

Vemuri et al. show that amyloid and vascular pathologies are independent processes, and that both are major drivers of cognitive decline in the elderly. Cognitive reserve as measured by educational/occupational level and mid/late-life cognitive activity seems to offset the deleterious effects of both pathologies on cognitive trajectories.
Keywords: ageing, cognitive neurology, neuroimaging, neuro protective strategies
Abstract
Our primary objective was to investigate a biomarker driven model for the interrelationships between vascular disease pathology, amyloid pathology, and longitudinal cognitive decline in cognitively normal elderly subjects between 70 and 90 years of age. Our secondary objective was to investigate the beneficial effect of cognitive reserve on these interrelationships. We used brain amyloid-β load measured using Pittsburgh compound B positron emission tomography as a marker for amyloid pathology. White matter hyperintensities and brain infarcts were measured using fluid-attenuated inversion recovery magnetic resonance imaging as a marker for vascular pathology. We studied 393 cognitively normal elderly participants in the population-based Mayo Clinic Study of Aging who had a baseline 3 T fluid-attenuated inversion recovery magnetic resonance imaging assessment, Pittsburgh compound B positron emission tomography scan, baseline cognitive assessment, lifestyle measures, and at least one additional clinical follow-up. We classified subjects as being on the amyloid pathway if they had a global cortical amyloid-β load of ≥1.5 standard uptake value ratio and those on the vascular pathway if they had a brain infarct and/or white matter hyperintensities load ≥1.11% of total intracranial volume (which corresponds to the top 25% of white matter hyperintensities in an independent non-demented sample). We used a global cognitive z-score as a measure of cognition. We found no evidence that the presence or absence of vascular pathology influenced the presence or absence of amyloid pathology and vice versa, suggesting that the two processes seem to be independent. Baseline cognitive performance was lower in older individuals, in males, those with lower education/occupation, and those on the amyloid pathway. The rate of cognitive decline was higher in older individuals (P < 0.001) and those with amyloid (P = 0.0003) or vascular (P = 0.0037) pathologies. In those subjects with both vascular and amyloid pathologies, the effect of both pathologies on cognition was additive and not synergistic. For a 79-year-old subject, the predicted annual rate of global z-score decline was −0.02 if on neither pathway, −0.07 if on the vascular pathway, −0.08 if on the amyloid pathway and −0.13 if on both pathways. The main conclusions of this study were: (i) amyloid and vascular pathologies seem to be at least partly independent processes that both affect longitudinal cognitive trajectories adversely and are major drivers of cognitive decline in the elderly; and (ii) cognitive reserve seems to offset the deleterious effect of both pathologies on the cognitive trajectories.
Introduction
Universally observed cognitive decline in the elderly due to the pathological ageing of the brain will have a significant impact on public health. Alzheimer’s disease pathophysiology and cerebrovascular disease are the leading causes of age-related cognitive decline. The hallmarks of Alzheimer’s disease are the presence of amyloid-β plaques and neurofibrillary tangles. The deposition of amyloid pathology in the brain and simultaneous progression of neurodegeneration are hypothesized to be the first change in the largely sequential pathological cascade of Alzheimer’s disease (Ingelsson et al., 2004; Jack et al., 2013). Cerebrovascular disease is the other most prevalent pathology that is associated with age-related cognitive decline (Jellinger, 2005; Schneider et al., 2009). The hallmarks of cerebrovascular disease are the presence of microvascular changes (white matter hyperintensities) and macrovascular changes (subcortical and cortical infarcts). Although the underlying pathologies that cause white matter hyperintensities as seen on MRI have been shown to be heterogeneous based on imaging–pathology studies, the vast majority are believed to be vascular resulting from ischaemic and hypoxic changes (Thomas et al., 2002, 2003; Gouw et al., 2008; Murray et al., 2012). The co-occurrence of both Alzheimer’s disease pathophysiology and cerebrovascular disease is also a common pathological finding in the elderly (Jellinger, 2007; Schneider et al., 2009; White, 2009). There is a suggested interrelationship between the presence of cerebrovascular disease and Alzheimer’s disease pathophysiology because dementia due to these pathologies shares several of the same risk factors, which include diabetes, hypertension, stroke and cholesterol (Breteler, 2000). There is also evidence that the presence of vascular risk factors may increase the risk of Alzheimer’s disease (Kalaria, 2000; Viswanathan et al., 2009; de la Torre, 2010) and subjects with Alzheimer’s disease are at increased risk of cerebrovascular disease (Gurol et al., 2006; Gomis et al., 2009; Iadecola, 2010).
The primary goal of this study was to provide a biomarker driven model for the interrelationships between Alzheimer’s disease pathophysiology, cerebrovascular disease, and cognitive decline in a population-based sample. A secondary goal was to understand the protective effect of cognitive reserve against the deleterious effects of Alzheimer’s disease and cerebrovascular disease pathologies. We used a three step approach to investigate our goals (Fig. 1): (i) Does the occurrence of one pathology influence the presence of the other? i.e. are they independent or co-dependent processes? (co-dependence is illustrated by arrow A in Fig. 1 and independence suggests the absence of arrow A); (ii) Is the overall effect of cerebrovascular disease and Alzheimer’s disease on cognitive outcomes additive (sum of arrows B in Fig. 1) or synergistic (arrow C in Fig. 1)? Synergistic effects of cerebrovascular disease and Alzheimer’s disease, as suggested by Iadecola (2010), would mean that the impact of cerebrovascular disease and Alzheimer’s disease on cognitive decline is worse through an interaction (arrow C in Fig. 1) than the impact of either one added together (sum of arrows B); and (iii) To what extent does high versus low cognitive reserve protect against the deleterious effects of Alzheimer’s disease pathophysiology and cerebrovascular disease pathologies (arrow D in Fig. 1). We investigated these questions in cognitively normal subjects from the Mayo Clinic Study of Aging (MCSA); a population-based sample. We ascertained the presence or absence of significant Alzheimer’s disease pathophysiology and cerebrovascular disease pathologies using imaging. We used PIB-PET (Pittsburgh compound B PET) as a surrogate for cerebral amyloidosis and FLAIR-MRI as a surrogate for cerebrovascular disease.
Figure 1.
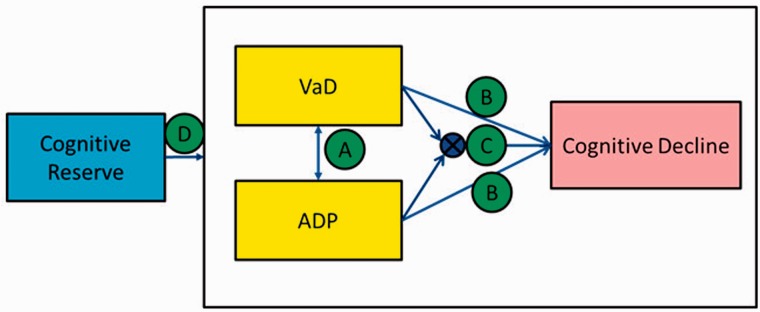
Illustration of the interrelationships investigated. We tested the following hypotheses: Arrow A investigates if the occurrence of one pathology influences the presence of the other (co-dependence is illustrated by the presence of arrow A and independence suggests the absence of arrow A). Arrow B indicates that the overall effect of cerebrovascular disease and Alzheimer’s disease on cognitive outcomes is additive (sum of arrows B), and Arrow C indicates that the overall effect of cerebrovascular disease and Alzheimer’s disease on cognitive outcomes is synergistic. Arrow D indicates the influence of high versus low cognitive reserve protect against the deleterious effects of Alzheimer’s disease pathophysiology and cerebrovascular disease pathologies.
Materials and methods
Selection of participants
Study subjects were participants in the Mayo Clinic Study of Aging (MCSA), an epidemiological study of the prevalence, incidence, and risk factors for mild cognitive impairment (MCI) and dementia among Olmsted County residents aged 70-89. We included all 393 healthy elderly participants in the MCSA who had a baseline 3 T FLAIR-MRI assessment, Amyloid PET scan, intellectual enrichment variables, complete neuropsychological assessments, and at least one additional clinical follow-up with complete neuropsychological assessments. The MCSA uses the Rochester Epidemiology Project records—linkage system infrastructure (St Sauver et al., 2011, 2012; Rocca et al., 2012). Complete details of the MCSA study design have been published elsewhere (Roberts et al., 2008, 2012; Petersen et al., 2010).
Standard protocol approvals, registrations and patient consents
The study was approved by the Mayo Clinic institutional review board and informed consent was obtained from all participants or their surrogates.
Imaging biomarkers
Amyloid pathology assessment from PIB-PET scans
PIB-PET images were acquired with a PET/CT operating in 3D mode (septa removed). The complete details of PET acquisition were described previously (Lowe et al., 2009). All PET quantitative image analysis including quality control were performed at the Mayo Clinic using the same fully automated image processing pipeline as described previously (Jack et al., 2008; Senjem et al., 2008). A global cortical PIB-PET retention ratio was computed by calculating the median uptake over voxels in the prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus regions of interest for each subject and dividing this by the median uptake over voxels in the cerebellar grey matter region of interest of the atlas (Lopresti et al., 2005). We classified subjects as being on the amyloid pathway (A+ or Alzheimer’s disease pathophysiology) if their global cortical PIB-PET value was ≥1.5.
Vascular pathology assessment from FLAIR-MRI scans
FLAIR-MRI images were acquired with 3 T MRI scanners and the complete details of the acquisition can be found elsewhere (Kantarci et al., 2013). Brain infarcts were assessed by a trained image analyst (G.M.P.) and confirmed by a radiologist (K.K.) blinded to all clinical information. Subcortical infarcts included infarcts in white matter, deep grey matter nuclei, cerebellum, and brainstem not involving the hemispheric infarcts. Cortical infarcts were ≥1 cm in largest diameter. Intra-rater reliability of this assessment is excellent (proportion in agreement 0.98 for cortical and 0.94 for subcortical infarcts) (Kantarci et al., 2008). White matter hyperintensities on FLAIR images were segmented using an automated slice-based seed initialization and region growing methods as previously described (Raz et al., 2013). We used white matter hyperintensities divided by the total intracranial volume as a measure of white matter disease. We used total intracranial volume instead of brain volume or total white matter to exclude the effect of atrophy due to other neurodegenerative pathologies.
To facilitate the combination of white matter hyperintensities with infarcts to represent cerebrovascular disease as well as describe vascular disease in a manner parallel to amyloidosis, we estimated cut-off for the white matter hyperintensities/total intracranial volume per cent (WMH/TIV%) to combine it with infarcts. We made a simplistic assumption that while the effect of an infarct on cognition may vary based on size and location, it is well known that the presence of a large cortical infarct or a subcortical infarct represents cerebrovascular disease. We estimated the degree of white matter hyperintensity burden that needs to be combined with infarcts using an independent subsample of 1082 non-demented subjects (869 cognitively normal, 213 mild cognitive impairment) from the MCSA who had vascular gradings available but did not have amyloid imaging. We took the following two approaches:
(i) We estimated the degree of white matter hyperintensity burden based on pathological criteria by assuming that the percentage of non-demented subjects with vascular disease in our population is about 33% based on existing published autopsy studies with similar populations (Schneider et al., 2003; Petrovitch et al., 2005; Longstreth et al., 2009). We estimated that with WMH/TIV% cut-point of 1.11, the total percentage of subjects with a brain infarct and/or ≥ the estimated WMH/TIV% cut-off was 33%.
(ii) We also estimated the degree of white matter hyperintensity burden using a statistical approach. As it is generally considered that the presence of a large cortical infarct or a subcortical infarct represents cerebrovascular disease, we estimated what amount of WMH/TIV% would cause the same annual rate of cognitive decline as seen by an infarct and found the cut-off point of 1.11.
Arriving at the same cut point by both independent methods supports the use of 1.11 as a cut-off point. We classified subjects in this study as being on the vascular pathway (V+) if they had a brain infarct (cortical or subcortical infarcts) and/or WMH/TIV% ≥ 1.11. The extent and distribution of white matter hyperintensities intensities in two subjects at the WMH/TIV% cut–off point of 1.11 are shown in the Supplementary material.
Cognitive reserve variables
In our previous work we found that intellectual lifestyle measures significantly predicted cognitive performance; therefore we included these variables as surrogates of cognitive reserve in the model (Vemuri et al., 2012). The primary intellectual activity variables of interest, assessed at baseline, included: (i) education, job-level score based on the primary occupation throughout life; and (ii) current weekly cognitive activity over the last 12 months and mid-life weekly cognitive activity (ages 50–65) (Geda et al., 2011). These intellectual enrichment data were recorded for all subjects at the enrolment visit into the MCSA. Details about the questionnaires used for recording and consolidating each individual measure are provided in our previous work (Vemuri et al., 2012). Education/occupation score and mid/late-life cognitive activity: using principal components applied to these four measures (i.e. education, job-level score, current cognitive activity, and mid-life cognitive activity), we separated the uncorrelated components of early life non-leisure activity and mid/late-life cognitive activity. The first two principal components explained 84% of the variance, and after a varimax rotation the data consolidated into two distinct composite measures of intellectual enrichment: (i) education/occupation score (i.e. lifelong non-leisure intellectual learning) assessed from years of education and job score (weighted contribution for education was 0.690 and for job score was 0.725); and (ii) mid/late-life cognitive activity from a self-report of cognitive activities in the previous 12 months and at mid-life (50–60 years) (weighted contribution for mid-life was 0.707 and for previous 12 months cognitive activities was 0.701).
Global cognition measure
The neuropsychological battery was constructed as previously described (Roberts et al., 2008, 2012; Petersen et al., 2010). Four cognitive domains were assessed by nine tests: executive function (Trail Making Test: Part B, Wechsler Adult Intelligence Scale-R Digit Symbol); language (Boston Naming Test, category fluency); memory [Wechsler Memory Scale (WMS)-R Logical Memory-II (delayed recall), WMS-R Visual Reproduction-II (delayed recall), Auditory Verbal Learning Test delayed recall]; and visuospatial performance (WAIS-R Picture Completion, WAIS-R Block Design). Individual test scores were first converted to z-scores using the mean and standard deviation from the MCSA 2004 enrolment cohort that consisted of non-demented subjects (n = 1969). A global cognitive summary score was estimated from the z-transformation of the average of the four domain z-scores and was used to assess cognitive impairment in our subjects. The baseline global z-score, and rate of decline, were the primary outcomes of interest. Of the 393 subjects in the study, 136 were test naive at the time of enrolment into this study and 257 subjects had previously completed the battery as part of an earlier study. Because of the influence of practice effects on the measured outcome variable (Duff et al., 2010; Dodge et al., 2011; Machulda et al., 2013), we controlled for the number of times the subject had the battery before receiving the MRI and PET scans using a variable named ‘baseline visit number’. A total of 136 patients had a baseline visit number of 1 (i.e. the first time they took the test was at baseline of the study), 65 patients had a baseline visit number of 2 (i.e. tested once before baseline), 37 patients had a baseline visit number of 3 (i.e. tested twice before baseline), and 155 patients had a baseline visit number of 4 or more.
Statistical analysis
When describing characteristics of the subjects, we categorized them as being on neither pathway (A−V−), vascular pathway only (A−V+), amyloid pathway only (A+V−), and vascular and amyloid pathway (A+V+). ANOVAs were performed to assess differences in continuous variables between groups whereas chi-squared tests were done to check for differences in proportions between groups for the categorical variables.
We performed two separate sets of analyses to investigate our aims. In the first set of analyses, we tested the independence of the cerebrovascular disease and Alzheimer’s disease pathophysiology processes by testing the association of the presence or absence of Alzheimer’s disease pathophysiology and cerebrovascular disease using a chi-square test of independence, and summarized using the odds ratio (OR) and P-value. We also investigated whether (i) the presence of cerebrovascular disease was associated with worse amyloid pathologies, i.e. A+V+ had worse amyloid load than A+V−, and whether A−V+ had worse amyloid load than A−V−; and (ii) the presence of Alzheimer’s disease pathophysiology was associated with worse vascular disease, i.e. A+V+ had worse white matter hyperintensities/total intracranial volume load and brain infarctions than A−V+, and whether A+V− had worse white matter hyperintensities/total intracranial volume load than A−V−.
In the second set of analyses, we tested the effect of cerebrovascular disease and Alzheimer’s disease pathophysiology on cognitive decline using linear mixed models fit by maximum likelihood. We examined demographic variables, cognitive reserve variables, and the presence or absence of Alzheimer’s disease pathophysiology and cerebrovascular disease as predictors of global cognitive z-scores in the model. In these models, we also examined the coefficients that were associated with time from baseline (i.e. related to rate of cognitive decline). The initial model included baseline age (years), sex, APOE carrier status, time from baseline (years), the intellectual enrichment variables, baseline visit number, all two-way interactions of these variables, a three-way interaction of the intellectual enrichment variables with time, and a three-way interaction of Alzheimer’s disease pathophysiology and cerebrovascular disease with time. The models were fit with random subject-specific intercepts and slopes. We tested for the statistical significance of these random terms using likelihood ratio tests. We also used likelihood ratio tests to compare independence (where the within-subject errors are independent) and continuous first-order autoregression. Both random terms were significant (P < 0.0001). The models also incorporated the continuous first-order autoregressive AR(1) correlation structures (estimated correlation for values 1 year apart = 0.41, P < 0.0001).
The three-way interactions were not significant, so we removed them from further consideration. We then used a backwards elimination procedure, respecting the need to retain nested terms, to remove predictors and to form the most parsimonious model. The final model contained baseline age (years), sex, time from baseline (years), baseline visit number, education/occupation score, being on the amyloid pathway, and being on the vascular pathway. The model also included three two-way interactions: baseline age with time from baseline, amyloid pathway with time, and vascular pathway with time. Significant interaction terms with time indicate that the variables are associated with the rate of cognitive decline. Both random intercepts (P < 0.0001) and random slopes (P < 0.0001) were deemed necessary.
Results
The demographics, clinical, intellectual enrichment variables, and imaging biomarkers of the subjects included in this analysis are shown in Table 1. Among the demographic, intellectual lifestyle, and clinical variables only age, APOE ε4 status, and global cognitive z-scores were significantly different between the four groups (P < 0.01). Subjects in the A+ groups were more likely to be APOE ε4 carriers and subjects with A+ and/or V+ were older than subjects in A−V−. Subjects in A+V+ had significantly lower global z-scores (P = 0.002) and were significantly more likely to progress to a diagnosis of mild cognitive impairment or dementia at follow-up (P = 0.003) compared to the other groups.
Table 1.
Patient characteristics
| A−V− | A−V+ | A+V+ | A+V− | P-value | |
|---|---|---|---|---|---|
| No. of subjects (%) | 178 (45) | 89 (23) | 45 (11) | 81 (21) | |
| No. of Females (%) | 85 (48) | 41 (46) | 19 (42) | 33 (41) | 0.73 |
| Age (years) | 76 (73, 81) | 78 (75, 83) | 82 (79, 83) | 78 (75, 81) | <0.001 |
| No. of ε4 carriers (%) | 34 (19) | 17 (19) | 15 (33) | 40 (49) | <0.001 |
| Education (years) | 13.5 (12, 16) | 14 (12, 16) | 13 (12, 16) | 14 (12, 16) | 0.48 |
| Short Test of mental status | 35.5 (34, 37) | 35 (34, 37) | 34 (32, 36) | 35 (33, 36) | 0.17 |
| Global z-score | 0.80 (0.19, 1.34) | 0.71 (0.24, 1.07) | 0.08 (−0.36, 0.59) | 0.64 (0.13, 1.26) | 0.002 |
| Job Score | 4 (3, 6) | 4 (3, 6) | 4 (3, 6) | 4 (3, 6) | 0.97 |
| Global cortical PIB | 1.33 (1.29, 1.38) | 1.34 (1.30, 1.38) | 1.93 (1.64, 2.22) | 1.86 (1.68, 2.11) | |
| Mid-life intellectual score | 21 (15.5, 27.5) | 20 (14.5, 28) | 21 (15.5, 27) | 21 (14, 24.5) | 0.84 |
| Late-life intellectual score | 23.5 (17.5, 30.5) | 23 (15, 30.5) | 23 (17.5, 30.5) | 21 (16, 28) | 0.35 |
| WMH/TIV % | 0.48 (0.36, 0.67) | 1.12 (0.59, 1.58) | 1.19 (0.87, 1.73) | 0.46 (0.35, 0.68) | |
| No. with cortical infarctions (%) | 0 | 11 (12) | 9 (20) | 0 | |
| No. with subcortical infarctions (%) | 0 | 55 (62) | 21 (47) | 0 | |
| Baseline visit number | 2 (1, 4) | 3 (1, 4) | 4 (3, 5) | 3 (1, 4) | <0.001 |
| Follow-up (years) | 2.7 (1.0, 7.7) | 2.8 (1.2, 6.7) | 2.7 (1.2, 6.9) | 2.7 (1.2, 6.6) | 0.49 |
| No of progressors to mild cognitive impairment/dementia (%) | 22 (12) | 14 (16) | 16 (36) | 14 (17) | 0.003 |
Testing independence of Alzheimer’s disease pathophysiology and cerebrovascular disease processes
The odds ratio for the chi-square test of independence was 1.31 (P = 0.745) providing evidence that the occurrence of Alzheimer’s disease pathophysiology and cerebrovascular disease are not dependent. The A+V+ subjects did not have worse amyloid than A+V− subjects (P = 0.26), and the A−V+ subjects did not have worse amyloid than A−V− subjects (P = 0.70). This finding suggests that the presence of vascular pathologies did not contribute to worse amyloid pathology. Similarly, A+V+ subjects did not have worse white matter hyperintensities/total intracranial volume (P = 0.28) or number of brain infarcts (P = 0.22) than A−V+ subjects, and A+V− subjects did not have worse white matter hyperintensities/total intracranial volume than A−V− subjects (P = 0.81). These findings suggest that the presence of amyloid pathologies did not contribute to worse vascular disease.
Effect of Alzheimer’s disease pathophysiology, cerebrovascular disease and cognitive reserve on longitudinal cognitive decline
The results of the linear mixed effects models are presented in Table 2. Baseline global z-scores were lower in males, older subjects and those with lower education/occupation scores (P < 0.001 for all). There was also evidence that subjects, on the amyloid pathway (A+) had slightly lower baseline global z-scores (P = 0.039). Subjects who had exposure to the neuropsychological battery before the baseline testing (practice effects discussed earlier) performed better (P < 0.001).
Table 2.
Parsimonious model, random subject-specific intercepts and slopes
| Coefficient | Standard error | P-value | |
|---|---|---|---|
| (Intercept) | 5.00 | 0.68 | <0.0001 |
| Baseline age (years) | −0.05 | 0.01 | <0.0001 |
| Males | −0.37 | 0.08 | <0.0001 |
| Time (years) | 0.56 | 0.12 | <0.0001 |
| Education/occupation score | 0.26 | 0.04 | <0.0001 |
| Baseline visit number | 0.10 | 0.02 | <0.0001 |
| Amyloid pathway | −0.17 | 0.08 | 0.0386 |
| Vascular pathway | −0.10 | 0.08 | 0.2223 |
| Baseline age × time | −0.0073 | 0.0016 | <0.0001 |
| Amyloid pathway × time | −0.06 | 0.02 | 0.0003 |
| Vascular pathway × time | −0.05 | 0.02 | 0.0037 |
From a mixed effects model that predicts longitudinal global cognitive z-scores with demographics and presence or absence of pathologies as predictors.
Among all the intellectual enrichment and demographic variables tested for interaction with time, only older age at baseline visit (P < 0.001), being on the vascular pathway (P = 0.0037), and being on the amyloid pathway (P = 0.0003) significantly predicted the rate of global cognitive decline subsequent to baseline. None of the other two-way interactions and three-way interactions were statistically significant. The interaction terms containing A+ and V+ together (i.e. being on the vascular pathway and amyloid pathway) were not significant. Our model therefore indicated that the effect of cerebrovascular disease and Alzheimer’s disease pathophysiology on cognition and rate of cognitive decline was additive and there was no significant interaction between the effect of both pathologies on cognition.
The cognitive z-score trajectories for subjects on each of the pathways are illustrated in Fig. 2. We separated the plot by sex because the baseline cognitive performance differed between these groups (intercept with the y-axis). For a 79-year-old subject (at the average age of the entire cohort), the predicted annual rate of global z-score decline was −0.02 if the subject were on neither pathway (A−V−), −0.07 if on vascular pathway only (A−V+), −0.08 if on amyloid pathway only (A+V−), and −0.13 if on both pathways (A+V+).
Figure 2.

Decrease in predicted cognitive scores with pathway in female (A) and male (B) subjects.
To illustrate the effect of intellectual lifestyle variables, we separated the plots in Fig. 2 by high (75th percentile) versus low (25th percentile) values of education/occupation scores in Fig. 3. The thicker lines indicate the subjects with high education/occupation scores (75th percentile) and thinner lines indicate the subjects with low high education/occupation scores (25th percentile). Because of the absence of interaction terms with the intellectual lifestyle variables, the only effect of intellectual lifestyle variables on cognition was to shift the baseline cognitive performance up or down (intercept on the y-axis). By drawing a horizontal line that corresponds to the baseline cognitive performance of a 79-year-old subject with low education/occupation score (at 0.44 for female and 0.07 for male); we were able to illustrate the relative contribution of intellectual lifestyle and Alzheimer’s disease and cerebrovascular disease pathologies to cognitive performance. We found that the predicted cognitive score to decrease to the baseline score of a 79-year-old A−V− subject with low education/occupation of a 79-year-old A−V+ subject with high education/occupation would take 7 years, a 79-year-old A+V− subject with high education/occupation would take 5 years, and a 79-year-old A+V+ subject with high education/occupation would take 2 years.
Figure 3.

Decrease in predicted cognitive scores with pathway stratified by subjects with high cognitive reserve (75th percentile of education/occupation scores) and low cognitive reserve (25th percentile of education/occupation scores).
Discussion
The major conclusions of this population-based study of cognitively normal individuals are: (i) cerebrovascular disease and Alzheimer’s disease pathophysiology were not predictive of each other (i.e. cerebrovascular disease and Alzheimer’s disease pathophysiology seem to be independent processes); (ii) the effect of cerebrovascular disease and Alzheimer’s disease pathophysiology on cognition and rate of cognitive decline was additive, and the magnitude of the effect on the rate of future cognitive decline was similar; and (iii) the protective effect of high cognitive reserve at baseline seems to offset the deleterious effect of both pathologies, not to alter the rate of cognitive decline.
Independence of Alzheimer’s disease pathophysiology and cerebrovascular disease processes
The first set of statistical analyses provided evidence that the presence of one pathological process does not influence the presence or absence of the other. The probability of Alzheimer’s disease pathophysiology and cerebrovascular disease occurring together (0.11) is a product of probability of each process alone (= 0.32 × 0.34), which is a necessary condition to prove the independence of the processes. Several pathological studies have found that vascular risk factors may independently contribute to the risk of dementia but does not influence the degree of Alzheimer’s disease pathophysiology (Peila et al., 2002; Arvanitakis et al., 2006; Wang et al., 2009; Ahtiluoto et al., 2010; Dolan et al., 2010; Launer et al., 2011). Biomarker studies have also shown poor correlation between vascular disease (white matter hyperintensities and/or brain infarcts) and amyloid load, further suggesting the independence of the two processes (Hedden et al., 2012; Marchant et al., 2012, 2013; Gurol et al., 2013). The combination of evidence with our work strongly suggests the non-existence of arrow A in Fig. 1 when the interrelationships between cerebrovascular disease and Alzheimer’s disease pathophysiology are studied using neuropathological evidence at autopsy or using imaging as a surrogate of pathology.
Additive effect of Alzheimer’s disease pathophysiology and cerebrovascular disease on cognitive trajectories
Age is a major risk factor for both vascular (Kozachuk et al., 1990) and amyloid (Braak and Braak, 1997) pathologies, and both pathologies are associated with poorer cognitive performance in cognitively normal controls. Although the effect of vascular disease, specifically white matter hyperintensities, is thought to be more strongly associated with executive function (DeCarli et al., 1995; Raz et al., 2007; Brickman et al., 2011), amyloid pathology is thought to be more strongly associated with memory, these generalizations are overly simplistic (Resnick et al., 2010; Pike et al., 2011; Hedden et al., 2013). In this study we found that both pathologies significantly influence the global cognitive trajectories in elderly normal individuals. Based on the mixed effects model, we found that both A+ and V+ subjects had a significantly faster rate of decline compared to A−V− subjects, with A+ subjects starting out significantly lower than A−V− subjects. The classification of subjects as A+/A− and V+/V− allowed us to discern the independent and combined effects of Alzheimer’s disease pathophysiology and cerebrovascular disease on cognition. The lack of interaction between Alzheimer’s disease pathophysiology and cerebrovascular disease terms provided evidence for the additive effect (the existence of arrows B in Fig. 1) and not a synergistic effect (arrow C) of Alzheimer’s disease pathophysiology and cerebrovascular disease on cognition. In elderly subjects who are performing at a cognitively normal level, we found that the magnitude of the independent effect of each of these pathologies on annual rate of cognitive decline was similar (A+V− subjects had an annual decline of −0.08 and A−V+ subjects had an annual decline of −0.07). Another interesting finding was that once the effect of Alzheimer’s disease pathophysiology and cerebrovascular disease on cognitive decline was removed, the annual rate of decline owing to typical aging processes or other unaccounted pathologies was only −0.02 per year compared to a total decline of −0.15 per year seen in subjects with both the pathologies. This suggests that cerebrovascular disease and Alzheimer’s disease pathophysiology are two major drivers of cognitive decline in the elderly.
Our findings of an additive effect are consistent with cross-sectional biomarker studies that have suggested that the effect of both pathologies are additive on cognitive performance (Launer et al., 2008, 2011; Lo and Jagust, 2012; Marchant et al., 2012). This additive effect explains why there is faster decline in those with both Alzheimer’s disease pathophysiology and cerebrovascular disease pathologies (Del Ser et al., 2005) and why a lower degree of Alzheimer’s disease pathophysiology is seen in cases with both Alzheimer’s disease pathophysiology and cerebrovascular disease at the same level of cognitive performance (Riekse et al., 2004). Therefore future analyses aimed at truly understanding the effect of Alzheimer’s disease pathophysiology on cognition will need to account for the presence and severity of cerebrovascular disease because cerebrovascular disease worsens the clinical expression of Alzheimer’s disease pathophysiology (Nagy et al., 1997; Snowdon et al., 1997; Zekry et al., 2002; Weller et al., 2009). A recent review by Chui et al. (2012) suggests that this lowering of the threshold for dementia diagnosis due to cerebrovascular disease explains why some epidemiological studies find an association between vascular risk factors and Alzheimer’s disease even though the two pathologies are not interactive.
Protective effect of cognitive reserve
Cognitive reserve as measured by intellectual lifestyle, independently predicted baseline cognitive performance and did not influence the rate of cognitive decline. These findings are consistent with previous studies showing that cognitive reserve exerts a protective effect on cognition that is independent of the underlying pathology (Vemuri et al., 2011, 2012; Wilson et al., 2013). This translates into a parallel shift in the cognitive trajectories with cognitive reserve as illustrated in Fig. 3. It was surprising to find that a 79-year-old at the 75th percentile of education/occupation and both cerebrovascular disease and Alzheimer’s disease pathophysiology pathologies still performed better cognitively than a 79-year-old at the 25th percentile of education/occupation with neither pathology. These results support the conclusion that engaging in intellectually enriching lifestyles may delay the onset of cognitive decline. As shown in Fig. 3, a 79-year-old person with higher education/occupation and amyloid pathology alone had better baseline cognitive performance than a 79-year-old person with lower education/occupation and no pathologies. It appears to take five additional years for the cognitive performance of the 79-year-old person with higher education/occupation and amyloid pathology to decline to the baseline cognitive performance of a 79-year-old person with lower education/occupation and no pathologies.
Among the demographic variables, older individuals and male subjects had lower baseline global z-scores. Additionally, age was significantly associated with future cognitive decline. Age is the strongest risk factor for cognitive decline. The finding that higher baseline age is associated with worse cognitive scores at baseline and faster cognitive decline is consistent with other studies (Nichols et al., 1994; Hickman et al., 2000; Salmon et al., 2013). The fact that males have lower cognitive performance at baseline is consistent with the literature that males are at higher risk for MCI, particularly at younger ages (Roberts et al., 2012). There is also evidence in the literature that there are sex differences in the cognitive performance in males and females due to neurodevelopmental factors (van Exel et al., 2001; van Hooren et al., 2007; Proust-Lima et al., 2008).
Although the dichotomous approach used in this study may seem to be an oversimplification and investigating the study questions using continuous variables is desirable in the future, there were three main reasons for employing a dichotomous approach for classification of variables in this paper. First, there is no established methodology that is available to generate a single variable for vascular disease based on the continuous variables (i.e. combination of infarcts and white matter hyperintensities). Second, when studying the interrelationships between three sets of variables as in this study (Alzheimer’s disease pathophysiology, cerebrovascular disease and cognitive decline), it is straightforward to estimate and interpret the cognitive outcomes in each of these groups instead of interpreting the effect of two separate continuous pathology variables on cognition. Finally, the latest diagnostic criteria for Alzheimer’s disease (NIA-AA as well as International Working Group criteria) propose the incorporation of biomarker-based cut-offs for diagnosis (Jack et al., 2011; Dubois et al., 2014). The fact that the field is moving towards the use of cut-off points for operationalization of biomarker-based diagnostic criteria supports the methodology used in this study.
The study has some limitations. First, the study results are pertinent to cognitively normal individuals and the interrelationships we investigated may be different in cognitively impaired individuals. Second, we used linear models that may or may not hold true over a 5-year period. Third, although practice effects were not a focus of this manuscript, our finding that greater past exposure to the cognitive tests resulted in better performance are consistent with the literature (Duff et al., 2010; Dodge et al., 2011; Machulda et al., 2013). Fourth, we do not have cerebral microbleed data in the majority of individuals in this study and therefore were unable to examine the possible role of vascular amyloid data in the context of this work. The study also has major strengths. First, the population-based nature of the sample makes the results of the study more generalizable and enhances their external validity. Non-representative samples such as the Alzheimer's Disease Neuroimaging Initiative, which excludes subjects with Hachinski score ≥4 or subjects with significant cerebrovascular disease on MRI, do not lend themselves to investigating the interrelationships between cerebrovascular disease and Alzheimer’s disease pathophysiology. Second, the inclusion of cognitive reserve variables along with pathology variables allowed us to measure the beneficial effect of cognitive reserve against cognitive decline due to ongoing pathological processes.
Funding
This work was supported by NIH grants R00 AG37573, R01 AG11378, P50 AG16574, U01 AG06786; the Gerald and Henrietta Rauenhorst Foundation grant, the Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Foundation, the Elsie and Marvin Dekelboum Family Foundation, U.S.A and Opus building NIH grant C06 RR018898. The funding sources were not involved in the manuscript review or approval.
References
- Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, et al. Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology. 2010;75:1195–202. doi: 10.1212/WNL.0b013e3181f4d7f8. [DOI] [PubMed] [Google Scholar]
- Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z, et al. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology. 2006;67:1960–5. doi: 10.1212/01.wnl.0000247053.45483.4e. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–7. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Brickman AM, Siedlecki KL, Muraskin J, Manly JJ, Luchsinger JA, Yeung LK, et al. White matter hyperintensities and cognition: testing the reserve hypothesis. Neurobiol Aging. 2011;32:1588–98. doi: 10.1016/j.neurobiolaging.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breteler MM. Vascular risk factors for Alzheimer's disease: an epidemiologic perspective. Neurobiol Aging. 2000;21:153–60. doi: 10.1016/s0197-4580(99)00110-4. [DOI] [PubMed] [Google Scholar]
- Chui HC, Zheng L, Reed BR, Vinters HV, Mack WJ. Vascular risk factors and Alzheimer's disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review. Alzheimers Res Ther. 2012;4:1. doi: 10.1186/alzrt98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCarli C, Murphy DG, Tranh M, Grady CL, Haxby JV, Gillette JA, et al. The effect of white matter hyperintensity volume on brain structure, cognitive performance, and cerebral metabolism of glucose in 51 healthy adults. Neurology. 1995;45:2077–84. doi: 10.1212/wnl.45.11.2077. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. The vascular hypothesis of Alzheimer's disease: bench to bedside and beyond. Neurodegener Dis. 2010;7:116–21. doi: 10.1159/000285520. [DOI] [PubMed] [Google Scholar]
- Del Ser T, Hachinski V, Merskey H, Munoz DG. Alzheimer's disease with and without cerebral infarcts. J Neurol Sci. 2005;231:3–11. doi: 10.1016/j.jns.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Dodge HH, Wang CN, Chang CC, Ganguli M. Terminal decline and practice effects in older adults without dementia: the MoVIES project. Neurology. 2011;77:722–30. doi: 10.1212/WNL.0b013e31822b0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan H, Crain B, Troncoso J, Resnick SM, Zonderman AB, Obrien RJ. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore longitudinal study of aging cohort. Ann Neurol. 2010;68:231–40. doi: 10.1002/ana.22055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–29. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- Duff K, Beglinger LJ, Moser DJ, Paulsen JS, Schultz SK, Arndt S. Predicting cognitive change in older adults: the relative contribution of practice effects. Arch Clin Neuropsychol. 2010;25:81–8. doi: 10.1093/arclin/acp105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geda YE, Topazian HM, Roberts LA, Roberts RO, Knopman DS, Pankratz VS, et al. Engaging in cognitive activities, aging, and mild cognitive impairment: a population-based study. J Neuropsychiatry Clin Neurosci. 2011;23:149–54. doi: 10.1176/appi.neuropsych.23.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis M, Sobrino T, Ois A, Millan M, Rodriguez-Campello A, Perez de la Ossa N, et al. Plasma beta-amyloid 1-40 is associated with the diffuse small vessel disease subtype. Stroke. 2009;40:3197–201. doi: 10.1161/STROKEAHA.109.559641. [DOI] [PubMed] [Google Scholar]
- Gouw AA, Seewann A, Vrenken H, van der Flier WM, Rozemuller JM, Barkhof F, et al. Heterogeneity of white matter hyperintensities in Alzheimer's disease: post-mortem quantitative MRI and neuropathology. Brain. 2008;131(Pt 12):3286–98. doi: 10.1093/brain/awn265. [DOI] [PubMed] [Google Scholar]
- Gurol ME, Irizarry MC, Smith EE, Raju S, Diaz-Arrastia R, Bottiglieri T, et al. Plasma beta-amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology. 2006;66:23–9. doi: 10.1212/01.wnl.0000191403.95453.6a. [DOI] [PubMed] [Google Scholar]
- Gurol ME, Viswanathan A, Gidicsin C, Hedden T, Martinez-Ramirez S, Dumas A, et al. Cerebral amyloid angiopathy burden associated with leukoaraiosis: A positron emission tomography/magnetic resonance imaging study. Ann Neurol. 2013;73:529–36. doi: 10.1002/ana.23830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedden T, Oh H, Younger AP, Patel TA. Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology. 2013;80:1341–8. doi: 10.1212/WNL.0b013e31828ab35d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedden T, Van Dijk KR, Shire EH, Sperling RA, Johnson KA, Buckner RL. Failure to modulate attentional control in advanced aging linked to white matter pathology. Cereb Cortex. 2012;22:1038–51. doi: 10.1093/cercor/bhr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Howieson DB, Dame A, Sexton G, Kaye J. Longitudinal analysis of the effects of the aging process on neuropsychological test performance in the healthy young-old and oldest-old. Dev Neuropsychol. 2000;17:323–37. doi: 10.1207/S15326942DN1703_3. [DOI] [PubMed] [Google Scholar]
- Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010;120:287–96. doi: 10.1007/s00401-010-0718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–31. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr., Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257–62. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr., Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131(Pt 3):665–80. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Understanding the pathology of vascular cognitive impairment. J Neurol Sci. 2005;229-230:57–63. doi: 10.1016/j.jns.2004.11.029. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. The enigma of mixed dementia. Alzheimers Dement. 2007;3:40–53. doi: 10.1016/j.jalz.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Kalaria RN. The role of cerebral ischemia in Alzheimer's disease. Neurobiol Aging. 2000;21:321–30. doi: 10.1016/s0197-4580(00)00125-1. [DOI] [PubMed] [Google Scholar]
- Kantarci K, Weigand SD, Przybelski SA, Preboske GM, Pankratz VS, Vemuri P, et al. MRI and MRS predictors of mild cognitive impairment in a population-based sample. Neurology. 2013;81:126–33. doi: 10.1212/WNL.0b013e31829a3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Petersen RC, Przybelski SA, Weigand SD, Shiung MM, Whitwell JL, et al. Hippocampal volumes, proton magnetic resonance spectroscopy metabolites, and cerebrovascular disease in mild cognitive impairment subtypes. Arch Neurol. 2008;65:1621–8. doi: 10.1001/archneur.65.12.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozachuk WE, DeCarli C, Schapiro MB, Wagner EE, Rapoport SI, Horwitz B. White matter hyperintensities in dementia of Alzheimer's type and in healthy subjects without cerebrovascular risk factors. A magnetic resonance imaging study. Arch Neurol. 1990;47:1306–10. doi: 10.1001/archneur.1990.00530120050009. [DOI] [PubMed] [Google Scholar]
- Launer LJ, Hughes TM, White LR. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol. 2011;70:774–80. doi: 10.1002/ana.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launer LJ, Petrovitch H, Ross GW, Markesbery W, White LR. AD brain pathology: vascular origins? Results from the HAAS autopsy study. Neurobiol Aging. 2008;29:1587–90. doi: 10.1016/j.neurobiolaging.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo RY, Jagust WJ. Vascular burden and Alzheimer disease pathologic progression. Neurology. 2012;79:1349–55. doi: 10.1212/WNL.0b013e31826c1b9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longstreth WT, Jr., Sonnen JA, Koepsell TD, Kukull WA, Larson EB, Montine TJ. Associations between microinfarcts and other macroscopic vascular findings on neuropathologic examination in 2 databases. Alzheimer Dis Assoc Disord. 2009;23:291–4. doi: 10.1097/WAD.0b013e318199fc7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med. 2005;46:1959–72. [PubMed] [Google Scholar]
- Lowe VJ, Kemp BJ, Jack CR, Jr., Senjem M, Weigand S, Shiung M, et al. Comparison of 18F-FDG and PiB PET in cognitive impairment. J Nucl Med. 2009;50:878–86. doi: 10.2967/jnumed.108.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machulda MM, Pankratz VS, Christianson TJ, Ivnik RJ, Mielke MM, Roberts RO, et al. Practice effects and longitudinal cognitive change in normal aging vs. incident mild cognitive impairment and dementia in the Mayo Clinic Study of Aging. Clin Neuropsychol. 2013;27:1247–64. doi: 10.1080/13854046.2013.836567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machulda MM, Pankratz VS, Christianson TJ, Ivnik RJ, Mielke MM, Roberts RO, et al. [Formula: see text]Practice effects and longitudinal cognitive change in normal aging vs. incident mild cognitive impairment and dementia in The Mayo Clinic Study of Aging. Clin Neuropsychol. 2013;27:1247–64. doi: 10.1080/13854046.2013.836567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NL, Reed BR, DeCarli CS, Madison CM, Weiner MW, Chui HC, et al. Cerebrovascular disease, beta-amyloid, and cognition in aging. Neurobiol Aging. 2012;33:1006 e25–36. doi: 10.1016/j.neurobiolaging.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NL, Reed BR, Sanossian N, Madison CM, Kriger S, Dhada R, et al. The aging brain and cognition: contribution of vascular injury and abeta to mild cognitive dysfunction. JAMA Neurol. 2013;70:488–95. doi: 10.1001/2013.jamaneurol.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray ME, Vemuri P, Preboske GM, Murphy MC, Schweitzer KJ, Parisi JE, et al. A quantitative postmortem MRI design sensitive to white matter hyperintensity differences and their relationship with underlying pathology. J Neuropathol Exp Neurol. 2012;71:1113–22. doi: 10.1097/NEN.0b013e318277387e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Z, Esiri MM, Jobst KA, Morris JH, King EM, McDonald B, et al. The effects of additional pathology on the cognitive deficit in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:165–70. doi: 10.1097/00005072-199702000-00007. [DOI] [PubMed] [Google Scholar]
- Nichols ME, Meador KJ, Loring DW, Poon LW, Clayton GM, Martin P. Age-related changes in the neurologic examination of healthy sexagenarians, octogenarians, and centenarians. J Geriatr Psychiatry Neurol. 1994;7:1–7. doi: 10.1177/089198879400700101. [DOI] [PubMed] [Google Scholar]
- Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–62. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Roberts RO, Knopman DS, Geda YE, Cha RH, Pankratz VS, et al. Prevalence of mild cognitive impairment is higher in men. The Mayo Clinic Study of Aging. Neurology. 2010;75:889–97. doi: 10.1212/WNL.0b013e3181f11d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovitch H, Ross GW, Steinhorn SC, Abbott RD, Markesbery W, Davis D, et al. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann Neurol. 2005;57:98–103. doi: 10.1002/ana.20318. [DOI] [PubMed] [Google Scholar]
- Pike KE, Ellis KA, Villemagne VL, Good N, Chetelat G, Ames D, et al. Cognition and beta-amyloid in preclinical Alzheimer's disease: Data from the AIBL study. Neuropsychologia. 2011;49:2384–90. doi: 10.1016/j.neuropsychologia.2011.04.012. [DOI] [PubMed] [Google Scholar]
- Proust-Lima C, Amieva H, Letenneur L, Orgogozo JM, Jacqmin-Gadda H, Dartigues JF. Gender and education impact on brain aging: a general cognitive factor approach. Psychol Aging. 2008;23:608–20. doi: 10.1037/a0012838. [DOI] [PubMed] [Google Scholar]
- Raz L, Jayachandran M, Tosakulwong N, Lesnick TG, Wille SM, Murphy MC, et al. Thrombogenic microvesicles and white matter hyperintensities in postmenopausal women. Neurology. 2013;80:911–8. doi: 10.1212/WNL.0b013e3182840c9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Kennedy KM, Acker JD. Vascular health and longitudinal changes in brain and cognition in middle-aged and older adults. Neuropsychology. 2007;21:149–57. doi: 10.1037/0894-4105.21.2.149. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology. 2010;74:807–15. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riekse RG, Leverenz JB, McCormick W, Bowen JD, Teri L, Nochlin D, et al. Effect of vascular lesions on cognition in Alzheimer's disease: a community-based study. J Am Geriatr Soc. 2004;52:1442–8. doi: 10.1111/j.1532-5415.2004.52405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RO, Geda YE, Knopman DS, Cha RH, Pankratz VS, Boeve BF, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30:58–69. doi: 10.1159/000115751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RO, Geda YE, Knopman DS, Cha RH, Pankratz VS, Boeve BF, et al. The incidence of MCI differs by subtype and is higher in men: The Mayo Clinic Study of Aging. Neurology. 2012;78:342–51. doi: 10.1212/WNL.0b013e3182452862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca WA, Yawn BP, St Sauver JL, Grossardt BR, Melton LJ., 3rd History of the Rochester Epidemiology Project: half a century of medical records linkage in a US population. Mayo Clin Proc. 2012;87:1202–13. doi: 10.1016/j.mayocp.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon DP, Ferris SH, Thomas RG, Sano M, Cummings JL, Sperling RA, et al. Age and apolipoprotein E genotype influence rate of cognitive decline in nondemented elderly. Neuropsychology. 2013;27:391–401. doi: 10.1037/a0032707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Wilson RS, Cochran EJ, Bienias JL, Arnold SE, Evans DA, et al. Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology. 2003;60:1082–8. doi: 10.1212/01.wnl.0000055863.87435.b2. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis. 2009;18:691–701. doi: 10.3233/JAD-2009-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senjem ML, Lowe V, Kemp B, Weigand S, Knopman D, Boeve B, et al. Automated ROI analysis of 11C Pittsburgh compound B images using structural magnetic resonance imaging atlases, Alzheimer's and Dementia. Alzheimer's Association International Conference on Alzheimer's Disease. 2008: Elsevier Inc; 2008. [Google Scholar]
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–7. [PubMed] [Google Scholar]
- St Sauver JL, Grossardt BR, Yawn BP, Melton LJ, 3rd, Pankratz JJ, Brue SM, et al. Data resource profile: the Rochester Epidemiology Project (REP) medical records-linkage system. Int J Epidemiol. 2012;41:1614–24. doi: 10.1093/ije/dys195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Sauver JL, Grossardt BR, Yawn BP, Melton LJ, 3rd, Rocca WA. Use of a medical records linkage system to enumerate a dynamic population over time: the Rochester epidemiology project. Am J Epidemiol. 2011;173:1059–68. doi: 10.1093/aje/kwq482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AJ, Perry R, Barber R, Kalaria RN, O'Brien JT. Pathologies and pathological mechanisms for white matter hyperintensities in depression. Ann N Y Acad Sci. 2002;977:333–9. doi: 10.1111/j.1749-6632.2002.tb04835.x. [DOI] [PubMed] [Google Scholar]
- Thomas AJ, O'Brien JT, Barber R, McMeekin W, Perry R. A neuropathological study of periventricular white matter hyperintensities in major depression. J Affect Disord. 2003;76:49–54. doi: 10.1016/s0165-0327(02)00064-2. [DOI] [PubMed] [Google Scholar]
- van Exel E, Gussekloo J, de Craen AJ, Bootsma-van der Wiel A, Houx P, Knook DL, et al. Cognitive function in the oldest old: women perform better than men. Journal of neurology, neurosurgery, and psychiatry. 2001;71:29–32. doi: 10.1136/jnnp.71.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hooren SA, Valentijn AM, Bosma H, Ponds RW, van Boxtel MP, Jolles J. Cognitive functioning in healthy older adults aged 64-81: a cohort study into the effects of age, sex, and education. Neuropsychology, development, and cognition Section B, Aging, neuropsychology and cognition. 2007;14:40–54. doi: 10.1080/138255890969483. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Roberts RO, Lowe VJ, et al. Effect of lifestyle activities on Alzheimer disease biomarkers and cognition. Ann Neurol. 2012;72:730–8. doi: 10.1002/ana.23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Weigand SD, Przybelski SA, Knopman DS, Smith GE, Trojanowski JQ, et al. Cognitive reserve and Alzheimer's disease biomarkers are independent determinants of cognition. Brain. 2011;134(Pt 5):1479–92. doi: 10.1093/brain/awr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan A, Rocca WA, Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology. 2009;72:368–74. doi: 10.1212/01.wnl.0000341271.90478.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Larson EB, Sonnen JA, Shofer JB, McCormick W, Bowen JD, et al. Blood pressure and brain injury in older adults: findings from a community-based autopsy study. J Am Geriatr Soc. 2009;57:1975–81. doi: 10.1111/j.1532-5415.2009.02493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller RO, Boche D, Nicoll JA. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer's disease and their potential impact on therapy. Acta Neuropathol. 2009;118:87–102. doi: 10.1007/s00401-009-0498-z. [DOI] [PubMed] [Google Scholar]
- White L. Brain lesions at autopsy in older Japanese-American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu-Asia aging study. J Alzheimers Dis. 2009;18:713–25. doi: 10.3233/JAD-2009-1178. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Boyle PA, Yu L, Barnes LL, Schneider JA, Bennett DA. Life-span cognitive activity, neuropathologic burden, and cognitive aging. Neurology. 2013;81:314–21. doi: 10.1212/WNL.0b013e31829c5e8a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekry D, Duyckaerts C, Moulias R, Belmin J, Geoffre C, Herrmann F, et al. Degenerative and vascular lesions of the brain have synergistic effects in dementia of the elderly. Acta Neuropathol. 2002;103:481–7. doi: 10.1007/s00401-001-0493-5. [DOI] [PubMed] [Google Scholar]