

Abstract
Background
Exposure to ambient particulate matter (PM) is known to be associated with increased morbidity and mortality in human populations. During the winter months in Fairbanks, Alaska, severe temperature inversions lead to elevated concentrations of ambient PM smaller than 2.5 microns (PM2.5). Sled dogs represent an easily accessible environmentally exposed population that may yield findings informative for human health risk assessment.
Objectives
In this pilot study, we evaluated whether ambient PM was associated with markers of global methylation in sled dogs.
Methods
Kennels were strategically recruited to provide a wide PM2.5 exposure gradient for the Fairbanks area. Continuous monitoring of ambient PM2.5 was conducted at each kennel during the winter of 2012/13 using a DustTrak 8530. Dogs received a physical examination and assessment of standard hematology and clinical chemistries. Global methylation was determined using the LUminometric Methylation Assay (LUMA) and 5-Methycytosine (5-mC) Quantification.
Results
Three sled dog kennels (n ~30 dogs/kennel) were evaluated and sampled. The average PM2.5 concentrations measured for kennels A, B, and C were 90 μg/m3, 48 μg/m3, 16 μg/m3 (p< 0.0001), respectively. The average (standard deviation) global methylation percentage for each kennel measured by LUMA was 76.22 (1.85), 76.52 (1.82), and 76.72 (2.26), respectively. The average (standard deviation) global methylation percentage for each kennel measured by 5-mC was 0.16 (0.04), 0.15 (0.04), and 0.15 (0.05), respectively. There was no statistically significant difference between the three kennels and their average global methylation percentage either by LUMA or 5-mC.
Conclusions
In this study we evaluated global methylation using LUMA and 5-mC and found no differences between kennels, though exposure to ambient PM2.5 was significantly different between kennels. As more information becomes available regarding immunologically-related canine genes and functionally active promoter subunits, the utility of this surrogate could increase.
INTRODUCTION
Ambient particulate matter (PM) is known to be associated with increased morbidity and mortality in many exposed human populations, with these adverse effects particularly evident among individuals with pre-existing respiratory or cardiovascular conditions (Sacks et al. 2011). However, the mechanisms whereby PM causes adverse health outcomes in the general population including those with pre-existing conditions remain unclear.
The inflammatory response generated from human exposure to airborne PM is largely determined by source and size of the particles (Kocbach et al. 2008). Source apportionment studies show that the Fairbanks airshed is dominated by wood smoke PM (Ward et al. 2012), which is at least partially due to the low cost and availability of wood relative to other popular fuel sources such as heating oil or natural gas. During the cold winter months in Fairbanks, severe temperature inversions (characterized by stagnant air) and extreme low temperatures lead to significant PM exposures for extended periods of time. It is not uncommon to see 24-hour average concentrations of ambient PM smaller than 2.5 microns (PM2.5) reach 50–60 μg/m3 for several consecutive days, significantly above the Environmental Protection Agency’s (EPA) 24-hour National Ambient Air Quality Standard (NAAQS) for PM2.5 (35 μg/m3). These elevated wintertime PM2.5 concentrations have resulted in Fairbanks having some of the highest measured PM2.5 concentrations throughout the entire EPA monitoring network, and a designation of nonattainment for PM2.5.
There is considerable evidence suggesting that traffic related pollution is a risk factor for poor respiratory and cardiovascular outcomes (Ruckerl et al. 2011). Previously, we have demonstrated that elevated ambient PM2.5 concentrations due to residential wood smoke was associated with respiratory conditions and symptoms (Noonan et al. 2012). Because immunological responses are likely specific to the source, size and composition of the inhaled PM (Miyata and van Eeden 2011), there is a need to evaluate health outcomes in geographical locations where high ambient PM2.5 occur.
There are often practical, logistical, and ethical considerations that make human community-based studies challenging. Additionally, humans often have practices that are potentially confounding such as tobacco use, legal and illegal use of drugs, varying transportation and movement. In such cases, there is utility in establishing a sentinel model for human health that could reduce the likelihood of potential confounding from these factors (Reif 2011). Groups such as the Morris Animal Foundation have highlighted the utility of the domestic dog as a biomedical model for researching the genetic and epigenetic etiologies of canine cancers (Morris Animal Foundation). Domestic dogs have been used previously to study the public health effects of air pollution both in lieu of human studies (Calderon-Garciduenas et al. 2001) and in parallel (Calderon-Garciduenas et al. 2008).
This study represents a novel application of sled dogs as sentinel animals for health impacts associated with exposure to ambient air pollution. The long winters and pristine mushing topography makes Fairbanks ideal for such a study. For example, Fairbanks is home to a large potential cohort of thousands of sled dogs, with these working domestic animals clustered in large groups of up to 100 per kennel. The relatively place-bound subjects, the spatial distribution of kennels, and the elevated but varied concentrations of ambient PM2.5 provide an ideal opportunity to accurately assess ambient air exposures in this setting.
Uniform diets, structured exercise routines, and multigenerational pedigrees add elements that are similar to experimental animal studies and a clear advantage over human subjects with high variability in these and other factors. Considering body size, life span, and behavior, which can all impact disease pathology, a dog may serve as a more biologically relevant model for human health compared to mice or rats. Dogs have also shared a living environment with humans longer than any other domestic animal and very likely also shared pathogens, which may have equally shaped the way both species’ immune systems defend against disease (Boyko 2011).
Epigenetics play an important role in developmental processes, cell proliferation, and the maintenance of genome stability (Jones and Baylin 2007). Global DNA hypomethylation is one type of epigenetic alteration and is hypothesized to play a role early in carcinogenesis by activating oncogenes and/or genomic instability (Feinberg 2004). Recently, epigenetic modifications such as DNA hypo- or hyper-methylation have been shown to occur in response to environmental exposures, and both human and animal studies suggest that such changes are relevant to risk for chronic disease. In particular, studies have found that exposure to air pollution can affect methylation patterns at the global (Madrigano et al. 2011; Baccarelli et al. 2009; Tarantini et al. 2009) and gene-specific levels (Liu et al. 2008; Salam et al. 2012; Nadeau et al. 2010). The current body of literature clearly indicates a role for methylation pattern changes in genes responsible for the regulation of the T-effector pathway, T-regulatory pathway, and airway inflammation (Lovinsky-Desir and Miller 2012). Deciphering the epigenetic landscape of exposed populations could lead to potential treatments because of the reversible nature of epigenetic markers (Wright 2013).
In this manuscript, we for the first time evaluate global methylation in Alaskan sled dogs in order to determine the effect of exposure to ambient PM (composed predominantly of residential wood smoke) on global DNA methylation.
METHODS
Kennel Recruitment
Data from both stationary and mobile PM2.5 air monitors were used to identify kennels located in “high”, “medium”, and “low” PM2.5 areas around the city of Fairbanks. Ultimately three kennels were strategically recruited with the intention of yielding a wide PM2.5 exposure gradient across sample locations. To be considered for inclusion, the kennel had to have greater than 30 sled dogs on site and access to an electrical outlet. Kennel A was an urban kennel located inside the Fairbanks EPA PM2.5 designated non-attainment area, Kennel B was an urban kennel located approximately 15 miles Southwest of Fairbanks, and Kennel C was a rural kennel located approximately 50 miles Southeast of Fairbanks. The sled dog owners were contacted and given information about the study. Those owners who agreed to participate were asked to sign a consent form. This study was approved by the University of Montana (UM) and University of Alaska Fairbanks (UAF) Institutional Animal Care ad Use Committee (IACUC #283615-4) before any onsite work was conducted.
PM2.5 Exposure Characterization
During the winter of 2012/13, a DustTrak 8530 (TSI, Shoreview, MN) that continuously measured PM2.5 mass was deployed to each of the three kennels. The DustTrak was housed in an insulated and heated enclosure to protect it from the extreme winter temperatures. While sampling, this monitor was placed approximately 1 meter off of the ground within 15 meters from the dog yard. The sled dogs in this study were all housed in very close proximity to each other at their respective kennel. The DustTrak was on a 60-second recording interval, and was zero calibrated prior to each sampling event. Field staff collected PM2.5 mass data at each kennel from November 21, 2012 through February 27, 2013.
Veterinary Examination, Hematology and Clinical Chemistry
Following the winter sampling period, field staff and a licensed veterinarian visited each dog kennel to conduct a standard physical examination and collect three blood samples from all study animals. Serum samples, collected in serum separator tubes, were used for comprehensive clinical chemistry panels to analyze general health markers. Whole blood samples were collected for hematocrit and complete blood counts. Whole blood and serum samples were processed and analyzed at the UAF Animal Resource Center clinical laboratory and the Wildlife Toxicology Laboratory (WTL). Blood samples for DNA analysis were collected in a PreAnalytix DNeasy blood tube (Qiagen, Germantown, MD) and shipped to the University of Montana for processing.
Global Methylation Analysis
DNA was extracted using a PreAnalytix DNeasy Kit (Qiagen, Germantown, MD). Concentration of DNA was assessed using a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE).
LUminometric Methylation Assay (LUMA)
Global methylation was determined using LUMA (Karimi et al. 2006). The assay was run in duplicate using 500ng of total DNA with samples analyzed in a randomized order to reduce the effect of inter-plate variation. Each plate included 21 DNA samples, as well as positive, negative, and water controls. The assay was visualized using a Pyromark Q96 MD (Qiagen, Germantown, MD). All samples used in the final analysis had an intra-assay coefficient of variation of ≤5%. Lambda DNA (NEB, Ipswich, MA) was used as a negative control, and was methylated using CpG methyltransferase (NEB, Ipswich, MA) and a New England Biolabs optimized protocol, which was then used as a positive control.
5-Methylcytosine (5-mC) Quantification
Global DNA methylation levels also were evaluated using the ELISA-based Methylflash™ Methylated DNA Quantification Kit (Colorimetric) (Epigentek, NY, USA). The Kit measures the 5-mC content as a percentage of the total cytosine in the DNA sample. The assay was performed in duplicate according to manufacturer’s instructions with 100ng of total DNA. The 5-mC in DNA was detected using capture and detection antibodies and then quantified colorimetrically by reading the absorbance in a microplate spectrophotometer. Absolute quantification standard curves were obtained by plotting the concentration of the positive control (methylated polynucleotide containing 50% of 5-methylcytosine (5-mC)) supplied with the assay kit against the optical density (OD) at 450nm. The assay was validated by calibrating the different amounts of 5-mC DNA in a commercially available standard (CpGenome™ 5-mC) (EMD Millipore, Billerica, USA). The percentage of 5-mC in genomic DNA was calculated using the following formula:
Statistical Analysis
Descriptive factors including dog sex, age, and weight were compared between kennels using analysis of variance or chi-square tests. Clinical chemistry and hematology variables were evaluated as pairwise comparisons relative to the lowest exposure group, Kennel C. Ambient PM2.5 concentrations at each kennel site were summarized for the total monitoring periods and by the two-week period prior to the day of data collection for dogs at the kennels. Differences in mean ambient PM2.5 concentrations and mean global methylation percentage between kennel sites were evaluated by analysis of variance, followed by post-hoc t-tests when appropriate. Pearson correlations were used to compare global methylation percentage and the 35 clinical chemistry variables across and within kennels.
RESULTS
In total, 87 sled dogs participated in this study, including 29 from Kennel A, 30 from Kennel B, and 28 from Kennel C. Two UAF veterinarians, who were blinded to the kennel designation, evaluated the clinical chemistry data in an effort to identify if animals should be removed based on health characteristics. Following this evaluation, the veterinarians advised that four animals should be excluded. An additional four animals were excluded because they had more than five clinical chemistry values that were greater than two standard deviations away from the mean. Therefore a population of 75 sled dogs was used in the following analysis. The weight and sex of the excluded animals were not significantly different from the included population of sled dogs (p = 0.94 and 0.38 respectively), however, the dogs that were removed from the study were more likely to be older compared to those included in the study (p= 0.07). With respect to kennel designation there was a significant difference in age and weight. Dogs in Kennel C were significantly younger than dog in the other kennels (p= 0.01), and dogs in kennel A weighed significantly less than dogs in the other kennels (p= 0.01; table 1).
Table 1.
Characteristics of study population.
| Characteristics a | Kennel A (n=27) | Kennel B (n=25) | Kennel C (n=23) |
|---|---|---|---|
| Age, years | 6.1 (3.2) | 6.0 (3.3) | 3.6 ( 2.8) |
| Weight (lbs) | 49.6 (6.3) | 55.5 (7.4) | 52.6 (6.6) |
| Sex | |||
| Female | 17 (63) | 13 (52) | 10 (43) |
| Male | 10 (37) | 12 (48) | 13 (57) |
| Pregnant b | 2 (12) | 1 (8) | 0 |
mean (sd) or n (%)
n (% female)
Ambient PM2.5 Concentrations at the Kennels
PM2.5 data were collected at each kennel from November 21, 2012 through February 27, 2013. Although samples were collected at each site the majority of the time, due to logistic issues including weather and power outages, each kennel was sampled for a subset of the total study period (see table 2). The average PM2.5 concentration (sd) measured during the study period for each site was, 90 (15) μg/m3 for Kennel A, 48 (22) μg/m3 for Kennel B, and 16 (8) μg/m3 for Kennel C (p< 0.0001). For the 2-week period prior to the dog sampling and examination at each kennel, the measured average PM2.5 concentration (sd) was 46 (32) μg/m3 for Kennel A, 42 (32) μg/m3 for Kennel B, and 9 (4) μg/m3 for Kennel C (p< 0.001).
Table 2.
Selected clinical chemistry mean (sd) values for subjects at the time of sample collection
| Variable a | Kennel A (n=27) | Kennel B (n=25) | Kennel C (n=23) |
|---|---|---|---|
| Na | 146.14 (1.23) b | 146.48 (1.66) | 147.13 (1.82) |
| K | 4.47 (0.34) | 4.31 (0.22) | 4.41 (0.32) |
| Cl | 110.00 (2.76) | 104.88 (3.66) b | 109.22 (3.19) |
| ALP | 44.58 (17.57) b | 44.60 (41.41) | 29.93 (11.98) |
| ALT | 63.62 (46.50) | 49.36 (29.23) | 47.73 (21.24) |
| BUN | 17.19 (4.14) | 21.48 (4.32) | 25.08 (19.66) |
| Ca | 9.15 (0.44) | 9.52 (0.45) | 9.89 (4.56) |
| CRE | 0.44 (0.09) | 0.50 (0.09) | 2.00 (4.71) |
| GGT | 14.88 (2.47) | 15.28 (3.80) | 19.09 (17.81) |
| GLU | 97.54 (6.51) | 110.52 (11.15) b | 94.12 (25.69) |
| TP | 5.87 (0.39) | 6.04 (0.37) | 5.63 (1.16) |
| TBIL | 0.22 (0.15) | 0.22 (0.17) | 1.60 (6.85) |
| ALB | 3.62 (0.23) b | 3.87 (0.26) | 3.91 (0.30) |
| TCHO | 246.92 (74.13) | 225.48 (74.92) | 219.13 (58.07) |
| IP | 4.05 (0.56) | 3.84 (0.59) | 15.94 (59.09) |
| GLOB | 2.25 (0.34) b | 2.17 (0.32) b | 1.96 (0.30) |
| NaKRatio | 32.89 (2.33) | 34.08 (1.87) | 33.57 (2.50) |
mean (sd)
p < 0.05 compared to Kennel C
Abbreviations (units): Na: Sodium (mEq/l), K: Potassium (mEq/l), CL: Chloride (mEq/l), ALP: Alkaline Phosphatase (U/l), ALT: Alanine Transferase (U/l), BUN: Blood Urea Nitrogen (mg/dl), Ca: Calcium (mg/dl), CRE: Creatinine (mg/dl), GGT: Gamma-Glutamyl Transpeptidase (U/l), GLU: Glucose (mg/dl), TP: Total Protein (g/dl), TBIL: Total Bilirubin (mg/dl), ALB: Albumin(g/dl), TCHO: Total Cholesterol (mg/dl), IP: Phosphorus (mg/dl), GLOB: Globulins (g/dl), NaKRatio: Sodium Potassium Ratio
The average daily temperature for the winter period was −20°C (max 0°C, min −37°C) and the relative humidity was 79% (max 100%, min 60%) (Fairbanks North Star Borough ; Western Regional Climate Center). From January 27, 2013 to February 27, 2013, the average daily temperature was −18°C (max −12°C, min −37°C) and the relative humidity was 81% (max 93%, min 62%), (Fairbanks North Star Borough ; Western Regional Climate Center).
Global Methylation
DNA was extracted and analyzed for 83 dogs out of the original population of 87. Figure 2 shows the percent global methylation (%gMe) for the dogs included in this study (n=75). The average %gMe (SD) was 76.22 (1.85) for Kennel A, 76.52 (1.82) for Kennel B, and 76.72 (2.26) for Kennel C (p= 0.66). While the PM2.5 exposure of Kennels A and B were different over the course of the study, there was no difference in exposure in the two-week period prior to blood draw. If Kennels A and B are combined, there is no difference between the average %gMe of Kennel A+B relative to Kennel C measured by LUMA or 5-mC (data not shown). Global methylation was not associated with age, weight, or sex. The average (standard deviation) global methylation percentage for each kennel measured by 5-mC was 0.16 (0.04) for Kennel A, 0.15 (0.04) for Kennel B, and 0.15 (0.05) for Kennel C (p= 0.24). The methylation percentages from the LUMA and 5-mC assay were not significantly correlated. The above findings were not influenced by the exclusion criteria based on aberrant veterinary examinations and outlying clinical chemistry values. Global methylation values among the excluded dogs (n=8) were similar to the other dogs. Inclusion of all dogs in the analyses did not result in any statistical differences in mean global methylation by kennel or other descriptive characteristics (data not shown).
Figure 2.
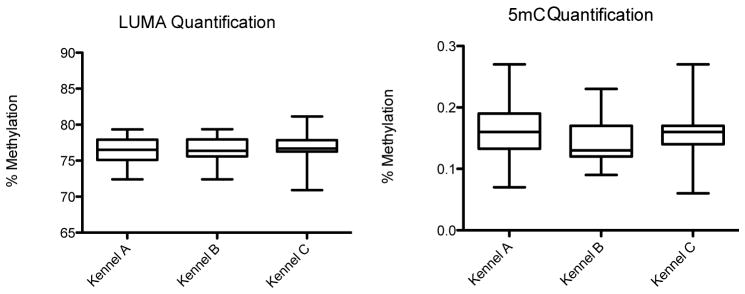
Box plots showing the % global methylation for the three kennels quantified by LUMA (p= 0.66) and 5mC (p= 0.24)
Standard Hematology and Clinical Chemistry
A total of 35 parameters were evaluated (see table 2 and 3). Sodium (Na) (p= 0.01), glucose (GLU) (p= 0.05), and granulocytes (GRAN) (p= 0.05), were significantly correlated and monocyte percentage (MPR) (p= 0.06) and white blood cell count (WBC) (p= 0.07) were marginally correlated with global methylation values analyzed by LUMA. White blood cell counts (WBC) (p= 0.004) and mid-sized cells (MID) (p= 0.01) were significantly correlated with global methylation values analyzed by 5-mC.
Table 3.
Selected hematology mean (sd) values for subjects at the time of sample collection
| Variable a | Kennel A (n=27) | Kennel B (n=25) | Kennel C (n=23) |
|---|---|---|---|
| WBC | 7.52 (1.96) | 8.98 (2.59) | 7.79 (1.97) |
| LYMF | 1.20 (0.61) b | 1.62 (0.62) | 1.56 (0.54) |
| MID | 0.76 (0.22) b | 0.77 (0.25) b | 0.60 (0.12) |
| GRAN | 5.56 (1.62) | 6.58 (2.39) | 5.63 (1.75) |
| LPR | 16.55 (7.44) b | 19.29 (6.56) | 20.90 (6.56) |
| MPR | 9.36 (1.45) b | 7.99 (1.93) | 7.54 (1.83) |
| GPR | 74.09 (7.82) | 72.72 (6.95) | 71.56 (6.99) |
| HCT | 44.26 (5.02) b | 46.04 (4.74) | 47.98 (4.18) |
| MCV | 64.48 (1.84) | 61.31 (2.99) b | 63.31 (2.31) |
| RDWR | 22.97 (1.08) b | 23.69 (1.25) b | 24.63 (1.80) |
| RBC | 6.86 (0.75) b | 7.51 (0.70) | 7.60 (0.84) |
| HGB | 16.02 (1.70) b | 16.68 (1.69) | 17.54 (1.46) |
| MCHC | 36.23 (0.55) b | 36.25 (0.51) | 36.57 (0.64) |
| MCH | 23.37 (0.67) | 22.22 (1.14) b | 23.14 (0.93) |
| PLT | 343.48 (92.54) b | 364.28 (107.51) b | 279.13 (62.89) |
| MPV | 8.75 (0.76) | 8.68 (0.69) | 9.14 (0.98) |
| TRBC | 12.69 (0.06) b | 12.68 (0.05) b | 12.53 (0.05) |
| TWBC | 12.88 (0.04) b | 12.82 (0.04) b | 12.66 (0.05) |
mean (sd)
p < 0.05 compared to Kennel C
Abbreviations (units): WBC: White Blood Cells (103/μl), LYMF: Lymphocytes (103/μl), MID: Mid-sized Cells (103/μl), GRAN: Granulocytes (103/μl), LPR: Lymphocyte Percentage (%), MPR: Monocyte Percentage (%), GPR: Granulocyte Percentage (%), HCT: Hematocrit (%), MCV: Red Blood Cell Count Derivative (fl), RDWR: Red Blood Cell Count Derivative (%), MCHC: Red Blood Cell Count Derivative (g/dl), MCH: Red Blood Cell Count Derivatives (pg), RBC: Red Blood Cells (106/μl), HGB: Hemoglobin (g/dl), PLT: Platelets (103/μl), MPV: Platelet Count Derivative (fl), TRBC: Red Blood Cell Count Time (sec), TWBC: White Blood Cell Count Time (sec)
DISCUSSION
In this study, two approaches were used to summarize genome-wide DNA methylation. Our results suggest that global methylation is not associated with elevated levels of ambient PM2.5 compared to relatively low levels when using sled dogs as sentinels. It is suggested that hypomethylation of the genome does occur with age in animals (Dolinoy and Faulk 2012). However, age was not associated with global methylation in this study. We had no a priori hypotheses with respect to clinical chemistries and global methylation. Thus, these finding should be interpreted with caution and perhaps warrant further investigation.
Global methylation quantified by LUMA for dogs appears to be consistent with recent work published by Head et al., which suggests that mammals have intermediate global methylation (Head et al. 2014). This publication found that fish had higher global methylation relative to mammals, while birds had lower.
There have been recent publications that suggest some genes may be up regulated in response to hypomethylation (Spruijt and Vermeulen 2014), but it is generally accepted that promoter region hypermethylation of a gene suppresses protein production and global hypomethylation causes genome instability. Several genes in the T-helper 2 (Th2) pathway have been found to be susceptible to methylation (Lovinsky-Desir and Miller 2012). It is hypothesized that there is interplay between environmental factors such as PM2.5 and respiratory phenotypes, which may be mediated by epigenetic factors such as DNA methylation (Zhang et al. 2014). A recent genome wide study investigated 19,000 genes in 141 subjects for the Normative Aging Study and concluded that airborne particulate matter was associated with the methylation pattern of genes involved in the asthma pathway (Sofer et al. 2013). The literature regarding the temporal nature and sustainability of these epigenetic changes in vivo is sparse (Lovinsky-Desir and Miller 2012). It is possible that the time course from exposure to the resulting epigenetic change ranges from minutes to months and is gene specific.
Two exposure sampling time frames are described in table 1: the total study period and the two-week period leading up to the physical exam and blood collection. Two weeks is an arbitrary cut-point, but it does draw attention to the idea that proximity of the clinical examination to an air pollution event should be considered. For example, differential methylation patterns of the iNOS gene, which is involved in asthma and other obstructive pulmonary conditions, have been found in welders after an 8-hour shift (Kile et al. 2013). The authors used Line-1 and Alu as proxies for global methylation but did not find differences relative to exposure. However, short-term exposure was associated with epigenetic changes to the iNOS gene promoter region.
In this study LUMA and 5mC were used to detect differential global methylation profiles between three kennels using samples collected at a single visit. Future studies could consider broadening the focus to include gene-specific methylation profiles and conduct more frequent blood collections, including a pre-winter blood collection, to increase the likelihood of capturing PM2.5 event-related changes in global and or gene-specific methylation. As more epigenetic information becomes available for the canine genome, genes responsible for the production of immuno-regulatory cytokines and methylation-related enzymes should be evaluated.
Human cell lineages by nature are epigenetically distinct and therefore data from studies that utilize whole blood samples should be evaluated with caution (Reinius et al. 2012). As strategies for partitioning cell populations are developed, the domestic dog model could be used to inform and validate these new methods.
CONCLUSION
In this study we identified PM2.5 micro climates in and around the Fairbanks non-attainment area for PM2.5. However, when global methylation was evaluated using LUMA and 5-mC, we found no differences between kennels. Several hematology and clinical chemistry variables were correlated with both LUMA and 5-mC, but these results are difficult to interpret and warrant further investigation.
The number of funded epigenetic studies has increased exponentially over the last twenty years. However, only a small percentage of those studies focused on environmental exposures (Burris and Baccarelli 2014). There is increasing interest in animal models for human diseases with environmental exposure and epigenetic components. With improvements in gene and whole-genome techniques, these sentinels can be valuable tools for approaches that are not feasible in humans (Dolinoy and Faulk 2012). Additional research to identify and characterize canine repetitive elements and specific genes that have human homologues will increase the utility of the domestic dog as a surrogate for environmental exposures and impacts on human health.
Figure 1.
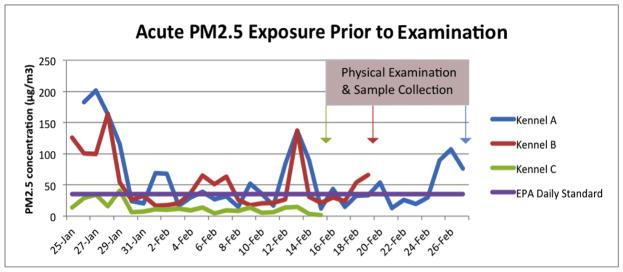
PM2.5 was measured at three sled dog kennels over the 2012/13 winter. Kennel A, Kennel B, and Kennel C were sampled for 78, 59, and 46 days respectively. This figure shows a subset of these days representing the acute exposure levels that were measured leading up to the sample collections.
Table 4.
Ambient PM2.5 Collection Data
| Kennel A | Kennel B | Kennel C | p value | |
|---|---|---|---|---|
| Sample Days | 78 | 59 | 46 | |
| Percentage of Day Above 35μg/m3 | 69 | 49 | 4 | |
| Percentage of Day Above 100μg/m3 | 35 | 12 | 0 | |
| Average PM2.5 (μg/m3) | ||||
| Nov 21 – Feb 27 mean (sd) | 90 (15) | 48 (22) | 16 (8) | < 0.0001 |
| 2 weeks prior to exam mean (sd) | 46 (32) | 42 (32) | 9 (4) | < 0.001 |
Highlights.
This is a novel pilot study evaluating PM2.5 exposure in sled dogs
PM2.5 microclimates were identified in the Fairbanks, Ak air shed
PM2.5 was not associated with two measures of global methylation
Acknowledgments
This work would not be possible with out participation from the sled dog owners and handlers. A special thanks to veterinarians Carla Willetto and Cristina Hansen as well as laboratory technician Virginia Porter. Thanks also to Jocelyn Krebs former director AK INBRE and Cathy Griseto. Research reported in this publication was partially supported by the Alaska INBRE with funding from the National Institute of General Medical Sciences of the National Institutes of Health under award number P20GM103395 as well as Montana COBRE 5P30GM103338.
Footnotes
2 is a factor to normalize 5-mC in the positive control to 100%, as the positive control contains only 50% of 5-mC
Competing financial interest declaration: The authors have no competing financial interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Sacks JD, Stanek LW, Luben TJ, Johns DO, Buckley BJ, Brown JS, et al. Particulate matter-induced health effects: who is susceptible? Environ Health Perspect. 2011;119(4):446–454. doi: 10.1289/ehp.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocbach A, Herseth JI, Lag M, Refsnes M, Schwarze PE. Particles from wood smoke and traffic induce differential pro-inflammatory response patterns in co-cultures. Toxicol Appl Pharmacol. 2008;232(2):317–326. doi: 10.1016/j.taap.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Ward T, Trost B, Conner J, Flanagan J, Jayanty RKM. Source Apportionment of PM2.5 in a Subarctic Airshed-Fairbanks, Alaska. Aerosol and Air Quality Research. 2012;(12):536–543. [Google Scholar]
- Ruckerl R, Schneider A, Breitner S, Cyrys J, Peters A. Health effects of particulate air pollution: A review of epidemiological evidence. Inhal Toxicol. 2011;23(10):555–592. doi: 10.3109/08958378.2011.593587. [DOI] [PubMed] [Google Scholar]
- Noonan CW, Ward TJ, Navidi W, Sheppard L. A rural community intervention targeting biomass combustion sources: effects on air quality and reporting of children’s respiratory outcomes. Occup Environ Med. 2012;69(5):354–360. doi: 10.1136/oemed-2011-100394. [DOI] [PubMed] [Google Scholar]
- Miyata R, van Eeden SF. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol Appl Pharmacol. 2011;257(2):209–226. doi: 10.1016/j.taap.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Reif JS. Animal sentinels for environmental and public health. Public Health Rep. 2011;126(Suppl 1):50–57. doi: 10.1177/00333549111260S108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris Animal Foundation. [accessed August 4, 2014]; Available: http://www.morrisanimalfoundation.org/
- Calderon-Garciduenas L, Mora-Tiscareno A, Fordham LA, Chung CJ, Garcia R, Osnaya N, et al. Canines as sentinel species for assessing chronic exposures to air pollutants: part 1. Respiratory pathology. Toxicol Sci. 2001;61(2):342–355. doi: 10.1093/toxsci/61.2.342. [DOI] [PubMed] [Google Scholar]
- Calderon-Garciduenas L, Mora-Tiscareno A, Ontiveros E, Gomez-Garza G, Barragan-Mejia G, Broadway J, et al. Air pollution, cognitive deficits and brain abnormalities: a pilot study with children and dogs. Brain Cogn. 2008;68(2):117–127. doi: 10.1016/j.bandc.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Boyko AR. The domestic dog: man’s best friend in the genomic era. Genome Biol. 2011;12(2):216. doi: 10.1186/gb-2011-12-2-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP. The epigenetics of cancer etiology. Semin Cancer Biol. 2004;14(6):427–432. doi: 10.1016/j.semcancer.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, et al. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119(7):977–982. doi: 10.1289/ehp.1002773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179(7):572–578. doi: 10.1164/rccm.200807-1097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, et al. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environ Health Perspect. 2009;117(2):217–222. doi: 10.1289/ehp.11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ballaney M, Al-alem U, Quan C, Jin X, Perera F, et al. Combined inhaled diesel exhaust particles and allergen exposure alter methylation of T helper genes and IgE production in vivo. Toxicol Sci. 2008;102(1):76–81. doi: 10.1093/toxsci/kfm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salam MT, Byun HM, Lurmann F, Breton CV, Wang X, Eckel SP, et al. Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children. J Allergy Clin Immunol. 2012;129(1):232–239. e231–237. doi: 10.1016/j.jaci.2011.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau K, McDonald-Hyman C, Noth EM, Pratt B, Hammond SK, Balmes J, et al. Ambient air pollution impairs regulatory T-cell function in asthma. J Allergy Clin Immunol. 2010;126(4):845–852. e810. doi: 10.1016/j.jaci.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Lovinsky-Desir S, Miller RL. Epigenetics, asthma, and allergic diseases: a review of the latest advancements. Curr Allergy Asthma Rep. 2012;12(3):211–220. doi: 10.1007/s11882-012-0257-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J. Epigenetics: reversible tags. Nature. 2013;498(7455):S10–11. doi: 10.1038/498S10a. [DOI] [PubMed] [Google Scholar]
- Karimi M, Johansson S, Stach D, Corcoran M, Grander D, Schalling M, et al. LUMA (LUminometric Methylation Assay)--a high throughput method to the analysis of genomic DNA methylation. Exp Cell Res. 2006;312(11):1989–1995. doi: 10.1016/j.yexcr.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Fairbanks North Star Borough. [accessed August 6, 2014]; Available: http://www.co.fairbanks.ak.us/
- Western Regional Climate Center. [accessed August 7, 2014]; Available: http://www.wrcc.dri.edu/
- Dolinoy DC, Faulk C. Introduction: The use of animals models to advance epigenetic science. Ilar J. 2012;53(3–4):227–231. doi: 10.1093/ilar.53.3-4.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head JA, Mittal K, Basu N. Application of the LUminometric Methylation Assay to ecological species: tissue quality requirements and a survey of DNA methylation levels in animals. Mol Ecol Resour. 2014;14(5):943–952. doi: 10.1111/1755-0998.12244. [DOI] [PubMed] [Google Scholar]
- Spruijt CG, Vermeulen M. DNA methylation: old dog, new tricks? Nat Struct Mol Biol. 2014;21(11):949–954. doi: 10.1038/nsmb.2910. [DOI] [PubMed] [Google Scholar]
- Zhang H, Tong X, Holloway JW, Rezwan FI, Lockett GA, Patil V, et al. The interplay of DNA methylation over time with Th2 pathway genetic variants on asthma risk and temporal asthma transition. Clin Epigenetics. 2014;6(1):8. doi: 10.1186/1868-7083-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofer T, Baccarelli A, Cantone L, Coull B, Maity A, Lin X, et al. Exposure to airborne particulate matter is associated with methylation pattern in the asthma pathway. Epigenomics. 2013;5(2):147–154. doi: 10.2217/epi.13.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile ML, Fang S, Baccarelli AA, Tarantini L, Cavallari J, Christiani DC. A panel study of occupational exposure to fine particulate matter and changes in DNA methylation over a single workday and years worked in boilermaker welders. Environ Health. 2013;12(1):47. doi: 10.1186/1476-069X-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7(7):e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris HH, Baccarelli AA. Environmental epigenetics: from novelty to scientific discipline. J Appl Toxicol. 2014;34(2):113–116. doi: 10.1002/jat.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]