

See Fagan (doi:10.1093/awu387) for a scientific commentary on this article.
CSF and PET biomarkers of β-amyloid are not always fully congruent. Mattsson et al. reveal that the markers provide partly independent information in Alzheimer's disease. CSF biomarkers may be more sensitive to early stages of disease pathogenesis, while PET imaging may reflect downstream pathology.
Keywords: Alzheimer's disease, biomarker, cerebrospinal fluid, positron emission tomography, amyloid
Abstract
Reduced cerebrospinal fluid amyloid-β42 and increased retention of florbetapir positron emission tomography are biomarkers reflecting cortical amyloid load in Alzheimer's disease. However, these measurements do not always agree and may represent partly different aspects of the underlying Alzheimer's disease pathology. The goal of this study was therefore to test if cerebrospinal fluid and positron emission tomography amyloid-β biomarkers are independently related to other Alzheimer's disease markers, and to examine individuals who are discordantly classified by these two biomarker modalities. Cerebrospinal fluid and positron emission tomography amyloid-β were measured at baseline in 769 persons [161 healthy controls, 68 subjective memory complaints, 419 mild cognitive impairment and 121 Alzheimer's disease dementia, mean age 72 years (standard deviation 7 years), 47% females] and used to predict diagnosis, APOE ε4 carriage status, cerebral blood flow, cerebrospinal fluid total-tau and phosphorylated-tau levels (cross-sectionally); and hippocampal volume, fluorodeoxyglucose positron emission tomography results and Alzheimer's Disease Assessment Scale-cognitive subscale scores (longitudinally). Cerebrospinal fluid and positron emission tomography amyloid-β were highly correlated, but adjusting one of these predictors for the other revealed that they both provided partially independent information when predicting diagnosis, APOE ε4, hippocampal volume, metabolism, cognition, total-tau and phosphorylated-tau (the 95% confidence intervals of the adjusted effects did not include zero). Cerebrospinal fluid amyloid-β was more strongly related to APOE ε4 whereas positron emission tomography amyloid-β was more strongly related to tau levels (P < 0.05). Discordance (mainly isolated cerebrospinal fluid amyloid-β positivity) differed by diagnostic group (P < 0.001) and was seen in 21% of cognitively healthy people but only 6% in dementia patients. The finding that cerebrospinal fluid and positron emission tomography amyloid-β provide partially independent information about a wide range of Alzheimer's measures supports the theory that these modalities represent partly different aspects of Alzheimer's pathology. The fact that mismatch, with positive cerebrospinal fluid amyloid-β but normal positron emission tomography amyloid-β, is relatively common in cognitively healthy people may be considered when using these biomarkers to identify early stage Alzheimer's disease. Reduced cerebrospinal fluid amyloid-β may be more strongly related to early stage Alzheimer's disease, whereas increased positron emission tomography amyloid-β may be more strongly related to disease progression.
Introduction
Reduced CSF amyloid-β42 (amyloid-β1–42, referred to as CSF amyloid-β) (Blennow et al., 2010) and increased uptake of amyloid tracers visualized by PET (referred to as PET amyloid-β) (Klunk et al., 2004) are used as in vivo measurements of brain amyloid-β accumulation. This is a key feature of Alzheimer's disease and biomarker evidence of brain amyloid-β have been included in Alzheimer's disease research criteria (Albert et al., 2011; McKhann et al., 2011; Sperling et al., 2011; Dubois et al., 2014).
Several studies have shown that low CSF, and high PET amyloid-β are correlated (Fagan et al., 2006, 2009; Forsberg et al., 2008; Koivunen et al., 2008; Grimmer et al., 2009; Tolboom et al., 2009), and they are often interchangeable for the purpose of diagnosing Alzheimer's disease dementia (Mattsson et al., 2014; Zwan et al., 2014). These biomarkers are therefore often assumed to provide the same information. In this study, we investigated the alternative theory that CSF and PET amyloid-β provide partly different and independent information. There are several reasons to suspect this. First, PET amyloid-β is directly related to brain fibrillar amyloid-β (Ikonomovic et al., 2008), whereas CSF amyloid-β is a marker of soluble amyloid-β and only indirectly related to fibrillar amyloid-β. In addition to fibrillar deposits, CSF amyloid-β may also be affected by variations in amyloid precursor protein-processing and amyloid-β production (May et al., 2011; Mattsson et al., 2012; Reiman et al., 2012; Potter et al., 2013) and non-fibrillar aggregation (Cairns et al., 2009; Schöll et al., 2012), and is for unknown reasons reduced in other medical conditions where plaques are not present, including neuroinflammation (Mattsson et al., 2009; Augutis et al., 2013), Creuzfeldt-Jakob's disease and amyotrophic lateral sclerosis (Blennow and Hampel, 2003). Second, although dichotomous CSF and PET amyloid-β classification often agree (Mattsson et al., 2014; Zwan et al., 2014), they classify a proportion of people discordantly (up to 25%, higher numbers in cognitively healthy people) (Cairns et al., 2009; Fagan et al., 2009; Schöll et al., 2012; Landau et al., 2013). Third, the overall correlation between CSF and PET amyloid-β is modest (Landau et al., 2013) and the correlation is very poor within the positive (low CSF, and high PET amyloid-β) and negative (high CSF amyloid-β and low PET amyloid-β) biomarker ranges. Fourth, it has been suggested that CSF amyloid-β is reduced before an increase in PET amyloid-β (Fagan et al., 2009), although not all studies have supported this (Landau et al., 2013).
If CSF and PET amyloid-β partly reflect different aspects of amyloid metabolism—which is a core aspect of Alzheimer's disease pathophysiology (Hardy and Selkoe, 2002)—they may be expected to provide partly independent information in Alzheimer's disease studies. We therefore tested the specific hypothesis that CSF and PET amyloid-β are independent predictors of other Alzheimer's disease measurements and features, including APOE ε4 carriage status, Alzheimer's disease dementia diagnosis, cognitive deficits, hippocampal atrophy, brain hypometabolism, brain hypoperfusion, and increased levels of CSF tau biomarkers. We also tested if CSF and PET amyloid-β were differently related to these outcomes (suggesting that the amyloid-β biomarkers had different sensitivity to pathological changes during the disease course). Finally, in an exploratory analysis, we tested if a categorical classification using both CSF and PET amyloid-β (classifying subjects as concordant positive, discordant, or concordant negative) was associated with different outcomes, and if the frequency of discordant classification differed by disease stage.
Materials and methods
Study design
The objective of this study was to compare CSF and PET amyloid-β to predict several different biomarkers and features of Alzheimer's disease. Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.ucla.edu). The Principal Investigator of this initiative is Michael W. Weiner, MD, VA Medical Centre and University of California San Francisco. ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the USA and Canada. The data used in this study included all subjects acquired in ADNI-2 with available baseline data on CSF and PET amyloid-β. For up-to-date information, see http://www.adni-info.org.
Cohort
Our study population consisted of cognitively healthy controls (cognitively normal) and people with subjective memory complaints (SMC), early mild cognitive impairment (MCI), late MCI (late MCI) or Alzheimer's disease dementia. Inclusion/exclusion criteria are described in detail at http://www.adni-info.org. Briefly, all subjects included in ADNI-2 were between the ages of 55 and 90 years, had completed at least 6 years of education, were fluent in Spanish or English, and were free of any significant neurological disease other than Alzheimer's disease. Cognitively normal subjects had Mini-Mental State Examination score ≥24, and Clinical Dementia Rating scale score 0. SMC subjects had normal test results but subjective memory impairment. Subjects with early MCI had Mini-Mental State Examination score ≥ 24, objective memory loss as shown on scores on delayed recall of the Wechsler Memory Scale Logical Memory II [>0.5 standard deviations (SD) below the normal mean], Clinical Dementia Rating scale 0.5, preserved activities of daily living, and absence of dementia. Late MCI subjects fulfilled early MCI criteria but had worse delayed recall (>1 SD below the normal mean). Patients with Alzheimer's disease dementia fulfilled the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria for probable Alzheimer's disease, had Mini-Mental State Examination scores between 20–26 and a Clinical Dementia Rating scale of 0.5 or 1.0.
CSF biomarkers
All subjects underwent CSF sampling at baseline. Amyloid-β42, total tau and phosphorylated tau were measured using the multiplex xMAP® Luminex platform (Luminex Corp) with the INNOBIA AlzBio3 kit (Innogenetics) as described previously (Olsson et al., 2005; Shaw et al., 2009). When indicated, subjects were classified as amyloid-β-positive or -negative using a previously defined cut-off (CSF amyloid-β42 < 192 ng/l), which has been shown to be discriminative of brain amyloid-β pathology in both PET imaging (Weigand et al., 2011) and two independent autopsy studies (Shaw et al., 2009; De Meyer et al., 2010). For this study, we used the data files ‘UPENNBIOMK5_10_31_13.csv’, ‘UPENNBIOMK6_07_02_13.csv’ and ‘UPENNBIOMK7.csv’.
Florbetapir PET
All subjects underwent florbetapir PET imaging at baseline. Images were acquired at baseline and processed as described previously (Landau et al., 2012). Images were acquired 50 to 70 min post-injection, reconstructed immediately following the scan, and repeat scans were acquired if motion artefacts were detected. For quantification of florbetapir, 3 T 3D magnetization prepared rapid gradient echo (MPRAGE) MRI scans were used. MRI images were segmented and parcellated into individual cortical regions with FreeSurfer, and used to extract mean florbetapir uptake (standardized uptake value ratio) from grey matter within lateral and medial frontal, anterior and posterior cingulate, lateral parietal, and lateral temporal regions relative to uptake in the whole cerebellum (white and grey matter). The overall cortical mean standardized uptake value ratio was used in this study. When indicated, subjects were classified as PET amyloid-β-positive or -negative using a previously defined cut-off (>1.11 standardized uptake value ratio) (Clark et al., 2011; Joshi et al., 2012; Landau et al., 2012).
Cognition
We used the Alzheimer's Disease Assessment Scale-cognitive subscale (ADAS-cog11) to assess cognitive function (numbers in brackets indicate data points in each diagnostic group, also presented below for other measurements) at baseline (cognitively normal 161, SMC 68, early MCI 269, late MCI 150, Alzheimer's disease 121) and follow-up at 0.5 (cognitively normal 155, SMC 62, early MCI 246, late MCI 147, Alzheimer's disease 109), 1 (cognitively normal 152, SMC 15, early MCI 254, late MCI 141, Alzheimer's disease 91), 2 (cognitively normal 131, early MCI 193, late MCI 96, Alzheimer's disease 29) and 3 years (cognitively normal 4, early MCI 86).
Fluorodeoxyglucose PET
FDG image data were acquired at baseline (cognitively normal 161, SMC 67, early MCI 268, late MCI 150, Alzheimer's disease 119) and after 2 years (cognitively normal 80, early MCI 125, late MCI 39, Alzheimer's disease 13) and processed as described previously (Landau et al., 2012). Images were acquired 30 to 60 min post-injection. The average FDG-PET from three regions (temporal regions, lateral parietal regions, and posterior cingulate cortex) was used in this study. Data were expressed relative to the mean in a reference region (pons and cerebellar vermis) (Landau et al., 2011).
Structural MRI
Structural MRI brain scans were acquired at baseline (cognitively normal 156, SMC 65, early MCI 261, late MCI 145, Alzheimer's disease 115) and follow-up at 0.25 (cognitively normal 133, SMC 31, early MCI 248, late MCI 140, Alzheimer's disease 90), 0.5 (cognitively normal 139, SMC 6, early MCI 221, late MCI 134, Alzheimer's disease 71), 1 (cognitively normal 127, SMC 5, early MCI 218, late MCI 118, Alzheimer's disease 60), 2 (cognitively normal 80, early MCI 137, late MCI 46, Alzheimer's disease 14) and 3 (cognitively normal 1, early MCI 37) years using 3 T MRI scanners with a standardized protocol including T1-weighted MRI scans using a sagittal volumetric MPRAGE sequence (Jack et al., 2008). Data on hippocampal volume (averaged right and left) were used in this study. Quantification was done in an automated pipeline using FreeSurfer software package version 5.1 (http://surfer.nmr.mgh.harvard.edu/fswiki) (Fischl et al., 2002, 2004).
Arterial spin labelling MRI
Cerebral blood flow was measured by arterial spin labelling MRI at baseline (cognitively normal 39, SMC 9, early MCI 61, late MCI 40, Alzheimer's disease 22) and processed as described online at www.loni.usc.edu and as reported previously (Mattsson et al., 2014). In short, arterial spin labelling MRI was performed on 3 T MRI machines using a pulsed method with echo-planar imaging (Luh et al., 1999). All ASL images were processed using a largely automated pipeline which included motion correction, computation of perfusion weighted images, co-registration to structural MRI and correction for partial grey/white matter volume effects. In this study, we used data on temporal cerebral blood flow (a combination of cerebral blood flow from all temporal lobe FreeSurfer regions, averaged between right and left hemisphere), as we previously found associations between amyloid-β and several temporal lobe regions (Mattsson et al., 2014).
Statistical analysis
Continuous CSF and PET amyloid-β were used as predictors of diagnosis, APOE ε4 carriage status, CSF total tau, CSF phosphorylated tau, cerebral blood flow, hippocampal volume, FDG-PET and ADAS-cog. Three models were generated for each response, using CSF amyloid-β, PET amyloid-β, or CSF and PET amyloid-β as predictors. Non-parametric 95% confidence intervals (CI) of coefficients were estimated from the percentiles of bootstrap samples. A 95% CI that excluded zero when CSF or PET amyloid-β was adjusted for the other modality indicated a partially independent association with the response. Because CSF and PET amyloid-β were correlated we used ridge regression, which provides stable estimates of coefficients of correlated predictors (Friedman et al., 2009). To test if the combination of CSF and PET amyloid-β improved model fits, we also reported the Akaike information criterion (AIC). AIC is a measure of the relative quality of a statistical model for a given set of data, and provides a means for model selection. AIC is defined as 2 k − 2 ln (L), where k is the number of parameters and L is the maximized value of the likelihood function for the model. A lower AIC represents a better fit (penalized for the number of predictors to avoid over-fitting). A difference in AIC (ΔAIC) of 0–2, 4–7, and >10 represent some evidence, considerable evidence, and very strong evidence, respectively, for differences between two models (Burnham and Anderson, 2002). All analyses were done first on all subjects and then separately on subjects within the concordant ‘positive’ (CSF amyloid-β42 < 192 and florbetapir PET > 1.11 standardized uptake value ratio) and ‘negative quadrants’. Except for models of diagnosis (cognitively normal versus Alzheimer's disease), all analyses were done on all diagnoses combined.
For all analyses described above, CSF and PET amyloid-β were used as continuous data. In a second step we combined the two amyloid-β modalities into four categories by dichotomizing CSF and PET amyloid-β data using a priori cut-offs and combining them into concordant negative (CSF−PET−), discordant with CSF positivity (CSF+PET−), discordant with PET positivity (CSF−PET+) and concordant positive (CSF+PET+). The chi-square test was used to test if the proportion of discordance differed by diagnostic group. We repeated the regression analyses described above using the categorical predictor, with CSF−PET− as the reference category. This was done within the diagnostic groups. These exploratory analyses were corrected for multiple comparisons using false discovery rate correction.
Logistic regression was used for diagnosis and APOE ε4 (cross-sectional dichotomous data). Ordinary least square regression was used for cerebral blood flow, CSF phosphorylated tau and total tau (cross-sectional continuous data). Linear mixed effects was used for hippocampal volume, FDG-PET and ADAS-cog (longitudinal continuous data). All models were adjusted for age and sex. Additionally, temporal cerebral blood flow was adjusted for cerebral blood flow in a reference region (precentral cerebral blood flow), hippocampal volume was adjusted for total intracranial volume, and ADAS-cog was adjusted for education (years). All linear mixed effects models included the interaction terms Time × CSF amyloid-β and/or Time × PET amyloid-β, the corresponding main effects, and random intercepts and slopes. Mixed effects models were fit using the maximum likelihood criterion, as this is preferred for model comparison (Bates, 2010) but the overall results were similar when refitting the models with the restricted maximum likelihood estimation criterion.
The model assumptions were assessed by evaluating normality and homoscedasticity of residuals with q-q plots and plots of residuals versus fitted values. Estimates from the full sample were compared to bootstrapped estimates. To test if variables were related to study drop-out (missing data for ADAS-cog, hippocampal volume and FDG-PET) we used a generalized mixed effects model with a binomial response, with age, sex, education, CSF amyloid-β, and PET amyloid-β as predictors of ‘missing data’ [a missing indicator (true/false) for each study visit]. All tests were two-sided, and significance was determined at P < 0.05. All statistics were done using R (v.3.0.1, The R Foundation for Statistical Computing).
Results
Study demographics are shown in Table 1. As explained above, the available follow-up data varied for longitudinal MRI, FDG-PET and ADAS-cog. Amyloid-β measures were not associated with missing data, except for an association between high PET amyloid-β and missing longitudinal MRI data (P = 0.039). This was driven mainly by the patients with Alzheimer's disease dementia, who had high PET amyloid-β levels and relatively short follow-up. Thus, the effects of PET amyloid-β on longitudinal atrophy might be slightly underestimated, although mixed-effect models are robust to this type of covariate associated missing data (Little and Rubin, 2002).
Table 1.
Study demographics
| Group | n | Sex (M/F) | Age (y) | Education (y) | APOE ε4 (−/+) |
|---|---|---|---|---|---|
| Cognitively normal | 161 | 78/83 (52%) | 74 (6) | 16.6 (2.5) | 117/43 (27%) |
| SMC | 68 | 26/42 (62%) | 72 (6) | 16.6 (2.6) | 46/20 (30%) |
| Early MCI | 269 | 153/116 (43%) | 71 (7) | 16.0 (2.7) | 154/114 (43%) |
| Late MCI | 150 | 78/72 (48%) | 72 (8) | 16.7 (2.6) | 63/87 (58%) |
| Alzheimer's disease dementia | 121 | 73/48 (40%) | 75 (9) | 15.8 (2.6) | 39/82 (68%) |
| All | 769 | 408/361 (47%) | 72 (7) | 16.2 (2.6) | 419/346 (45%) |
Data for continuous measurements are mean (SD).
CSF and PET amyloid-β as predictors of diagnosis, cognition and biomarkers
CSF and PET amyloid-β provided partially independent information for prediction of Alzheimer's disease diagnosis, APOE ε4, high CSF total tau and phosphorylated tau, small hippocampal volume, hypometabolism and low ADAS-cog. For cerebral blood flow, only CSF amyloid-β was an independent predictor. Figure 1 shows coefficients for all outcomes (log-odds ratios for binary outcomes, diagnosis and APOE ε4). For interpretation, note that CSF and PET amyloid-β and all continuous responses were standardized (centred and scaled), and that the sign of CSF amyloid-β was changed to facilitate comparisons between amyloid-β modalities. Therefore, a positive coefficient indicates a positive association between high amyloid-β load (by any modality) and the response of interest.
Figure 1.

Effects of CSF amyloid-β42 and florbetapir PET on diagnosis, cognition and biomarkers. The graph shows the effects (regression coefficients) of CSF amyloid-β (circles) and PET amyloid-β (triangles) to predict different Alzheimer's disease features. For Alzheimer's disease diagnosis we compared Alzheimer's disease dementia versus cognitively normal. For APOE ε4 we compared subjects with the APOE ε4 allele versus subjects without the APOE ε4 allele. For cerebral blood flow (CBF), ADAS-cog, hippocampal volume and FDG-PET we used these parameters as continuous outcomes. Coefficients are shown both for unadjusted models (black, closed symbols) and for models adjusted (red, open symbols) for the other modality. The error bars are 95% CI. If the adjusted 95% CI excluded zero it indicated a significant independent effect for that modality on the response. The relative change when adjusting for the other modality is shown on the right y-axis. All measures were scaled (centred around the mean and standardized) and CSF amyloid-β was polarized to facilitate comparisons with PET amyloid-β. Top: Cross-sectional responses (including baseline ADAS-cog, hippocampal volume and FDG-PET); Bottom: Longitudinal responses. Note that the scales are different for cross-sectional and longitudinal responses.
To interpret the results for binary outcomes, consider for example PET amyloid-β and Alzheimer's disease diagnosis. The adjusted log-odds ratio was 0.9 (95% CI 0.8 to 1.1), indicating that (i) high PET amyloid-β was associated with increased risk of Alzheimer's disease dementia (the coefficient was positive); (ii) high PET amyloid-β provided information that was partially independent from CSF amyloid-β (the 95% CI adjusted for CSF amyloid-β, did not include zero); and (iii) the risk (log-odds ratio) of Alzheimer's disease dementia increased on average 0.9 per standard deviation increase in PET amyloid-β. Exponentiation of the log-odds ratio gives the odds ratio (OR = e0.9) 2.5, which can be interpreted as a 150% increase in the odds for having Alzheimer's disease dementia for each standard deviation increase in PET amyloid-β. To interpret the results for continuous outcomes, consider for example CSF amyloid-β and baseline hippocampal volume. The adjusted β-coefficient was −0.17 (95% CI −0.23 to −0.11), indicating that (i) low CSF amyloid-β (recall the sign of CSF amyloid-β was changed) was associated with small baseline hippocampi; (ii) low CSF amyloid-β provided partially independent information (the 95% CI adjusted for PET amyloid-β, did not include zero); and (iii) the mean hippocampal volume was 0.17 SD smaller for each standard deviation decrease in CSF amyloid-β.
These analyses were done without adjusting for diagnostic group. When adjusting for diagnosis the effects of CSF and PET amyloid-β were slightly reduced as expected, because diagnosis is associated with amyloid-β. However, CSF and PET amyloid-β remained partially independent predictors of APOE ε4, CSF total tau, CSF phosphorylated tau, hippocampal volume (at baseline and over time), ADAS-cog (at baseline and over time) and FDG-PET (at baseline) (Supplementary Fig. 1).
Change in effects of CSF and PET amyloid-β when combining them as predictors
Figure 1 also shows the relative change in coefficients when adjusting CSF and PET amyloid-β for each other (right y-axis). The adjusted coefficients were always reduced, as CSF and PET amyloid-β partly provide the same information. However, the magnitude of the changes differed markedly between modalities and responses. For example, for APOE ε4, the PET amyloid-β coefficient was reduced 52% by adjusting for CSF amyloid-β, while the CSF amyloid-β coefficient was reduced 25% by adjusting for PET amyloid-β, therefore the difference in reduction was 27% (95% CI 3.3 to 49), indicating a stronger relationship between CSF amyloid-β and APOE ε4. Interpreting the data this way, CSF amyloid-β was more closely related to APOE ε4; and PET amyloid-β was more closely related to CSF total tau (difference 50%, 95% CI 25 to 78), phosphorylated tau (37%, 95% CI 15 to 64) and ADAS-cog (baseline 24%, 95% CI −5.4 to 53; slope 25%, 95% CI −2.5 to 53; and the two modalities were about equally related to diagnosis (14%, 95% CI −12 to 45), hippocampal volume (baseline 0%, 95% CI −30 to 37; slope 9%, 95% CI −22 to 45) and FDG-PET (baseline 9%, 95% CI −22 to 41; slope −5%, 95% CI −146 to 108). For cerebral blood flow, the CSF amyloid-β coefficient was only reduced by 4% whereas the PET amyloid-β coefficient was reduced by 91%, giving a difference in reduction of 87%, but the 95% CI for this difference was wide (−113 to 550).
Improved model fits when combining CSF and PET amyloid-β as predictors
The left columns of Table 2 show AIC for all models applied to subjects in all biomarker quadrants, and ΔAIC for comparisons of models with the same responses. Combining the two amyloid-β modalities was preferable for most responses (ΔAIC ≥ 4), and there was very strong support (ΔAIC ≥ 10) for the combination to predict APOE ε4, hippocampal volume, ADAS-cog and CSF phosphorylated tau.
Table 2.
CSF amyloid-β42 and florbetapir PET to predict diagnosis, cognition and biomarkers
| All amyloid-β quadrants |
Positive amyloid-β quadrant |
||||
|---|---|---|---|---|---|
| Test | Amyloid-β biomarker | AIC | ΔAIC | AIC | ΔAIC |
| Diagnosis (cognitively normal versus Alzheimer's disease) | CSF | 285 | 27 | 168 | 10 |
| PET | 266 | 8 | 160 | 2 | |
| CSF and PET | 258 | 0 | 158 | 0 | |
| APOE ε4 carriage status | CSF | 840 | 12 | 449 | −2 |
| PET | 889 | 61 | 465 | 14 | |
| CSF and PET | 828 | 0 | 451 | 0 | |
| Hippocampal volume | CSF | 1289 | 23 | 945 | 16 |
| PET | 1280 | 14 | 942 | 13 | |
| CSF and PET | 1266 | 0 | 929 | 0 | |
| FDG-PET | CSF | 2422 | 16 | 1232 | 2 |
| PET | 2414 | 8 | 1228 | −2 | |
| CSF and PET | 2406 | 0 | 1230 | 0 | |
| ADAS-cog | CSF | 4399 | 58 | 2244 | 25 |
| PET | 4354 | 13 | 2229 | 10 | |
| CSF and PET | 4341 | 0 | 2219 | 0 | |
| CSF total tau | CSF | 1940 | 97 | 1017 | 13 |
| PET | 1848 | 5 | 1008 | 4 | |
| CSF and PET | 1843 | 0 | 1004 | 0 | |
| CSF phosphorylated tau | CSF | 1978 | 105 | 1063 | 16 |
| PET | 1889 | 16 | 1050 | 3 | |
| CSF and PET | 1873 | 0 | 1047 | 0 | |
| Cerebral blood flow | CSF | 312 | −2 | 92 | 1 |
| PET | 316 | 2 | 95 | 4 | |
| CSF and PET | 314 | 0 | 91 | 0 | |
The table shows AIC for models with only CSF amyloid-β, only PET amyloid-β, or both CSF amyloid-β, only PET amyloid-β as predictors. AIC is a measure of model fit and is penalized for including additional predictors (it thereby protects against overfitting). A small AIC is preferable when comparing models (but the absolute AIC cannot be compared between models with different data). The smallest AIC for each response is indicated in bold in the table. For each model, the ΔAIC is the difference between that model's AIC and the AIC of the model when including both CSF and PET amyloid-β as predictors. ΔAIC 0–2, 4–7, and >10 represent some evidence, considerable evidence, and very strong evidence, respectively, for difference between two models (Burnham and Anderson, 2002). For example, there was very strong evidence that the combination of CSF and PET amyloid-β provided a better model for ADAS-cog than any of the individual amyloid-β biomarkers. The table shows results for models built on all data (left columns) and models restricted to subjects in the ‘positive quadrant’ (right columns, CSF amyloid-β42 < 192 ng/l and florbetapir PET > 1.11 standardized uptake value ratio).
Effects of CSF and PET amyloid-β in the positive biomarker range
We repeated the analyses within the ‘positive biomarker quadrant’ (CSF amyloid-β42 < 192 ng/l and florbetapir PET > 1.11 standardized uptake value ratio). Here, PET amyloid-β was no longer an independent predictor of APOE ε4, but the two amyloid-β modalities were partially independent predictors of diagnosis, CSF total tau, CSF phosphorylated tau, hippocampal volume (baseline and follow-up), ADAS-cog (baseline and follow-up), and FDG-PET (baseline) (Supplementary Fig. 2). As seen in Table 2 the AIC analyses demonstrated that the combination of the two amyloid-β modalities was preferred before either measure alone to predict hippocampal volume, ADAS-cog and total tau within the ‘positive biomarker quadrant’ (ΔAIC ≥ 4). The relative change when adjusting CSF and PET amyloid-β for each other did not differ between modalities (comparing β-ratios on the secondary y-axis of Supplementary Fig. 2).
Effects of CSF and PET amyloid-β in the negative biomarker range
Within the ‘negative quadrant’ (CSF amyloid-β42 > 192 ng/l and florbetapir PET < 1.11 standardized uptake value ratio) there were no significant effects of amyloid-β except that low CSF amyloid-β was associated with high CSF total tau (β = −0.36, P < 0.0001) and phosphorylated tau (β = −0.24, P < 0.0001).
Differences between amyloid-β groups in brain structure, cognition and CSF tau
When examining concordant negative, discordant and concordant positive individuals, we found that 21% of cognitively normal/SMC, 13% of early MCI, 11% of late MCI and 6% of Alzheimer's disease dementia patients were discordant (Fig. 2). Most discordant subjects had isolated low CSF amyloid-β. Concordance increased significantly with disease stage (P < 0.001).
Figure 2.
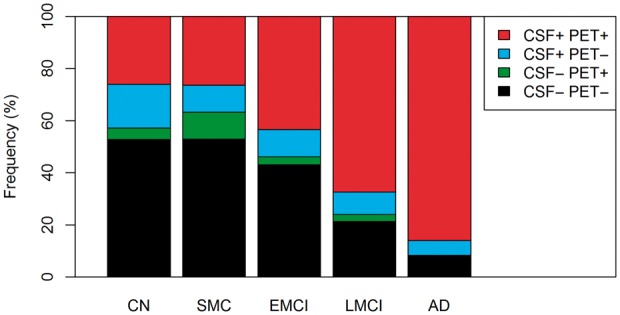
Frequencies of different CSF and PET amyloid-β profiles. The graph shows the frequency of different amyloid-β groups in different diagnostic groups. We used combinations of dichotomous CSF amyloid-β42 (positive < 192 ng/l) and dichotomous florbetapir PET (positive > 1.11 standardized uptake value ratio) to classify subjects as concordant negative (CSF−PET−), discordant (CSF+PET− and CSF−PET+) and concordant positive (CSF+PET+). Concordance increased significantly with disease stage (P < 0.001). AD = Alzheimer's disease; CN = cognitively normal; EMCI = early MCI; LMCI = late MCI.
The concordant negative often differed from the concordant positive group on several measures, but not from any of the discordant groups (when adjusting for multiple comparisons). All of the following comparisons use the CSF−PET− group as reference category. APOE ε4 carriage status was more common in CSF+PET+ cognitively normal (OR = 1.71, P = 0.0037), SMC (OR = 3.36, P = 0.0037), early MCI (OR = 2.51, P < 0.0001), late MCI (OR = 3.43, P < 0.0001) and Alzheimer's disease (OR = 3.42, P = 0.014), and in CSF+PET− early MCI (OR = 1.65, P = 0.0055). For ADAS-cog, baseline scores were elevated in CSF+PET+ late MCI (β = 3.35, P = 0.0020), and deficits accelerated over time in CSF+PET+ early MCI (β = 0.55, P = 0.034) and late MCI (β = 2.60, P = 0.00011). For hippocampus, baseline volumes were reduced in CSF+PET+ late MCI (β = −384, P = 0.0018) and shrinkage accelerated over time in CSF+PET+ early MCI (β = −33.1, P = 0.034). For CSF phosphorylated tau, levels were increased in CSF+PET+ cognitively normal (β = 11.0, P = 0.0047), SMC (β = 30.5, P < 0.0001), early MCI (β = 25.8, P < 0.0001) and late MCI (β = 30.8, P < 0.0001). CSF total tau levels were similar to CSF phosphorylated tau but with slightly less pronounced group differences (data not shown). For FDG-PET, the signal was lower in CSF+PET+ at baseline in late MCI (β = −0.085, P = 0.036), and Alzheimer's disease (β = −0.17, P = 0.021) and over time in late MCI (β = −0.037, P = 0.034). Cerebral blood flow did not differ by amyloid-β group in any diagnostic group. These tests were corrected for multiple comparisons (using false discovery rate).
Discussion
Our main findings were that (i) CSF and PET amyloid-β provided partially independent information when predicting most Alzheimer's disease-associated measures (even within amyloid-β positive subjects, defined by combined CSF and PET amyloid-β positivity); (ii) CSF and PET amyloid-β were related to multiple measures in different ways (for example, APOE ε4 was preferentially associated with CSF amyloid-β); (iii) CSF and PET amyloid-β discordance (mainly consisting of isolated CSF amyloid-β positivity) was common in cognitively normal and SMC subjects; and (iv) concordant CSF and PET amyloid-β positivity was common in late stage disease (late MCI and Alzheimer's disease) and associated with most other Alzheimer's disease measures. The results were in agreement with our hypothesis that the two amyloid-β modalities provide partially independent information. The results may support CSF amyloid-β as a more sensitive marker of very early disease and PET amyloid-β as a more sensitive marker of disease progression and down-stream pathology.
The first finding was that CSF and PET amyloid-β were partially independent predictors of diagnosis, APOE ε4, high CSF total tau and phosphorylated tau, hypometabolism, hippocampal atrophy and (for CSF) reduced cerebral blood flow. Most of these are well established characteristics of Alzheimer's disease. The fact that the two amyloid-β modalities were partially independent predictors across most outcomes supports our underlying theory that they partly represent different aspects of Alzheimer's disease pathology. This likely includes several different but important aspects of amyloid metabolism, and could for example be related to differences across disease stages in the composition of amyloid plaques (Yamaguchi et al., 1989; Schmidt et al., 1994). CSF and PET amyloid-β predicted several Alzheimer's disease features even when restricting the analysis to people with strong evidence of amyloid-β pathology (combined CSF and PET amyloid-β positivity). The strongest relationships were between amyloid-β biomarkers and hippocampal volume and ADAS-cog, suggesting that amyloid-β metabolism continues to be related to brain injury and cognitive deficits even after the first appearance of fibrillar amyloid-β. This is in line with recent reports of dynamic changes in amyloid-β biomarkers also in demented amyloid-β-positive patients (Toledo et al., 2013; Villemagne et al., 2013).
The second finding was that CSF amyloid-β was more strongly related to APOE ε4, whereas PET amyloid-β was more strongly related to ADAS-cog, CSF total tau and CSF phosphorylated tau. There was also some evidence that CSF amyloid-β was more strongly related to reduced cerebral blood flow, as CSF amyloid-β (but not PET amyloid-β) remained a significant predictor of cerebral blood flow when both modalities were included as predictors. There are several possible interpretations of these findings. First, although amyloid-β load as measured by PET correlates with reduced cerebral blood flow (Mattsson et al., 2014), the fact that CSF amyloid-β rather than PET amyloid-β was preferentially associated with cerebral blood flow suggests that this association goes beyond the presence of overt plaque pathology. For example, it could be related to arterial blood flow regulating the CSF-brain interstitial fluid exchange (Iliff et al., 2013). Second, the strong relationship between APOE ε4 and CSF amyloid-β suggests a role for APOE ε4 in amyloid-β metabolism that goes beyond fibrillar amyloid-β, perhaps including formation of other types of aggregates, such as amyloid-β oligomers, or epitope masking due to binding of amyloid-β with chaperoning APOE protein (Verghese et al., 2011; Slemmon et al., 2012). Third, the strong relationship between PET amyloid-β and total tau, phosphorylated tau and ADAS-cog suggests that PET amyloid-β may be a better measurement than CSF amyloid-β of late stage progression of pathology.
The third finding was that CSF and PET amyloid-β mismatch classification was dependent on diagnostic category and seen in 21% of cognitively normal and SMC subjects, 10–14% in MCI, and 6% of Alzheimer's disease dementia. For the cognitively normal group, the discordance was slightly higher than in a previous analysis of this cohort (including about half the subjects, n = 374) (Landau et al., 2013), but was in agreement with other studies where 15–25% of healthy controls were discordantly classified (Fagan et al., 2009; Zwan et al., 2014). Most mismatched people in our study had low CSF amyloid-β without increased PET amyloid-β [this may vary between cohorts and cut-offs (Fagan et al., 2009; Zwan et al., 2014)]. A number of factors could cause isolated low CSF amyloid-β, including non-fibrillar brain deposits (Cairns et al., 2009; Schöll et al., 2012), reduced amyloid-β42 production (Mattsson et al., 2012), other inflammatory and degenerative diseases reducing CSF amyloid-β42 without formation of fibrillar deposits (Blennow and Hampel, 2003; Mattsson et al., 2010; Augutis et al., 2013), and false low values due to technical variability (Mattsson et al., 2011, 2013). Another possibility is that CSF amyloid-β could be sensitive to amyloid pathology primarily in specific regions of the brain, although several studies argue against this (Grimmer et al., 2009; Mattsson et al., 2014). Isolated increased PET amyloid-β may be due to unspecific retention of the amyloid ligand, false high levels due to technical issues (processing errors) and perhaps also physiologically high CSF amyloid-β levels in individuals who remain above the CSF cut-off even in the presence of fibrillar amyloid-β. Note that we propose that CSF-PET discordance contains relevant information, but it may be possible to construct a CSF measurement that is more specific for fibrillar amyloid-β (with less mismatches) by adjusting CSF amyloid-β42 for CSF amyloid-β40, removing some of the variance due to overall amyloid-β production and clearance (Hansson et al., 2007; Wiltfang et al., 2007). The development of novel assays for CSF amyloid-β42 will likely reduce errors due to technical variability (Korecka et al., 2014; Leinenbach et al., 2014). One study found very high concordance between low CSF amyloid-β and increased PET amyloid-β in patients with MCI (Palmqvist et al., 2014), although it is possible that the concordance may vary depending on pre-analytical and analytical factors for CSF amyloid-β42 measurements (Oskar Hansson, personal communication). The fact that the proportion of discordant cases dropped with disease stage argues against technical variability in CSF or PET measurements as the primary source of discordance in this study. Irrespective of cause, this and previous reports point to a CSF-PET mismatch frequency of ∼20% in cognitively healthy subjects, which is relevant to consider when designing preclinical trials using amyloid-β biomarkers to identify early stage brain pathology. Mismatch was not associated with increased risk of longitudinal brain atrophy or cognitive decline over the time period studied here. However, the finding that low CSF amyloid-β but normal PET amyloid-β was most common in cognitively normal subjects and less common in advanced disease stages is compatible with a model where low CSF amyloid-β appears earlier than high PET amyloid-β. This is also supported by the finding that the risk factor APOE ε4 was preferentially associated with CSF amyloid-β whereas downstream measures, including cognitive deficits and CSF tau biomarkers, were preferentially associated with PET amyloid-β.
The final finding was that concordant amyloid-β classification was associated with high risk of most other Alzheimer's disease measures. This is in agreement with the previous literature, which has linked amyloid-β pathology as measured by either modality to many features of Alzheimer's disease. By examining CSF and PET amyloid-β simultaneously we increased our power to detect effects as mismatch cases likely add noise to studies only including one modality. In Alzheimer's disease dementia, most patients were concordant positive. Reassuringly, the few amyloid negative Alzheimer's disease dementia patients (unlikely to have Alzheimer's disease according to modern criteria) were stable in ADAS-cog. Curiously, they showed the same rate of hippocampal shrinkage as amyloid-positive patients (data not shown), but note that the number of negative Alzheimer's disease dementia patients was small.
A possible limitation was the influence of data points close to the cut-offs, but the results were stable when excluding individuals with CSF or PET amyloid-β within 5% of the respective cut-offs (data not shown). Another possible limitation when comparing different models is the risk of over-fitting models with multiple predictors. However, AIC, which was used for model comparison, penalizes for additional predictors and thereby protects against this cause of over-fitting. Another limitation to the generalizability of the results of this study is that medical conditions where CSF amyloid-β42 is known to be reduced without presence of plaques [including neuroinflammation (Mattsson et al., 2009; Augutis et al., 2013) Creuzfeldt-Jakob's disease and amyotrophic lateral sclerosis (Blennow and Hampel, 2003)], where not included in this cohort, although patients with such conditions would have been interesting to investigate with a combination of CSF and PET amyloid-β. Finally, we performed a large number of tests of the effects of continuous CSF amyloid-β and PET amyloid-β on different outcomes without correcting for multiple comparisons. Our results pointed strongly at associations between amyloid-β measures and deleterious effects (this was seen in 41 of 44 comparisons in the main analysis, presented in Fig. 1). Taken together with the previous literature that also supports associations between amyloid-β and brain injury and impaired cognition, we find it exceedingly unlikely that these significant associations are due to type I error. An adjustment for multiple comparisons would therefore risk an overcorrection. In contrast, the more exploratory comparisons within diagnostic groups using the classification of concordant and discordant CSF-PET amyloid-β as a categorical predictor were adjusted for multiple comparisons.
In sum, we found that CSF and PET amyloid-β show partially independent associations with a wide range of Alzheimer's disease measures, supporting our theory that CSF and PET amyloid-β to some extent represent different aspects of Alzheimer's disease-relevant amyloid metabolism. Combining CSF and PET amyloid-β may improve prediction of clinical and pathological aspects of Alzheimer's disease. CSF-PET mismatch was dominated by isolated reduced CSF amyloid-β and was most common in controls. Furthermore, reduced CSF amyloid-β was more strongly associated with APOE ε4 and increased PET amyloid-β was more strongly associated with CSF total tau and phosphorylated tau, which are markers of neurodegeneration, and ADAS-cog, which is an indicator of clinical disease progression. Taken together, this suggests that reduced CSF amyloid-β may represent an earlier signal than increased brain amyloid-β detected by PET, and that PET amyloid-β may be a more sensitive marker of disease progression during the development of Alzheimer's disease pathology.
Funding
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, Glaxo-SmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace Inc., Merck and Co. Inc., Novartis AG, Pfizer Inc., F. Hoffman-La Roche, Schering-Plough, Synarc Inc., as well as non-profit partners the Alzheimer's Association and Alzheimer's Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514, the Swedish Research Council, Goteborgs Lakaresallskap, Svenska Lakaresallskapet, Sahlgrenska Universitetssjukhuset, Carl-Bertil Laurells fond, and Klinisk Biokemi i Norden. N.M. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. No sponsor had any role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Conflict of interest
N.M., P.I., M.D., W.J., S.L. and H.Z. report no conflicts of interest. K.B. has served at Advisory Boards for Pfizer, Roche, Kyowa Kirin Pharma and Innogenetics. J.Q.T. may accrue revenue in the future on patents submitted by the University of Pennsylvania wherein he is co-Inventor and he received revenue from the sale of Avid to Eli Lily as co-inventor on imaging related patents submitted by the University of Pennsylvania; and is the William Maul Measey-Truman G. Schnabel, Jr., M.D. Professor of Geriatric Medicine and Gerontology. L.M.S. previously was consultant for Innogenetics and collaborates on quality assessment activities as part of the Alzheimer's Disease Neuroimaging Initiative; and serves as a consultant to Janssen AI R & D on biomarker studies. M.W. has been on scientific advisory boards for Pfizer and BOLT Inter-national; has been a consultant for Pfizer Inc., Janssen, KLJ Associates, Easton Associates, Harvard University, inThought, INC Research, Inc., University of California, Los Angeles, Alzheimer's Drug Discovery Foundation and Sanofi-Aventis Groupe; has received funding for travel from Pfizer, Alzheimer's disease PD meeting, Paul Sabatier University, Novartis, Tohoku University, MCI Group, France, Travel eDreams, Inc., Neuroscience School of Advanced Studies (NSAS), Danone Trading, BV, CTAD ANT Congres; serves as an associate editor of Alzheimer's & Dementia; has received honoraria from Pfizer, Tohoku University, and Danone Trading, BV; has research support from Merck, Avid, DOD and VA; and has stock options in Synarc and Elan.
Supplementary material
Supplementary material is available from Brain online.
Glossary
Abbreviations
- ADAS-cog
Alzheimer's Disease Assessment Scale-cognitive subscale
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- AIC
Akaike information criterion
- FDG
fluorodeoxyglucose
- MCI
mild cognitive impairment
- SMC
subjective memory complaint
Appendix 1
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu/ As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
References
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augutis K, Axelsson M, Portelius E, Brinkmalm G, Andreasson U, Gustavsson MK, et al. Cerebrospinal fluid biomarkers of β-amyloid metabolism in multiple sclerosis. Mult Scler. 2013;19:543–52. doi: 10.1177/1352458512460603. [DOI] [PubMed] [Google Scholar]
- Bates D. lme4: Mixed-effects modeling with R. New York: Springer; 2010. [Google Scholar]
- Blennow K, Hampel H. CSF markers for incipient Alzheimer's disease. Lancet Neurol. 2003;2:605–13. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–44. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR. Model Selection and Multimodal Inference. 2 edn. New York: Springer; 2002. [Google Scholar]
- Cairns NJ, Ikonomovic MD, Benzinger T, Storandt M, Fagan AM, Shah A, et al. Absence of PIttsburgh compound B detection of cerebralamyloid beta in a patient with clinical, cognitive, and cerebrospinal fluidmarkers of alzheimer disease. Arch Neurol. 2009;66:1557–62. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011;305:275–83. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer G, Shapiro F, Vanderstichele H, Vanmechelen E, Engelborghs S, De Deyn PP, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67:949–56. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–29. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer's disease. EMBO Mol Med. 2009;1:371–80. doi: 10.1002/emmm.200900048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–55. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Ségonne F, Salat DH, et al. Automatically parcellating the human cerebral cortex. Cereb Cortex. 2004;14:11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2008;29:1456–65. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. The elements of statistical learning. New York: Springer Series in Statistics; 2009. [Google Scholar]
- Grimmer T, Riemenschneider M, Forstl H, Henriksen G, Klunk WE, Mathis CA, et al. Beta amyloid in Alzheimer's disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry. 2009;65:927–34. doi: 10.1016/j.biopsych.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson O, Zetterberg H, Buchhave P, Andreasson U, Londos E, Minthon L, et al. Prediction of Alzheimer's disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;23:316–20. doi: 10.1159/000100926. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008;131:1630–45. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. 2013;33:18190–9. doi: 10.1523/JNEUROSCI.1592-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–91. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AD, Pontecorvo MJ, Clark CM, Carpenter AP, Jennings DL, Sadowsky CH, et al. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer's disease and cognitively normal subjects. J Nucl Med. 2012;53:378–384. doi: 10.2967/jnumed.111.090340. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Koivunen J, Pirttilä T, Kemppainen N, Aalto S, Herukka S-K, Jauhianen AM, et al. PET amyloid ligand [11C]PIB uptake and cerebrospinal fluid beta-amyloid in mild cognitive impairment. Dementia and Geriatr Cogn Disord. 2008;26:378–83. doi: 10.1159/000163927. [DOI] [PubMed] [Google Scholar]
- Korecka M, Waligorska T, Figurski M, Toledo JB, Arnold SE, Grossman M, et al. Qualification of a surrogate matrix-based absolute quantification method for amyloid-β42in human cerebrospinal fluid using 2D UPLC-tandem mass spectrometry. J Alzheimers Dis. 2014;41:441–51. doi: 10.3233/JAD-132489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32:1207–18. doi: 10.1016/j.neurobiolaging.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Lu M, Joshi AD, Pontecorvo M, Mintun MA, Trojanowski JQ, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of β-amyloid. Ann Neurol. 2013;74:826–36. doi: 10.1002/ana.23908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012;72:578–86. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinenbach A, Pannee J, Dülffer T, Huber A, Bittner T, Andreasson U, et al. Mass spectrometry-based candidate reference measurement procedure for quantification of amyloid-β in cerebrospinal fluid. Clin Chem. 2014;60:987–94. doi: 10.1373/clinchem.2013.220392. [DOI] [PubMed] [Google Scholar]
- Little R, Rubin D. Statistical analysis with missing data. 2 edn. Hoboken, New Jersey: Wiley; 2002. [Google Scholar]
- Mattsson N, Andreasson U, Persson S, Arai H, Batish SD, Bernardini S, et al. The Alzheimer's association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 2011;7:386–95, e6. doi: 10.1016/j.jalz.2011.05.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Andreasson U, Persson S, Carrillo MC, Collins S, Chalbot S, et al. CSF biomarker variability in the Alzheimer's Association quality control program. Alzheimers Dement. 2013;9:251–261. doi: 10.1016/j.jalz.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Axelsson M, Haghighi S, Malmestrom C, Wu G, Anckarsater R, et al. Reduced cerebrospinal fluid BACE1 activity in multiple sclerosis. Mult Scler. 2009;15:448–54. doi: 10.1177/1352458508100031. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Bremell D, Anckarsater R, Blennow K, Anckarsater H, Zetterberg H, et al. Neuroinflammation in Lyme neuroborreliosis affects amyloid metabolism. BMC Neurol. 2010;10:51. doi: 10.1186/1471-2377-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Insel PS, Landau S, Jagust W, Donohue M, Shaw LM, et al. Diagnostic accuracy of CSF Ab42 and florbetapir PET for Alzheimer's disease. Ann Clin Transl Neurol. 2014;1:534–43. doi: 10.1002/acn3.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Rajendran L, Zetterberg H, Gustavsson M, Andreasson U, Olsson M, et al. BACE1 Inhibition induces a specific cerebrospinal fluid beta-amyloid pattern that identifies drug effects in the central nervous system. PLoS One. 2012;7:e31084. doi: 10.1371/journal.pone.0031084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Tosun D, Insel PS, Simonson A, Jack CR, Beckett LA, et al. Association of brain amyloid-β with cerebral perfusion and structure in Alzheimer's disease and mild cognitive impairment. Brain. 2014;137:1550–61. doi: 10.1093/brain/awu043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, Boggs LN, et al. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J Neurosci. 2011;31:16507–16. doi: 10.1523/JNEUROSCI.3647-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, et al. Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005;51:336–45. doi: 10.1373/clinchem.2004.039347. [DOI] [PubMed] [Google Scholar]
- Palmqvist S, Zetterberg H, Blennow K, Vestberg S, Andreasson U, Brooks DJ, et al. Accuracy of brain amyloid detection in clinical practice using cerebrospinal fluid β-Amyloid 42: a cross-validation study against amyloid positron emission tomography. JAMA Neurol. 2014;71:1282–1289. doi: 10.1001/jamaneurol.2014.1358. [DOI] [PubMed] [Google Scholar]
- Potter R, Patterson BW, Elbert DL, Ovod V, Kasten T, Sigurdson W, et al. Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers. Sci Transl Med. 2013;5:189ra77. doi: 10.1126/scitranslmed.3005615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11:1048–1056. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, DiDario AG, Lee VM, Trojanowski JQ. An extensive network of PHF tau-rich dystrophic neurites permeates neocortex and nearly all neuritic and diffuse amyloid plaques in Alzheimer disease. FEBS Lett. 1994;344:69–73. doi: 10.1016/0014-5793(94)00259-2. [DOI] [PubMed] [Google Scholar]
- Schöll M, Wall A, Thordardottir S, Ferreira D, Bogdanovic N, Långström B, et al. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012;79:229–36. doi: 10.1212/WNL.0b013e31825fdf18. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slemmon JR, Meredith J, Guss V, Andreasson U, Andreasen N, Zetterberg H, et al. Measurement of Abeta1-42 in cerebrospinal fluid is influenced by matrix effects. J Neurochem. 2012;120:325–33. doi: 10.1111/j.1471-4159.2011.07553.x. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolboom N, van der Flier WM, Yaqub M, Boellaard R, Verwey NA, Blankenstein MA, et al. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med. 2009;50:1464–70. doi: 10.2967/jnumed.109.064360. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Xie SX, Trojanowski JQ, Shaw LM. Longitudinal change in CSF Tau and Aβ biomarkers for up to 48 months in ADNI. Acta Neuropathol. 2013;126:659–70. doi: 10.1007/s00401-013-1151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 2011;10:241–52. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12:357–67. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- Weigand SD, Vemuri P, Wiste HJ, Senjem ML, Pankratz VS, Aisen PS, et al. Transforming CSF A?42 measures into calculated Pittsburgh Compound B (PIBcalc) units of brain A? amyloid. Alzheimers Dement. 2011;7:133–41. doi: 10.1016/j.jalz.2010.08.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltfang J, Esselmann H, Bibl M, Hüll M, Hampel H, Kessler H, et al. Amyloid beta peptide ratio 42/40 but not A beta 42 correlates with phospho-Tau in patients with low- and high-CSF A beta 40 load. J Neurochem. 2007;101:1053–9. doi: 10.1111/j.1471-4159.2006.04404.x. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Nakazato Y, Hirai S, Shoji M, Harigaya Y. Electron micrograph of diffuse plaques. Initial stage of senile plaque formation in the Alzheimer brain. Am J Pathol. 1989;135:593–7. [PMC free article] [PubMed] [Google Scholar]
- Zwan M, van Harten A, Ossenkoppele R, Bouwman F, Teunissen C, Adriaanse S, et al. Concordance between cerebrospinal fluid biomarkers and [11C]PIB PET in a memory clinic cohort. J Alzheimers Dis. 2014;41:801–7. doi: 10.3233/JAD-132561. [DOI] [PubMed] [Google Scholar]