

Abstract
As a biomarker of beta-amyloid, positron emission tomography (PET) amyloid imaging offers a unique opportunity to detect the presence of this protein in the human body during life. Besides Alzheimer’s disease (AD), deposits of beta-amyloid in the brain are also present in other neurodegenerative diseases associated to dementia, such as Parkinson’s disease and dementia with Lewy bodies, as well as in other processes affecting brain function, such as cerebral amyloid angiopathy, brain trauma, Down’s syndrome and meningiomas, as shown by post-mortem pathology studies. Furthermore, in systemic amyloidosis other organs besides the brain are affected, and amyloid PET imaging may be suitable for the identification of these extra-cerebral amyloid depositions. Finally, the potential use of amyloid PET tracer accumulation in cerebral white matter (WM) as a marker of myelin is being investigated, leading to some promising results in patients with WM lesions and multiple sclerosis. In this article, a review of the ongoing research pointing to a broader application of amyloid PET imaging in clinical practice beyond AD is provided.
Keywords: Amyloid PET, Brain trauma, Cardiac amyloidosis, Multiple sclerosis, Cerebral amyloid angiopathy, Down’s syndrome
Introduction
The development of 11C-PIB allowed for the first time detection of beta-amyloid plaques in the human brain during life using positron emission tomography (PET) imaging. Subsequent development of 18F-labeled amyloid PET tracers, such as 18F-florbetapir, 18F-florbetaben, 18F-flutemetamol (all three FDA and EMA approved) and 18F-AZD4694 has contributed to a widespread use of amyloid PET imaging. Except for the latter, which still is an investigational compound, the approved indication is for PET imaging of beta-amyloid neuritic plaque density in the brains of adult patients with cognitive impairment who are being evaluated for Alzheimer’s disease (AD) and other causes of cognitive impairment. However, as a biomarker of beta-amyloid, PET amyloid imaging offers a unique opportunity to detect the presence of this protein in the human body during life. Hence, active research has been already initiated to investigate the potential clinical relevance of the in vivo diagnosis of other conditions beyond AD, which are known to present with beta-amyloid depositions from post-mortem pathology studies. This is the case of other neurodegenerative diseases associated to dementia, such as Lewy body diseases, including dementia with Lewy bodies (DLB) and Parkinson’s disease (PD), as well as in other processes affecting brain function, such as cerebral amyloid angiopathy (CAA), brain trauma including traumatic brain injury (TBI) and chronic traumatic encephalopathy (CTE), Down’s syndrome (DS), and meningiomas. Furthermore, in systemic amyloidosis other organs besides the brain are affected, and amyloid PET imaging may be suitable for the identification of these extra-cerebral amyloid depositions. Cardiac amyloidosis (CA) is of particular interest, since it is the main cause of mortality and morbidity in systemic amyloidosis. Finally, the potential use of amyloid PET tracer accumulation in cerebral white matter (WM) as a marker of myelin is being investigated, leading to some promising results in patients with WM lesions and multiple sclerosis (MS). In this article, a review of the ongoing research pointing to a broader application of amyloid PET imaging in clinical practice beyond AD is provided. For this purpose, a PubMed search was performed using the combination of the terms “Amyloid PET” or “beta-amyloid” and each of the following: “Parkinson’s disease”, “DLB”, “CAA”, “TBI”, “Down’s syndrome”, “meningiomas”, “cardiac amyloidosis”, “multiple sclerosis”. Publications including amyloid PET imaging data in humans were selected. Additional publications that came out from the references of the retrieved articles were also included. 11C-PIB was by large the most used tracer. Despite further data with 18F-labeled amyloid PET tracers is definitively needed, the reported relationship between 11C-PIB and 18F-labeled amyloid PET tracers behavior [1–4], supports the hypothesis that similar results could be obtained with these other tracers.
Lewy body diseases: dementia with Lewy bodies and Parkinson’s disease
Co-pathology of beta-amyloid, tau and alpha-synuclein is present in several neurodegenerative diseases, including AD, PD, and DLB, and some cases of multiple system atrophy (MSA). DLB is the second most common neurodegenerative dementia, after AD [5]. Moreover, together with PD, it is the most common synucleinopathy associated with dementia. Both PD and DLB patients share pathology findings, including cortical Lewy body accumulation/alpha-synuclein toxicity and beta-amyloid plaques [6], and show clinically similar manifestations. It has therefore been hypothesized that PD patients with dementia (PDD) and DLB may be different phenotypic expressions of the same underlying process [7]. Some differences include the time elapsed between the onset of Parkinsonism and the manifestation of dementia, which is shorter for DLB than for PD [8]. According to post-mortem data, beta-amyloid can be found in about one-fourth of patients with alpha-synuclein, with the highest proportion in DLB, followed by PDD [9]. It has been hypothesized that early and significant amyloid burden may accelerate cognitive decline in patients with DLB [10]. However, the relative timing of these events cannot be determined by postmortem studies, and the clinical significance of the beta-amyloid deposits in DLB is still unclear. In agreement with this hypothesis, Villemagne et al. [11] found a trend to a significant correlation between neocortical 18F-florbetaben standardized uptake value ratio (SUVR) and time elapsed between onset of cognitive impairment and development of the diagnostic clinical features in DLB, which was not found in AD or fronto-temporal lobe degeneration patients. Therefore, amyloid PET imaging may provide some light into the consequences of beta-amyloid deposition and its relationship with cognitive decline and other clinical, demographic, genetic, and biochemical characteristics. The level of amyloid PET tracer binding, as well as its regional localization and the relationship with clinical symptomatology has been investigated in a series of studies including PDD and DLB patients, in addition to AD and normal controls. Amyloid PET imaging findings in PD are subject of an independent article in this journal issue (Frey K). Hence, data focused on DLB studies is provided here.
The finding of increased amyloid PET tracer uptake in DLB patients and PDD varies across series (Table 1). Patient characteristics and recruitment sources may account for this variability. Patients recruited from dementia centers suffer primarily from cognitive decline and have a higher risk for AD, and therefore a higher frequency of positive PET scans in DLB (85–88 %) [12] than those presenting to movement disorder centers (33–44 %), who suffer mainly from motor disabilities [13, 14]. No differences in 11C-PIB uptake between PDD and DLB were found in one study [13], but a lower 11C-PIB retention in PDD than in DLB has been more often reported [12]. Actually, whereas it may be difficult to differentiate PDD patients from PD patients without dementia, who show mild 11C-PIB uptake or even overlapping distribution volume ratios (DVRs) with controls, a majority of DLB patients show similar or slightly lower levels of 11C-PIB binding than AD, and clearly higher than PD and PDD [12–16]. This finding has been replicated in one study using 18F-florbetaben [11], although the proportion of DLB patients with positive scan was lower (2 out of 7 patients) (Fig. 1). However, some DLB patients do not show beta-amyloid plaques at autopsy. In agreement with this finding, a minority of DLB patients have shown negative 11C-PIB scans (e.g. 1 out of 8 DLB patients in [15]), and five out of seven DLB patients in a study with 18F-florbetaben [11]. Differences in the prevalence of neuritic and diffuse plaques may partially explain the differential 11C-PIB retention in DLB and PDD. Both neuritic dense core plaques and diffuse plaques are associated with DLB [6] whereas in PDD, diffuse plaques are common and more prevalent [17]. It has been postulated that a “pure” diffuse Lewy body changes without accompanying AD neuropathology changes may explain the negative scan findings in DLB. A case of a 11C-PIB negative patient with DLB who showed sparse beta-amyloid levels at autopsy in some brain regions has been reported [18]. Likewise, a minority of PDD subjects may show high cortical 11C-PIB retention, similar to AD patients [12–14, 19, 20]. In a study comparing characteristics of 11C-PIB positive and negative DLB patients, Maetzler et al. [14] reported lower mini-mental score examination (MMSE) scores, higher prevalence of ApoE4 and females, and higher age at inclusion, onset of parkinsonism, and onset of dementia in 11C-PIB positive patients.
Table 1.
Summary of clinical amyloid PET results in Lewy body diseases (focus on Lewy body dementia)
| References | Study objective | Subject sample (n, clinical diagnosis) | Main amyloid PET results |
|---|---|---|---|
| Gomperts et al. [15] | 11C-PIB in DLB and PDD | 8, DLB 7, PDD 11, PD 15, AD 37, HC |
DLB higher uptake than PDD, comparable to AD PDD lower uptake, comparable to PD and HC Occipital uptake lower in AD than in DLB Relationship of parietal uptake with visuospatial impairment in DLB, PDD and PD Striatal uptake related to less impaired motor function in DLB and PDD |
| Association of 11C-PIB with motor or cognitive symptoms | |||
| Maetzler et al. [14] | Differences in PIB + and PIB − Lewy body disease patients | 9, DLB 12, PDD 14, PD |
11C-PIB − in all PD (non-demented) 11C-PIB + in 4PDD and 4 DLB 11C-PIB + associated with MMSE scores, ApoE4, age, and onset of parkinsonism and dementia |
| Claassen et al. [16] | MSA and DLB comparison with 11C-PIB and 18F-FDG | 3, MSA 3, DLB 12, HC |
All DLB increased 11C-PIB uptake No 11C-PIB uptake in MSA In DLB, correspondence between areas with hypometabolism and high 11C-PIB uptake |
| Villemagne et al. [11] | 18F-florbetaben PET comparison in MCI, FTLD, DLB, VaD, PD, AD | 20, MCI 30, AD 11, FTLD 7, DLB 5, PD 4, VaD 32, HC |
18F-florbetaben + in 29 % DLB, cortical binding distribution similar to AD, but cortical uptake lower than in AD 18F-florbetaben − in all PD Trend for SUVR correlation time elapsed between onset of cognitive impairment and diagnostic clinical features only in DLB |
| Goemperts et al. [19] | Relationship between 11C-PIB and cognitive impairment in DLB and PD | 29, PD (cognitively normal) 14, PD-MCI 18, DLB 12, PDD 85, HC |
11C-PIB uptake higher in DLB than in any of the other groups No differences in 11C-PIB uptake across PDD, PD-MCI, PD and HC 11C-PIB increased with age and ApoE4 in all patient groups DLB only PIB association with impaired cognition |
| Ikonomovic et al. [18] | 11C-PIB in DLB and AD with autopsy findings | 1, Probable DLB and possible AD 1, Probable AD |
11C-PIB − in probable DLB, with scares beta-amyloid plaques in less than 2 % cortical area at autopsy 11C-PIB + in probable AD with plaques in up to 12 % cortical area |
| Shimada et al. [89] | Relationship between 11C-PIB and cortical atrophy in DLB. | 8, DLB (with dementia) 7, PDD 13, AD 17, HC |
11C-PIB + in forty percent of DLB/PDD 11C-PIB + in all AD 11C-PIB − in all HC 11C-PIB + DLB/PDD/AD had similar pattern of cortical atrophy 11C-PIB − DLB/PDD had no significant cortical atrophy |
| Kim et al. [24] | 11C-PIB in MSA | 9, MSA (without dementia) 6, MSA (with dementia) 10, HC |
11C-PIB − in MSA, comparable to controls |
+ Positive scan, − negative scan, AD Alzheimer’s disease, DLB dementia with Lewy bodies, PD Parkinson’s disease, PDD Parkinson’s disease dementia, MCI mild cognitive impairment, HC healthy controls, FTLD frontotemporal lobe degeneration, VaD vascular dementia, MSA multiple system atrophy, MMSE mini-mental state examination
Fig. 1.
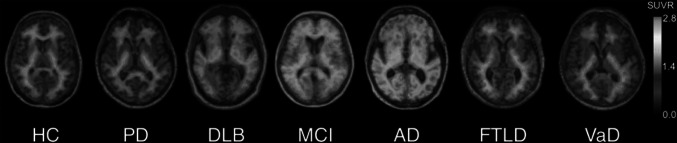
Representative 18F-florbetaben PET transaxial images overlaid on individual coregistered MR images of a healthy control (HC) and patients with Parkinson’s disease (PD), dementia with Lewy bodies (DLB), mild cognitive impairment (MCI), Alzheimer’s disease (AD), frontotemporal lobar degeneration (FTLD), and vascular dementia (VaD). Only the DLB, MCI and AD patients show cortical 18F-florbetaben uptake in addition to the non-specific white matter uptake visible in negative scans of HC, PD, FTLD, and VaD. The cortical tracer uptake in DLB is lower than in AD. All images are scaled to same SUVR maximum. This research was originally published in JNM. Villemagne et al. [11] © by the Society of Nuclear Medicine and Molecular Imaging, Inc
With regard to the regional localization of amyloid PET tracer uptake, Gomperts et al. [15] reported a higher accumulation of 11C-PIB in the occipital lobe in DLB, PDD and PD patients than in AD, suggesting that this localized amyloid burden might be related to, or may be the cause of the occipital hypometabolism frequently found in 18F-FDG PET scans in these patients [21]. Regional 11C-PIB binding in the occipital and primary visual cortex in DLB patients, as well as in other areas such as prefrontal and parieto-temporal, have reported to correspond to regions with hypometabolism shown in 18F-FDG PET images [14, 16]. Some relationships of 11C-PIB uptake patterns and clinical symptoms have been reported. Regional amyloid deposition in the parietal (lateral and precuneus)/posterior cingulate region in DLB, PDD, and PD was related to visuospatial impairment [15]. Foster et al. [13] reported an association between global and caudate 11C-PIB increased uptake and severity of global cognitive impairment in both DLB and PDD, but not with other neuropsychological features. Striatal 11C-PIB retention in the DLB and PDD groups has been associated with less impaired motor function, suggesting that the presence of beta-amyloid in striatum of DLB and PDD patients at autopsy [22] is not a marker of motor dysfunction, but could be an early site for amyloid deposition in DLB [12, 14]. Interestingly, a higher 11C-PIB binding in striatum seems to be a present in familial than in sporadic AD cases [23]. These findings point to a clinical meaning of the topography of beta-amyloid accumulation, hence encouraging the regional assessment in front of a global assessment of amyloid PET tracer uptake, to gather more information on the potential implications for diagnosis and prognosis.
Amyloid PET imaging can also be useful in the differentiation of patients with DLB and MSA, since both may present with rapid eye movement sleep behavior disorder and severe autonomic dysfunction, and in some cases, MSA patients may present cognitive impairment and dementia [24, 25]. Although the pathology hallmarks are different for each disease (i.e. alpha-synuclein in DLB and oligodendroglial cellular inclusions in MSA), co-pathology coexists in 10–20 % of MSA cases. However, while amyloid pathology is frequent in DLB, it is very rare in MSA [26]. Preliminary amyloid PET imaging results using 11C-PIB in DLB and MSA patients have been reported. Claassen et al. [16] found no 11C-PIB uptake in any of the three MSA patients included in their study, but in all the three DLB patients included. Kim et al. [24] reported no statistically significant differences in global 11C-PIB binding between normal controls and MSA patients, neither when comparing the subgroups of MSA patients without and with dementia with normal controls (Table 1). These findings suggest that amyloid PET imaging could be helpful in the differential diagnosis in cases where DLB and MSA may present with similar clinical symptoms. However, one MSA patient without dementia and two MSA patients with dementia in Kim et al. [24] series, were found to show quantitative 11C-PIB values above the cut-off threshold of 1.5 for 11C-PIB positivity, which might be explained by the rare MSA cases reported with amyloid pathology [26], and contributing to dementia in the two latter cases. Authors proposed a longitudinal follow-up of the 11C-PIB positive MSA patient without dementia, considering that amyloid burden is detected even in the presymptomatic phase of AD [27]. Further studies with larger patient samples and 18F-labeled amyloid tracers are needed to establish the usefulness of amyloid PET imaging in the differential diagnosis of DLB and MSA, and the role of beta-amyloid deposition in the pathogenesis and clinical course of dementia in these diseases. Future research should be aiming to determine whether amyloid PET imaging can help identifying which MCI patients will progress to AD or DLB, and to clarify the potential clinical implications of the topography of the different regional beta-amyloid depositions in these neurodegenerative diseases. Moreover, multimodal molecular imaging, allowing the in vivo mapping of dopamine transporters, glucose metabolism, neuroinflammation and tau, besides amyloid, can be used to achieve an accurate pathology diagnosis and classification in vivo.
Cerebral amyloid angiopathy
Perivascular amyloid depositions are a hallmark of CAA, a major cause of spontaneous hemorrhagic stroke [microhemorrhages (MH), intracerebral hemorrhage (ICH)) and vascular cognitive impairment in the elderly [28]. CAA is present in about one-third of subjects 60 years or older, and in up to 90 % of AD patients in post-mortem studies [29]. Similarly to what happens in AD, the clinical diagnosis of “possible” and “probable” CAA is currently based on clinical guidelines such as the Boston operational criteria [30], but brain tissue pathology is needed for confirmation. An accurate and early diagnosis is of importance given the high morbidity and mortality rates associated with ICHs [28], and the risk of vasogenic edema associated with MH reported in clinical trials with anti-beta-amyloid immunotherapy for AD [31]. An accurate, non-invasive biomarker would also be useful in monitoring novel drugs for CAA.
Amyloid PET tracer binding to fibrillar amyloid in perivascular beta-amyloid depositions (hallmark of CAA) in addition to beta-amyloid plaques in the cerebral parenchyma (hallmark of AD), has been reported using 11C-PIB [32, 33] which hass been the most extensively used tracer to investigate the potential clinical utility of amyloid imaging in CAA (Table 2). Since the PET signal cannot distinguish between binding in parenchymal beta-amyloid plaques and beta-amyloid in vessel walls, and given the high incidence of CAA in AD patients, the inclusion of non-demented CAA patients in the amyloid PET studies is an indirect way to minimize the effect of accompanying AD pathology [34]. This may explain the lack of significant differences in average cortical 18F-florbetapir SUVR found in one study between AD cases with and without higher CAA density [35], in contrast to the replicated finding of a higher 11C-PIB uptake in non-demented CAA patients, which has been consistently reported to be intermediate between healthy controls and AD [34, 36, 37]. Interestingly, the amyloid accumulation pattern seems to differ from the AD pattern in that the occipital lobe is more frequently affected in CAA (Fig. 2). Quantification may be required to pick up better this different pattern than visual assessment [38], and a higher occipital to global 11C-PIB ratio in CAA than in AD has been a replicated finding [34, 36, 37]. Further in-vivo data supporting the feasibility for the identification on 11C-PIB binding in vascular amyloid include the co-localization of 11C-PIB uptake with MH as detected by magnetic resonance imaging (MRI) [39], the correlation with WM hyperitensities (WMH) volume in probable CAA patients but not in AD or cognitively normal controls [40] and the higher 11C-PIB uptake in cognitively normal controls with lobar MH than in those without lobar MH [41].
Table 2.
Summary of clinical amyloid PET results in cerebral amyloid angiopathy
| References | Study objective | Subject sample (n, clinical diagnosis) | Main amyloid PET results |
|---|---|---|---|
| Johnson et al. [90] | 11C-PIB detection of CAA | 6, CAA non-demented 15, HC 9, Probable AD |
All CAA and AD PIB + (DVR, visual) 11C-PIB global cortical retention in CAA higher than HCs, lower than AD Occipital to global 11C-PIB ratio higher in CAA than AD |
| Dierksen et al. [36] | Spatial relationship between CAA and microbleeds | 16, Probable CAA non demented-580 microbleed regions on T2-MRI |
11C-PIB global cortical retention in CAA higher than HCs, lower than AD Occipital to global 11C-PIB ratio higher in CAA than AD Mean DVR at microbleed loci greater than at sites of simulated lesions DVR declined with increasing distance from the microbleed center |
| Ly et al. [37] | 11C-PIB in CAA and AD | 12, CAA 22, NC 13, AD |
11C-PIB global cortical retention in CAA higher than HVs, lower than AD Occipital to global PIB ratio higher in CAA than AD |
| Yates et al. [41] | 11C-PIB in asymptomatic elderly is associated with lobar MH | 84, HC 28, MCI 26, Probable AD |
HC with LMH higher SUVR than HC without LMH Positive correlation between number of LMH and SUVR Positive correlation between number of LMH and age Both SUVR and age predicted the occurrence of LMH using logistic regression |
| Gurol et al. [39] | 11C-PIB prediction of location and number of future hemorrhages in CAA (longitudinal study) | 11, CAA non demented | Mean DVR was greater at sites of incident bleeds than simulated lesions DVR declines with increasing distance from the center Mean DVR in superior frontal/parasagittal ROI correlated with future hemorrhages after adjusting for covariates |
| Gurol et al. [40] | Contribution of vascular amyloid to chronic brain ischemia [11C-PIB correlates with MRI WMH (leukoaraiosis)] | 42, CAA non-demented 50, HC 43, AD/MCI |
Higher amounts of WMH in CAA than HC and AD/MCI, despite CAA pts were younger Strong correlation between global 11C-PIB retention and WMH in CAA but not in HC, AD |
| Chao et al. [91] | 11C-PIB association with FA (DTI) | 57, Non (n = 29)-mild (n = 28) cognitive impairment subjects with PIB PET and DTI | Ab deposition significantly associated with FA in fornix and splenium of corpus callosum |
| Baron et al. [38] | Diagnostic utility of 11C-PIB in CAA-related symptomatic lobar intracerebral hemorrhage (sl-ICH) | 11, Non-demented probable CAA-related sl-ICH 20, HC without cognitive impairment (9 elderly age-matched) |
No significant difference in whole cortex or regional DVRs between CAA and HC 4/9 aged HC and 10/11 CAA had whole cortex DVR above 95 % CL (sensitivity 91 %, specificity 55 %) Region/frontal or occipital ratios did not have better discriminative value Similar but less accurate results by visual inspection |
| Dugger et al. [34] | 18F-florbetapir in CAA, postmortem correlates | 11, Probable AD with severe CAA | No significant differences in average cortical SUVR between AD cases with and without higher CAA density |
| Sengoku et al. [42] | CAA and encephalopathy | 2, Encephalopathy with CAA | Case 1: 11C-PIB + in AD-like pattern (fronto-parieto-temporal, precuneus) Case 2: 11C-PIB + focal uptake corresponding to subarachnoid hemosiderosis and microbleeds |
+ Positive scan, − negative scan, CAA cerebral amyloid angiopathy, MH microhemorrhages, LMH lobar microhemorrhages, HC healthy controls, MCI mild cognitive impairment, AD Alzheimer’s disease, WMH white matter hyperintensities, FA fractional anisotropy, DTI diffusor tensor imaging, SUVR standard uptake value ratio, DVR distribution volume ratio
Fig. 2.

Representative 11C-PIB PET images at two transaxial levels from normal control (NC) (11C-PIB-negative), Alzheimer’s disease (AD), and cerebral amyloid angiopathy (CAA) subjects. Compared with AD and NC, CAA subjects had an intermediate level of global 11C-PIB retention, but compared with AD, had relatively increased occipital retention. Microbleeds seen in this patient, shown in coregistered gradient echo magnetic resonance images, at times appear proximal to foci of amyloid deposition (small arrows). Reproduced from Ann Neurol, Johnson et al. [34] ©2007 with permission from the American Neurological Association
Amyloid PET imaging seems to have potential to predict future hemorrhages, since new CAA-related hemorrhages occur preferentially at sites of increased amyloid deposition as detected by 11C-PIB PET images [39]. It could also be a marker of CAA severity, given the reported strong correlation between global 11C-PIB retention and WMH in CAA but not in healthy controls or AD, supporting the hypothesis that vascular amyloid burden directly contributes to chronic cerebral ischemia [40]. Moreover, 11C-PIB imaging could allow investigating atypical clinical courses of patients with CAA, as suggested by the positive 11C-PIB scan findings in two reported cases of CAA-associated encephalopathy [42]. The diagnostic utility of amyloid PET imaging in CAA-related symptomatic lobar intracerebral hemorrhage has been recently investigated using 11C-PIB [38]. A low specificity (55 %) to differentiate probable CAA from age-matched healthy controls was found, in consistency with the high frequency of 11C-PIB positive scans in healthy aged controls [43, 44]. In contrast, a high sensitivity (91 %) and negative predictive value (83 %) were found, indicating that a negative 11C-PIB scan allows ruling out CAA with good confidence. Replication of these 11C-PIB findings with 18F-labeled amyloid PET tracers would foster the clinical use of amyloid PET imaging in CAA. In particular, pathology studies similar to those reported with 11C-PIB, to confirm a similar selectivity for fibrillary amyloid in CAA are still needed with 18F-labeled tracers. Future studies aiming to further define the relationship between CAA and its clinical consequences, and investigation of the performance of novel radioligands, selective for beta-amyloid deposit in vessel walls [45, 46] in CAA are required to better understand the potential benefit of amyloid imaging in clinical practice.
Brain trauma
Brain trauma is a common cause of permanent neurological deficits, most often occurring in young people as a consequence of a single, acute head contusion. Beta-amyloid deposition has been found in 30 % of fatal TBI patients [47], thus increasing the risk of AD. Chronic repetitive TBI, as observed in athletes and military personnel, first identified in boxers as dementia pugilistica [48], may increase the risk of CTE. Neuropathologically, CTE is a tangle-predominant disease or tauopathy, but variable degrees of diffuse beta-amyloid deposition have been found in about 47 % of pathology-verified CTE cases [49].
There is preliminary evidence that amyloid PET imaging is suitable to detect cerebral beta-amyloid deposits in patients with TBI, using 11C-PIB as a tracer [50, 51] (Table 3). However, limited sample sizes and differences in study design, including differences in the patient population [i.e. severity of TBI, neuropsychological status, other brain lesions causing abnormal structural findings in computerized tomography (CT) and MRI, age], time of scanning after TBI, quantification and regions assessed, highlight the need of replication in further studies. Kawai et al. [50] examined for the first time the cerebral beta-amyloid deposition in a total of 12 patients at the chronic stage after TBI (range = 5–129 months, median 54 months). TBI was severe in nine patients and moderate in three. All patients showed some degree of neuropsychological impairment, but none of them had clinical signs of dementia. No patients had a history of repetitive mild TBI such as contact sports or boxing. Only 3 out of the 12 patients showed positive scans consistent with presence of beta-amyloid, thus questioning the hypothesis of progressive beta-amyloid accumulation over time in the traumatized brain. This finding is however consistent with pathology studies reporting extensive beta-amyloid deposition within a day, as earliest as 2 h after injury in surgically excised brain tissue after TBI [52], and absence of beta-amyloid in long-term survivors after TBI [53]. 11C-PIB uptake was moderate, with lower binding potential values than those observed in two age- and MMSE score-matched AD patient controls. There was no correlation between 11C-PIB deposition and the severity of injury, initial CT findings, elapsed time from the injury, and neuropsychological test scores. Hong et al. [51] studied 15 patients with moderate to severe TBI from 3 days to 1 year after injury. No data on patient cognitive status was provided. Significantly increased 11C-PIB DVRs and SUVRs were found in cortical gray matter and striatum of TBI patients as a group, but not in thalamus or WM. The difference in the results from Kawai et al. [50] study may be explained by the shorter time elapsed in this case between the TBI and the scan, which is further supported by the finding of neocortical gray matter plaques found with immunohistochemistry and 3H-PIB autoradiography performed in the same study in an additional independent cohort of 16 patients who died between 3 h and 56 days after TBI. Data also suggested substantial spatial and temporal variations in binding (Fig. 3), although sample size and numbers at late time points were too small to draw robust conclusions, and subjects were not tested longitudinally. Two recently published case reports using 18F-florbetapir scan in the clinical evaluation of one patient after 1 year of severe TBI and another patient with CTE, found negative scan results in both patients [54].
Table 3.
Summary of clinical amyloid PET results in brain trauma
| References | Study objective | Subject sample (n, clinical diagnosis) | Main amyloid PET results |
|---|---|---|---|
| Kawai et al. [50] | 11C-PIB in patients with neuropsychological impairment after TBI | 12, Post-traumatic neuropsychological impairment (PET at the chronic stage up to 129 months after TBI) |
11C-PIB + in 3/11C-PIB − in 9 No correlation of 11C-PIB deposition with severity of injury |
| Hong et al. [51] | 11C-PIB in TBI | 15, TBI (within 1 year after TBI) 11, HC |
11C-PIB uptake higher in TBI than HC in cortex and striatum |
| Mitsis et al. [54] | Two case reports with 18F-florbetapir | 1 TBI 1 CTE |
18F-florbetapir − in the TBI patient 18F-florbetapir − in the CTE patient, but focal accumulation at the site of impact in the occipital region |
+ Positive scan, − negative scan, HC healthy controls, TBI traumatic brain injury, CTE chronic traumatic encephalopathy
Fig. 3.

11C-PIB distribution volume ratio (DVR) maps from a control and three patients scanned at different times after TBI. Note the progressive decrease in 11C-PIB uptake as time after TBI increases. Reproduced from JAMA Neurol, Hong YT et al. (doi:10.1001/jamaneurol.2013.4847) with permission from American Medical Association
Although the use of amyloid PET in TBI and CTE has not been yet established, these initial findings suggest that amyloid PET imaging can offer the opportunity to early detect the neuropathology during life, and therefore to understand the pathophysiology of TBI and the mechanistic drivers of disease progression or suboptimal recovery after TBI. Identification of different beta-amyloid deposition patterns may improve etiological likelihood and help in the differential diagnosis, given the overlap in neurocognitive and behavioral symptoms across AD, TBI/CTE, and frontotemporal dementia [55]. For example, the early binding in striatum found in Hong et al. [51] is consistent with the atypical early binding reported in patients with familiar AD (presenilin-1 mutations), implying worse prognosis than sporadic forms of AD [23]. Future studies are needed to determine the influence of age and genetic factors, the prognostic value of the presence of beta-amyloid in the brain after TBI, and to longitudinally address the time course of amyloid deposition and clearance, as well as the topography of beta-amyloid depositions during life. The existence and pattern of beta-amyloid depositions over time in mild TBI, which is difficult to study pathologically in post-mortem studies, is now possible. Furthermore, amyloid imaging can contribute to the study of the relationships between TBI and development of neuropsychological symptoms, including the risk to develop AD. This will have a major impact when disease modifying therapies will be available, allowing to prevent or at least delay the clinical manifestations of neurodegenerative disease after TBI.
Down’s syndrome
DS is a developmental disorder caused by a chromosome 21 triplication, which leads to an over expression of the APP gene and cerebral beta-amyloid deposition [56, 57]. Patients with DS have an extremely high incidence of early-onset AD [58], which has reported to be age dependent. Histopathology studies demonstrate beta-amyloid plaques in all individuals with DS 40 years and older with neurofibrillary tangles occurring generally after the age of 40 [59]. The incidence of dementia by age 69 may be as high as 75 % [60]. The current mean life expectancy of DS exceeds 50 years, with 20 % or more of the DS population now aged older than 55 years [61, 62], with the subsequent increase in the incidence of dementia in this population. Amyloid PET imaging has been used in DS as a model of the natural history of beta-amyloid deposition in the brain. Early detection of beta-amyloid deposition using imaging prior to cognitive symptoms is critical as disease-modifying therapies such as amyloid immunotherapy are developed [63–65]. Subjects from DS might benefit from novel anti-amyloid disease modifying drugs, and inclusion of this population in clinical trials would in addition facilitate targeting early phases of beta-amyloid deposition.
Beta-amyloid accumulation in DS has been reported in two PET studies using 11C-PIB PET imaging with limited sample sizes [66, 67], one study using 18F-florbetaben [68] in a larger sample, and a case report using 18F-florbetapir [69] (Table 4). These studies have replicated the finding of an age dependence of beta-amyloid accumulation in the brain in DS. In the 11C-PIB studies, which included young and older DS, all DS subjects 45 years or older (n = 5) [66], and only the two oldest (38 and 44 years) from seven non-demented DS [67] showed positive 11C-PIB scans. Positive 18F-florbetapir uptake was found in a 55-year-old end of life DS patient with AD, with post-mortem pathology confirmation [69]. Using 18F-florbetaben, Jennings et al. [68] reported a positive correlation between 18F-florbetaben SUVR and age in non-demented DS subjects. 18F-florbetaben scans were positive in 7 % of DS aged 40–44 years, in 53 % of DS aged 45–49 years, and in 90 % of DS subjects ≥50 years. No relationship with age was found in an independent cohort of 70 young cognitively normal healthy volunteers (range 21–40 years), assumed to be devoid of brain beta-amyloid (unpublished data) (Fig. 4). Both visual and quantitative assessments in the sample of n = 70 healthy volunteers and n = 39 DS were significantly and strongly related (χ2 = 50.2, p < 0.0001; Cohen’s κ = 0.71), with 92.7 % of the scans equally classified as positive or negative by both the visual read and the quantitative approach (SUVR cutoff of ≥1.45) (unpublished data). These results provided in vivo evidence of brain beta-amyloid deposition in non-demented DS subjects. Moreover, subtle cognitive changes were associated with beta-amyloid deposition. The topography of beta-amyloid depositions reported in these PET studies seems to be overall matching the cortical AD pattern, although a marked striatal uptake has been found in some DS subjects [67]. Since striatal uptake has also been reported in presymptomatic presenilin-1 mutation carriers [23], a link between beta-amyloid overproduction and early striatal deposition has been postulated [67].
Table 4.
Summary of clinical amyloid PET results in Down’s syndrome
| References | Study objective | Subject sample (n, clinical diagnosis) | Main amyloid PET results |
|---|---|---|---|
| Sabbagh et al. [69] | 18F-florbetapir in end-of-life DS with neuropathology assessment (case report) | 1, DS with AD (55 years) |
18F-florbetapir uptake similar to AD Confirmed Ab in pathology (and diffuse plaques in cerebellum) |
| Landt et al. [66] | 11C-PIB in DS with and without AD | 9, Mild to moderate demented DS (5 with AD) (25–64 years) 14, HC (33–69 years) |
11C-PIB in DS older than 45 years independently of AD |
| Handen et al. [67] | 11C-PIB in non-demented young adults with DS | 7, DS (20–44 years) | 11C-PIB + in two oldest subjects (38 and 44 years) |
| Jennings et al. [68] | Correlation of 18F-florbetaben in DS with age | 39, Non-demented DS (40–56 years) | Age-dependent 18F-florbetaben + from 7 % in 4th decade to 90 % in 5th decade Non-demented DS subjects show 18F-florbetaben + Subtle cognitive changes are associated with 18F-florbetaben uptake |
+ Positive scan, − negative scan, HC healthy controls, DS Down’s syndrome
Fig. 4.

18F-florbetaben SUVR versus age in young cognitively normal healthy controls (n = 70; SUVR = 0.001 × age + 1.173, R 2 = −0.01) (left graph) (Piramal Imaging, unpublished data) and subjects with Down’s syndrome (n = 39; SUVR = 0.030 × age + 0.045, R 2 = 0.39) (middle graph). A graph showing the percentage of positive 18F-florbetaben scans grouped by age clusters is shown in the right hand side
Altogether, these studies further replicate the ability of amyloid PET imaging to detect beta-amyloid deposition in the brain independently of the presence of dementia, and provide evidence of an age-dependence of beta-amyloid deposition in DS subjects. Since the incidence of concomitant diseases such as vascular dementia is much lower in DS than in general aged population, the development of dementia is much more likely to be beta-amyloid driven [66]. Thus, amyloid PET imaging in this population would be useful to study the role of the APP and beta-amyloid in the pathogenesis of AD [70]. Elucidating the interval between beta-amyloid deposition and the onset of dementia provides the opportunity to initiate interventions during the optimal period prior to substantial beta-amyloid deposition and clinical decline. Longitudinal studies conducted in individuals with DS would provide crucial data to establish the association of amyloid deposition with the threshold of cognitive dysfunction and further enhance our understanding of change in beta-amyloid deposition over time.
The higher incidence of dementia in DS as compared to the general population, the beta-amyloid deposition at younger ages, and the shortest time of conversion to dementia make longitudinal studies easier, which are also needed to determine the potential prognostic value of amyloid PET imaging in DS.
Meningioma
Two incidental findings of amyloid PET tracer uptake in meningiomas using PET have been reported [71, 72]. Amyloid stroma in meningiomas has been reported in a pathology study [73], therefore supporting the hypothesis of specific binding to amyloid fibrils in these tumors. However, none of the two reported cases had histologic confirmation of amyloid deposition in the lesions. Therefore, further studies are needed to replicate these findings, confirm with pathology, and investigate the mechanism of amyloid PET tracer binding in meningiomas.
Systemic/cardiac amyloidosis
Amyloidosis is characterized by the extracellular deposition of insoluble amyloid fibrils in different organs, leading to progressive organ dysfunction. In AD, the brain is affected. However, in systemic amyloidosis the brain is almost always spared, but any other organ (e.g. lungs, liver, kidneys, bones, etc.) can be affected. The disease is life-threatening and of insidious nature, thus diagnosis is frequently delayed. Advanced organ dysfunction is usually present at the time of diagnosis. Heart involvement, leading to cardiac insufficiency or arrhythmias, is the main cause of mortality and morbidity in amyloidosis, and is a differential diagnosis of heart failure. Several types of amyloid can infiltrate the heart, but the most frequent ones are systemic light chain (AL) and transthyretin (ATTR, wild type or mutant) amyloidosis [74], which have different clinical, therapeutic and prognostic implications. Cardiac AL amyloidosis is characterized by a rapidly progressive clinical course and high mortality, while ATTR wild-type disease (senile amyloidosis) typically has a slower clinical course with a much longer overall survival [75]. Although non-invasive imaging techniques such as echocardiography and MRI can support the diagnosis, the findings are non-specific and can be found in other cardiac diseases. Clinical manifestations also overlap with other heart diseases, and diagnosis is made in an advanced stage, when anatomic changes have occurred and response to therapy is slow and not easily measurable [76]. The definitive diagnosis requires endomyocardial biopsy with immunohistochemistry or mass spectroscopy to identify the type of amyloidosis. However, this is an invasive procedure with high risk of complications, and sampling errors and the lack of information about amyloid distribution, extent and progression are limitations. Therefore, non-invasive diagnostic tools allowing early detection, quantitative assessment and distribution of amyloid proteins, and monitoring disease progression and response to treatments are needed. Ability to identify potential amyloid deposition in several organs (whole body imaging) is an additional advantage. Despite SPECT imaging of CA has also been investigated (for a review, see [77]), amyloid PET may be the nuclear imaging diagnostic modality of choice in the future, given the higher resolution of PET and the high target selectivity of amyloid PET tracers, together with the growing availability of PET in most nuclear medicine departments.
However, there is still limited evidence pointing to a potential utility of amyloid PET imaging in CA. In our search, only two original studies were found, one using 11C-PIB [78], another using 18F-florbetapir [76] (Table 5). Both included limited sample of subjects, but results were quite in agreement. Independency of tracer uptake from blood flow was shown in Antoni et al. [78], and both studies reported a diffuse uptake that was significantly higher in patients, and higher amyloid tracer uptake relative to blood flow (Fig. 5). Both tracers seem to bind to amyloid fibrils of any type, since cardiac uptake was detected in all patients independently of the type of amyloidosis (AL or ATTR). A trend to higher uptake in AL was described in Antoni et al. [78], but no specific comparison between AL and ATTR was made in Dorbala et al. [76]. Distribution of tracer uptake seems to be homogeneous in all cases, including uptake in left and right ventricles and atriums. Local accumulation of tracer in the septum was reported in one of the patients in each study. Quantification was performed by using SUV, target to background ratio, and myocardial retention index (myocardial tissue radiotracer concentration divided by the integral of the blood pool time–activity curve from 0 to 20 min after injection). A threshold to separate patients from controls was found in Dorbala et al. [76]. Diffuse bilateral 18F-florbetapir uptake in the lungs of a patient with AL amyloidosis of the lungs and pleural effusions, but not in another patient with ATTR congestive heart failure and pleural effusions was reported [76]. Antoni et al. [78] found no significant differences between patients with and without melphalan treatment (6–54 months before PET). Myocardial uptake with 18F-flutemetamol has also been reported in a case report of a 56-year-old patient with well documented cardiac AL amyloidosis, compared to no myocardial uptake in an elderly healthy control [79]. In contrast, an amyloid cardiac imaging mismatch between 99mTc-aprotinin SPECT (positive) and 11C-PIB PET (negative) was reported in a 82-year-old patient with suspected CA who showed 11C-PIB positive brain scan [80] (Table 5).
Table 5.
Summary of clinical amyloid PET results in cardiac amyloidosis
| References | Study objective | Subject sample (n, clinical diagnosis) | Main amyloid PET results |
|---|---|---|---|
| Antoni et al. [78] | 11C-PIB in CA | 10, CA (AL and ATTR) 5, HC |
11C-PIB + in all CA patients 11C-PIB − in all HC Significant difference in 11C-PIB uptake in the heart between patients and controls No correlation between PIB and myocardial blood flow |
| Dorbala et al. [76] | 18F-florbetapir in CA | 9, CA (5 AL and 4 ATTR) 5, HC |
18F-florbetapir + in all CA patients 18F-florbetapir − in all HC Trend to a higher uptake in the AL subjects compared to the ATTR subjects without reaching statistical significance |
| Minamimoto et al. [80] | Case report with 11C-PIB | 1, Suspected cardiac amyloidosis |
11C-PIB + in brain 11C-PIB – in heart, 99mTc-aprotinin + in heart |
| Lhommel et al. [79] | Case report with 18F-flutemetamol | 1, Well documented cardiac AL amyloidosis 1, HC |
18F-flutemetamol + in myocardium compared with no uptake in the heart of the HC |
+ Positive scan, − negative scan, HC healthy controls, CA cardiac amyloidosis, AL light chain amyloidosis, ATTR transthyretin amyloidosis
Fig. 5.

Short-axis images of 11C-PIB retention index (RI) and myocardial blood flow (MBF) in (left to right) patients with high, intermediate, and partially increased 11C-PIB retention and a healthy control. Liver is clearly visible in 11C-PIB images of one patient (second column images) patient and healthy control, and is just outside PET field of view for other two patients. Liver uptake is due to biliary excretion of 11C-PIB and is likely not related to amyloid binding. This research was originally published in JNM. Antoni et al. [78] © by the Society of Nuclear Medicine and Molecular Imaging, Inc
These preliminary results suggest that amyloid PET can have potential utility in CA. Replication of these findings are needed from studies in larger sample sizes, including controls with heart failure to evaluate usefulness in this current differential diagnosis need. Studies powered to investigate the possibility to differentiate between AL and ATTR CA, which have different prognosis and therapies are also required. Selectivity of different amyloid PET tracer binding sites for each type of amyloidosis from pathology studies, potential clinical implications of the pattern of accumulation in different regions of the heart, as well as influence of therapy in the uptake pattern are still to be investigated.
Amyloid PET imaging as myelin biomarker
All amyloid PET tracers show binding to the WM in more or less degree, independently of the presence or absence of cortical beta-amyloid deposition (positive and negative scans, respectively). The mechanism of this binding is poorly understood, and it is considered to be non-specific, mainly due to the non-displaceable and non-saturable characteristics, which have been reported using 11C-PIB [81, 82]. The high lipid content of the WM and the lipophilic nature of the amyloid PET tracers can contribute to this non-specific binding. The hypothesis that amyloid PET tracers could bind to myelin has been investigated and led to controversial results in in vitro studies using 11C-PIB autoradiography and autofluorescence in human brain tissue, some supporting staining or binding in WM tracts [83] but not others [82]. Given that some Congo red derivatives originally described as amyloid PET markers show affinity for myelin, it has been suggested that the beta-sheet structure that is present in both the AD amyloid peptide and in the myelin binding protein, which is a major protein component of myelin, can be a common target for amyloid PET tracers [82–85]. Hence, there is increasing interest in investigating whether amyloid PET imaging could be a suitable biomarker for WM and myelin lesions, and therefore be potentially useful in detecting WM lesions and demyelination–remyelination processes as occur in MS. Again, there is very limited experience in this regard (Table 6). Stankoff et al. [83] reported average 30 % (5–57 %) reduction in 11C-PIB uptake compared to the adjacent normal-appearing WM, in a total of eight demyelinated lesions in two patients with relapsing-remitting MS. The high variation in the reduced 11C-PIB uptake among lesions was consistent with the different degrees of demyelination identified in MRI T2 sequences [86]. Interestingly, the decreased uptake was higher in MRI (3D MP-RAGE) non-active than in active lesions, suggesting that gadolinium-enhanced lesions are less demyelinated (Fig. 6). Glodzik et al. [87] reported a significant mean of 14 % reduction in 11C-PIB uptake in WM lesions than in normal-appearing WM defined by FLAIR MRI in a group of 73 cognitively healthy elderly subjects. The reduced uptake was higher in periventricular WM lesions (17.8 %) than in deep WM lesions (7.5 %), in consistency with the greater myelin binding protein loss reported using immunohistochemistry in periventricular WM lesions [88]. Although these are very preliminary results, it has been postulated that the lower reduction percentages found in WM lesions from elderly subjects compared to MS patients may indicate that demyelination associated with age is less pronounced than in MS.
Table 6.
Summary of clinical amyloid PET results in myelin lesions
| References | Study objective | Subject sample (n, clinical diagnosis) | Main amyloid PET results |
|---|---|---|---|
| Stankoff et al. [84] | 11C-PIB as a myelin marker | 2, MS (relapsing remitting) |
11C-PIB uptake reduction (5-57 %) in MS lesions Quantitative assessment of demyelination with dynamic PET analysis |
| Glodzik et al. [87] | 11C-PIB in white matter lesions | 73, Elderly subjects cognitively normal | Reduced 11C-PIB uptake in white matter lesions compared to normal appearance white matter |
+ Positive scan, − negative scan, MS multiple sclerosis
Fig. 6.

MP-RAGE 3T MRI (a, d, f) and corresponding co-registered 11C-PIB slices (b, c, e, g) from two representative patients from Stankoff et al. [83] (d–g correspond to the same patient). c Shows superposition of MRI and PET images. Arrows point to MA plaques, and arrowheads point to gray matter structures, both appearing as a loss of uptake on PET images. Reproduced from Ann Neurol, Stankoff et al. [83] with permission from the American Neurological Association
A surrogate marker to assess potential therapies in MS, by identifying the demyelination–remyelination process in the different brain lesions is an unmet need. These initial, preliminary results reported to date with amyloid PET imaging are promising. However, replication of these findings in studies with larger patient samples and different amyloid tracers are still needed. Scanner resolution limitations in the detection of small size lesions, MRI sequences used to identify lesions, region of interest method to quantify tracer uptake in PET images, potential partial volume effects, and effects of blood–brain barrier disruption or blood flow influence are methodological challenges to be overcome in future studies.
Conclusion
Investigation of amyloid PET imaging beyond AD is subject of current active research. Amyloid PET imaging offers a unique possibility to explore the relationships between beta-amyloid depositions and clinical outcomes during life. As it happens in AD, for non-AD diseases where brain amyloid is present and may be involved in the disease pathogenesis (e.g. DLB, TBI/CTE, CAA, DS), development of efficacious disease-modifying drugs will be needed in order to have a direct impact in patient treatment. In the meanwhile, the major clinical benefit in these cases would be related to early identification and most likely prognosis. Longitudinal studies are clearly needed to progress in this direction. Despite the good correlation between visual assessments and quantitative PET measurements, amyloid PET quantification becomes crucial for assessing changes in brain amyloid load over time, as well as for some research purposes, e.g. patient group comparisons or relationships with clinical features.
Current evidence of the potential usefulness of amyloid PET imaging in CA and MS is very limited. However, if the reported results are confirmed in future studies, therapeutic implications of amyloid PET imaging would be of immediate application in CA, since treatments are already available and both therapy and prognosis are different for AL and ATTR types. Early diagnosis would also have beneficial impact in treatment efficacy. Similarly, an accurate and prompt identification of the demyelination–remyelination process in cerebral lesions by a non-invasive myelin biomarker would improve management of patients with MS.
The potential clinical implications of different patterns of regional brain amyloid PET tracer uptake deserve further investigation, as the localization of brain beta-amyloid depositions is a unique benefit of amyloid PET imaging compared with other pathology biomarkers such as cerebrospinal fluid or blood tests. Most of the currently available data has been obtained using 11C-PIB. Further studies replicating 11C-PIB findings using 18F-labeled amyloid tracers, with larger sample sizes, and designed to overcome the limitations identified to date in the process, are needed and would be an important step towards the extended clinical use of amyloid PET imaging in clinical practice.
Conflict of interest
Ana M. Catafau and Santiago Bullich are employees of Piramal Imaging GmbH.
Ethical standard
This article does not contain any study with human or animal rights subjects performed by any of the authors.
References
- 1.Villemagne VL, Mulligan RS, Pejoska S, Ong K, Jones G, O’Keefe G, Chan JG, Young K, Tochon-Danguy H, Masters CL, Rowe CC. Comparison of 11C-PiB and 18F-florbetaben for Abeta imaging in ageing and Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2012;39(6):983–989. doi: 10.1007/s00259-012-2088-x. [DOI] [PubMed] [Google Scholar]
- 2.Landau SM, Breault C, Joshi AD, Pontecorvo M, Mathis CA, Jagust WJ, Mintun MA. Amyloid-beta imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med Off Publ Soc Nucl Med. 2013;54(1):70–77. doi: 10.2967/jnumed.112.109009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatashita S, Yamasaki H, Suzuki Y, Tanaka K, Wakebe D, Hayakawa H. [18F]Flutemetamol amyloid-beta PET imaging compared with [11C]PIB across the spectrum of Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2014;41(2):290–300. doi: 10.1007/s00259-013-2564-y. [DOI] [PubMed] [Google Scholar]
- 4.Rowe CC, Pejoska S, Mulligan RS, Jones G, Chan JG, Svensson S, Cselenyi Z, Masters CL, Villemagne VL. Head-to-head comparison of 11C-PiB and 18F-AZD4694 (NAV4694) for beta-amyloid imaging in aging and dementia. J Nucl Med Off Publ Soc Nucl Med. 2013;54(6):880–886. doi: 10.2967/jnumed.112.114785. [DOI] [PubMed] [Google Scholar]
- 5.Vann Jones SA, O’Brien JT. The prevalence and incidence of dementia with Lewy bodies: a systematic review of population and clinical studies. Psychol Med. 2014;44(4):673–683. doi: 10.1017/S0033291713000494. [DOI] [PubMed] [Google Scholar]
- 6.Harding AJ, Halliday GM. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol. 2001;102(4):355–363. doi: 10.1007/s004010100390. [DOI] [PubMed] [Google Scholar]
- 7.Aarsland D, Ballard CG, Halliday G. Are Parkinson’s disease with dementia and dementia with Lewy bodies the same entity? J Geriatr Psychiatry Neurol. 2004;17(3):137–145. doi: 10.1177/0891988704267470. [DOI] [PubMed] [Google Scholar]
- 8.Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF, Brooks DJ, Dickson DW, Dubois B, Emre M, Fahn S, Farmer JM, Galasko D, Galvin JE, Goetz CG, Growdon JH, Gwinn-Hardy KA, Hardy J, Heutink P, Iwatsubo T, Kosaka K, Lee VM, Leverenz JB, Masliah E, McKeith IG, Nussbaum RL, Olanow CW, Ravina BM, Singleton AB, Tanner CM, Trojanowski JQ, Wszolek ZK. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68(11):812–819. doi: 10.1212/01.wnl.0000256715.13907.d3. [DOI] [PubMed] [Google Scholar]
- 9.Jellinger KA, Seppi K, Wenning GK. Clinical and neuropathological correlates of Lewy body disease. Acta Neuropathol. 2003;106(2):188–189. doi: 10.1007/s00401-003-0721-2. [DOI] [PubMed] [Google Scholar]
- 10.Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM, Marsh L, Troncoso JC. Abeta deposition is associated with enhanced cortical alpha-synuclein lesions in Lewy body diseases. Neurobiol Aging. 2005;26(8):1183–1192. doi: 10.1016/j.neurobiolaging.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Villemagne VL, Ong K, Mulligan RS, Holl G, Pejoska S, Jones G, O’Keefe G, Ackerman U, Tochon-Danguy H, Chan JG, Reininger CB, Fels L, Putz B, Rohde B, Masters CL, Rowe CC. Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J Nucl Med Off Publ Soc Nucl Med. 2011;52(8):1210–1217. doi: 10.2967/jnumed.111.089730. [DOI] [PubMed] [Google Scholar]
- 12.Edison P, Rowe CC, Rinne JO, Ng S, Ahmed I, Kemppainen N, Villemagne VL, O’Keefe G, Nagren K, Chaudhury KR, Masters CL, Brooks DJ. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry. 2008;79(12):1331–1338. doi: 10.1136/jnnp.2007.127878. [DOI] [PubMed] [Google Scholar]
- 13.Foster ER, Campbell MC, Burack MA, Hartlein J, Flores HP, Cairns NJ, Hershey T, Perlmutter JS. Amyloid imaging of Lewy body-associated disorders. Mov Disord Off J Mov Disord Soc. 2010;25(15):2516–2523. doi: 10.1002/mds.23393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maetzler W, Liepelt I, Reimold M, Reischl G, Solbach C, Becker C, Schulte C, Leyhe T, Keller S, Melms A, Gasser T, Berg D. Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiol Dis. 2009;34(1):107–112. doi: 10.1016/j.nbd.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Gomperts SN, Rentz DM, Moran E, Becker JA, Locascio JJ, Klunk WE, Mathis CA, Elmaleh DR, Shoup T, Fischman AJ, Hyman BT, Growdon JH, Johnson KA. Imaging amyloid deposition in Lewy body diseases. Neurology. 2008;71(12):903–910. doi: 10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Claassen DO, Lowe VJ, Peller PJ, Petersen RC, Josephs KA. Amyloid and glucose imaging in dementia with Lewy bodies and multiple systems atrophy. Parkinsonism Relat Disord. 2011;17(3):160–165. doi: 10.1016/j.parkreldis.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 17.Braak H, Braak E. Cognitive impairment in Parkinson’s disease: amyloid plaques, neurofibrillary tangles, and neuropil threads in the cerebral cortex. J Neural Transm Parkinson’s Dis Dement Sect. 1990;2(1):45–57. doi: 10.1007/BF02251245. [DOI] [PubMed] [Google Scholar]
- 18.Ikonomovic MD, Abrahamson EE, Price JC, Hamilton RL, Mathis CA, Paljug WR, Debnath ML, Cohen AD, Mizukami K, DeKosky ST, Lopez OL, Klunk WE. Early AD pathology in a [C-11]PiB-negative case: a PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol. 2012;123(3):433–447. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomperts SN, Locascio JJ, Marquie M, Santarlasci AL, Rentz DM, Maye J, Johnson KA, Growdon JH. Brain amyloid and cognition in Lewy body diseases. Mov Disord Off J Mov Disord Soc. 2012;27(8):965–973. doi: 10.1002/mds.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edison P, Ahmed I, Fan Z, Hinz R, Gelosa G, Ray Chaudhuri K, Walker Z, Turkheimer FE, Brooks DJ. Microglia, amyloid, and glucose metabolism in Parkinson’s disease with and without dementia. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2013;38(6):938–949. doi: 10.1038/npp.2012.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yong SW, Yoon JK, An YS, Lee PH. A comparison of cerebral glucose metabolism in Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies. Eur J Neurol Off J Eur Fed Neurol Soc. 2007;14(12):1357–1362. doi: 10.1111/j.1468-1331.2007.01977.x. [DOI] [PubMed] [Google Scholar]
- 22.Jellinger KA, Attems J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 2006;112(3):253–260. doi: 10.1007/s00401-006-0088-2. [DOI] [PubMed] [Google Scholar]
- 23.Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, DeKosky ST. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci Off J Soc Neurosci. 2007;27(23):6174–6184. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HJ, Jeon BS, Kim YE, Kim JY, Kim YK, Sohn CH, Yun JY, Jeon S, Lee JM, Lee JY. Clinical and imaging characteristics of dementia in multiple system atrophy. Parkinsonism Relat Disord. 2013;19(6):617–621. doi: 10.1016/j.parkreldis.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Kitayama M, Wada-Isoe K, Irizawa Y, Nakashima K. Assessment of dementia in patients with multiple system atrophy. Eur J Neurol Off J Eur Fed Neurol Soc. 2009;16(5):589–594. doi: 10.1111/j.1468-1331.2009.02544.x. [DOI] [PubMed] [Google Scholar]
- 26.Jellinger KA. More frequent Lewy bodies but less frequent Alzheimer-type lesions in multiple system atrophy as compared to age-matched control brains. Acta Neuropathol. 2007;114(3):299–303. doi: 10.1007/s00401-007-0227-4. [DOI] [PubMed] [Google Scholar]
- 27.Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain J Neurol. 2009;132(Pt 5):1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry. 2012;83(2):124–137. doi: 10.1136/jnnp-2011-301308. [DOI] [PubMed] [Google Scholar]
- 29.Nicoll JA, Yamada M, Frackowiak J, Mazur-Kolecka B, Weller RO (2004) Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer’s disease. Pro-CAA position statement. Neurobiol Aging 25(5):589–597. doi:10.1016/j.neurobiolaging.2004.02.003. (discussion 603–584) [DOI] [PubMed]
- 30.Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Dichgans M, van Buchem MA, Bruckmann H, Greenberg SM. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology. 2010;74(17):1346–1350. doi: 10.1212/WNL.0b013e3181dad605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgan D. Immunotherapy for Alzheimer’s disease. J Intern Med. 2011;269(1):54–63. doi: 10.1111/j.1365-2796.2010.02315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L, Libri V, Leppert D, Beach TG. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain J Neurol. 2007;130(Pt 10):2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 33.Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain J Neurol. 2008;131(Pt 6):1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, Smith EE, Rosand J, Rentz DM, Klunk WE, Mathis CA, Price JC, Dekosky ST, Fischman AJ, Greenberg SM. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62(3):229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- 35.Dugger BN, Clark CM, Serrano G, Mariner M, Bedell BJ, Coleman RE, Doraiswamy PM, Lu M, Fleisher AS, Reiman EM, Sabbagh MN, Sadowsky CH, Schneider JA, Zehntner SP, Carpenter AP, Joshi AD, Mintun MA, Pontecorvo MJ, Skovronsky DM, Sue LI, Beach TG. Neuropathologic heterogeneity does not impair florbetapir-positron emission tomography postmortem correlates. J Neuropathol Exp Neurol. 2014;73(1):72–80. doi: 10.1097/NEN.0000000000000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dierksen GA, Skehan ME, Khan MA, Jeng J, Nandigam RN, Becker JA, Kumar A, Neal KL, Betensky RA, Frosch MP, Rosand J, Johnson KA, Viswanathan A, Salat DH, Greenberg SM. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann Neurol. 2010;68(4):545–548. doi: 10.1002/ana.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ly JV, Donnan GA, Villemagne VL, Zavala JA, Ma H, O’Keefe G, Gong SJ, Gunawan RM, Saunder T, Ackerman U, Tochon-Danguy H, Churilov L, Phan TG, Rowe CC. 11C-PIB binding is increased in patients with cerebral amyloid angiopathy-related hemorrhage. Neurology. 2010;74(6):487–493. doi: 10.1212/WNL.0b013e3181cef7e3. [DOI] [PubMed] [Google Scholar]
- 38.Baron JC, Farid K, Dolan E, Turc G, Marrapu ST, O’Brien E, Aigbirhio FI, Fryer TD, Menon DK, Warburton EA, Hong YT. Diagnostic utility of amyloid PET in cerebral amyloid angiopathy-related symptomatic intracerebral hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2014;34(5):753–758. doi: 10.1038/jcbfm.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gurol ME, Dierksen G, Betensky R, Gidicsin C, Halpin A, Becker A, Carmasin J, Ayres A, Schwab K, Viswanathan A, Salat D, Rosand J, Johnson KA, Greenberg SM. Predicting sites of new hemorrhage with amyloid imaging in cerebral amyloid angiopathy. Neurology. 2012;79(4):320–326. doi: 10.1212/WNL.0b013e31826043a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurol ME, Viswanathan A, Gidicsin C, Hedden T, Martinez-Ramirez S, Dumas A, Vashkevich A, Ayres AM, Auriel E, van Etten E, Becker A, Carmasin J, Schwab K, Rosand J, Johnson KA, Greenberg SM. Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann Neurol. 2013;73(4):529–536. doi: 10.1002/ana.23830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yates PA, Sirisriro R, Villemagne VL, Farquharson S, Masters CL, Rowe CC. Cerebral microhemorrhage and brain beta-amyloid in aging and Alzheimer disease. Neurology. 2011;77(1):48–54. doi: 10.1212/WNL.0b013e318221ad36. [DOI] [PubMed] [Google Scholar]
- 42.Sengoku R, Matsushima S, Murakami Y, Fukuda T, Tokumaru AM, Hashimoto M, Suzuki M, Ishiwata K, Ishii K, Mochio S (2014) (11)C-PiB PET imaging of encephalopathy associated with cerebral amyloid angiopathy. Intern Med (Tokyo, Jpn) 53(17):1997–2000 [DOI] [PubMed]
- 43.Chetelat G, La Joie R, Villain N, Perrotin A, de La Sayette V, Eustache F, Vandenberghe R. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. NeuroImage Clin. 2013;2:356–365. doi: 10.1016/j.nicl.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathis CA, Kuller LH, Klunk WE, Snitz BE, Price JC, Weissfeld LA, Rosario BL, Lopresti BJ, Saxton JA, Aizenstein HJ, McDade EM, Kamboh MI, DeKosky ST, Lopez OL. In vivo assessment of amyloid-beta deposition in nondemented very elderly subjects. Ann Neurol. 2013;73(6):751–761. doi: 10.1002/ana.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han BH, Zhou ML, Vellimana AK, Milner E, Kim DH, Greenberg JK, Chu W, Mach RH, Zipfel GJ. Resorufin analogs preferentially bind cerebrovascular amyloid: potential use as imaging ligands for cerebral amyloid angiopathy. Mol Neurodegener. 2011;6:86. doi: 10.1186/1750-1326-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zha Z, Choi SR, Ploessl K, Lieberman BP, Qu W, Hefti F, Mintun M, Skovronsky D, Kung HF. Multidentate (18)F-polypegylated styrylpyridines as imaging agents for Abeta plaques in cerebral amyloid angiopathy (CAA) J Med Chem. 2011;54(23):8085–8098. doi: 10.1021/jm2009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57(4):419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodwin L (2006) Dementia pugilistica. J Insur Med (N Y, NY) 38(4):300–302 [PubMed]
- 49.McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC. The spectrum of disease in chronic traumatic encephalopathy. Brain J Neurol. 2013;136(Pt 1):43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawai N, Kawanishi M, Kudomi N, Maeda Y, Yamamoto Y, Nishiyama Y, Tamiya T. Detection of brain amyloid beta deposition in patients with neuropsychological impairment after traumatic brain injury: PET evaluation using Pittsburgh compound-B. Brain Inj [BI] 2013;27(9):1026–1031. doi: 10.3109/02699052.2013.794963. [DOI] [PubMed] [Google Scholar]
- 51.Hong YT, Veenith T, Dewar D, Outtrim JG, Mani V, Williams C, Pimlott S, Hutchinson PJ, Tavares A, Canales R, Mathis CA, Klunk WE, Aigbirhio FI, Coles JP, Baron JC, Pickard JD, Fryer TD, Stewart W, Menon DK. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 2014;71(1):23–31. doi: 10.1001/jamaneurol.2013.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190(1):192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 53.Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19(2):214–223. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mitsis EM, Riggio S, Kostakoglu L, Dickstein DL, Machac J, Delman B, Goldstein M, Jennings D, D’Antonio E, Martin J, Naidich TP, Aloysi A, Fernandez C, Seibyl J, DeKosky ST, Elder GA, Marek K, Gordon W, Hof PR, Sano M, Gandy S. Tauopathy PET and amyloid PET in the diagnosis of chronic traumatic encephalopathies: studies of a retired NFL player and of a man with FTD and a severe head injury. Transl Psychiatry. 2014;4:e441. doi: 10.1038/tp.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitsis EM, Bender HA, Kostakoglu L, Machac J, Martin J, Woehr JL, Sewell MC, Aloysi A, Goldstein MA, Li C, Sano M, Gandy S. A consecutive case series experience with [18 F] florbetapir PET imaging in an urban dementia center: impact on quality of life, decision making, and disposition. Mol Neurodegener. 2014;9:10. doi: 10.1186/1750-1326-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del-Favero J, Cruts M, van Duijn CM, Van Broeckhoven C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain J Neurol. 2006;129(Pt 11):2977–2983. doi: 10.1093/brain/awl203. [DOI] [PubMed] [Google Scholar]
- 57.Zhou ZD, Chan CH, Ma QH, Xu XH, Xiao ZC, Tan EK. The roles of amyloid precursor protein (APP) in neurogenesis: implications to pathogenesis and therapy of Alzheimer disease. Cell Adhes Migr. 2011;5(4):280–292. doi: 10.4161/cam.5.4.16986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mann DM, Esiri MM. The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci. 1989;89(2–3):169–179. doi: 10.1016/0022-510X(89)90019-1. [DOI] [PubMed] [Google Scholar]
- 59.Mann DM. Alzheimer’s disease and Down’s syndrome. Histopathology. 1988;13(2):125–137. doi: 10.1111/j.1365-2559.1988.tb02018.x. [DOI] [PubMed] [Google Scholar]
- 60.Visser FE, Aldenkamp AP, van Huffelen AC, Kuilman M, Overweg J, van Wijk J. Prospective study of the prevalence of Alzheimer-type dementia in institutionalized individuals with Down syndrome. Am J Ment retard AJMR. 1997;101(4):400–412. [PubMed] [Google Scholar]
- 61.Malone Q. Mortality and survival of the Down’s syndrome population in Western Australia. J Ment Defic Res. 1988;32(Pt 1):59–65. doi: 10.1111/j.1365-2788.1988.tb01388.x. [DOI] [PubMed] [Google Scholar]
- 62.Strauss D, Eyman RK. Mortality of people with mental retardation in California with and without Down syndrome, 1986–1991. Am J Ment Retard AJMR. 1996;100(6):643–653. [PubMed] [Google Scholar]
- 63.Cutler NR, Sramek JJ. Review of the next generation of Alzheimer’s disease therapeutics: challenges for drug development. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25(1):27–57. doi: 10.1016/S0278-5846(00)00147-0. [DOI] [PubMed] [Google Scholar]
- 64.Solomon A, Mangialasche F, Richard E, Andrieu S, Bennett DA, Breteler M, Fratiglioni L, Hooshmand B, Khachaturian AS, Schneider LS, Skoog I, Kivipelto M. Advances in the prevention of Alzheimer’s disease and dementia. J Intern Med. 2014;275(3):229–250. doi: 10.1111/joim.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lannfelt L, Relkin NR, Siemers ER. Amyloid-ss-directed immunotherapy for Alzheimer’s disease. J Intern Med. 2014;275(3):284–295. doi: 10.1111/joim.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Landt J, D’Abrera JC, Holland AJ, Aigbirhio FI, Fryer TD, Canales R, Hong YT, Menon DK, Baron JC, Zaman SH. Using positron emission tomography and carbon 11-labeled Pittsburgh compound B to image brain fibrillar beta-amyloid in adults with down syndrome: safety, acceptability, and feasibility. Arch Neurol. 2011;68(7):890–896. doi: 10.1001/archneurol.2011.36. [DOI] [PubMed] [Google Scholar]
- 67.Handen BL, Cohen AD, Channamalappa U, Bulova P, Cannon SA, Cohen WI, Mathis CA, Price JC, Klunk WE. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimer’s Dement J Alzheimer’s Assoc. 2012;8(6):496–501. doi: 10.1016/j.jalz.2011.09.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jennings D, Seibyl J, Sabbagh M, Lai F, Hopkins W, Bullich S, Gimenez M, Reininger C, Putz B, Stephens A, Catafau AM, Marek K (2015) Age dependence of brain β-amyloid deposition in Down syndrome: an [18F]florbetaben PET study. Neurology [Epub ahead of print] [DOI] [PubMed]
- 69.Sabbagh MN, Fleisher A, Chen K, Rogers J, Berk C, Reiman E, Pontecorvo M, Mintun M, Skovronsky D, Jacobson SA, Sue LI, Liebsack C, Charney AS, Cole L, Belden C, Beach TG. Positron emission tomography and neuropathologic estimates of fibrillar amyloid-beta in a patient with Down syndrome and Alzheimer disease. Arch Neurol. 2011;68(11):1461–1466. doi: 10.1001/archneurol.2011.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem. 2009;110(4):1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 71.Kim HY, Kim J, Lee JH. Incidental finding of meningioma on C11-PIB PET. Clin Nucl Med. 2012;37(2):e36–e37. doi: 10.1097/RLU.0b013e318238f25a. [DOI] [PubMed] [Google Scholar]
- 72.Yamamoto Y, Maeda Y, Kawai N, Kudomi N, Nishiyama Y. Unexpected finding of cerebral meningioma on (11)C-PiB PET. Clin Nucl Med. 2013;38(4):292–293. doi: 10.1097/RLU.0b013e3182817c8f. [DOI] [PubMed] [Google Scholar]
- 73.Foschini MP, D’Adda T, Bordi C, Eusebi V. Amyloid stroma in meningiomas. Virchows Archiv A Pathol Anat Histopathol. 1993;422(1):53–59. doi: 10.1007/BF01605133. [DOI] [PubMed] [Google Scholar]
- 74.Sachchithanantham S, Wechalekar AD. Imaging in systemic amyloidosis. Br Med Bull. 2013;107:41–56. doi: 10.1093/bmb/ldt021. [DOI] [PubMed] [Google Scholar]
- 75.Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–1300. doi: 10.1161/CIRCULATIONAHA.111.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dorbala S, Vangala D, Semer J, Strader C, Bruyere JR, Jr, Di Carli MF, Moore SC, Falk RH. Imaging cardiac amyloidosis: a pilot study using (1)(8)F-florbetapir positron emission tomography. Eur J Nucl Med Mol Imaging. 2014;41(9):1652–1662. doi: 10.1007/s00259-014-2787-6. [DOI] [PubMed] [Google Scholar]
- 77.Chen W, Dilsizian V. Molecular imaging of amyloidosis: will the heart be the next target after the brain? Curr Cardiol Rep. 2012;14(2):226–233. doi: 10.1007/s11886-011-0239-5. [DOI] [PubMed] [Google Scholar]
- 78.Antoni G, Lubberink M, Estrada S, Axelsson J, Carlson K, Lindsjo L, Kero T, Langstrom B, Granstam SO, Rosengren S, Vedin O, Wassberg C, Wikstrom G, Westermark P, Sorensen J. In vivo visualization of amyloid deposits in the heart with 11C-PIB and PET. J Nucl Med Off Publ Soc Nucl Med. 2013;54(2):213–220. doi: 10.2967/jnumed.111.102053. [DOI] [PubMed] [Google Scholar]
- 79.Lhommel R, Sempoux C, Ivanoiu A, Michaux L, Gerber B. Is 18F-flutemetamol PET/CT able to reveal cardiac amyloidosis? Clin Nucl Med. 2014;39(8):747–749. doi: 10.1097/RLU.0000000000000492. [DOI] [PubMed] [Google Scholar]
- 80.Minamimoto R, Ishii K, Kubota K, Morooka M, Okasaki M, Ito K, Mitsumoto T, Nakajima K, Sato T, Mochizuki M, Okazaki O. Amyloid imaging mismatch. Clin Nucl Med. 2012;37(8):807–809. doi: 10.1097/RLU.0b013e318251e1d3. [DOI] [PubMed] [Google Scholar]
- 81.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol. 2004;55(3):306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 82.Fodero-Tavoletti MT, Rowe CC, McLean CA, Leone L, Li QX, Masters CL, Cappai R, Villemagne VL. Characterization of PiB binding to white matter in Alzheimer disease and other dementias. J Nucl Med Off Publ Soc Nucl Med. 2009;50(2):198–204. doi: 10.2967/jnumed.108.057984. [DOI] [PubMed] [Google Scholar]
- 83.Stankoff B, Freeman L, Aigrot MS, Chardain A, Dolle F, Williams A, Galanaud D, Armand L, Lehericy S, Lubetzki C, Zalc B, Bottlaender M. Imaging central nervous system myelin by positron emission tomography in multiple sclerosis using [methyl-(1)(1)C]-2-(4′-methylaminophenyl)-6-hydroxybenzothiazole. Ann Neurol. 2011;69(4):673–680. doi: 10.1002/ana.22320. [DOI] [PubMed] [Google Scholar]
- 84.Ito H, Shimada H, Shinotoh H, Takano H, Sasaki T, Nogami T, Suzuki M, Nagashima T, Takahata K, Seki C, Kodaka F, Eguchi Y, Fujiwara H, Kimura Y, Hirano S, Ikoma Y, Higuchi M, Kawamura K, Fukumura T, Boo EL, Farde L, Suhara T. Quantitative analysis of amyloid deposition in Alzheimer Disease using PET and the radiotracer 11C-AZD2184. J Nucl Med Off Publ Soc Nucl Med. 2014;55(6):932–938. doi: 10.2967/jnumed.113.133793. [DOI] [PubMed] [Google Scholar]
- 85.Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci Off J Soc Neurosci. 2003;23(6):2086–2092. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moll NM, Cossoy MB, Fisher E, Staugaitis SM, Tucky BH, Rietsch AM, Chang A, Fox RJ, Trapp BD, Ransohoff RM. Imaging correlates of leukocyte accumulation and CXCR4/CXCL12 in multiple sclerosis. Arch Neurol. 2009;66(1):44–53. doi: 10.1001/archneurol.2008.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Glodzik L, Kuceyeski A, Rusinek H, Tsui W, Mosconi L, Li Y, Osorio RS, Williams S, Randall C, Spector N, McHugh P, Murray J, Pirraglia E, Vallabhajosula S, Raj A, de Leon MJ. Reduced glucose uptake and Abeta in brain regions with hyperintensities in connected white matter. NeuroImage. 2014;100:684–691. doi: 10.1016/j.neuroimage.2014.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murray ME, Vemuri P, Preboske GM, Murphy MC, Schweitzer KJ, Parisi JE, Jack CR, Jr, Dickson DW. A quantitative postmortem MRI design sensitive to white matter hyperintensity differences and their relationship with underlying pathology. J Neuropathol Exp Neurol. 2012;71(12):1113–1122. doi: 10.1097/NEN.0b013e318277387e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shimada H, Shinotoh H, Hirano S, Miyoshi M, Sato K, Tanaka N, Ota T, Fukushi K, Irie T, Ito H, Higuchi M, Kuwabara S, Suhara T. β-Amyloid in Lewy body disease is related to Alzheimer’s disease-like atrophy. Mov Disord. 2013;28(2):169–175. doi: 10.1002/mds.25286. [DOI] [PubMed] [Google Scholar]
- 90.Johnson KA, Fox NC, Sperling RA, Klunk WE. Brain imaging in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(4):a006213. doi: 10.1101/cshperspect.a006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chao LL, Decarli C, Kriger S, Truran D, Zhang Y, Laxamana J, Villeneuve S, Jagust WJ, Sanossian N, Mack WJ, Chui HC, Weiner MW. Associations between white matter hyperintensities and beta amyloid on integrity of projection, association, and limbic fiber tracts measured with diffusion tensor MRI. PLoS One. 2013;8(6):e65175. doi: 10.1371/journal.pone.0065175. [DOI] [PMC free article] [PubMed] [Google Scholar]