

Graphical abstract

Keywords: 8-Amino imidazo[1,2-a]pyrazine; Bacterial type IV secretion system; HP0525; ATPase inhibitor
Abstract
A novel series of 8-amino imidazo[1,2-a]pyrazine derivatives has been developed as inhibitors of the VirB11 ATPase HP0525, a key component of the bacterial type IV secretion system. A flexible synthetic route to both 2- and 3-aryl substituted regioisomers has been developed. The resulting series of imidazo[1,2-a]pyrazines has been used to probe the structure–activity relationships of these inhibitors, which show potential as antibacterial agents.
1. Introduction
Microorganisms have evolved a number of macromolecular secretion machineries to translocate proteins and nucleoprotein complexes from the bacterial cytosol to the host cell. Seven types of secretion systems (I–VII) have so far been identified, with a diverse range of functions including: the transfer of plasmid DNA from one cell to another (the major mechanism for the spread of antibiotic resistance genes between pathogenic bacteria); the secretion of proteins toxic to host cells; and the secretion of effector molecules required for the propagation of the microorganism within the host cell.1
Bacterial secretion systems represent attractive targets for the development of novel antibacterial agents.2 As these systems are not required for bacterial growth, it is believed that bacteria would be slow to develop resistance to drugs targeting these system. Several groups have developed promising small molecule inhibitors that are effective against the type III secretion systems (T3SS) found in Gram-negative pathogens such as Yersinia, Salmonella and Chlamydia.2
Type IV secretion systems (T4SS) are vital for the pathogenicity of a number of important Gram-negative bacteria, such as Helicobacter pylori, Legionella pneumophilia and Bordetella pertussis, which cause serious infections in both animals and plants.3, 4 H. pylori utilizes the type IV secretion system to translocate the toxic protein CagA into gastric epithelial cells, and in doing so induces a number of changes in the host cell.5, 6 To date, little attention has been paid to T4SS as targets for antibacterial agents, although a VirB8 dimerisation inhibitor has recently been described as a T4SS inhibitor.7 Type IV secretion systems require ATP as an energy source to drive this transport and therefore require a class of ATPases known as VirB11 ATPases, which are associated with the inner membrane. The crystal structure of the VirB11 ATPase HP0525 has been solved, with the apo-,8 ADP bound9 and ATPγS bound8 forms of HP0525 being studied. The HP0525 forms double hexameric ring structures where each subunit monomer consists of 328 amino acid residues comprising the N-terminal domain (NTD) and C-terminal domain (CTD). Each domain forms a hexameric ring with the CTDs forming a closed ring mounted onto the dynamic open hexameric ring formed by the NTDs. The result is a dome-like structure, open at the NTD end and closed at the CTD end, which is large enough to accommodate a macromolecule such as CagA. The nucleotide-free (apo-HP0525) form exists as an asymmetric hexamer with the NTDs displaying mobility and the CTDs maintaining the scaffold. When three molecules of ATP bind, three of the subunits are locked into rigid conformations. Hydrolysis of these three ATP molecules to give ADP, together with binding of a further three ATP molecules to the remaining three nucleotide-free subunits results in a perfect hexameric rigid form. When all ATP molecules are hydrolysed and released the symmetric hexameric ring returns to its asymmetric form. The structure of HP1451, an inhibitory factor of HP0525 which regulates Cag-mediated secretion, bound to HP0525, has also been studied.10
Whilst targeting the ATPase activity of HP0525 would represent an attractive approach to generating novel antibacterial agents,11 so far only one group have previously published a series of inhibitors of this enzyme.12 In this paper, we describe a novel series of imidazo[1,2-a]pyrazine derivatives which act as inhibitors of the HP0525 ATPase from H. pylori. In the course of this work we have developed a flexible synthetic route to the core heterocycle, which can deliver either 2-aryl or 3-aryl imidazo[1,2-a]pyrazines.
2. Results and discussion
2.1. Lead identification
Structures from the kinase-directed SoftFocus library (BioFocus) were modeled into ATP-binding site of HP0525 ATPase7 (1NLY) and scored using DOCK 6.13 A group of 3-aryl 8-amino imidazo[1,2-a]pyrazines 1–6 (Fig. 1) was identified among the best-scoring compounds. These were selected for experimental study, along with the regioisomeric 2-aryl 8-amino imidazo[1,2-a]pyrazines 7–13 (Fig. 1).
Figure 1.

Initial lead structures of 2- and 3-substituted 8-amino imidazo[1,2-a]pyrazines.
2.2. Synthesis of first generation imidazo[1,2-a]pyrazine inhibitors
The classical synthetic route to substituted imidazo[1,2-a]pyrazines involves a simple condensation between 2-amino pyrazine and chloroacetaldehyde. Previously reported methods14 using DMF as a solvent proved to be low yielding, but with methanol as solvent, the product was isolated in 98% yield. However, all subsequent attempts to functionalise the imidazo[1,2-a]pyrazine core via bromination14, 15 gave extremely poor yields and inseparable mixtures of dibrominated regioisomers. Furthermore, these could not be further transformed using telesubstitution with ammonia,15, 16 to give 8-amino imidazo[1,2-a]pyrazines as planned.
We therefore planned to prepare the core heterocycle with functional groups already installed at the 2- or 3-positions, and with a leaving group at the 8-position to allow amino or other functionality to be added. In order to access the 3-substituted heterocycles 1–6, we adapted the procedure of MacCoss et al.17 to give 3-aryl substituted 8-chloroimidazo[1,2-a]pyrazines from 2-aryl-2-hydroxy amines 14a–e (Scheme 1). In each case the synthesis of the amino alcohol was achieved via α-bromination18 of the aryl ketone with pyridinium tribromide to give 15a–e. This was followed by substitution of the α-bromine with an azide,19 giving 16a–e, which were then reduced20 to give alcohols 17a–e. Hydrogenation21 to give amino alcohols 14a–e was performed at atmospheric pressure, with the exception of the thiophene analogue which required 3 bar pressure19 to go to completion. Coupling with 2,3-dichloropyrazine in 1,4-dioxane afforded the pyrazinyl-amino alcohols 18a–e in good yields. Swern oxidation22 to the ketones 19a–e was followed by acid-induced cyclisation to form the 3-aryl-8-chloroimidazo[1,2-a]pyrazines 20a–e.
Scheme 1.

Synthesis of 3-aryl-8-chloroimidazo[1,2-a]pyrazines 20a–e.
3-substituted imidazo[1,2-a]pyrazine derivatives have previously been reported as kinase inhibitors,23, 24, 25 and a range of strategies is available for synthesis of the key heterocyclic core. However, 2-aryl imidazo[1,2-a]pyrazine derivatives have been less frequently explored,26 and consequently fewer synthetic approaches to these compounds have been reported. Using the α-bromo aryl ketones 15a–e, the key 2-aryl-8-chloroimidazo[1,2-a]pyrazine intermediates 21a–e were readily prepared by condensation with 2-amino-3-chloropyrazine (Scheme 2).
Scheme 2.
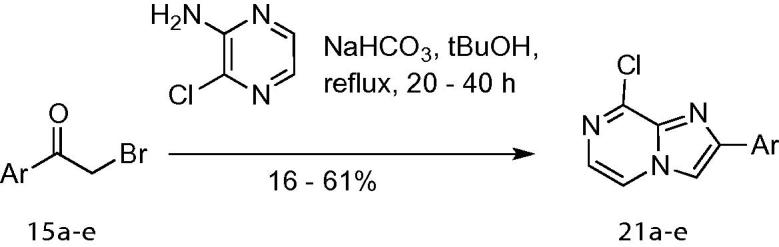
Synthesis of 2-aryl-8-chloroimidazo[1,2-a]pyrazines 21a–e.
To install the sulfonamido and sulfonamidoaniline groups, Buchwald–Hartwig coupling chemistry was employed.27 tBu-XPhos28 has been previously used successfully in the Pd-catalysed amidation of aryl bromides with tert-butyl carbamate.29 The combination of tBu-XPhos/Pd(dba)2/K2CO3/tBuOH gave low to moderate yields for all couplings with p-toluene sulfonamide 22 and N-(4-aminophenyl)-4-methylbenzenesulfonamide 23 (Table 1), with the exception of 21d, for which Pd(dppf)Cl2 had to be used30 as an alternative. As the coupling of sulfonamides with aryl chlorides had been reported31 to give high yields using DavePhos32 a complete range of precatalyst, base, solvent and temperature was screened for the conversion of 21a to 7 and 11 (Table S1, Supporting information). The combination of Pd2(dba)3/DavePhos/NaOtBu/toluene gave 7 in excellent yield, however this reaction is clearly highly substrate-dependent and these optimised coupling conditions did not give such good results with other 2- and 3-aryl-8-chloroimidazo[1,2-a]pyrazines. Likewise, conditions for microwave coupling were explored, which gave excellent yields of 7 but which were less successful for the synthesis of 11 (Table 1).
Table 1.
| Starting Material | Ar1 | Ar2 | Ar3NH2 | Conditions | Product (Yield) |
|---|---|---|---|---|---|
| 20a | H | 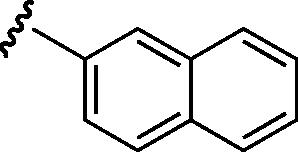 |
22 | tBu-XPhos (5 mol %) Pd(dba)2 (1 mol %) K2CO3 (1.2 equiv) tBuOH, reflux, 40 h | 1 (41%) |
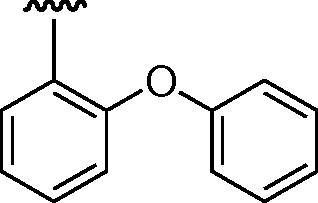
| 20b | H | 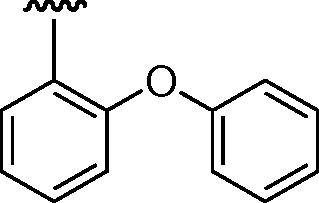 |
22 | tBu-XPhos (5 mol %) Pd(dba)2 (1 mol %) K2CO3 (1.2 equiv) tBuOH, reflux, 40 h | 2 (26%) |
| 20c | H | 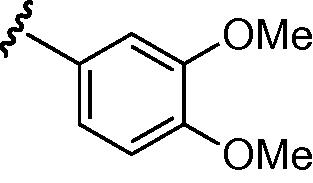 |
22 | tBu-XPhos (5 mol %) Pd(dba)2 (1 mol %) K2CO3 (1.2 equiv) tBuOH, reflux, 40 h | 3 (30%) |
| 20d | H | 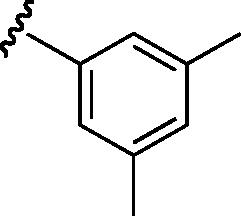 |
22 | tBu-XPhos (5 mol %) Pd(dba)2 (1 mol %) K2CO3 (1.2 equiv) tBuOH, reflux, 40 h | 4 (67%) |
| 20a | H | 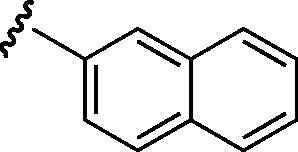 |
23 | tBu-XPhos (5 mol %) Pd(dba)2 (1 mol %) K2CO3 (1.2 equiv) tBuOH, reflux, 46 h | 5 (35%) |
| 20e | H | 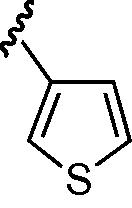 |
23 | DavePhos (3 mol %), Pd2(dba)3 (1 mol %) NaOtBu (1.4 equiv) toluene, reflux, 24 h | 6 (30%) |
| 21a | 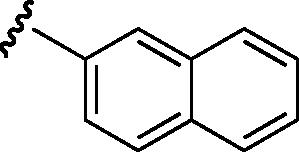 |
H | 22 | DavePhos (3 mol %), Pd2(dba)3 (1 mol %) NaOtBu (1.4 equiv) toluene, reflux, 24 h | 7 (93%) |
| 21a | 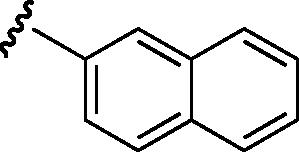 |
H | 22 | DavePhos (3 mol %), Pd2(dba)3 (1 mol %) NaOtBu (1.4 equiv) toluene, 160 °C (μW), 10 min | 7 (46%) |
| 24 | 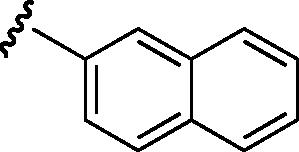 |
H | 22 | NaH (2 equiv), DMF, 100 °C, 20 h | 7 (72%) |
| 21b | 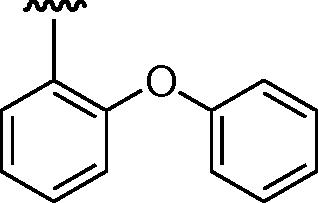 |
H | 22 | tBu-XPhos (5 mol %) Pd(dba)2 (1 mol %) K2CO3 (1.2 equiv) tBuOH, reflux, 48 h | 8 (13%) |
| 21c | 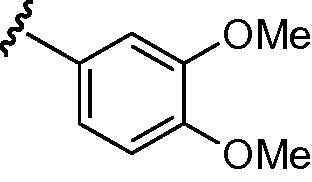 |
H | 22 | tBu-XPhos (5 mol %) Pd(dba)2 (1 mol %) K2CO3 (1.2 equiv) tBuOH, reflux, 48 h | 9 (2%) |
| 21d | 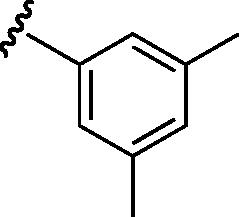 |
H | 22 | Pd(dppf)2Cl2 (2 mol %), K2CO3 (1.2 equiv), tBuOH, reflux, 21 h | 10 (29%) |
| 21a | 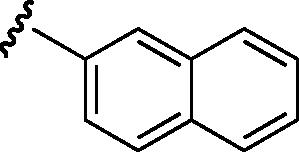 |
H | 23 | DavePhos (3 mol %), Pd2(dba)3 (1 mol %) NaOtBu (1.4 equiv) toluene, reflux, 24 h | 11 (22%) |
| 21a | 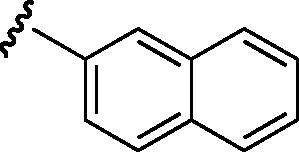 |
H | 23 | DavePhos (3 mol %), Pd2(dba)3 (1 mol %) NaOtBu (1.4 equiv) toluene, 160 °C (μW), 10 min | 11 (47%) |
| 21e | 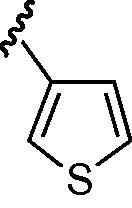 |
H | 23 | tBu-XPhos (5 mol %) Pd(dba)2 (1 mol %) K2CO3 (1.2 equiv) tBuOH, reflux, 48 h | 12 (8%) |
| 21b | 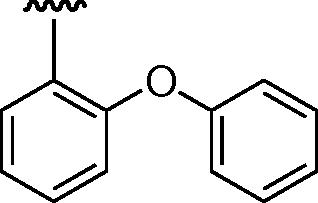 |
H | 23 | DavePhos (3 mol %), Pd2(dba)3 (1 mol %) NaOtBu (1.4 equiv) toluene, reflux, 24 h | 13 (9%) |
Methyl sulfones have previously been used to attach a variety of nucleophiles onto imidazo[1,2-a]pyrazine rings.24, 33 As an alternative to the Buchwald–Hartwig coupling, 21a was converted to methyl sulfone 24 (Scheme 3). Treatment with 4-toluene sulfonamide/NaH/DMF gave 7 in good yield, however for this substrate no improvement over the optimised Buchwald–Hartwig conditions could be effected via this route.
Scheme 3.
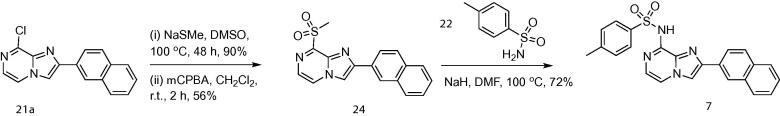
Alternative route to 7 via methylsulfone 24.
The regiochemistry of the 2- and 3-substituted imidazo[1,2-a]pyrazines was confirmed by 2D NMR. For the 2-substituted imidazo[1,2-a]pyrazine 21a, HMBC (Fig. S1, Supporting information) showed correlation between the protons and carbons in the 3- and 5-positions of the imidazo[1,2-a]pyrazine rings. Conversely, with the 3-substituted imidazo[1,2-a]pyrazines no correlation was observed between the protons in the corresponding 2- and 5-positions.
2.3. Biochemical evaluation of first generation imidazo[1,2-a]pyrazine inhibitors
The HP0525 protein was produced recombinantly in Escherichia coli and purified to high purity as described previously.10 The ATPase activity of HP0525 was measured by monitoring the release of inorganic phosphate (Pi) using an in vitro ATPase assay (see Section 4 and SI). Initially, the inhibitory effects of the compounds were evaluated by performing the ATPase assay with and without compound present at concentrations of 500 μM (or 250 μM), 50 μM and 5 μM (data not shown). The compounds with inhibitory activities were further analyzed for dose-dependency by estimating their IC50s (Fig. 2). Those with the highest inhibitory effect, 11, 5 and 6 with IC50s of 6, 20 and 48 μM, respectively, showed similarities in their chemical structures, see below.
Figure 2.

Dose-dependent and steady-state inhibition of HP0525. Dose–response curves used for IC50 estimations of compounds 11 (A) and 32 (B). Michaelis–Menten (C) and Lineweaver–Burk (D) plots, corresponding to ✠ without inhibitor, and ✠ with 11. Error bars represent standard deviations using triplicate data.
To test our hypothesis that the inhibitors bind in the substrate pocket, as suggested by the molecular docking, we tested the mode of inhibition of 11. Steady-state kinetic data displayed Michaelis–Menten behavior, and 11 unambiguously behaved as a competitive inhibitor (Fig. 2).
We verified that our analogues displayed suitable physicochemical profiles by calculating log P and log S using A log P2.1 applet.34 The results presented in Table 2 suggest that our first generation of compounds display limited solubility (0.1 to 10 mg/L) and log P values that are high but within limits described by the Lipinski’s ‘rule of five’ (i.e., ⩽5).35
Table 2.
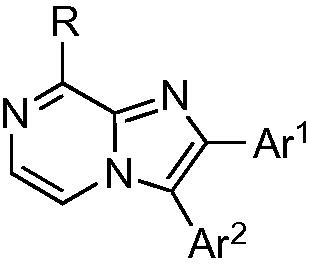
| Compound | R | Ar1 | Ar2 | IC50/μM | A log sP | A log pS |
|---|---|---|---|---|---|---|
| 1 |  |
H |  |
77 ± 16 | 3.7 | −5.7 |
| 2 |  |
H |  |
96 ± 42 | 4.3 | −5.8 |
| 3 | 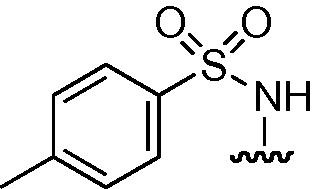 |
H | 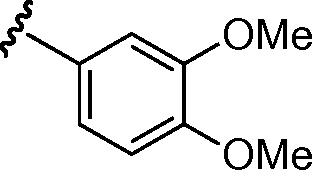 |
144 ± 14 | 3.1 | −4.7 |
| 4 | 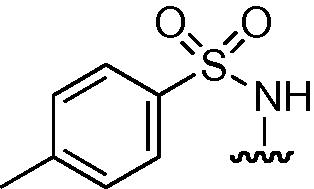 |
H | 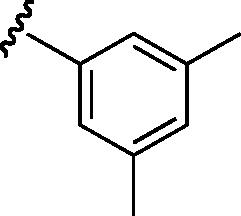 |
154 ± 22 | 3.3 | −5.4 |
| 5 | 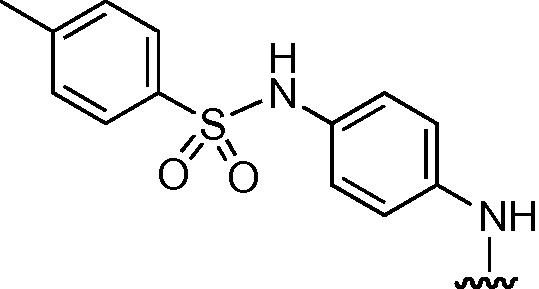 |
H | 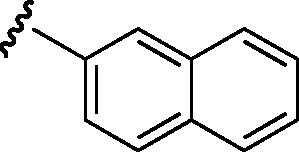 |
20 ± 3 | 4.6 | −6.0 |
| 6 | 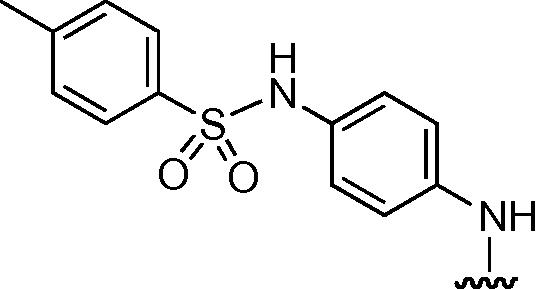 |
H | 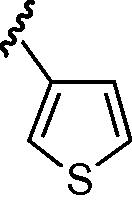 |
48 ± 7 | 3.5 | −5.1 |
| 7 | 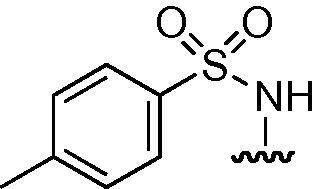 |
 |
H | 88 ± 14 | 4.3 | −5.7 |
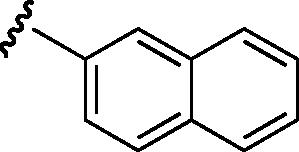
| 8 | 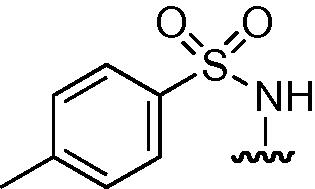 |
 |
H | 82 ± 14 | 4.5 | −5.8 |
| 9 | 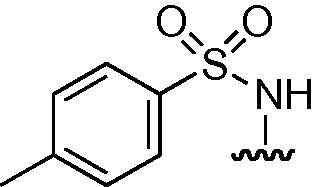 |
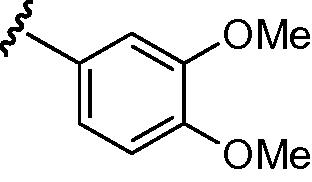 |
H | 167 ± 43 | 3.5 | −4.7 |
| 10 | 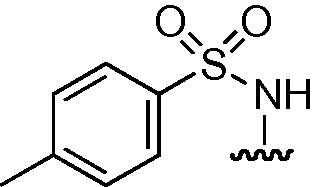 |
 |
H | 133 ± 16 | 3.5 | −5.4 |
| 11 | 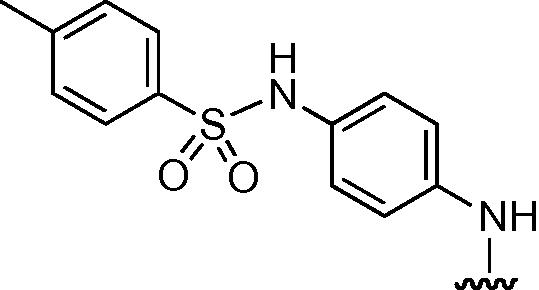 |
 |
H | 6 ± 1 | 5.0 | −6.0 |
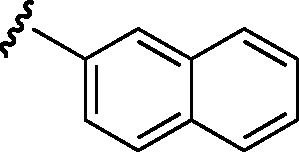
| 12 | 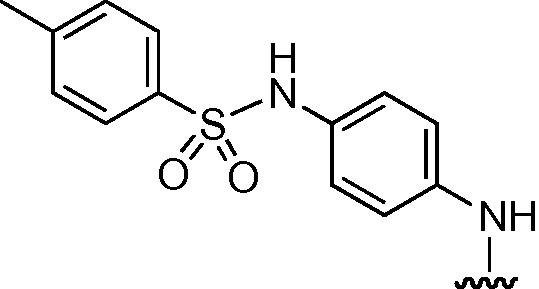 |
 |
H | 99 ± 18 | 3.9 | −5.1 |
| 13 | 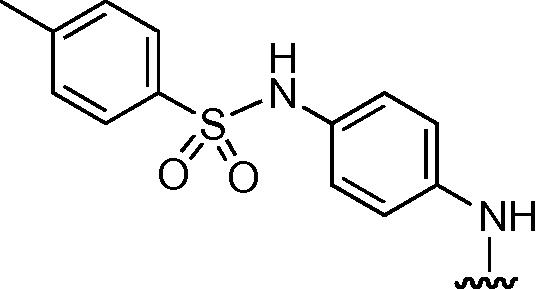 |
 |
H | 18 ± 4 | 5.5 | −6.2 |
2.4. Second generation imidazo[1,2-a]pyrazine inhibitors
The promising IC50 values observed for these compounds, and especially for 11, prompted a further investigation of related structures. Table 2 shows that compounds with substituents in the 8-position where the sulfonamide moiety was remote from the core heterocycle showed the greatest potency. In particular, 11 showed the most promise as a lead for further investigation. However, the physicochemical properties (partition coefficient and solubility) of this series of compounds, were poor, and we therefore sought to explore structural modifications that would give candidates suitable for preclinical drug development with improved solubility as well as potency. As the sulfonamide could potentially act as a bioisostere of one of the phosphate groups of ATP,36 we aimed to evaluate analogues with different spacing and flexible or rigid linkers between the core heterocycle and the sulfonamide, and also analogues with the sulfonamide group absent. Analogues with heterocyclic groups at position 2, or lacking the aryl group altogether, were also evaluated, in a further effort to improve the solubility of this series. Finally, substitution at position 6 was also investigated.
A series of analogues of 11, differing at the 8-position, were prepared (Table 3) via either Buchwald–Hartwig coupling from 21a or nucleophilic substitution of the methyl sulfone 24, in moderate to good yields. Deleting the arylsulfonamide group completely (25) resulted in complete loss of activity, further suggesting that this region of the 8-substituted imidazo[1,2-a]pyrazines is important for binding to the active site. The majority of variants of the N-(4-aminophenyl)-4-methylbenzenesulfonamido group (26–30) showed a decrease in potency compared to 11 when the sulfonamido group was placed at a greater distance from the imidazo[1,2-a]pyrazine ring, modified, or deleted completely. However, when the aniline group was replaced by an ethyl linker (31) comparable inhibition with better solubility and log P were obtained. Replacing the p-toluene sulfonamidyl group with quinoline-8-sulfonamide (33, 34) also resulted in compounds of high potency. Surprisingly, when the N-(4-aminophenyl)-4-methylbenzenesulfonamido group was replaced by 3-(pyridin-3-yl)aniline (32) a further slight improvement in potency was seen (Table 3). Unfortunately, the most active compounds in this series (32, 33 and 34) all exhibited comparable solubility and log P to 11.
Table 3.
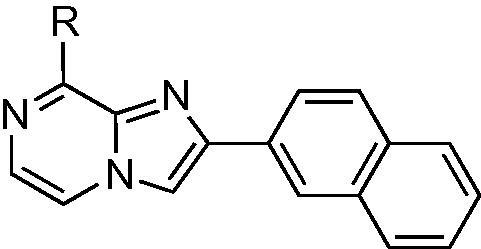
| Compound | R | Synthesis method | IC50/μM | A log sP | A log pS |
|---|---|---|---|---|---|
| 25 | NH2 | — | >1000 | 3.2 | −4.3 |
| 26 | 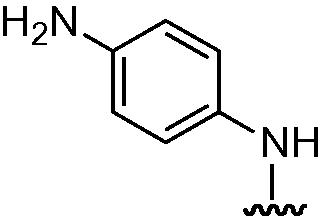 |
A | 18 ± 5 | 4.4 | −5.3 |
| 27 | 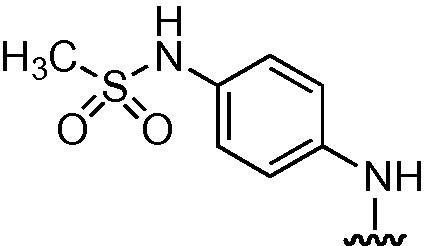 |
A | 61 ± 28 | 4.0 | −5.4 |
| 28 | 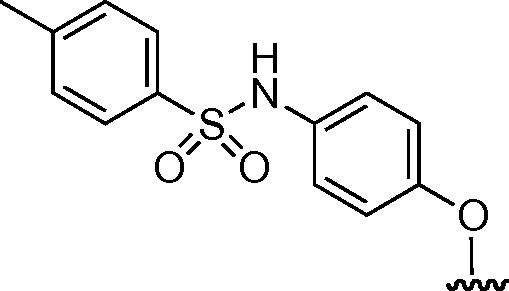 |
A | 29 ± 9 | 5.0 | −6.0 |
| 29 | 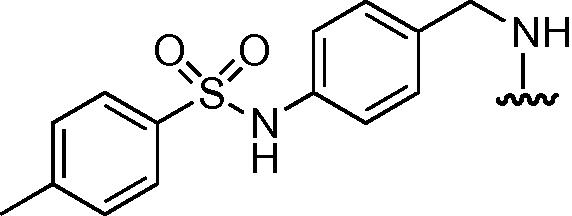 |
A | 75 ± 35 | 5.3 | −6.0 |
| 30 | 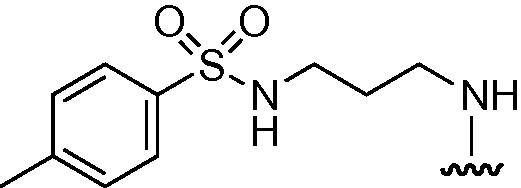 |
B | 58 ± 38 | 4.3 | −5.7 |
| 31 | 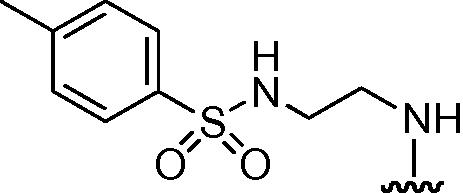 |
B | 7 ± 2 | 4.1 | −5.6 |
| 32 | 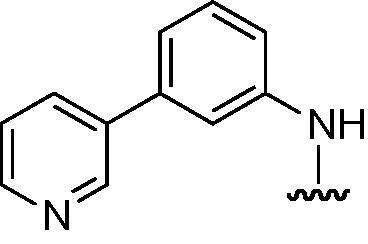 |
A | 4 ± 1 | 5.0 | −5.8 |
| 33 | 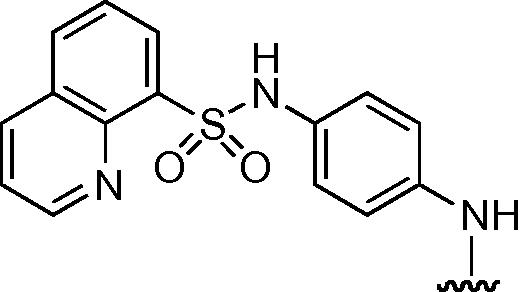 |
A | 7 ± 2 | 5.1 | −6.0 |
| 34 | 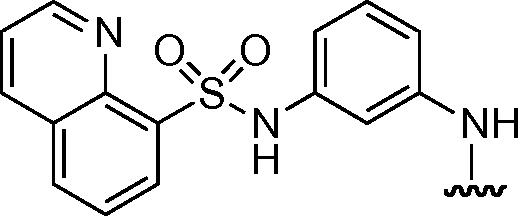 |
A | 7 ± 2 | 5.0 | −6.0 |
Method A: Buchwald–Hartwig coupling from 21a. Method B: NaH, 24. Section 4 are given in the Supporting information.
The importance of the aryl substituent in position 2 was further reinforced by compound 35; deleting the aryl substituent resulted in a complete loss of activity (Table 4). As the naphthalene substituent is highly lipophilic and is a major contributor to the insolubility of this series of inhibitors, we replaced this with a quinoxaline (36). However, this gave no appreciable improvement in either potency or solubility.
Table 4.
Variants of the lead compound 11 at the 2- and 6-positions
| Compound | IC50/μM | A log sP | A log pS | |
|---|---|---|---|---|
| 35 | 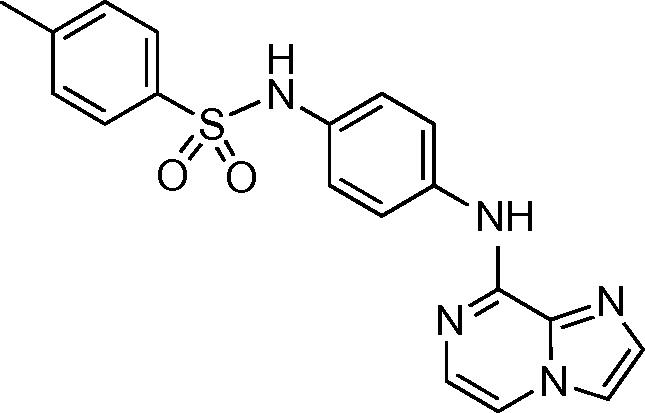 |
>1000 | 2.7 | −4.8 |
| 36 | 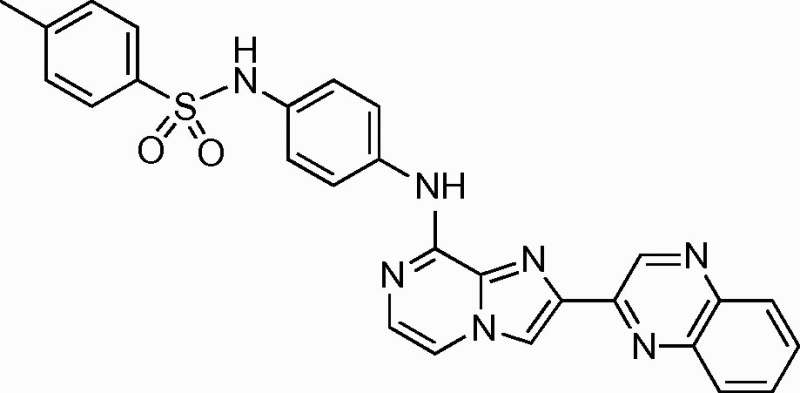 |
28 ± 6 | 4.2 | −5.3 |
| 37 | 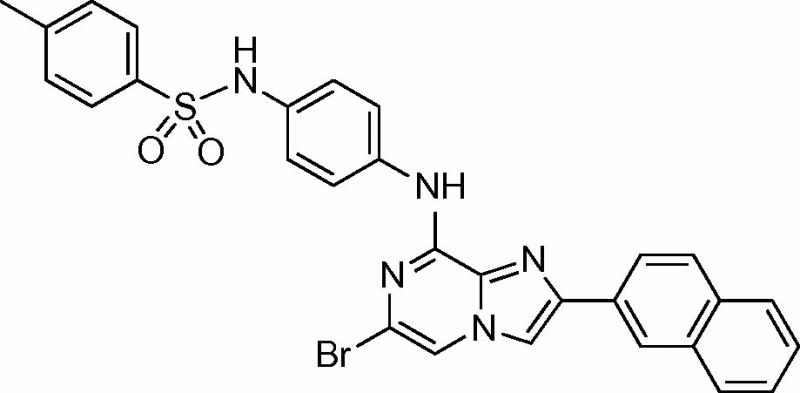 |
6 ± 2 | 6.6 | −6.3 |
| 38 | 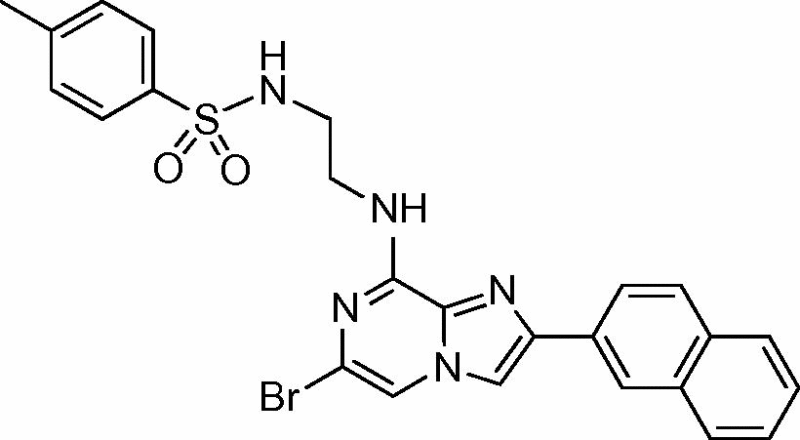 |
77 ± 20 | 5.0 | −5.8 |
In order to explore the effects of substitution at the 6-position of the imidazo[1,2-a]pyrazine, 37 and 38 were synthesized. The key 6,8-dibromoimidazo[1,2-a]pyrazine 39 was prepared by condensation of 4,6-dibromo-2-aminopyrazine with 2-bromoacetyl naphthalene (Scheme 4). Nucleophilic reaction with the appropriate monotosylated diamine proceeded smoothly and exclusively at the 8-position to afford 37 or 38 in good yield. However, 38 showed poorer physicochemical properties and IC50 compared to the analogue 31. Likewise, 37 showed poorer solubility and log P compared with 11, which lacks the 6-substitutent, but showed comparable potency (Table 4). Overall, this suggests that the bromide substituent at the 5-position does not improve potency and leads to a poorer physicochemical profile.
Scheme 4.
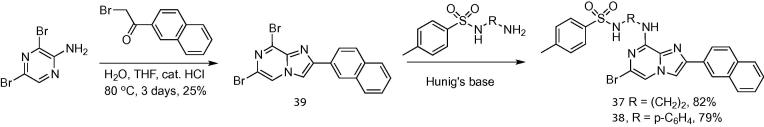
Synthesis of 37 and 38 via 6,8-dibromoimidazo[1,2-a]pyrazine 39.
2.5. Docking studies
In order to further understand the binding of these imidazo[1,2-a]pyrazines to HP0525, and to direct the design of more potent inhibitors, molecular docking studies were carried out using AutoDock Vina.37 Structural studies showed that the conformation of the apo form of each unit is variable, but the two structures 1G6O9 and 1NLY8 which possess ADP, or the ATP mimic ATP-γS, respectively, are structurally highly similar with an RMS of 0.50 Å for CA atoms and 0.78 Å for all atoms. Both of these structures contain two identical chains, A and B. For drug design purposes any of the four chains from the two crystal structures above are acceptable for use in modelling/screening. Investigation of the ligand–protein interactions of each, in particular the hydrogen bonding, reveals a more extensive network of interactions in 1NLY and the A chain also includes the active site metal (Mg). This might therefore be taken to be the more physiological representative structure. However, the resolution of 1G6O is 2.50 Å compared to 2.80 Å for 1NLY, and a Ramachandran38 analysis gives 4% of residues in more favourable regions for 1G6O. The crystal structure of the ADP-bound HP0525, 1G6O, with heteroatoms and ADP removed, was therefore used for the docking studies. Examination of the ADP/ATP binding pocket shows that it adopts a conical topology in which the entrance is wide open and the bottom of the cavity very narrow and very likely to tolerate small groups only. In accordance with kinases and phosphorylases topologies the entrance to the active side is highly lipophilic and the end of the cavity highly hydrophilic. In the entrance to the active site (adenosine binding region), lipophilicity is governed by three aromatic residues (Tyr140, Phe144 and Phe145). Hydrophilicity within the cavity (triphosphate binding region) is created by a tetrad Gly181/Ser182/Gly183/Lys184 along with Arg133. A third binding region, not exploited by ADP/ATP, is located in the direction of the 2′-ribose hydroxyl and is likely to tolerate small aliphatic moieties (Fig. 3a).
Figure 3.

(a) Binding of ADP to HP0525 (PDB 1G6O). Key residues are labelled. (b) Comparison of the binding of 11 and ADP (Yellow). (c) Binding of 11 to HP0525, showing regions that could be explored to increase the potency of the series of inhibitors. (d) Compounds 11 (white), 30 (magenta), 31 (violet-brown) and 34 (turquoise) are overlaid, when docked in HP0525. Polar bonds are indicated corresponding to interactions involving compound 11. (e) Comparison of the docking of 32 (white/blue) and ADP in HP0525.
Docking of the lead compound 11 (Fig. 3b), and comparison with the binding of ADP, showed a binding mode in which the inhibitor is deeply buried within the enzymatic cavity. In this orientation, the naphthalene group occupies the purine-binding region of the active site, possibly making π-stacking interactions with Phe145. The core imidazo[1,2-a]pyrazine ring sits in place of the ribose moiety of ADP, with the sulfonamide occupying the phosphate binding region and making polar contacts with Gly181, Lys184 and Thr185. Examining the surface of the ADP binding site with the docked 11 (Fig. 3c) suggested that these two binding regions could be explored to increase the potency of this series of inhibitors. For example, in order to optimize the predicted interaction of the sulfonamide moiety with the phosphate binding site, a series of analogues (30, 31, 33, 34) in which different spacer lengths and orientations between the sulfonamide and the 8-position were synthesized and tested. Docking studies on 30 (IC50 58 μM), 31 (IC50 7 μM), and 34 (IC50 7 μM) suggest that the propyl chain of 30 may position the sulfonamide moiety too far from the phosphate binding region (Fig. 3d). Intriguingly, the most potent lead compound in this series, 32, which is also the most ligand efficient, lacks the sulfonamide group completely. Here the docking (Fig. 3e) suggests a similar orientation, with the naphthalene group occupying the purine-binding region, and the imidazo[1,2-a]pyrazine ring occupying the ribose-binding area. However, the 3-(pyridin-3-yl)aniline moiety in this case occupies the phosphate binding region. This may indicate that the sulfonamide group is not always necessary for binding if other H-bond donor or acceptor groups are present in the correct orientation.
3. Conclusions
Following a virtual high throughput screen, a novel series of 8-amino imidazo[1,2-a]pyrazine derivatives have been developed, using a flexible synthetic route to deliver 2- and 3-aryl regioisomers. Biochemical evaluation showed moderate to good potency highlighting this class of compound as competitive inhibitors of the HP0525 ATPase from H. pylori, with potential as antibacterial agents. The structure–activity relationships of these 8-amino imidazo[1,2-a]pyrazines has been explored through docking studies, however co-crystallisation of these inhibitors with HP0525 is imperative to fully understand the interaction within the nucleotide binding site and aid in the development of more potent inhibitors. Furthermore, development of these compounds will require improvements in their aqueous solubility to enable a more suitable physicochemical profile.
4. Experimental section
4.1. Lead identification and molecular docking
The initial lead compounds were identified through screening of the SoftFocus kinase-targeted compound library (BioFocus). The pharmacophore alignment function of MOE39 was used to produce a rough initial alignment to ATP of the diverse input set based on common chemical features. The aligned structures were then energy-minimised in the context of the nucleotide binding site of HP0525 (PDB ID 1NLY8), with the atoms of the protein frozen, using NAMD.40 Ligand force field parameters were assigned according to the General Amber Force Field.41 The resulting binding poses were scored using the Hawkins GB/SA function of DOCK 6.13
Further molecular docking studies were carried out using AutoDock Vina.37 The crystal structure of ADP-HP0525 (PDB entry 1G60) was used to define a docking grid around the nucleotide binding site, with a size of 14 × 16 × 24 and a grid center of −12.034, 24.627 and 22.363 in the x, y, and z coordinates, respectively. An ‘exhaustiveness’ parameter of 8 was used. Ligand structures were generated using chem3D pro and further prepared using AutoDock Tools (ADT)42 as recommended in the documentation.
4.2. General chemistry
Melting points (Mp) were recorded on a Gallenkamp Melting Point Apparatus and are uncorrected. 1H and 13C NMR were recorded using Bruker AV400 (400 and 100 MHz, respectively), AV500 (500 and 125 MHz, respectively) and AV600 (600 and 150 MHz, respectively) spectrometers as indicated. Chemical shifts are quoted on the δ scale in units of ppm using TMS as an internal standard. Spectra were obtained using CDCl3, CD3OD, CD2Cl2 and DMSO-d6 as solvents and coupling constants (J) are reported in Hz with the following splitting abbreviations: s (singlet), d (doublet), t (triplet), dd (doublet of doublets), bs (broad singlet). Infra-Red (IR) spectroscopy was carried out using a PerkinElmer Spectrum 100 FT-IR Spectrometer using thin films. Absorption maxima (νmax) are reported in wavenumbers (cm−1).
Solvents and reagents were obtained from commercial sources and were used as received unless otherwise stated. Petroleum ether refers to the fraction of light petroleum ether boiling in the range 40–60 °C.
Representative examples of each of the synthetic routes shown in Scheme 1, Scheme 2, Scheme 3 are given. Full experimental for the preparation of the remaining compounds, and full compound characterisation, is given in the Supplementary data.
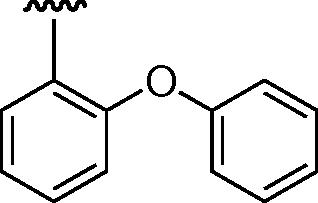
4.3. General method for synthesis of α-bromo aryl ketones, illustrated for the preparation of 2-bromo-1-(2-phenoxyphenyl)ethanone, 15b
1-(2-Phenoxyphenyl)ethanone (2.00 g, 9.42 mmol) was dissolved in chloroform (60 mL) and ethanol (60 mL). Pyridinium tribromide (7.50 g, 23.6 mmol) was added and the reaction was stirred at 50 °C for 16 h. The reaction mixture was cooled to room temperature and the solvents removed in vacuo. The resulting orange slurry was suspended in H2O (30 mL) and extracted with EtOAc (4 × 30 mL). The combined organic extracts were washed with H2O (2 × 20 mL) and brine (1 × 20 mL), dried (Na2SO4), filtered and concentrated in vacuo to give a yellow oil. Flash chromatography was carried out (applied in petroleum ether; eluted 0% to 10% to 33% CH2Cl2) to afford the title compound as a pale yellow oil (2.30 g, 7.90 mmol, 84%). Rf = 0.68 (CH2Cl2); IR (νmax/cm−1, thin film): 1677, 1598, 1574; 1H NMR (500 MHz, CDCl3): δH = 4.65 (s, 2H), 6.86 (d, J = 8.4 Hz, 1H), 7.09 (d, J = 7.7 Hz, 2H), 7.17 (t, J = 7.6 Hz, 1H), 7.22 (t, J = 7.3 Hz, 1H), 7.40–7.47 (m, 3H), 7.92 (dd, J = 7.6, 1.5 Hz, 1H); 13C NMR (125 MHz, CDCl3): δC = 36.8, 117.6, 119.5, 123.0, 124.4, 126.2, 129.9, 131.3, 134.2, 155.0, 156.5, 191.6; LRMS m/z (EI+): 292 [M (81Br)]+, 290 [M (79Br)]+, 212 [M−Br]+, 197 [M−CH2Br]+; HRMS m/z (EI+): Found 289.99403 [M(79Br)]+; C14H11BrO2 requires 289.99369.
4.4. General method for synthesis of α-azido aryl ketones, illustrated for the preparation of 2-azido-1-(2-naphthyl)ethanone 16a
2-(Bromoacetyl)naphthalene (2.00 g, 8.03 mmol) was dissolved in DMSO (10 mL) and the mixture was cooled on ice such that the temperature was kept below 10 °C. Sodium azide (0.630 g, 9.64 mmol) was added in one portion and the reaction was stirred under argon at room temperature for 90 min. The reaction was quenched with H2O (20 mL), and extracted with EtOAc (3 × 30 mL). The organic layers were combined, washed with H2O, dried (Na2SO4) and filtered. The solvent was removed in vacuo to give the title compound as a brown/orange oil (1.69 g, 8.01 mmol, 100%) with NMR consistent with literature values.43 Rf = 0.63 (5:1 petroleum ether/EtOAc); IR (νmax/cm−1, thin film): 2105, 1690; 1H NMR (600 MHz, CDCl3): δH = 4.73 (s, 2H), 7.59–7.62 (m, 1H), 7.65–7.68 (m, 1H), 7.91 (d, J = 8.1 Hz, 1H), 7.95 (d, J = 8.6 Hz, 1H), 7.99–8.01 (m 2H), 8.42 (s, 1H); 13C NMR (150 MHz, CDCl3): δC = 55.0, 123.3, 127.2, 127.9 129.0, 129.1, 129.6, 129.8, 131.7, 132.4, 136.0, 193.2; LRMS m/z (EI+): 211 [M]+, 155 [M−CH2N3]+, 127 [Naphthalene]+.
4.5. General method for synthesis of α-azido aryl alcohols, illustrated for the preparation of 2-azido-1-(2-naphthyl)ethanol 17a
Azidoketone 16a (2.11 g, 10.0 mmol) was dissolved in anhydrous MeOH (100 mL) and cooled on ice. Sodium borohydride (568 mg, 15.0 mmol) was added portion wise and the mixture was stirred on ice under argon for 1 h until the reaction had gone to completion by TLC. The solvent was removed and the resulting residue was taken up in CH2Cl2 (100 mL) and carefully washed with H2O (2 × 60 mL) followed by brine (60 mL). The organic extracts were dried over Na2SO4, filtered and concentrated in vacuo to give the title compound as a brown oil (2.14 g, 10.0 mmol, 100%). Spectroscopic data (for the racemic material) was consistent with that previously reported44 (for the (S)-enantiomer): Rf = 0.65 (3:1 petroleum ether/EtOAc); IR (νmax/cm−1, thin film): 3398, 2100; 1H NMR (500 MHz, CDCl3): δH = 2.70 (br s, 1H), 3.46–3.58 (m, 2H), 5.02 (dd, J = 8.1, 3.9 Hz, 1H), 7.44 (dd J = 8.4, 1.6 Hz, 1H), 7.49–7.52 (m, 2H), 7.83–7.86 (m, 4H); 13C NMR (125 MHz, CDCl3): δC = 58.1, 73.6, 123.7, 125.1, 126.4, 126.5, 127.8, 128.1, 128.6, 133.3 (2 signals), 138.0; LRMS m/z (EI+): 221, 157 [M−CH2N3]+, 147, 129.
4.6. General method for synthesis of α-amino aryl alcohols, illustrated for the preparation of 2-amino-1-(2-naphthyl)ethanol 14a
Azidoalcohol 17a (2.18 g, 10.2 mmol) was dissolved in anhydrous MeOH (50 mL) and 10% palladium on carbon (218 mg, 10% w/w) was added. The vessel was evacuated and purged with Ar (3×) and under static vacuum a balloon of hydrogen was added. The reaction mixture was stirred under hydrogen atmosphere until completion as determined by TLC and disappearance of N3 peak by IR. After 3½ h, the hydrogen was carefully released, the vessel evacuated and purged argon (3×), and the reaction mixture was filtered through Celite (pre-washed with MeOH). Solvent removal in vacuo gave the crude compound as a orange oil (1.91 g, 10.2 mmol, 100%). Spectroscopic data was consistent with that previously reported.44 Rf = 0.06 (5:1 EtOAc/MeOH); IR (νmax/cm−1, thin film): 3290, 3054, 2916, 1599; 1H NMR (500 MHz, CDCl3): δH = 2.82 (m, 2H), 4.59–4.75 (m, 1H), 7.36–7.40 (m, 3H), 7.74–7.76 (m, 4H); 13C NMR (125 MHz, CDCl3): δC = 49.8, 74.3, 123.8, 124.5, 125.7, 126.0, 127.5, 128.1, 132.8, 133.1 (2 signals), 139.7; LRMS m/z (ESI+): 229.2 [M+MeCN]+, 211.2 [M+Na]+, 188.1 [M+H]+, 170 [M−OH]+.
4.7. General method for synthesis of 2-[(3-chloropyrazin-2-yl)amino]-1-(2-aryl)ethanol, illustrated for the preparation of 2-[(3-chloropyrazin-2-yl)amino]-1-(2-naphthyl)ethanol 18a
Amino alcohol 14a (289 mg, 1.55 mmol), 2,3-dichloropyrazine (177 μL, 1.70 mmol) and Et3N (301 μL, 2.16 mmol) were dissolved in 1,4-dioxane (3 mL) and the reaction was stirred under reflux, under argon. After 19 h, the reaction was cooled to room temperature and the solvent removed in vacuo. The residue was taken up in CH2Cl2 and washed with H2O (3 × 20 mL) and brine (1 × 20 mL). The organic extracts were dried (Na2SO4), filtered and concentrated to give the crude product as an amber oil. Purification was carried out via flash chromatography (applied in CH2Cl2; eluted 0% to 33% EtOAc) to afford the title compound as a yellow oil (295 mg, 0.983 mmol, 63%). Rf = 0.64 (2:1 CH2Cl2/EtOAc); IR (νmax/cm−1, thin film): 3419, 3054, 2922, 1523; 1H NMR (500 MHz, CDCl3): δH = 3.65–3.70 (m, 1H), 3.88 (br s, 1H), 3.92–3.97 (m, 1H), 5.12 (dd, J = 7.5, 2.8 Hz), 5.67 (t, J = 5.3 Hz), 7.47–7.50 (m, 3H), 7.59 (d, J = 2.7 Hz, 1H), 7.81–7.84 (m, 3H), 7.86 (s, 1H), 7.91 (d, J = 2.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δC = 49.4, 73.9, 123.9, 124.8, 126.1, 126.4, 127.8, 128.0, 128.4, 131.3, 133.2, 133.3, 135.1, 139.4, 140.2, 151.5; LRMS m/z (ESI+): 300.1 [M(35Cl)+H]+, 284.2 [M(37Cl)−OH]+, 282.2 [M(35Cl)−OH]+; HRMS m/z (ESI−): Found 298.0731 [M(35Cl)−H]−; C16H13ClN3O requires 298.0747.
4.8. General method for synthesis of 2-[(3-chloropyrazin-2-yl)amino]-1-(2-aryl)ethanones, illustrated for the preparation of 2-[(3-chloropyrazin-2-yl)amino]-1-(2-naphthyl)ethanone 19a
DMSO (982 μL, 13.9 mmol) was dissolved in anhydrous CH2Cl2 (60 mL) and the reaction mixture was cooled to and maintained at −78 °C. Oxalyl chloride (586 μL, 6.93 mmol) was added drop wise and the mixture was stirred for 20 min. 17a (1.60 g, 5.33 mmol), dissolved in anhydrous CH2Cl2 (40 mL) was added dropwise, and stirred for 20 min. Et3N (3.54 mL, 26.6 mmol) was added dropwise and the reaction mixture was allowed to warm to room temperature over a period of 2.5 h. The reaction was then quenched with H2O (50 mL) and organics extracted, which were then washed with 2.0 M HCl (2 × 40 mL), NaHCO3 (satd aq 40 mL), H2O (40 mL) and brine (40 mL). The organic layer was dried (MgSO4), filtered and the solvent removed in vacuo to give a yellow/orange solid. Flash chromatography (applied in CH2Cl2; eluted 100:1 to 30:1 CH2Cl2/EtOAc) afforded the title compound as a yellow solid (903 mg, 3.03 mmol, 57%). Mp: 160 °C; Rf = 0.30 (30:1 CH2Cl2/EtOAc); IR (νmax/cm−1, thin film): 1680; 1H NMR (500 MHz, CDCl3): δH = 5.10 (d, J = 4.3, 2H), 6.54 (s, 1H), 7.58–7.62 (m, 1H), 7.64–7.67 (m, 1H), 7.68 (d, J = 6.1 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.96 (d, J = 8.6 Hz, 1H), 8.00–8.02 (m, 2H), 8.10 (dd, J = 8.6, 1.8 Hz, 1H), 8.62 (s, 1H); 13C NMR (125 MHz, CDCl3): δC = 48.4, 123.4, 127.3, 128.0, 129.0, 129.2, 129.8, 130.1, 131.3, 131.8, 132.6, 136.2, 139.7, 193.8; LRMS m/z (ESI+): 300 [M(37Cl)+H]+, 298 [M(35Cl)+H]+, 282 [M(37Cl)−OH]+, 280 [M(35Cl)−OH]+; HRMS m/z (ESI−): Found 296.0591 [M(35Cl)−H]−; C16H11ClN3O requires 296.0591.
4.9. General method for synthesis of 3-aryl-8-chloro-imidazo[1,2-a]pyrazines, illustrated for the preparation of 8-chloro-3-(2-naphthyl)imidazo[1,2-a]pyrazine 20a
Compound 19a (903 mg, 3.03 mmol) was dissolved in anhydrous toluene (40 mL) and the mixture was cooled on ice. Trifluoroacetic acid (1.64 mL, 21.2 mmol) was added and the reaction was allowed to stir on ice for 30 min, followed by the addition of trifluoroacetic anhydride (2.95 mL, 21.2 mmol). The reaction mixture was then stirred on ice for a further 30 min and then at room temperature for 65 h. The reaction was then diluted with toluene (20 mL) and washed with NaHCO3 solution (10% w/v, 3 × 20 mL) and brine (20 mL). The organics were dried (MgSO4), filtered and concentrated to give the crude product as an amber oil. Purification was carried out via flash chromatography (applied in CH2Cl2; eluted 80:1 to 10:1 CH2Cl2/EtOAc) to afford the title compound as an off white solid (386 mg, 1.38 mmol, 45%). Mp: 166 °C; Rf = 0.21 (10:1 CH2Cl2/EtOAc); IR (νmax/cm−1, thin film): 3102, 3052; 1H NMR (500 MHz, CDCl3): δH = 7.58–7.62 (m, 2H), 7.64 (dd, J = 8.6, 1.7 Hz, 1H), 7.73 (d, J = 4.6 Hz, 1H), 7.92–7.95 (m, 2H), 8.02 (s, 1H), 8.04–8.05 (m, 1H), 8.04 (s, 1H), 8.30 (d, J = 4.6 Hz, 1H); 13C NMR (125 MHz, CDCl3): δC = 116.4, 124.7, 125.2, 127.3, 127.4, 127.6, 128.0, 128.2, 128.6, 129.4, 129.7, 133.4, 133.5, 134.8, 138.4, 144.5; LRMS m/z (ESI+): 282 [M(37Cl)+H]+, 280 [M(35Cl)+H]+; HRMS m/z (ESI+): Found 280.0646 [M(35Cl)+H]+; C16H11ClN3 requires 280.0642.
4.10. General method for the synthesis of 4-methyl-N-[4-[3-arylimidazo[1,2-a]pyrazine-8-yl]]-sulfonamides, illustrated for the preparation of 4-methyl-N-[4-[3-(2-naphthyl)imidazo[1,2-a]pyrazine-8-yl]aminophenyl]benzenesulfonamide 5
All glassware was evacuated and flushed with argon prior to use. Compound 20a (283 mg, 1.01 mmol), N-(4-aminophenyl)-4-methylbenzenesulfonamide 23 (318 mg, 1.21 mmol), K2CO3 (167 mg, 1.21 mmol), Pd(dba)2 (5.80 mg, 1 mol %) and tert-butyl XPhos (21.5 mg, 5 mol %) were taken up in tBuOH (6 mL) and the reaction was stirred under reflux under Ar for 46 h. The reaction mixture was cooled to room temperature, diluted with MeOH (100 mL) and filtered through Celite (pre-washed with MeOH). Flash chromatography (applied in CH2Cl2; eluted 100:1 to 50:1 to 8:1 CH2Cl2/EtOAc) was carried out to give the title compound as a yellow solid (181 mg, 0.355 mmol, 35%). Mp: >200 °C; Rf = 0.12 (10:1 CH2Cl2/EtOAc); IR (νmax/cm−1, thin film): 3240, 3057, 1623, 1500, 1330, 1154; 1H NMR (600 MHz, CDCl3): δH = 2.37 (s, 3H), 7.08–7.10 (m, 2H), 7.17 (s, 1H), 7.21 (d, J = 8.3 Hz, 2H), 7.52 (d, J = 4.7 Hz, 1H), 7.57–7.58 (m, 2H), 7.64 (dd, J = 8.5, 1.6 Hz, 1H), 7.65 (d, J = 8.3 Hz, 2H), 7.73 (s, 1H), 7.79–7.80 (m, 3H), 7.91–7.92 (m, 2H), 8.00 (d, J = 8.5 Hz, 1H), 8.03 (s, 1H), 8.39 (br s, 1H); 13C NMR (150 MHz, CDCl3): δC = 21.6, 109.4, 120.3, 123.7, 125.2, 125.4, 127.1, 127.3, 127.9, 128.1, 128.9, 129.0, 129.3, 129.7, 130.4, 131.2, 133.1, 133.4, 136.1, 137.2, 143.7, 146.2; LRMS m/z (ESI−): 504 [M−H]−; HRMS m/z (ESI−): Found 504.1503 [M−H]−; C29H22N5O2S requires 504.1494.
4.11. General method for synthesis of 2-aryl-8-chloro-imidazo[1,2-a]pyrazines, illustrated for the preparation of 8-chloro-2-(2-naphthyl)imidazo[1,2-a]pyrazine 21a
2-(Bromoacetyl)naphthalene 15a (3.14 g, 12.6 mmol), 2-amino-3-chloropyrazine (1.63 g, 12.6 mmol), NaHCO3 (1.32 g, 16.7 mmol) and tBuOH (60 mL) were stirred under reflux for 40 h. The reaction mixture was cooled to room temperature and the solvent removed in vacuo. The resulting orange solid was taken up in H2O (100 mL) and extracted with CH2Cl2 (3 × 150 mL). The combined organic layers were washed with H2O (75 mL) and brine (75 mL), dried (MgSO4), filtered and concentrated in vacuo to give crude orange solid. On addition of CH2Cl2 and MeOH (∼1:1), insoluble material filtered off to give the title compound as a cream fluffy solid (1.05 g). Purification of the remaining filtrate via flash chromatography (applied in petroleum ether; eluted 10% to 20% to 33% EtOAc) afforded the title compound as a pale orange/brown solid (1.38 g, 4.95 mmol, 39%). Mp: Decomposed before melting; Rf = 0.34 (1:1 petroleum ether/EtOAc); IR (νmax/cm−1, thin film): 3026, 2921, 1495; 1H NMR (600 MHz, (CD3)2SO): δH = 7.55–7.58 (m, 2H), 7.76 (d, J = 4.4 Hz, 1H), 7.95–7.97 (m, 1H), 8.04 (d, J = 8.6 Hz, 1H) 8.07–8.09 (m, 1H), 8.16 (dd, J = 8.6, 1.7 Hz, 1H), 8.64 (s, 1H), 8.67 (d, J = 4.4 Hz, 1H), 8.87 (s, 1H,); 13C NMR (150 MHz, CDCl3): δC = 113.7, 120.6, 124.1, 124.9, 126.6, 126.7, 127.7 (2 signals), 128.4, 128.6, 129.9, 133.1, 133.2, 137.5, 141.2, 146.4; LRMS m/z (ESI+): 282 [M(37Cl)+H]+, 280 [M(35Cl)+H]+; HRMS m/z (EI+): Found 279.05574 [M(35Cl)]+; C16H10N3Cl requires 279.05578.
4.12. General method for the synthesis of 4-methyl-N-[4-[2-arylimidazo[1,2-a]pyrazine-8-yl]]-sulfonamides, illustrated for the preparation of 4-methyl-N-[4-[2-(2-naphthyl)imidazo[1,2-a]pyrazine-8-yl]aminophenyl]benzenesulfonamide 11
All glassware was evacuated and flushed with argon prior to use. 21a (50.0 mg, 0.178 mmol), N-(4-aminophenyl)-4-methylbenzenesulfonamide 23 (56.3 mg, 0.215 mmol), NaOtBu (24.1 mg, 0.250 mmol, 1.4 equiv), 1 mol % Pd2(dba)3 (1.6 mg) and 3 mol % DavePhos (2.1 mg) were weighed into a 25 mL round bottom flask. Toluene (2 mL) was added and the reaction was stirred under reflux for 24 h. The reaction mixture was cooled to room temperature and the solvents removed in vacuo. The residue was taken up in CH2Cl2 (30 mL) and washed with water (3 × 30 mL). The combined aqueous extracts were washed with CH2Cl2 (30 mL). The CH2Cl2 layers were combined, washed with brine (30 mL), dried over MgSO4 and the solvents removed in vacuo to give an off-white solid. Flash chromatography (applied in toluene; eluted 3:1 toluene/EtOAc) gave the title compound as an off-white solid (20 mg, 0.039 mmol, 22%). Mp: decomposed before melting; Rf = 0.50 (1:1 petroleum ether/EtOAc); IR (νmax/cm−1, thin film): 3126, 2923, 2853, 1507, 1325, 1143; 1H NMR (600 MHz, CD3OD): δH = 2.42 (s, 3H), 7.20 (d, J = 5.2 Hz, 1H), 7.29 (d, J = 8.9 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 7.52–7.56 (m, 2H), 7.58 (d, J = 8.9 Hz, 2H), 7.75 (d, J = 8.2 Hz, 2H), 7.92 (d, J = 7.1, 1H), 7.96–7.99 (m, 3H), 8.13 (dd, J = 8.5, 1.7 Hz, 1H), 8.53 (s, 1H), 8.56 (s, 1H); 13C NMR (150 MHz, CD3OD): δC = 21.4, 113.9, 115.9 (2 signals), 122.9, 124.9, 125.8, 126.1, 127.6, 127.7, 128.4, 128.9, 129.3, 129.7, 130.7, 131.0, 133.5 (2 signals) 135.0 (2 signals), 137.7, 138.2, 145.2, 146.1, 148.1; LRMS m/z (ESI+): 506 [M+H]+, (ESI−): 504 [M−H]−; HRMS m/z (ESI+): Found 506.1651 [M+H]+; C29H24N5O2S requires 506.1651.
4.13. 8-(Methylsulfonyl)-2-(naphthalen-2-yl)imidazo[1,2-a]pyrazine 24
Compound 21a (1.12 g, 4.01 mmol) was dissolved in anhydrous DMSO (16 mL). NaSMe (337 mg, 4.81 mmol) was added portionwise and the reaction was stirred at 100 °C for 16 h. The mixture was then cooled to room temperature, diluted with brine (50 mL) and extracted with CH2Cl2 (100 mL). The organic layer was washed with H2O (5 × 30 mL) and brine (30 mL), dried (MgSO4), filtered and solvent removed in vacuo. Flash chromatography (applied in CH2Cl2; eluted 0% to 1% to 2% EtOAc) afforded 8-(methylthio)-2-(naphthalen-2-yl)imidazo[1,2-a]pyrazine as an off white/yellow solid (989 mg, 3.40 mmol, 85%). Mp: 168 °C; Rf = 0.47 (5% EtOAc/CH2Cl2); IR (νmax/cm−1, thin film): 3055; 1H NMR (600 MHz, CDCl3): δH = 2.71 (s, 3H), 7.47–7.52 (m, 2H), 7.72 (d, J = 4.5 Hz, 1H), 7.82 (d, J = 4.5 Hz, 1H), 7.85 (d, J = 7.4 Hz, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.94 (d, J = 7.4 Hz, 1H), 7.98 (s, 1H), 8.04 (dd, J = 8.5, 1.6 Hz, 1H), 8.55 (s, 1H); 13C NMR (150 MHz, CDCl3): δC = 12.3, 110.3, 115.1, 124.3, 125.6, 126.4, 126.5, 127.9, 128.5, 128.6, 129.0, 130.2, 133.5 (2 signals), 138.9, 146.4, 154.4; LRMS m/z (ES+): 292 [M+H]+; HRMS m/z (ES+): Found 292.0909 [M+H]+; C17H14N3S requires 292.0908.
8-(Methylthio)-2-(naphthalen-2-yl)imidazo[1,2-a]pyrazine (1.77 g, 6.07 mmol) was dissolved in anhydrous CH2Cl2 (50 mL) and the mixture was cooled on ice. mCPBA (5.23 g, 30.3 mmol) was added in one portion and the reaction continued to stir at room temperature for 5 h. The reaction was partitioned with NaHCO3 (40 mL) and extracted with CH2Cl2 (3 × 30 mL); the combined organic extracts were then washed with brine (30 mL), dried (MgSO4), filtered and concentrated in vacuo. Flash chromatography (applied in CH2Cl2; eluted 2–5% EtOAc) afforded the title compound as a yellow solid (1.10 g, 3.40 mmol, 56.0%). Mp: >200 °C; Rf = 0.22 (10% EtOAc/CH2Cl2); IR (νmax/cm−1, thin film): 3121, 3010, 1312, 1138; 1H NMR (600 MHz, CDCl3): δH = 3.84 (s, 3H), 7.51–7.54 (m, 2H), 7.86–7.87 (m, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.95–7.96 (m, 1H), 8.02 (d, J = 4.3 Hz, 1H), 8.08 (dd, J = 8.5, 1.4 Hz, 1H), 8.26 (s, 1H), 8.32 (d, J = 4.3 Hz, 1H), 8.59 (s, 1H); 13C NMR (150 MHz, CDCl3): δC = 41.7, 110.9, 122.1, 124.3, 126.7, 126.8, 127.0, 127.9, 128.1, 128.7, 128.8, 129.1, 133.5, 134.0, 136.3, 148.6, 149.8; LRMS m/z (EI+): 323 [M]+; HRMS m/z (EI+): Found 323.07266 [M]+; C17H13N3O2S requires 323.07230.
4.14. Conversion of 24 to 4-methyl-N-[2-(2-naphthyl)imidazo[1,2-a]pyrazine-8-yl]benzenesulfonamide 7
NaH (7.1 mg of a 60% suspension in mineral oil, 4.6 mg, 0.186 mmol, 2.0 equiv) was washed by stirring in anhydrous hexane (3 mL), syringing out the solvent and drying. Anhydrous DMF (0.5 mL) was added to the flask under Ar, followed by 4-toluene sulfonamide 22 (31.8 mg, 0.186 mmol, 2.0 equiv) in anhydrous DMF (0.5 mL). The contents were then stirred at rt for 20 min before 24 (30 mg, 0.093 mmol) in DMF (2 mL) was added dropwise, and the reaction was stirred at 100 °C for 20 h. The reaction was cooled to room temperature and quenched with satd aq NH4Cl (30 mL) and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with H2O (5 × 30 mL) and brine, dried (MgSO4), filtered and solvent removed. Flash chromatography (applied in petroleum ether; gradient 20% to 66% EtOAc) afforded 7 (27.8 mg, 0.669 mmol, 72%). Mp: >200 °C; Rf = 0.32 (1:1 CH2Cl2/EtOAc); IR (νmax/cm−1, thin film): 3253; 1H NMR (600 MHz, DMSO-d6): δH = 2.37 (s, 3H), 7.16 (br d, J = 5.2 Hz, 1H), 7.39 (d, J = 8.2 Hz, 2H), 7.51–7.54 (m, 2H), 7.86 (br d, J = 5.2 Hz, 1H), 7.89 (d, J = 8.0 Hz, 2H), 7.92 (d, J = 7.7 Hz, 1H), 7.98 (d, J = 8.6 Hz, 1H), 8.01–8.05 (m, 2H), 8.52 (s, 1H), 8.59 (s, 1H), 11.69 (s, 1H, NH); 13C NMR (150 MHz, DMSO-d6): δC = 21.0, 111.0, 115.3, 116.8, 123.8, 124.2, 126.2, 126.3, 126.6, 127.7, 128.3, 128.4, 129.5, 130.0, 132.8, 133.2, 135.6, 140.0, 142.7, 144.5, 145.3; LRMS m/z (ESI+): 415 [M+H]+, (ESI−): 413 [M−H]−, HRMS m/z (ESI+): Found 415.1219 [M+H]+; C23H19N4O2S requires 415.1229.
4.15. Enzyme activity measurements
Assays for the activity of the HP0525 inhibitors were performed using a colorimetric ATPase assay (Innova Biosciences), see SI for details. The enzymatic reactions were performed in 96-well format for 30 min at 37 °C followed by measuring the absorbance at 620 nm, detecting the presence of inorganic phosphate product. For the IC50 measurements, each reaction contained 100 mM Tris–HCl (pH 7.5), 2.5 mM MgCl2, 125 μM ATP, 5% DMSO, 25 mM NaCl, 0.5 mM DTT 0.053 μM HP0525 and various concentrations of inhibitors (0–50 μM or 0–250 μM). Michaelis–Menten kinetics were performed under the same conditions as above but with various concentrations of ATP ranging from 0 to 500 μM, with and without 10 μM of compound 11. Both IC50 and Michaelis–Menten kinetics measurements were made in triplicate.
Acknowledgments
We thank the BBSRC for a PhD studentship (BBS/SE/2006/1326/9) to JS, a PhD studentship (BBS/K/2005/12145) to F.B., and a Research Grant (BB/D005469/1) to T.P.B. The Wellcome Trust are also thanked for a Research Grant (082227) to K.W. and for a PhD studentship (096617/Z/11/Z) to H.K. We would also like to thank UCLB for HEIF Proof of Concept Funding (T.P., F.C., P.G. and M.S.) and BioFocus for access to their SoftFocus library.
Footnotes
Supplementary data (experimental details for the synthesis of compounds 1–4, 6–10, 12, 13, 25–38, details of the optimization of the conversion of 21a to 7, NMR data confirming the regiochemistry of 21a, 1H NMR spectra for all novel compounds, full description of the ATPase assay, IC50 curves for all compounds) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2014.09.036.
Supplementary data
Experimental details for the synthesis of compounds 1–4, 6–10, 12, 13, 25–38, details of the optimization of the conversion of 21a to 7, NMR data confirming the regiochemistry of 21a, 1H NMR spectra for all novel compounds, full description of the ATPase assay, IC50 curves for all compounds.
References and notes
- 1.Gerlach R.G., Hensel M. Int. J. Med. Microbiol. 2007;297:401. doi: 10.1016/j.ijmm.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 2.Baron C. Curr. Opin. Microbiol. 2010;13:100. doi: 10.1016/j.mib.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Wallden K., Rivera-Calzada A., Waksman G. Cell. Microbiol. 2010;12:1203. doi: 10.1111/j.1462-5822.2010.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selbach M., Moese S., Backert S., Jungblut P.R., Meyer T. Proteomics. 2004;4:2961. doi: 10.1002/pmic.200400915. [DOI] [PubMed] [Google Scholar]
- 5.Christie P.J., Vogel J.P. Trends Microbiol. 2000;8:354. doi: 10.1016/s0966-842x(00)01792-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fronzes R., Christie P.J., Waksman G. Nat. Rev. Microbiol. 2009;7:703. doi: 10.1038/nrmicro2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paschos A., den Hartigh A., Smith M.A., Atluri V.L., Sivanesan D., Tsolis R.M., Baron C. Infect. Immun. 2011;1033:79. doi: 10.1128/IAI.00993-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savvides S.N., Yeo H.-J., Beck M.R., Blaesing F., Lurz R., Lanka E., Buhrdorf R., Fischer W., Haas R., Waksman G. EMBO J. 1969;2003:22. doi: 10.1093/emboj/cdg223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeo H.-J., Savvides S.N., Herr A.B., Lanka E., Waksman G. Mol. Cell. 2000;6:1461. doi: 10.1016/s1097-2765(00)00142-8. [DOI] [PubMed] [Google Scholar]
- 10.Hare S., Fischer W., Williams R., Terradot L., Bayliss R., Haas R., Waksman G. EMBO J. 2007;26:4926. doi: 10.1038/sj.emboj.7601904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skedelj V., Tomasic T., Masic L.P., Zega A. J. Med. Chem. 2011;54:915. doi: 10.1021/jm101121s. [DOI] [PubMed] [Google Scholar]
- 12.Hilleringmann M., Pansegrau W., Doyle M., Kaufman S., MacKichan M.L., Gianfaldoni G., Ruggiero P., Covacci A. Microbiology. 2006;152:2919. doi: 10.1099/mic.0.28984-0. [DOI] [PubMed] [Google Scholar]
- 13.Lang P.T., Brozell S.R., Mukherjee S., Pettersen E.F., Meng E.C., Thomas V., Rizzo R.C., Case D.A., James T.L., Kuntz I.D. RNA. 2009;15:1219. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sablayrolles C., McNeill J.H. J. Med. Chem. 1984;27:206. doi: 10.1021/jm00368a018. [DOI] [PubMed] [Google Scholar]
- 15.Bradac J., Furek Z. J. Org. Chem. 1977;42:4197. [Google Scholar]
- 16.Bonnet P.A., Chapat J.P. J. Med. Chem. 1992;35:3353. doi: 10.1021/jm00096a008. [DOI] [PubMed] [Google Scholar]
- 17.MacCoss M., Meurer L.C. J. Heterocycl. Chem. 1993;30:1213. [Google Scholar]
- 18.Koufaki M., Kiziridi C., Nikoloudaki F., Alexis M.N. Bioorg. Med Chem. Lett. 2007;17:4223. doi: 10.1016/j.bmcl.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 19.Shang G., Liu D., Allen S.E., Yang Q., Zhang X. Chem. Eur. J. 2007;13:7780. doi: 10.1002/chem.200700594. [DOI] [PubMed] [Google Scholar]
- 20.Benaissa T., Hamman S., Beguin C.G. J. Fluorine Chem. 1998;38:163. [Google Scholar]
- 21.Kamal A., Shaik A.A., Sandbhor M., Malik M.S. Tetrahedron: Asymmetry. 2004;15:3939. [Google Scholar]
- 22.Omura K., Swern D. Tetrahedron. 1978;34:1651. [Google Scholar]
- 23.Matthews T.P., McHardy T., Klair S., Boxall K., Fisher M., Cherry M., Allen C.E., Addison G.J., Ellard J., Aherne G.W., Westwood I.M., van Montfort R., Garrett M.D., Reader J.C., Collins I. Bioorg. Med. Chem. Lett. 2010;20:4045. doi: 10.1016/j.bmcl.2010.05.096. [DOI] [PubMed] [Google Scholar]
- 24.Belanger D.B., Curran P.J., Hruza A., Voigt J., Meng Z., Mandal A.K., Siddiqui M.A., Basso A.D., Gray K. Bioorg. Med. Chem. Lett. 2010;20:5170. doi: 10.1016/j.bmcl.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 25.Bouloc N., Large J.M., Kosmopoulou M., Sun C., Faisal A., Matteucci M., Reynisson J., Brown N., Atrash B., Blagg J., McDonald E., Linardopoulos S., Bayliss R., Bavetsias V. Bioorg. Med. Chem. Lett. 2010;20:5988. doi: 10.1016/j.bmcl.2010.08.091. [DOI] [PubMed] [Google Scholar]
- 26.Adib M., Sheibani E., Bijanzadeh H.R., Zhu L.-G. Tetrahedron. 2008;64:10681. [Google Scholar]
- 27.Surry D.S., Buchwald S.L. Chem. Sci. 2011;2:27. doi: 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchwald S.L., Ikawa T., Barder T.E., Biscoe M.R. J. Am. Chem. Soc. 2007;129:13001. doi: 10.1021/ja0717414. [DOI] [PubMed] [Google Scholar]
- 29.Bhagwanth S., Waterson A.G., Adjabeng G.M., Hornberger K.R. J. Org. Chem. 2009;74:4634. doi: 10.1021/jo9004537. [DOI] [PubMed] [Google Scholar]
- 30.Driver M.A., Hartwig J.F. J. Am. Chem. Soc. 1996;118:7217. [Google Scholar]
- 31.Burton G., Cao P., Li G., Rivero R. Org. Lett. 2003;5:4373. doi: 10.1021/ol035655u. [DOI] [PubMed] [Google Scholar]
- 32.Old D.W., Wolfe J.P., Buchwald S.L. J. Am. Chem. Soc. 1998;120:9722. [Google Scholar]
- 33.Voss M.E., Rainka M.P., Fleming M., Peterson L.H., Belanger D.B., Siddiqui M.A., Hruza A., Voigt J., Gray K., Basso A.D. Bioorg. Med. Chem. Lett. 2012;22:3544. doi: 10.1016/j.bmcl.2012.03.051. [DOI] [PubMed] [Google Scholar]
- 34.Tetko I.V., Gasteiger J., Todeschini R., Mauri A., Livingstone D., Ertl P., Palyulin V.A., Radchenko E.V., Zefirov N.S., Makarenko A.S., Tanchuk V.Y., Prokopenko V.V. J. Comput. Aid. Mol. Des. 2005;19:453. doi: 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- 35.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Adv. Drug Deliv. Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 36.Elliott T.S., Slowey A., Ye Y., Conway S.J. Med. Chem. Commun. 2012;3:735. [Google Scholar]
- 37.Trott O., Olson A.J. J. Comput. Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. J. App. Cryst. 1993;26:283. [Google Scholar]
- 39.MOE, Chemical Computing Group Inc., Montreal, Canada.
- 40.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. J. Comput. Chem. 2005;26:1781. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J., Wolf R.M., Caldwell J.W., Kollmann P.A., Case D.A. J. Comput. Chem. 2004;25:1157. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 42.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Goodsell D.S., Olson A.J. J. Comput. Chem. 2009;30:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moumne R., Larue V., Seijo B., Lecourt T., Micouin L., Tisne C. Org. Biomol. Chem. 2010;8:1154. doi: 10.1039/b921232a. [DOI] [PubMed] [Google Scholar]
- 44.Cho B.T., Kang S.K., Shin S.H. Tetrahedron: Asymmetry. 2002;13:1209. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details for the synthesis of compounds 1–4, 6–10, 12, 13, 25–38, details of the optimization of the conversion of 21a to 7, NMR data confirming the regiochemistry of 21a, 1H NMR spectra for all novel compounds, full description of the ATPase assay, IC50 curves for all compounds.