Knock-in, Ubiquitous expression | NS (D61G) | Enlarged cushions, ASD, VSD, DORV, myocardial thinning | Araki et al. 2004 | |
|
| ||||
| Inducible knock-in, Ubiquitous expression (Mox2-Cre) | NS (D61Y) | Enlarged cushions, ASD, VSD, DORV, myocardial thinning | Araki et al. 2009 | |
|
| ||||
| Inducible knock-in, Tie2-Cre driven expression | NS (D61Y and N308D) | Enlarged cushions, VSD, DORV, myocardial thinning | Araki et al. 2009 | |
| Inducible knock-in, βMHC-Cre driven expression | Absence of cardiac phenotype | |||
| Inducible knock-in, Wnt1-Cre driven expression | Absence of cardiac phenotype | |||
|
| ||||
| Transgenic overexpression of βMHC-SHP2NS | NS (Q79R) | Ventricular non compaction, VSD | Nakamura et al. 2007 | |
|
| ||||
| Tie2-Cre driven Transgenic overexpression of SHP2NS | NS (Q79R) | Enlarged cushions | Krenz et al. 2008 | |
|
| ||||
| Inducible knock-in, Ubiquitous expression (EIIA-Cre) | LS (Y279C) | Adult onset hypertrophic cardiomyopathy | Marin et al. 2011 | |
|
| ||||
| Transgenic overexpression of βMHC-SHPLS | LS (Q510E) | Hypertrophic cardiomyopathy, enlarged atria, thickened IVS, membranous and muscular VSD | Schramm et al. 2012 | |
| Transgenic overexpression of αMHC-SHPLS | Absence of cardiac phenotype | |||
Similar to what happens in trophoblastic and ES cells, SHP2 is required for FGF signaling during specification of cardiac progenitors. Studies on Xenopus embryos, using cardiac explant assays, have shown that SHP2 is required to maintain the population of cardiac progenitors. In the absence of SHP2, cardiac precursors remain in two unfused bilateral populations, and exhibit reduced expression of early cardiac markers NKX2.5, GATA4, and TBX5 [37]. As such, cardiac cells fail to initiate differentiation, as assessed by lack of MLC and MHC expressions. Co-immunoprecipitation experiments further suggest that SHP2 might play a role in FGF/FRS signaling, although inhibition of FGFR did not give a phenotype as strong as inhibition of SHP2 [37]. Interestingly, later FGF signaling inhibition (FGF3, 8, 18, 19) by BMP4 in cardiac progenitors is also necessary to reduce proliferation and initiate differentiation [38], suggesting that the SHP2-mediated FGF signaling mechanism in cardiac development, particularly during cardiac progenitor differentiation, might be more complex than it appears and may require a specific time-dependent regulation. In addition to the failure to differentiate, increased cell death is observed in the absence of SHP2 in Xenopus embryo heart explants, showing once again a prosurvival role for SHP2 in-vivo [37]. Coinciding with increased apoptosis, the failure of cardiac cells to fuse at the ventral midline might also be a secondary consequence of decreased cell survival (Figure 3). However, and alternatively, SHP2 might be involved in cell adhesion and/or migration, two processes necessary for morphogenetic movements.
Morphogenetic events: gastrulation, acquisition of embryonic asymmetry, and cardiac looping
Evidence of a role for SHP2 in cell migration in-vivo exists. Morpholino-mediated knock-down of SHP2 in zebrafish leads to defective convergence-extension movement during gastrulation, without affecting cell specification [39] (Table 2). It appears that this effect is not mediated by the MAPK/ERK pathway, which might be dispensable during gastrulation. Rather, the phenotype is rescued by the use of active SFKs or active RhoA, indicating that SHP2 is involved in gastrulation cell movements through its actions on these downstream pathways [39]. Interestingly, the expression of NS or LS mutant SHP2 in zebrafish embryos also induced similar convergence and extension cell movement defects as SHP knock-down and displayed craniofacial and cardiac defects, reminiscent of human symptoms. Moreover, NS and LS mutants did not show additive or synergistic effects, consistent with having activating and inactivating roles, respectively, within the same signaling pathway and likely through regulation of SFKs [39]. In addition, a parallel study comparing SHP2-deficient zebrafish embryos with those injected with mRNA encoding LS indicate both a phosphatase- and MAPK/ERK-dependent and -independent function for SHP2; phosphatase- and Erk-dependent function of SHP2 is required for neural crest specification and migration, whereas a phosphatase- and Erk-independent function for SHP2 is required to prevent apoptosis [40]. Further elucidation for the role of specific domains of SHP2 in cardiac development and/or in differing cardiac cell lineages remains to be elucidated.
Rightward looping of the heart tube is the first morphological manifestation of embryonic laterality. The mechanisms driving elongation, bending, and coiling of the tubular heart are controversial. The rapid elongation of the heart tube, by addition of progenitors cells from another cardiogenic area, termed second heart field [35, 41], at both sides of the tube is thought to create compressive loads resulting from growth differences between the heart and the confined space of the embryonic pericardial cavity [42]. Several computational models have supported the idea that these external physical forces drive the deformation of the straight-shaped heart tube into a helicoidal configuration [43, 44]. However, the fact that explanted embryonic hearts are able to undergo partial looping in-vitro suggests that other intrinsic mechanisms play a role during this process [45]. In fact, embryonic laterality is, in part, governed by the asymmetric expression of signaling molecules and transcription factors in any one side of the embryo. In the mouse, an organizing center called node, a distinct group of cells located at the anterior end of the primitive streak [46], is responsible for the left/right (LR) patterning of the embryo. It is proposed that FGF8 signaling triggers the expression of vesicles containing signaling factors, transported to one side of the embryo due to a leftward flow of fluid generated by motile cilia in the node [47], allowing for the transient expression of Nodal, a TGFβ family ligand. There, on the left side of the early developing embryo, Nodal controls the expression of further downstream regulators of LR patterning, Lefty1/2 and Pitx2 [48] (Figure 4). In the chick embryo, disruption of FGF8 leads to randomization of the direction of heart looping [49], whereas mice conditionally deficient in Fgf8 lack Nodal expression in the lateral plate mesoderm and exhibit right isomerism [50].
Figure 4.

Putative roles for SHP2 in embryonic Left-Right (LR) patterning and cardiac looping.
Embryonic cardiac laterality and related cardiac looping are governed by both tissue-extrinsic (top panel) and tissue-intrinsic (bottom panel) mechanisms. In the mouse node, a leftward nodal flow is generated by coordinated movements of cilia. This flow creates an asymmetric distribution of calcium and morphogens to the left side of the embryo, driving the expression of Nodal and subsequent effectors of LR patterning. SHP2 is involved in both FGF-mediated signaling and regulation of calcium. Therefore, SHP2 may not only participate in both cilia biogenesis and function, but also in the release of vesicles containing the morphogens.
Given the role for SHP2 in FGF signaling, it is not surprising to find that zebrafish embryos expressing NS or LS SHP2 mutants have impaired leftward heart displacement and randomization of LR asymmetry markers [51] (Table 2). In addition, SHP2 mutants display fewer and shorter cilia in Kupffer’s vesicle, the functional homologue of the mouse node, with additional loss of coordinated flow, suggesting that SHP2 may also play a role in cilia physiology itself. MAPK/ERK signaling, downstream of SHP2, is involved in this phenotype [51]. In addition, recent evidence suggests an important role for calcium (Ca2+) signaling in both cilia formation and sensing, possibly also explaining the heart defects observed in zebrafish mutants of SHP2. Indeed, disruption of receptor-mediated calcium release from the endoplasmic reticulum (ER) leads to randomization of heart and brain laterality by affecting the initial development of the Kupffer’s vesicle [52]. Furthermore, leftward fluid flow generated by the centrally located motile cilia is sensed by the peripherally located immotile cilia via the Ca2+-permeable cation channel Polycystin-2 (Pkd2). Flexing of these cilia results in an intracellular Ca2+ elevation on the left side of the node, and the elevated Ca2+ level in turn activates asymmetric patterns of gene expression [53]. The precise mechanistic role of the asymmetric calcium signaling on LR patterning is unclear. It is proposed that calcium induced calcium release between cells in the endoderm via gap junctions can relay the message from the node to the lateral plate mesoderm and therefore activate Nodal signaling [54]; how this occurs and whether SHP2 is involved in this process remains to be proven. Nonetheless, there is evidence linking SHP2 function to calcium signaling, modulating calcium flux either indirectly by MAPK/ERK mediated phosphorylation of the cav(1.2) subunit [55] or by direct association with IP3 receptors [56], thereby suggesting a possible additional role for SHP2 in LR patterning and cardiac looping (Figure 4, upper panels).
While SHP2 seems to play a role in LR patterning through its regulation of cilia physiology, it might also be involved in cardiac looping through a newly discovered tissue intrinsic mechanism requiring actin polymerization and non-muscle myosin II activity. In the presence of asymmetric Nodal signaling, actin polymerization in the heart is elevated, and clockwise rotation of the cardiac disc and leftward displacement of the linear heart tube are observed, resulting in robust dextral looping [57]. When actomyosin activity is blocked, even in the presence of asymmetric Nodal signaling, heart looping is impaired, indicating that Nodal is instructive in promoting laterality; however, other molecules, intrinsic to cardiac cells, are also thought to be required [57]. Interestingly, NS SHP2 mutations, through hyperactivation of ROCK, the downstream effector of RhoA, lead to defective formation and polarity of cardiac actin fibers and F-actin deposition in Xenopus embryos (Table 2). This, in turn, results in smaller hearts that fail to undergo complete looping and chamber formation and have delayed morphogenetic movements [58], suggesting that SHP2’s actions on actin polymerization in the myocyte itself might be important for cardiac looping (Figure 4, lower panels).
Complete defect in the LR symmetry, called situs inversus, is rare; however, it has been argued that even minor situs defects can cause cardiac defects, suggesting that the heart is more sensitive to situs defects than other organs [59, 60]. In this regard, minor impaired LR patterning evokes atrial and ventricular septation defects as well as causes alignment abnormalities of the great arteries. Therefore, these types of cardiac phenotypes, like NS and LS, might also be a direct consequence of SHP2 functional dysregulation in cardiac looping and embryonic laterality. Alternatively, SHP2 may also be involved in the intrinsic development of cardiac chamber myocytes and valvulogenesis, independently of these early developmental embryonic events.
Endocardial cushions and valve development
Following looping, the heart is segmented into the atrium, the atrioventricular canal (AVC), forming the junction between developing atria and ventricles, the ventricle, and the outflow tract (OFT), the region that develops into the left and right ventricular outlets, as well as the aorta and pulmonary trunk [61]. At the level of the AVC and proximal OFT, local tissue swellings, termed endocardial cushions, are formed by the accumulation of cardiac jelly between the endocardium and myocardium. Endocardial cells subsequently invade the cardiac jelly after undergoing endocardial-to-mesenchymal transition (EMT). At the level of the distal OFT, additional cushions (termed truncal cushions) arise and are later populated by mesenchymal cells originating from the neural crest, which also participate in OFT septation into the aorta and pulmonary trunk [61]. AVC endocardial cushions later develop into atrioventricular (mitral and tricuspid) valves, whereas the OFT endocardial cushions give rise to semilunar (aortic and pulmonic) valves, both forming thin, tapered leaflets with a single endothelial layer and a central matrix comprised of collagen, elastin, glycosaminoglycans, and fibroblastic valve interstitial cells [61]. Cell lineage tracking analysis has demonstrated the endothelial origin of resident cells throughout the leaflets of aortic and pulmonic valves, as well as the AV valves [62], although a contribution of epicardially-derived cells is also proposed for the parietal leaflet of AV valves [63]. The final transformation of endocardial cushions into valves and septa is a critical step in cardiac morphogenesis, as it initiates the development of the four-chambered heart. This transformation results from a region-specific balance between cellular proliferation, apoptosis, and differentiation and is orchestrated by progenitor cells derived from various origins, interacting with each other through multiple signaling pathways [64–66]. These pathways, which include VEGF, NFATc1, Notch, Wnt/β-catenin, BMP/TGF-β, ERBB, and Neurofibromin (NF1), are all known to involve SHP2; therefore it is possible that SHP2, and mutations therein, affect the various stages of valvulogenesis and mediate the formation of the chambers in the heart.
In particular, Epidermal Growth Factor (EGF) signaling inhibits endocardial cushion cell proliferation through antagonism of BMP-mediated activation of SMAD1/5/8 in the AVC and OFT [67]. Therefore, mice bearing an EGFR-hypomorphic allele (wa2/wa2) exhibit semilunar valve enlargement resulting from over-abundant mesenchymal cells. Further, heterozygous deletion of SHP2 in this reduced EGFR signaling background enhances the valve defects [68] (Table 2), thereby identifying a key role for SHP2 in valvulogenesis. Interestingly, GOF mutations of SHP2 in NS also lead to increased valve size. Ex-vivo analysis of endocardial cushions indicates that the observed valvulogenic abnormalities in NS are a consequence of prolonged EMT and increased ERK signaling [69] (Table 2). Indeed, hyaluronic acid signaling through ERBB2/3 heterodimers mediates the migratory process of endocardial cells into the cardiac jelly [70]. Therefore, it is possible that inhibition and over-activation of SHP2 can both lead to similar valve phenotypes by acting on different stages of valvulogenesis and/or through different signaling pathways (Figure 5).
Figure 5.

Putative roles for SHP2 in endocardial cushion and ventricular chamber development.
Cardiac valve and chamber development and maturation require the combined actions of multiple signaling pathways controlling proliferation, migration, differentiation, and apoptosis. SHP2 has proven to be involved in many of the pathways important for valvulogenesis and ventricular development, including signaling through EGF, ERBBs, and VEGF. While SHP2 involvement in endothelial cell proliferation and migration through ERK have previously been demonstrated, additional mechanisms involving other pathways in endocardium, as well as in cardiac chamber development, have yet to be validated.
VEGF signaling through NFATc1 increases endothelial cell proliferation [71], which might contribute to repopulation of the endothelial layer during EMT. In addition, VEGF also prevents excess endothelial cell transdifferentiation into mesenchymal cells by maintaining an endothelial cell fate [70]. It is possible that SHP2 may also be involved with the control of endocardial cell proliferation and differentiation through its direct actions downstream of VEGF and calcineurin/NFATc1. Indeed, SHP2 is directly involved in the regulation of calcium transients, as discussed above. Moreover, NS mutant expression of SHP2 in adult mouse hearts leads to increased oscillatory frequency of intracellular Ca2+ transients, perturbed calcineurin function, and decreased NFAT activation [72] (Figure 5).
The pathways controlling the final events of valve remodeling are not known, but are thought to implicate apoptotic signals, which might be affected by NS and LS mutations in SHP2. Indeed, anti-apoptotic functions of SHP2 are thought to be regulated in part by ERK-independent mechanisms involving non-phosphatase domains of SHP2 in zebrafish [40] (Table 2). However, SHP2 catalytic functions that control apoptosis, through AKT-mediated mechanisms, have also been implicated [73]. Further studies are needed to evaluate whether these functions and/or which domains of SHP2 are significantly important in valvulogenesis.
Importantly, the cardiac abnormalities observed in NS are found to be a consequence of cell-autonomous endocardial derived GOF SHP2 activity. By utilizing Cre-recombinase-expressing mouse lines that drive the expression of the D61Y NS allele in cardiomyocyte (αMHC-Cre), endocardium (Tie2-Cre) or neural crest (Wnt1-Cre), Araki, et. al., showed that only the endothelial-specific expression of NS caused thickening of valves, VSD, atrioseptal defect (ASD), double outlet right ventricle (DORV), and myocardium thinning [69]. Overexpression of a different NS mutation, Q79R, in the endocardial lineage [74], also validated these findings (Table 2).
In contrast, however, use of a βMHC promoter to drive expression of NS-SHP2 in myocardial ventricles, as opposed to the αMHC-Cre driver which expresses only in atria during development, showed cardiac muscle defects in mice, including ventricular noncompaction and septal defects, questioning the solely cell autonomous role for SHP2 in cardiomyocyte development [75]. Nevertheless, genetic modulation of ERK expression rescued both endocardial cushion enlargement induced by Tie2-Cre expression of NS and ventricular noncompaction in βMHC-NS mice [74, 75], confirming the importance of the MAPK/ERK pathway of regulation in both endocardial and myocardial development in NS mutants (Table 2).
Interestingly, while NS mutations in neural crest cells do not lead to cardiac defects [69], pre-migratory neural crest cells deleted for SHP2, initially exhibiting normal migratory and proliferative patterns, fail to migrate into the developing outflow tract. The resulting embryos display persistent truncus arteriosus, abnormalities of the great vessels, ventricular septal defects and elongation and thickening of semilunar valves, more closely phenocopying a 22q11 deletion rather than the LS phenotype and raising into question a role for phosphatase-independent functions of SHP2 in neural crest [76] (Table 2).
Whether the above-stated observations are similar for SHP2 deletion or LS LOF mutants remain, as yet, unknown. However, given that the mechanism leading to increased valve size in NS and EGFR(wa2/wa2);PTPTN11+/− is distinct, and that the biochemical consequences of NS and LS mutations are also divergent, it is likely that the mechanism of regulation here is also unique. Careful studies assessing valvulogenesis in LS, and the molecular mechanisms and cell lineages involved here are still needed, and are currently underway.
Cardiac chamber maturation and septation
The formation of the heart chambers occurs concomitantly with cardiac looping. Each chamber swells from the outer curvature of the looped heart in a segmental fashion. As the chamber matures, trabecular projections are formed in the inner curvature, by differentiation and migration of myocardial cells into the cardiac jelly, while the outer curvature wall proliferates and thickens forming the compact zone. Separation of pulmonary and aortic circulation occurs by septation of ventricles, atria, and OFT. In mammals, the interventricular septum is formed due to expansive growth of the apexes of the left and right ventricle; this muscular part of the septum then fuses with AV cushions to separate the ventricular chambers into left and right ventricles. With the completion of ventricular septation, further growth of the ventricle leads to the compression of trabecular projections within the ventricular wall, resulting in an increase in the thickness of the compact myocardium [61, 77] [78].
Regulation of ventricular development and maturation is controlled by complex cell-autonomous and non-autonomous processes involving cross-talk between the different cardiac cell lineages, particularly between endocardium and myocardium [79]. Briefly, Notch signaling is activated in the endocardium by unknown signals that stem from the developing myocardium. This leads to the transcription of EphrinB2, which in turn activates the transcriptional activation of Neuregulin1 (NRG1) [80]. As a secreted factor, NRG1 signals to adjacent cardiomyocytes to promote differentiation into trabecular myocytes [80]. In a parallel, Notch activity in the endocardium also activates BMP10 expression in the adjacent myocytes, promoting proliferation [80]. Similarly, expression of FOXP1 in the endocardium activates the Wnt/β cat pathway by inhibiting SOX17/18 expression [81]. This leads to the paracrine production of FGFs, which also activate cardiomyocyte proliferation [81].
While NS mouse embryos show diminished ventricular wall thickness and VSDs, as well have noncompaction phenotypes when expressed specifically in the myocardium [75], LS mice develop HCM [27] (Table 2). Surprisingly, no studies have been performed to explain the role of SHP2 specifically in the developing myocardium. Although the importance of the MAPK/ERK pathway has been emphasized in studies using transgenic overexpression of NS driven by the βMHC promoter followed by genetic modulation of ERK1 lineage [74, 75], no specific mechanism explaining the myocardial defects in NS and LS have been described. In addition, mouse models of LS have proven a central role for mTOR/AKT, not MAPK/ERK, in the development of HCM [27]. SHP2 can act downstream of both NRG1 and FGF, respectively through ERBB- and FGFR-mediated ERK/MAPK signaling. Indeed, mice lacking a functional NRG1 gene product or having deletion of ErbB2 or ErbB4 genes, fail to form trabeculae [82]. Moreover, mouse embryos lacking the retinoic acid receptor gene RXRa, having substantially reduced ERK1/2 activity, show thin-walled noncompacted ventricles with a poorly formed interventricular septum [83]. Consequently, NS mutants, with increased ERK activation, should have increased NRG1- and FGF1-mediated signals and therefore, should have had increased proliferation of the myocardial wall and induced differentiation of cells into trabecular myocytes, resulting in increased wall thickness; however, this is not the case. One possible explanation for this discrepency is that the ERK pathway may only operate within narrow limits in normal development. Another possibility is that additional targets of SHP2 might also be involved in the regulation of the cardiac phenotype. In particular, a recent study proposed an interesting ERK-independent and phosphatase-independent mechanism for SHP2’s role in adhesion and migration during gastrulation movement in zebrafish [84]. Briefly, Protein Zero Related (PZR) protein is hyper-tyrosyl-phosphorylated in both NS and LS zebrafish embryos, most likely indicating that a kinase phosphorylates PZR in an SHP2 catalytically-independent manner [84] (Table 2). This adhesion-responsive glycoprotein, once tyrosyl-phosphorylated, recruits SHP2, and promotes pro-migratory and cell adhesive effects [85]. It is likely that the kinase responsible for PZR hyper-phosphorylation in NS and LS is c-Src, which is shown to be increasingly associated with SHP2 in both NS and LS zebrafish embryos [84]. The open conformation of NS and LS mutants presumably exposes the PTP and SH2 domains, presenting binding surfaces for Src that might otherwise be inaccessible. Indeed, SHP2 binds to Src through its SH3 domains [86]. While the focus of this new study involves elucidating the Src/PZR/SHP2 axis in gastrulation movement and convergence and extension in zebrafish embryos, it would be valuable to further study the contribution of this pathway in other adhesion/migration events, notably cardiomyocyte migration into trabecular protrusions. Nevertheless, the open conformation of NS and LS mutants might represent an important molecular mechanism for the pathogenesis of these alleles and continuing studies focusing on the potential SHP2 catalytically independent targets might help uncover new mechanistic roles for SHP2 in cardiac chamber maturation.
Conclusions
In conclusion, while most of the work on SHP2 has focused on its role in the regulation of the MAPK/ERK signaling cascade, recent studies have emphasized the importance of additional mechanisms and pathways that may be involved in essential SHP2 functions, particularly regarding heart development and disease. Many targets of SHP2 phosphatase activity have not been uncovered as yet. Moreover, phosphatase-independent functions of SHP2 may have important roles during several steps and stages of cardiac development. Further studies are required to evaluate the precise functions of SHP2 in these processes, especially given the wide variety of signaling pathways and cellular events it has proven to be involved in mediating.
Highlights.
SHP2 structure and mechanism of action
SHP2 mutations and human disease
SHP2 and early development
Cardiac progenitor specification and survival
Morphogenetic events: gastrulation, acquisition of embryonic asymmetry, and cardiac looping
Endocardial cushions and valve development
Cardiac chamber maturation and septation
Acknowledgments
This work was supported by National Institutes of Health Grants R01-HL102368 and R01-HL114775 to M.I.K, Children’s Cardiomyopathy Foundation Grant to M.I.K., the Harvard Stem Cell Institute Seed Grant to M.I.K., and by an American Heart Association Postdoctoral Fellowship to J.L. This work was also supported in part by the Beth Israel Deaconess Medical Center Division of Cardiology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weismann CG, Gelb BD. The genetics of congenital heart disease: a review of recent developments. Curr Opin Cardiol. 2007;22(3):200–6. doi: 10.1097/HCO.0b013e3280f629c7. [DOI] [PubMed] [Google Scholar]
- 2.Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77(1):1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Hof P, et al. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92 (4):441–50. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 4.Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 1998;6(3):249–54. doi: 10.1016/s0969-2126(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 5.Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends in biochemical sciences. 2003;28 (6):284–93. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 6.Feng GS. Shp-2 tyrosine phosphatase: signaling one cell or many. Experimental cell research. 1999;253(1):47–54. doi: 10.1006/excr.1999.4668. [DOI] [PubMed] [Google Scholar]
- 7.Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine phosphatases. Current opinion in cell biology. 2001;13(2):182–95. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 8.Van Vactor D, O’Reilly AM, Neel BG. Genetic analysis of protein tyrosine phosphatases. Current opinion in genetics & development. 1998;8(1):112–26. doi: 10.1016/s0959-437x(98)80070-1. [DOI] [PubMed] [Google Scholar]
- 9.Zhang SQ, et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Molecular cell. 2004;13(3):341–55. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]
- 10.Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Molecular and cellular biology. 2003;23(21):7875–86. doi: 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanafusa H, et al. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. The Journal of biological chemistry. 2004;279(22):22992–5. doi: 10.1074/jbc.M312498200. [DOI] [PubMed] [Google Scholar]
- 12.Zhang SQ, et al. Receptor-specific regulation of phosphatidylinositol 3′-kinase activation by the protein tyrosine phosphatase Shp2. Molecular and cellular biology. 2002;22(12):4062–72. doi: 10.1128/MCB.22.12.4062-4072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehmann U, et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. The Journal of biological chemistry. 2003;278(1):661–71. doi: 10.1074/jbc.M210552200. [DOI] [PubMed] [Google Scholar]
- 14.You M, et al. Modulation of the nuclear factor kappa B pathway by Shp-2 tyrosine phosphatase in mediating the induction of interleukin (IL)-6 by IL-1 or tumor necrosis factor. The Journal of experimental medicine. 2001;193(1):101–10. doi: 10.1084/jem.193.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fornaro M, et al. SHP-2 activates signaling of the nuclear factor of activated T cells to promote skeletal muscle growth. J Cell Biol. 2006;175(1):87–97. doi: 10.1083/jcb.200602029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kontaridis MI, et al. SHP-2 positively regulates myogenesis by coupling to the Rho GTPase signaling pathway. Molecular and cellular biology. 2004;24(12):5340–52. doi: 10.1128/MCB.24.12.5340-5352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marino B, et al. Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. The Journal of pediatrics. 1999;135(6):703–6. doi: 10.1016/s0022-3476(99)70088-0. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida R, et al. Protein-tyrosine phosphatase, nonreceptor type 11 mutation analysis and clinical assessment in 45 patients with Noonan syndrome. J Clin Endocrinol Metab. 2004;89(7):3359–64. doi: 10.1210/jc.2003-032091. [DOI] [PubMed] [Google Scholar]
- 19.Tartaglia M, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. American journal of human genetics. 2002;70(6):1555–63. doi: 10.1086/340847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishikawa T, et al. Hypertrophic cardiomyopathy in Noonan syndrome. Acta Paediatr Jpn. 1996;38(1):91–8. doi: 10.1111/j.1442-200x.1996.tb03445.x. [DOI] [PubMed] [Google Scholar]
- 21.Digilio MC, et al. Noonan syndrome and aortic coarctation. Am J Med Genet. 1998;80(2):160–2. [PubMed] [Google Scholar]
- 22.Limongelli G, et al. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. The American journal of cardiology. 2007;100(4):736–41. doi: 10.1016/j.amjcard.2007.03.093. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Quintana E, Rodriguez-Gonzalez F. LEOPARD Syndrome: Clinical Features and Gene Mutations. Mol Syndromol. 2012;3(4):145–57. doi: 10.1159/000342251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keilhack H, et al. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. The Journal of biological chemistry. 2005;280(35):30984–93. doi: 10.1074/jbc.M504699200. [DOI] [PubMed] [Google Scholar]
- 25.Kontaridis MI, et al. PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. The Journal of biological chemistry. 2006;281(10):6785–92. doi: 10.1074/jbc.M513068200. [DOI] [PubMed] [Google Scholar]
- 26.Hanna N, et al. Reduced phosphatase activity of SHP-2 in LEOPARD syndrome: consequences for PI3K binding on Gab1. FEBS Lett. 2006;580(10):2477–82. doi: 10.1016/j.febslet.2006.03.088. [DOI] [PubMed] [Google Scholar]
- 27.Marin TM, et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. The Journal of clinical investigation. 2011;121(3):1026–43. doi: 10.1172/JCI44972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu ZH, et al. Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J Biol Chem. 2013;288(15):10472–82. doi: 10.1074/jbc.M113.450023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiu W, et al. Structural insights into Noonan/LEOPARD syndrome-related mutants of protein-tyrosine phosphatase SHP2 (PTPN11) BMC Struct Biol. 2014;14:10. doi: 10.1186/1472-6807-14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang W, et al. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Developmental cell. 2006;10(3):317–27. doi: 10.1016/j.devcel.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Anneren C, Cowan CA, Melton DA. The Src family of tyrosine kinases is important for embryonic stem cell self-renewal. J Biol Chem. 2004;279(30):31590–8. doi: 10.1074/jbc.M403547200. [DOI] [PubMed] [Google Scholar]
- 32.Wu D, et al. A conserved mechanism for control of human and mouse embryonic stem cell pluripotency and differentiation by shp2 tyrosine phosphatase. PLoS ONE. 2009;4(3):e4914. doi: 10.1371/journal.pone.0004914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dehaan RL. Migration patterns of the precardiac mesoderm in the early chick embrvo. Exp Cell Res. 1963;29:544–60. doi: 10.1016/s0014-4827(63)80016-6. [DOI] [PubMed] [Google Scholar]
- 34.Warkman AS, Krieg PA. Xenopus as a model system for vertebrate heart development. Semin Cell Dev Biol. 2007;18(1):46–53. doi: 10.1016/j.semcdb.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nature reviews. Genetics. 2005;6(11):826–35. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 36.Brade T, et al. Embryonic heart progenitors and cardiogenesis. Cold Spring Harb Perspect Med. 2013;3(10):a013847. doi: 10.1101/cshperspect.a013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langdon YG, et al. SHP-2 is required for the maintenance of cardiac progenitors. Development. 2007;134(22):4119–30. doi: 10.1242/dev.009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tirosh-Finkel L, et al. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development. 2010;137(18):2989–3000. doi: 10.1242/dev.051649. [DOI] [PubMed] [Google Scholar]
- 39.Jopling C, van Geemen D, den Hertog J. Shp2 knockdown and Noonan/LEOPARD mutant Shp2-induced gastrulation defects. PLoS genetics. 2007;3(12):e225. doi: 10.1371/journal.pgen.0030225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stewart RA, et al. Phosphatase-dependent and -independent functions of Shp2 in neural crest cells underlie LEOPARD syndrome pathogenesis. Developmental cell. 2010;18(5):750–62. doi: 10.1016/j.devcel.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Berg G, et al. A caudal proliferating growth center contributes to both poles of the forming heart tube. Circ Res. 2009;104(2):179–88. doi: 10.1161/CIRCRESAHA.108.185843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bayraktar M, Manner J. Cardiac looping may be driven by compressive loads resulting from unequal growth of the heart and pericardial cavity. Observations on a physical simulation model. Front Physiol. 2014;5:112. doi: 10.3389/fphys.2014.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y, et al. Bending and twisting the embryonic heart: a computational model for c-looping based on realistic geometry. Front Physiol. 2014;5:297. doi: 10.3389/fphys.2014.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramasubramanian A, et al. Computational model for early cardiac looping. Ann Biomed Eng. 2006;34(8):1655–69. doi: 10.1007/s10439-005-9021-4. [DOI] [PubMed] [Google Scholar]
- 45.Manning A, McLachlan JC. Looping of chick embryo hearts in vitro. J Anat. 1990;168:257–63. [PMC free article] [PubMed] [Google Scholar]
- 46.Davidson BP, Tam PP. The node of the mouse embryo. Curr Biol. 2000;10 (17):R617–9. doi: 10.1016/s0960-9822(00)00675-8. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka Y, Okada Y, Hirokawa N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature. 2005;435(7039):172–7. doi: 10.1038/nature03494. [DOI] [PubMed] [Google Scholar]
- 48.Hirokawa N, et al. Nodal flow and the generation of left-right asymmetry. Cell. 2006;125(1):33–45. doi: 10.1016/j.cell.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Boettger T, Wittler L, Kessel M. FGF8 functions in the specification of the right body side of the chick. Curr Biol. 1999;9(5):277–80. doi: 10.1016/s0960-9822(99)80119-5. [DOI] [PubMed] [Google Scholar]
- 50.Sun X, et al. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13(14):1834–46. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonetti M, et al. Noonan and LEOPARD syndrome Shp2 variants induce heart displacement defects in zebrafish. Development. 2014;141(9):1961–70. doi: 10.1242/dev.106310. [DOI] [PubMed] [Google Scholar]
- 52.Kreiling JA, et al. Suppression of the endoplasmic reticulum calcium pump during zebrafish gastrulation affects left-right asymmetry of the heart and brain. Mech Dev. 2008;125(5–6):396–410. doi: 10.1016/j.mod.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 53.Yoshiba S, et al. Cilia at the node of mouse embryos sense fluid flow for left-right determination via Pkd2. Science. 2012;338(6104):226–31. doi: 10.1126/science.1222538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saund RS, et al. Gut endoderm is involved in the transfer of left-right asymmetry from the node to the lateral plate mesoderm in the mouse embryo. Development. 2012;139(13):2426–35. doi: 10.1242/dev.079921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hagiwara Y, et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent I CaL, [Ca2+]i transient, and APD increase in cardiomyocytes. J Mol Cell Cardiol. 2007;43(6):710–6. doi: 10.1016/j.yjmcc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 56.Wang Q, et al. Phosphorylation of SHP-2 regulates interactions between the endoplasmic reticulum and focal adhesions to restrict interleukin-1-induced Ca2+ signaling. J Biol Chem. 2006;281(41):31093–105. doi: 10.1074/jbc.M606392200. [DOI] [PubMed] [Google Scholar]
- 57.Noel ES, et al. A Nodal-independent and tissue-intrinsic mechanism controls heart-looping chirality. Nat Commun. 2013;4:2754. doi: 10.1038/ncomms3754. [DOI] [PubMed] [Google Scholar]
- 58.Langdon Y, et al. SHP-2 acts via ROCK to regulate the cardiac actin cytoskeleton. Development. 2012;139(5):948–57. doi: 10.1242/dev.067579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kathiriya IS, Srivastava D. Left-right asymmetry and cardiac looping: implications for cardiac development and congenital heart disease. Am J Med Genet. 2000;97(4):271–9. doi: 10.1002/1096-8628(200024)97:4<271::aid-ajmg1277>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 60.Franco D, Campione M. The role of Pitx2 during cardiac development. Linking left-right signaling and congenital heart diseases. Trends Cardiovasc Med. 2003;13(4):157–63. doi: 10.1016/s1050-1738(03)00039-2. [DOI] [PubMed] [Google Scholar]
- 61.Lin CJ, et al. Partitioning the heart: mechanisms of cardiac septation and valve development. Development. 2012;139(18):3277–99. doi: 10.1242/dev.063495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Lange FJ, et al. Lineage and morphogenetic analysis of the cardiac valves. Circulation research. 2004;95(6):645–54. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- 63.Wessels A, et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev Biol. 2012;366(2):111–24. doi: 10.1016/j.ydbio.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abdelwahid E, Pelliniemi LJ, Jokinen E. Cell death and differentiation in the development of the endocardial cushion of the embryonic heart. Microsc Res Tech. 2002;58(5):395–403. doi: 10.1002/jemt.10159. [DOI] [PubMed] [Google Scholar]
- 65.Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95(5):459–70. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105(5):408–21. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- 68.Chen B, et al. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nature genetics. 2000;24(3):296–9. doi: 10.1038/73528. [DOI] [PubMed] [Google Scholar]
- 69.Araki T, et al. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(12):4736–41. doi: 10.1073/pnas.0810053106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circulation research. 2004;95(5):459–70. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson EN, et al. NFATc1 mediates vascular endothelial growth factor-induced proliferation of human pulmonary valve endothelial cells. J Biol Chem. 2003;278(3):1686–92. doi: 10.1074/jbc.M210250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uhlen P, et al. Gain-of-function/Noonan syndrome SHP-2/Ptpn11 mutants enhance calcium oscillations and impair NFAT signaling. Proc Natl Acad Sci U S A. 2006;103(7):2160–5. doi: 10.1073/pnas.0510876103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ivins Zito C, et al. SHP-2 regulates the phosphatidylinositide 3′-kinase/Akt pathway and suppresses caspase 3-mediated apoptosis. J Cell Physiol. 2004;199(2):227–36. doi: 10.1002/jcp.10446. [DOI] [PubMed] [Google Scholar]
- 74.Krenz M, et al. Role of ERK1/2 signaling in congenital valve malformations in Noonan syndrome. Proc Natl Acad Sci U S A. 2008;105(48):18930–5. doi: 10.1073/pnas.0806556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakamura T, et al. Mediating ERK 1/2 signaling rescues congenital heart defects in a mouse model of Noonan syndrome. The Journal of clinical investigation. 2007;117(8):2123–32. doi: 10.1172/JCI30756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakamura T, et al. Protein tyrosine phosphatase activity in the neural crest is essential for normal heart and skull development. Proc Natl Acad Sci U S A. 2009;106(27):11270–5. doi: 10.1073/pnas.0902230106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anderson RH, et al. Development of the heart: (2) Septation of the atriums and ventricles. Heart. 2003;89(8):949–58. doi: 10.1136/heart.89.8.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wagner M, Siddiqui MA. Signal transduction in early heart development (II): ventricular chamber specification, trabeculation, and heart valve formation. Exp Biol Med (Maywood) 2007;232(7):866–80. [PubMed] [Google Scholar]
- 79.Tian Y, Morrisey EE. Importance of myocyte-nonmyocyte interactions in cardiac development and disease. Circ Res. 2012;110(7):1023–34. doi: 10.1161/CIRCRESAHA.111.243899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grego-Bessa J, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12(3):415–29. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Y, et al. Foxp1 coordinates cardiomyocyte proliferation through both cell-autonomous and nonautonomous mechanisms. Genes Dev. 2010;24(16):1746–57. doi: 10.1101/gad.1929210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Odiete O, Hill MF, Sawyer DB. Neuregulin in cardiovascular development and disease. Circ Res. 2012;111(10):1376–85. doi: 10.1161/CIRCRESAHA.112.267286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sucov HM, et al. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 1994;8(9):1007–18. doi: 10.1101/gad.8.9.1007. [DOI] [PubMed] [Google Scholar]
- 84.Paardekooper Overman J, et al. PZR coordinates Shp2 Noonan and LEOPARD syndrome signaling in zebrafish and mice. Mol Cell Biol. 2014;34(15):2874–89. doi: 10.1128/MCB.00135-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roubelakis MG, et al. The murine ortholog of the SHP-2 binding molecule, PZR accelerates cell migration on fibronectin and is expressed in early embryo formation. J Cell Biochem. 2007;102(4):955–69. doi: 10.1002/jcb.21334. [DOI] [PubMed] [Google Scholar]
- 86.Walter AO, Peng ZY, Cartwright CA. The Shp-2 tyrosine phosphatase activates the Src tyrosine kinase by a non-enzymatic mechanism. Oncogene. 1999;18(11):1911–20. doi: 10.1038/sj.onc.1202513. [DOI] [PubMed] [Google Scholar]