

Abstract
We previously showed that mouse alpha-fetoprotein enhancer E3 activity is highly restricted to pericentral hepatocytes in the adult liver. Here, using transgenic mice, we show that the upstream enhancer of the rat glutamine synthetase gene is also active specifically in pericentral regions. Activity of both enhancers is lost in the absence of β-catenin, a key regulator of zonal gene expression in the adult liver. Both enhancers contain a single highly conserved TCF/LEF binding site that is required for responsiveness to β-catenin. We also show that endogenous AFP mRNA levels in the perinatal liver are lower when β-catenin is reduced. These data identify the first distinct zonally-active regulatory regions required for β-catenin responsiveness in the adult liver and suggest that postnatal AFP repression and the establishment of zonal regulation are controlled, at least in part, by the same factors.
Keywords: zonal gene regulation, TCF/LEF, development, transcription, hepatocellular carcinoma
The liver performs numerous metabolic and homeostatic functions in the body, including xenobiotic metabolism, energy storage/production, urea formation, glutamine synthesis and cholesterol homeostasis (1). Many of these functions require the unique architecture of the liver, which is comprised of periportal and pericentral regions (2). Certain hepatic enzymes are expressed solely in periportal regions along this porto-centro axis, whereas other enzymes are synthesized in pericentral regions. This compartmentalization of function, or “liver zonation”, enables the liver to perform multiple, and sometimes opposing, metabolic pathways in distinct hepatocyte subpopulations (3).
The pericentral expression of glutamine synthetase (GS) was the first example of zonal gene regulation in the adult liver (4). Since this discovery, numerous additional enzymes were found to exhibit zonal patterns of expression in the liver (5). It is generally believed that certain blood-borne compounds (i.e., oxygen, nutrients) that form a gradient along the porto-centro axis provide signals that establish the heterogeneity of gene expression in the liver, although the nature of such signals is not fully understood (6). Regardless of the stimulus, intracellular signaling pathways must link extracellular events to the nucleus to govern zonal gene regulation. An elegant study by Benhamouche, et al., demonstrated that signaling through β-catenin, a downstream activator of Wnt, governs zonal gene regulation in the adult liver (7). In the absence of Wnt signaling, cytosolic β-catenin is complexed with adenomatous polyposis coli (APC), Axin and the kinases GSK-3β and CK1. This inhibitory complex phosphorylates β-catenin at specific serine residues that mark it for ubiquitin-mediated proteolysis. In the absence of this phosphorylation (i.e., when blocked by Wnt signaling), β-catenin enters the nucleus and regulates target gene expression. Regarding zonal regulation, activated β-catenin (through expression of a non-degradable form of β-catenin or loss of APC) is associated with increased expression of GS (and other pericentral genes) and decreased expression of periportal genes in periportal regions (7–9). In contrast, blocking β-catenin signaling results in a loss of pericentral enzymes and increased periportal enzyme expression (7).
β-catenin does not bind DNA directly, but can regulate target genes through several pathways. In the canonical pathway, β-catenin controls target genes via interactions with the T Cell Factor/Lymphoid Enhancer Factor (TCF/LEF) family of transcription factors (10). In the absence of β-catenin, TCF/LEF proteins are bound to consensus motifs and silence target genes by recruitment of co-repressors such as Groucho (11). Upon nuclear entry, β-catenin interacts with TCF/LEF proteins, dissociating repressors and recruiting the co-activators CBP/p300, leading to target gene activation (12).
Using transgenic mice, we showed previously that the activity of alpha-fetoprotein (AFP) enhancer 3 (E3), one of the three AFP enhancers, is highly restricted to a single layer of pericentral hepatocytes in the adult liver (13, 14), a pattern identical to GS and other highly restricted pericentral enzymes. AFP is expressed abundantly in the fetal liver and silenced at birth, but can be transiently reactivated during liver regeneration and is often activated in hepatocellular carcinoma (HCC)(15). Here, we show that pericentral activity of E3 is β-catenin dependent. Using transgenic mice, we also show that the upstream enhancer of the rat GS gene can confer pericentral activity to a linked reporter gene, and that the activity of this enhancer is also β-catenin-dependent. Both enhancers contain a highly conserved TCF site that binds TCF4 in vitro and mutation of these TCF sites results in a loss of β-catenin responsiveness in cultured cells. We also show that E3 activity and endogenous AFP expression in the perinatal liver are reduced in the absence of β-catenin, suggesting that β-catenin regulates AFP during liver development. These data identify the first defined zonally-regulated cis-acting control regions that confer β-catenin responsiveness.
Materials and Methods
Plasmids
E3 was excised from E3-βgl-Dd (14) and cloned into pGL3-promoter (Promega) to generate E3-Luc. RGSe was amplified from rat DNA using primers RGSeU and RGSeL (Table I) and cloned into βgl-Dd to generate RGSe-βgl-Dd or into pGL3-promoter to generate RGSe-Luc. Megaprimer mutagenesis (16) using primers listed in Table I generated TCF site mutations in E3-Luc and RGSe-Luc; mutations were confirmed by DNA sequencing. TOP-Flash and FOP-Flash were provided by H. Clevers (17). Expression vectors for wild-type β-catenin, βcatS37A, and TCF4 were provided by S. Byers (18) and Chunming Lui (19).
Table I.
Oligonucleotides used in this study
| RGSeU: | 5’-GCTAGGATCCAAGCTTCTTGTTTACCCCTG |
| RGSeL: | 5’-TCAAGGATCCGAGTTTCAGATGGCAGCTTC |
| mE3 TFCU: | 5’-AGATAAAATTCCTTTGATGAAGGAAAA |
| mE3 TFCL: | 5’-TTTTCCTTCATCAAAGGAATTTTATCT |
| mE3 TFCMutU: | 5’-AGATAAAATTCCCGGGATGAAGGAAA |
| mE3 TFCMutL: | 5’-TTTTCCTTCATCCCGGGAATTTTATCT |
| RGSe TFCU: | 5’-CATGGAAGGATCAAAGCAAGCCTGC |
| RGSe TFCL: | 5’-GCAGGCTTGCTTTGATCCTTCCATG |
| RGSe TFCMutU: | 5’-CATGGAAGGACCCGGGCAAGCCTGC |
| RGSe TFCMutL: | 5’-GCAGGCTTGCCCGGGTCCTTCCATG |
| Control TCFU: | 5’-GGTACTGGCCCTTTGATCTTTCTGG |
| Control TCF L: | 5’-CCAGAAAGATCAAAGGGCCAGTACC |
| Control TCFMutU: | 5’-GGTACTGGCCCGGGGATCTTTCTGG |
| Control TCFMutL: | 5’-CCAGAAAGATCCCCGGGCCAGTACC |
| AFP5’: | 5' CCGGAAGCCACCGAGGAGGA |
| AFP3’: | 5’ TGGGACAGAGGCCGGAGCAG |
| bCat5’: | 5’ CTCTTCAGGACAGAGCCAATG |
| bCat3’: | 5’ ATGCTCCATCATAGGGTCCA |
| L305’: | 5’ ATGGTGGCCGCAAAGAAGACGAA |
| L303’: | 5’ CCTCAAAGCTGGACAGTTGTTGGCA |
Underlined sequences indicate the TCF binding motif. Nucleotides in bold indicate mutations that were incorporated to eliminate TCF binding.
Cell culture and luciferase assays
Hep3B human hepatoma cells and HEK293 cells were maintained and transfections were performed as described (20). Hep3B cells were seeded into 12-well dishes and transfected in duplicate with 500 ng of luciferase reporter plasmid, 1µg of expression plasmid and 12.5 ng of renilla luciferase plasmid. To prepare nuclear extracts, HEK293 cells were seeded onto 10 cm plates and transfected with 15 µg of the Flag-TCF4 expression plasmid (or mock transfected). For both cell types, media was changed after six hours and cells harvested 48 hours later. Transfected Hep3B cells were harvested into Glo-lysis buffer (Promega Corp.) and firefly/renilla luciferase levels were determined in duplicate. All transfections were repeated at least twice. Results were analyzed using the Student’s t-test with p values < 0.05 considered to be statistically significant.
Electrophoretic mobility shift assay (EMSA)
Nuclear lysates were collected from HEK293 cells using the NE-PER extraction kit (Thermo-Scientific). Protein concentrations were determined using the BCA protein assay (Pierce). Labeling of annealed oligonucleotides (Integrated DNA Technologies, Table I) with 32P and EMSAs were carried out as described (21).
Mouse studies
The E3-βgl-Dd transgenic mice were described (14). RGSe-βgl-Dd founder mice were generated at the University of Kentucky Transgenic Mouse facility. Mice were screened for the transgene by PCR analysis of tail DNA. Mice containing Albumin-Cre (Alb-Cre)(22) and the floxed β-catenin (Ctnnb1) gene (23) were purchased from The Jackson Labs. Standard breeding was performed to obtain mice of the appropriate genotype. Oligonucleotides and PCR conditions for screening genetically modified mice are available upon request. All mouse experiments were approved by the University of Kentucky Institutional Animal Care and Use Committee, following guidelines established by the NIH.
Immunohistochemistry
Frozen livers were sectioned at 10 µm thickness. For transgene analysis, slides were incubated overnight with FITC-conjugated anti-H2-Dd monoclonal antibodies (BD-Pharmingen 553579). For β-catenin and GS, antibodies against active β-catenin (Millipore 05-665) and GS (Sigma G2781) were used. Staining details are available upon request.
Hydrodynamic Tail Vein Injections/Flow Cytometry
Eight-week old E3-βgl-Dd mice were injected with 2.5 ml of 0.9% saline containing 50 µg of plasmid (24). After 48 hours, mice were sacrificed and livers harvested. Livers, along with 5 ml of RPMI containing 10% FBS, were placed in a stomacher bag and compressed for 60 seconds using Stomacher-80 Laboratory blender (Seward Lab Systems). Cells were transferred to 15 ml conical tubes along with DNaseI (160U/ml) and collagenase (400U/ml) and incubated at 37°C for 20 min, passaged through a 40 micron strainer, centrifuged, and washed with 2 ml ice-cold PBS with 0.1% BSA/0.1% Azide (PBS-BSA-Az). Hepatocytes were purified by percoll centrifugation, and stained with FITC-anti-H2-Dd or control IgG followed by flow cytometry at the University of Kentucky Flow Cytometry Facility.
RNA analysis
RNA was prepared using 2 rounds of Trizol (Life Technologies) and processed into cDNA using qscript (Quanta Biosciences). Quantitative PCR was carried out using Sybr Green (Quanta Biosciences) using the Bio-Rad MyiQ thermal cycler. AFP and β-catenin mRNA levels were normalized to ribosomal gene L30, using primers shown in Table I.
Results
The upstream GS enhancer exhibits pericentral activity in transgenic mice
Pericentral GS expression was first described in 1983 (4, 25), and GS continues to be the most extensively studied pericentral gene. Several elements controlling GS activity have been identified in tissue culture cells, including a single upstream enhancer centered 2.2 kb upstream of exon 1 and several regulatory regions within the first intron (26–30). Previous studies indicated that the 3.2 kb region upstream of the rat GS gene (−3150 to +59) could confer pericentral expression of a linked reporter gene in transgenic mice (31). To test whether the upstream enhancer of the rat GS gene exhibited pericentral activity by itself, this enhancer was cloned as a 400 bp fragment {as defined in (21) and which will be referred to as RGSe} and fused to the heterologous human β-globin promoter linked to the mouse H-2Dd reporter gene (βgl-Dd). We have used βgl-Dd extensively to monitor gene expression in cells and transgenic mice; this cassette is inactive in all mouse tissues but is highly responsive to linked enhancers (14). Furthermore, transgenes with βgl-Dd fused to the albumin enhancer are expressed throughout the adult liver, demonstrating that the β-globin promoter can be activated in all adult hepatocytes (unpublished data). Several founder mice containing the RGSe-βgl-Dd transgene were generated. Immunofluorescence staining using anti-Dd antibodies indicated that transgene expression was restricted to a single layer of pericentral hepatocytes in the adult liver (Fig. 1). This pericentral activity is identical to the expression of endogenous GS and to E3- βgl-Dd transgenes, which we previously showed to exhibit pericentral expression in the adult liver (13, 14). This data demonstrates that the upstream GS enhancer, RGSe, is sufficient to confer highly restricted pericentral activity in the adult liver in the absence of other GS regulatory regions.
Figure 1. Expression of RGSe-βgl-Dd transgenes is restricted to hepatocytes directly surrounding the central vein in adult liver.

The rat glutamine synthetase upstream enhancer centered at −2.2 kb (RGSe) was amplified as a 400 bp fragment and fused to βgl-Dd. Offspring of two different transgenic founders containing RGSe-βgl-Dd were sacrified at ~8 weeks of age. Frozen sections were prepared and stained with a monoclonal FITC-anti-Dd antibody. Dd expression was restricted to hepatocytes directly surrounding the central veins. The same pattern of staining was observed with lines from two other RGSe-βgl-Dd founder mice (data not shown). Magnification, 20X.
E3 and RGSe enhancer activities are dependent on β-catenin
Numerous genes expressed in pericentral regions are controlled by β-catenin signaling, with active β-catenin being found only in pericentral hepatocytes of the adult liver (7). Since E3 activity is highly restricted to this population of hepatocytes, we predicted that pericentral activity of this enhancer would co-localize with active β-catenin. To test this, we co-stained liver sections of adult E3-βgl-Dd mice with antibodies against Dd and active (unphosphorylated) β-catenin (Fig. 2A). Consistent with previous studies, we found active β-catenin localized to pericentral hepatocytes (7). E3-βgl-Dd expression and active β-catenin overlap in these pericentral cells. This co-localization of Dd and active β-catenin supports the possibility that active β-catenin is required for E3 activity.
Figure 2. E3-βgl-Dd transgene expression and active β-catenin are co-localized in pericentral hepatocytes.

(A) Transgenic mice containing E3-βgl-Dd were sacrificed at ~8 weeks of age. Sections were stained for Dd expression (left panel, green) and active β-catenin (middle panel, red) as described. Overlay of these (right panel, yellow) demonstrates co-location of Dd and active β-catenin in the same population of hepatoctyes. (B) Hydrodynamic tail-vein injection was used to transfer control plasmid (pcDNA3.1, left panels) and the constitutively active βcatS37A (right panels) into adult E3-βgl-Dd mice. After two days, hepatocytes were isolated, stained with control IgG antibodies (upper two panels) or FITC-anti-Dd antibodies (lower two panels). The percentage of cells gated as positive for Dd expression (those in the lower right area of each panel) are shown.
The phosphoryation and subsequent degradation of β-catenin accounts for its absence in non-pericentral hepatocytes. In contrast to the wild-type protein, a mutant form of β-catenin in which the serine at position 37 is changed to alanine (βcatS37A) cannot be phosphorylated and is less susceptible to degradation (18). Previous studies showed that adenovirus-mediated βcatS37A overexpression resulted in increased GS expression throughout the liver lobule (8). We used hydrodynamic tail-vein injection to express βcatS37A in the liver of adult E3-βgl-Dd mice. Three days after injection, hepatocytes were purified and analyzed by flow cytometry with anti-Dd antibodies. In control mice, roughly 3% of hepatocytes, presumably representing pericentral cells, stained positive for Dd (Fig. 2B). In contrast, 30% of hepatocytes expressed Dd in livers from βcatS37A-injected mice. This provides evidence that AFP enhancer E3 can be activated in non-pericentral hepatocytes by constitutively active β-catenin.
If β-catenin is required for the activity of pericentral enhancers, its absence should result in the loss of enhancer activity in the adult liver. To explore this possibility, we crossed E3- βgl-Dd and RGSe-βgl-Dd transgenic mice to mice that were homozygous for a floxed allele of the β-catenin gene (Ctnnb1) and expressed the Alb-Cre transgene. Previous studies showed that Alb-Cre transgene expression leads to a loss of β-catenin in essentially all hepatocytes of adult mice (32). Expression of both E3-βgl-Dd (Fig. 3A) and RGSe-βgl-Dd (Fig. 3B) transgenes was completely absent in hepatocytes lacking β-catenin (βCatΔliv), whereas both transgenes continued to be expressed in hepatocytes of βCatfl mice that did not contain Alb-Cre. As expected, endogenous GS proteins were also absent in β-catenin deficient livers (Fig. 3).
Figure 3. E3-βgl-Dd and RGSe-βgl-Dd expression is lost in the absence of β-catenin in adult hepatocytes.

Several rounds of breeding were performed to obtain that contained E3-βgl-Dd (Panel A) or RGSe-βgl-Dd(Panel B) transgenes, were homozygous for the floxed Ctnnb1 allele and contained (βcatΔliv; right panels) or did not contain (βcatfl; left panels) the Alb-Cre transgene. Frozen adult liver sections were stained with FITC-anti-Dd antibodies (upper panels) or TRITC-anti-GS (lower panels). In the presence of β-catenin, Dd-containing transgenes and the endogenous GS gene were expressed in pericentral hepatocytes. In the absence of β-catenin, no transgene-derived Dd or endogenous GS expression was observed.
β-catenin regulates E3 and RGSe through conserved TCF/LEF sites
β-catenin does not bind DNA directly but can regulate target gene expression through several mechanisms. In the canonical pathway, nuclear β-catenin replaces repressors interacting with bound TCF/LEF factors with coactivators. To explore further the control of E3 and RGSe by β-catenin, we searched for TCF sites in these two defined enhancers. AFP enhancer E3 contains a strong TCF/LEF site located towards the 3’ end of this 340 bp element. Previous studies had identified a single TCF/LEF site in the 3’ end of RGSe (33); our analysis indicates that this is the only TCF/LEF site in this 400 bp enhancer (33). The TCF sites in these two enhancers are highly conserved, suggesting that they are important for the activities of these elements (Table II).
Table II.
TCF sites in AFP E3 and GS upstream enhancer
 |
TCF consensus: A/T A/T C A A A G (G) where middle “A” is indispensable and (G) improves LEF binding (55). Residues that are different from the mouse are highlighted in grey.
EMSAs were used to determine whether the conserved TCF/LEF sites in E3 and RGSe could bind TCF4. Nuclear extracts were prepared from HEK293 cells that were mock-transfected or transfected with a FLAG-tagged TCF4 expression plasmid. Using extracts from TCF4-transfected cells, TCF4 binding could be detected using radiolabeled oligonucleotides containing the conserved TCF sites from E3 and RGSe (Fig. 4A and 4B, respectively); these complexes were not present in mock-transfected cell extracts. In both cases, oligonucleotides containing a consensus TCF site, as well as unlabeled self-fragments, could effectively compete for TCF4 binding. In contrast, mutant forms of these oligonucleotides could no longer act as cold competitors. The ability of anti-FLAG antibodies to supershift the bands with E3 and RGSe oligonucleotides confirmed the presence of TCF4 in these complexes. When the consensus TCF oligonucleotide was used as a radiolabeled probe, wild-type E3 and RGSe oligonucleotides could effectively compete for binding whereas mutant forms of these oligonucleotides could no longer compete (data not shown).
Figure 4. TCF4 binds TCF sites found in E3 and RGSe.

Radiolabeled oligonucleotides corresponding to TCF sites from E3 (Panel A) or GS upstream enhancer (Panel B) were used in EMSAs containing no extract (lane 1) or extracts from HEK293 cells that were mock-transfected (lane 2) or transfected with a FLAG-tagged TCF4 expression vector (lanes 3–8). Samples contained no cold competitor (lane 3) or 100-fold excess of cold competitors {oligonucleotides containing the TCF site of E3 (Panel A) or RGSe (panel B) that is wild-type (self-wt, lane 4) or mutant (self-mt, lane 5) or oligonucleotides containing a consensus TCF site that is wild-type (Con-wt, lane 6) or mutant (con-mt, lane 7)} or anti-FLAG antibody (lane 8). Bands corresponding to free probe (F.P.) and TCF4-DNA complex (TCF4) are designated. With both probes, the wild-type versions of the cold competitors can effectively compete for binding to TCF4 whereas the corresponding mutant forms of these oligonucleotides could not compete. The addition of the FLAG antibody resulted in a supershifted complex. Sequences of wild-type and mutant oligonucleotides are shown in Table I.
TCF4 regulation of E3 and RGSe was explored further using transient transfections. Both enhancers were linked to pGL3-Luciferase and transfected into Hep3B cells along with wild-type β-catenin or βcatS37A. Hep3B cells were used since they have low endogenous β-catenin levels. TOP-Flash and FOP-Flash vectors, which contain 3 copies of consensus or mutated TCF sites, respectively, upstream of the minimal c-fos promoter, were included as controls (17). As expected, TOP-Flash responded to increasing βcatS37A, as determined by normalized luciferase levels, whereas FOP-Flash was unresponsive to co-transfected βcatS37A (Fig. 5). Similar results were seen when wild-type β-catenin was transfected, although the extent of TOP-Flash activation was less than that seen with βcatS37A (data not shown). Both E3-Luc and RGSe-Luc were activated by βcatS37A (Fig. 5) and to a lesser extent by β-catenin (data not shown). Enhancers containing mutated TCF/LEF sites were also assayed for responsiveness to β-catenin. Mutation of the TCF4 site in both enhancers resulted in increased basal activity compared to wild-type enhancers, which is likely due to a release of repression from TCF4-associated co-repressors. The mutant E3-Luc reporter gene showed a ~2-fold induction in response to βcatS37A (Fig. 5) or β-catenin (data not shown); this modest activation was less than the wild-type enhancer and did not change with increasing amounts of βcatS37A. RGSe-Luc with the mutated TCF4 site showed no responsiveness to βcatS37A or β-catenin (Fig. 5 and data not shown).
Figure 5. β-catenin activates E3 and RGSe through their conserved TCF sites.
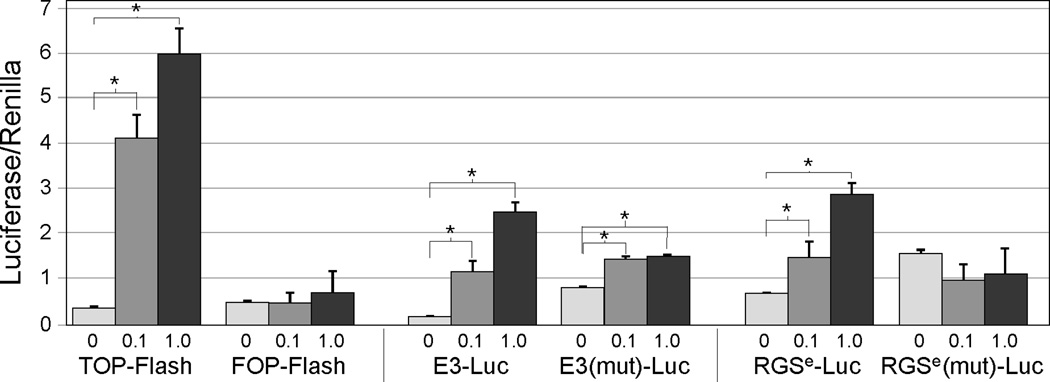
Hep3B cells were transfected with luciferase reporter constructs (TOP-Flash, FOP-Flash and E3-Luc and RGSe-Luc with wild-type and mutated TCF sites), pcDNA alone (0 µg) or in conjunction with increasing amounts of βcatS37A (0.1, 1.0 µg), and Renilla luciferase. Cells were harvested after 48 hours and luciferase levels were determined; firefly luciferase was normalized to renilla. TOP-Flash, E3-Luc and RGSe-Luc were activated by βcatS37A in a dose-dependent manner; FOP-Flash and RGSe-Luc with a mutated TCF site did not respond wherease E3-Luc with a mutated TCF site showed a modest response to βcatS37A. An asterisk (*) indicates significance over cells transfected with pcDNA alone (p < 0.05).
Control of E3 activity and endogenous AFP expression by β-catenin in perinatal livers
In contrast to the pericentral expression seen in the adult liver, E3-βgl-Dd transgenes are expressed in all hepatocytes in the fetal liver (14). A gradual loss of E3 activity in periportal hepatocytes occurs during the perinatal period, which led us to consider whether E3 activity during this time is also dependent on β-catenin. To test this, we monitored E3-βgl-Dd expression by immunofluorescence at postnatal day 1 (p1) in βCatΔliv mice (Fig. 6A). We chose p1 rather than prenatal timepoints since Alb-Cre expression begins later during fetal development and we wanted to allow maximal time for Cre to delete the floxed Ctnnb1 allele (22). In the presence of β-catenin, E3-regulated transgenes are zonally expressed, although activity is not yet fully restricted to a single layer of hepatocytes (Fig. 6A). Similarly to what was seen in the adult liver, E3-βgl-Dd transgenes were not expressed in p1 βCatΔliv livers; the small number of cells that still express Dd likely represents hepatocytes in which the Ctnnb1 gene had not yet been deleted (Fig. 6A). Since endogenous AFP expression in the developing liver requires the AFP enhancer region (34), we also analyzed hepatic AFP mRNA levels in p1 βCatΔliv mice. While AFP mRNA levels varied between mice, we found a significant reduction in AFP mRNA levels when β-catenin levels were low (Fig. 6B). This data suggests that β-catenin is required for normal AFP expression in the developing liver.
Figure 6. β-catenin is required for E3 activity and AFP expression in the perinatal liver.

Livers were removed from E3-βgl-Dd at postnatal day 1 (p1) that did (βcatfl) or did not (βcatΔliv) express β-catenin in hepatocytes. (A) The p1 livers were cryosectioned and stained with FITC-anti-Dd antibodies. E3-regulated transgenes were expressed in pericentral cells from β-catenin-positive livers, although expression is not as highly restricted as it is in adult livers. Transgene expression in βCatΔliv p1 livers was dramatically reduced. Magnification, 20X. (B) Total RNA was prepared from p1 livers from two independent litters and analyzed for β-catenin (left panel) and AFP (right panel) expression (normalized to L30) by real-time RT-PCR. Levels of both transcripts are significantly reduced in βcatΔliv livers (n= 7) compared to transcript levels in βcatfl livers (n=5). An asterisk (*) indicates a significant difference between βcatfl and βcatΔliv cohorts (p < 0.05).
Discussion
The compartmentalization of function enables the adult liver to carry out a variety of different and in some cases opposing functions. Previous studies showed that the β-catenin signaling pathway has an important role in regulating zonally-expressed genes in the adult liver, although the mechanism by which β-catenin regulates target genes is not fully understood. Here, we have shown that two defined enhancer elements that exhibit pericentral activity in the adult liver, E3 and RGSe, are regulated by β-catenin. This was accomplished by demonstrating overlapping E3 activity and active β-catenin expression in the adult liver, increased activation of E3-regulated transgenes in βcatS37A-overexpressing livers, and a loss of E3- and RGSe-regulated transgene expression in the absence of β-catenin. Furthermore, we identified evolutionarily conserved TCF/LEF sites in these enhancers that are required for β-catenin responsiveness. We also showed that endogenous AFP expression is reduced in the perinatal liver in the absence of β-catenin, indicating that this pathway also contributes to developmental AFP regulation.
Our data indicate that the canonical pathway involving β-catenin and TCF4 contributes to E3 and RGSe activity in cultured cells. The 340 bp E3 and 400 bp RGSe enhancers were both found to contain a single, highly conserved consensus TCF/LEF site that could bind TCF4. Both enhancers were activated by wild-type and the constitutively active S37A variant of β-catenin. This data provides strong evidence that the TCF/LEF sites in E3 and RGSe are essential for β-catenin-mediated regulation. When the TCF/LEF sites of E3 and RGSe were mutated, their basal activities increased in the absence of co-transfected β-catenin. Since TCF/LEF factors can bind the Groucho family of co-repressors in the absence of β-catenin, it is not surprising that we saw a de-repression of enhancer activity when TCF/LEF proteins and associated co-repressors could no longer bind their cognate sites. These results raise the question whether these co-repressors also contribute to zonal gene regulation in the adult liver. In this regard, it is interesting that overexpression of the Groucho-related co-repressor Grg3 in H2.35 liver cells reduced endogenous AFP mRNA levels (35). At least one groucho-related protein, Grg5, is expressed in the adult mouse liver (36, 37).
Mouse AFP enhancer E3 was originally defined as a 340 bp element by deletion analysis (38). Since then, research has focused on the 5’ end of E3 since it contains three important cis-acting sites in close proximity, one that binds Foxa and HNF6 proteins, a second that binds C/EBP proteins, and a third site that binds several orphan nuclear receptors, including COUP-TFs, RORα, Rev-erbα and Rev-erbβ (39, 40). In contrast to the 5’ end of E3, the role of the 3’ end has remained elusive. The TCF/LEF site identified here represents the fourth important factor binding site in E3 and the first functional site in the 3’ end of this enhancer. In contrast to E3, RGSe has not been well characterized. Purification of rat liver nuclear proteins bound to RGSe identified STAT5 and TCF (33); the TCF site identified in this earlier report is the one analyzed here.
While our studies demonstrate an important role for β-catenin in the zonal activity of E3 and RGSe, they cannot rule out a role for other factors in the pericentral activity of these enhancers. Using transgenic mice, we have found that mutating the E3 orphan receptor site resulted in increased E3 activity throughout the adult liver, suggesting that orphan receptors bound to this site might repress E3 activity in non-pericentral hepatocytes (JEB, ELC and BTS, manuscript in preparation). Consistent with this result, deleting the orphan receptor HNF4α gene in adult hepatocytes led to elevated expression of GS and other pericentral genes in periportal regions (41). This study, which also identified an HNF4α site in the mouse GS upstream enhancer, argues that HNF4α suppresses GS and other pericentral genes in periportal regions. This is consistent with studies in RLSC mouse liver cells showing a correlation between HNF4α expression and a periportal phenotype (42). How orphan receptors contribute to zonal control, and the possible interplay between these factors and β-catenin, will require further investigation.
The ability of β-catenin to control E3 activity raises the question of whether it also regulates AFP expression during liver development and in HCC. When the β-catenin gene was deleted early during hepatogenesis, AFP mRNA levels were reduced 4-fold (43). However, because liver development was severely disrupted in this study, changes in AFP mRNA could not be clearly attributed to direct or indirect effects of the absence of β-catenin. We found that AFP mRNA levels were significantly reduced in p1 livers when Alb-Cre was used to delete the β-catenin gene late during gestation. Since liver development occurs normally in these mice, our data indicates that β-catenin is required for normal developmental AFP expression. Several clinical studies have evaluated β-catenin and AFP levels in human HCC samples and have found no association between elevated AFP and β-catenin (44–46). In contrast, many pericentral genes, including GS, are highly expressed in liver tumors where β-catenin is activated (8, 47). Thus, AFP reactivation during hepatocarcinogenesis is likely due to mechanisms that do not require β-catenin.
E3 is active in all hepatocytes in the fetal liver, and pericentral expression of E3-regulated transgenes is established during the perinatal period in a periportal-pericentral direction. This developmental transition is similar to what is seen with many pericentral enzymes, including GS, which are also expressed in all hepatocytes throughout the fetal liver. Interestingly, there are parallels between AFP shut-off and establishment of pericentral gene expression; postnatal AFP silencing occurs in a periportal-pericentral direction with pericentral hepatocytes being the last cells to express AFP before the gene is completely silenced (48). Our data indicating that the activity of zonal enhancers in the adult liver and AFP expression in the perinatal liver are both regulated by β-catenin is consistent with the idea that postnatal AFP silencing and establishment of pericentral gene expression are controlled by similar factors. Future studies on the regulation of well-defined zonally active enhancers will further elucidate this unique aspect of hepatic gene regulation.
Acknowledgements
This work is supported by Public Health Service Grant DK-074816 to B.T.S.
The authors thank members of the Spear lab, Catherine Mao and Martha Peterson for helpful discussions and review of the manuscript, Drs. Steven Byers, Hans Clevers, Catherine Mao and Chunming Lui for plasmids, and Dr. Jeffrey Davidson for assistance with TCF/LEF site alignments.
Abbreviations
- GS
glutamine synthetase
- APC
adenomatous polyposis coli
- TCF/LEF
T Cell Factor/Lymphoid Enhancer Factor
- AFP
alpha-fetoprotein
- HCC
hepatocellular carcinoma
- RGSe
rat glutamine synthetase upstream enhancer
- βgl
β-globin
- EMSA
electrophoretic mobility shift assay
References
- 1.Gebhardt R. Metabolic zonation of the liver: regulation and implications for liver function. Pharmacol. Ther. 1992;53:275–354. doi: 10.1016/0163-7258(92)90055-5. [DOI] [PubMed] [Google Scholar]
- 2.Spear BT, Jin L, Ramasamy S, Dobierzewska A. Transcriptional control in the mammalian liver: liver development, perinatal repression, and zonal gene regulation. Cell Mol Life Sci. 2006;63:2922–2938. doi: 10.1007/s00018-006-6258-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jungermann K, Katz N. Functional specialization of different hepatocyte subpopulations. Physiol. Rev. 1989;69:708–763. doi: 10.1152/physrev.1989.69.3.708. [DOI] [PubMed] [Google Scholar]
- 4.Gebhardt R, Mecke D. Heterogenous distribution of glutamine synthetase among rat liver parenchymal cells in situ and in primary culture. EMBO J. 1983;2:5678–5700. doi: 10.1002/j.1460-2075.1983.tb01464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in the liver. Annu. Rev. Nutr. 1996;16:179–203. doi: 10.1146/annurev.nu.16.070196.001143. [DOI] [PubMed] [Google Scholar]
- 6.Jungermann K, Kietzmann T. Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology. 2000;31:255–260. doi: 10.1002/hep.510310201. [DOI] [PubMed] [Google Scholar]
- 7.Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, Vasseur-Cognet M, et al. Apc tumor suppressor gene is the "zonation-keeper" of mouse liver. Dev Cell. 2006;10:759–770. doi: 10.1016/j.devcel.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, Kitajewski J, et al. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002;21:8293–8301. doi: 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- 9.Hailfinger S, Jaworski M, Braeuning A, Buchmann A, Schwarz M. Zonal gene expression in murine liver: lessons from tumors. Hepatology. 2006;43:407–414. doi: 10.1002/hep.21082. [DOI] [PubMed] [Google Scholar]
- 10.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 11.Jennings BH, Ish-Horowicz D. The Groucho/TLE/Grg family of transcriptional co-repressors. Genome Biol. 2008;9:205. doi: 10.1186/gb-2008-9-1-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Y, Kolligs FT, Hottiger MO, Mosavin R, Fearon ER, Nabel GJ. Regulation of beta -catenin transformation by the p300 transcriptional coactivator. Proc Natl Acad Sci U S A. 2000;97:12613–12618. doi: 10.1073/pnas.220158597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peyton DK, Ramesh T, Spear BT. Position-dependent activity of α-fetoprotein enhancer element III in the adult liver is due to negative regulation. Proc.Nat.Acad.Sci.,USA. 2000;97:10890–10894. doi: 10.1073/pnas.200290397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramesh T, Ellis AW, Spear BT. Individual mouse α-fetoprotein enhancer elements exhibit different patterns of tissue-specific and hepatic position-dependent activity. Molec. and Cell. Biol. 1995;15:4947–4955. doi: 10.1128/mcb.15.9.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belayew A, Tilghman SM. Genetic analysis of α-fetoprotein synthesis in mice. Mol. Cell. Biol. 1982;2:1427–1435. doi: 10.1128/mcb.2.11.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarkar G, Sommer SS. The "megaprimer" method of site-directed mutagenesis. Biotechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 17.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 18.Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J Biol Chem. 1997;272:24735–24738. doi: 10.1074/jbc.272.40.24735. [DOI] [PubMed] [Google Scholar]
- 19.Evans PM, Chen X, Zhang W, Liu C. KLF4 interacts with beta-catenin/TCF4 and blocks p300/CBP recruitment by beta-catenin. Mol Cell Biol. 2010;30:372–381. doi: 10.1128/MCB.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spear BT, Tilghman SM. Role of α-fetoprotein regulatory elements in transcriptional activition in transient heterokaryons. Mol. Cell. Biol. 1990;10:5047–5054. doi: 10.1128/mcb.10.10.5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H, Ren H, Spear BT. The Mouse Alpha-Albumin (Afamin) Promoter Is Differentially Regulated by Hepatocyte Nuclear Factor 1alpha and Hepatocyte Nuclear Factor 1beta. DNA Cell Biol. 2010 doi: 10.1089/dna.2010.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26:149–150. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 23.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 24.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum. Gene Therapy. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 25.Gebhardt R, Williams GM. Glutamine synthetase and hepatocarcinogenesis. Carcinogenesis. 1995;16:1673–1681. doi: 10.1093/carcin/16.8.1673. [DOI] [PubMed] [Google Scholar]
- 26.Fahrner J, Labruyere WT, Gaunitz C, Moorman AFM, Gebhardt R, Lamers WH. Identification and functional characterization of regulatory elements of the glutamine synthetase gene from rat liver. Eur. J. Biochem. 1993;213:1067–1073. doi: 10.1111/j.1432-1033.1993.tb17854.x. [DOI] [PubMed] [Google Scholar]
- 27.Gaunitz F, Heise K, Gebhardt R. A silencer element in the first intron of the glutamine synthetase gene represses induction by glucocorticoids. Mol Endocrinol. 2004;18:63–69. doi: 10.1210/me.2003-0062. [DOI] [PubMed] [Google Scholar]
- 28.Gaunitz F, Weber S, Scheja L, Gebhardt R. Identification of a cis-acting element and a novel trans-acting factor of the glutamine synthetase gene in liver cells. Biochem. Biophys. Res. Comm. 2004;284:377–383. doi: 10.1006/bbrc.2001.4967. [DOI] [PubMed] [Google Scholar]
- 29.Garcia de Veas Lovillo RM, Ruijter JM, Labruyere WT, Hakvoort TB, Lamers WH. Upstream and intronic regulatory sequences interact in the activation of the glutamine synthetase promoter. Eur J Biochem. 2003;270:206–212. doi: 10.1046/j.1432-1033.2003.03424.x. [DOI] [PubMed] [Google Scholar]
- 30.Lie-Venema H, de Boer PA, Moorman AF, Lamers WH. Organ-specific activity of the 5' regulatory region of the glutamine synthetase gene in developing mice. Eur J Biochem. 1997;248:644–659. doi: 10.1111/j.1432-1033.1997.00644.x. [DOI] [PubMed] [Google Scholar]
- 31.Lie-Venema H, Labruyere WT, van Roon MA, de Boer PA, Moorman AF, Berns AJ, Lamers WH. The spatio-temporal control of the expression of glutamine synthetase in the liver is mediated by its 5'-enhancer. J. Biol. Chem. 1995;270:28251–28256. doi: 10.1074/jbc.270.47.28251. [DOI] [PubMed] [Google Scholar]
- 32.Braeuning A, Singh Y, Rignall B, Buchmann A, Hammad S, Othman A, von Recklinghausen I, et al. Phenotype and growth behavior of residual beta-catenin-positive hepatocytes in livers of beta-catenin-deficient mice. Histochem Cell Biol. 2010;134:469–481. doi: 10.1007/s00418-010-0747-1. [DOI] [PubMed] [Google Scholar]
- 33.Werth M, Gebhardt R, Gaunitz F. Hepatic expression of glutamine synthetase in rats is controlled by STAT5 and TCF transcription factors. Hepatology. 2006;44:967–975. doi: 10.1002/hep.21322. [DOI] [PubMed] [Google Scholar]
- 34.Jin L, Long L, Green MA, Spear BT. The alpha-fetoprotein enhancer region activates the albumin and alpha-fetoprotein promoters during liver development. Dev Biol. 2009;336:294–300. doi: 10.1016/j.ydbio.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sekiya T, Zaret KS. Repression by Groucho/TLE/Grg proteins: genomic site recruitment generates compacted chromatin in vitro and impairs activator binding in vivo. Mol Cell. 2007;28:291–303. doi: 10.1016/j.molcel.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mallo M, Franco del Amo F, Gridley T. Cloning and developmental expression of Grg, a mouse gene related to the groucho transcript of the Drosophila Enhancer of split complex. Mech Dev. 1993;42:67–76. doi: 10.1016/0925-4773(93)90099-j. [DOI] [PubMed] [Google Scholar]
- 37.Miyasaka H, Choudhury BK, Hou EW, Li SS. Molecular cloning and expression of mouse and human cDNA encoding AES and ESG proteins with strong similarity to Drosophila enhancer of split groucho protein. Eur J Biochem. 1993;216:343–352. doi: 10.1111/j.1432-1033.1993.tb18151.x. [DOI] [PubMed] [Google Scholar]
- 38.Godbout R, Ingram RS, Tilghman SM. Fine-structure mapping of the three mouse α-fetoprotein enhancers. Mol. Cell. Biol. 1988;8:1169–1178. doi: 10.1128/mcb.8.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomassin H, Bois-Joyeux B, Delille R, Ikonomova R, Danan J-L. Chicken Ovalbumin Upstream Promoter-Transcription Factor, Hepatocyte Nuclear Factor 3, an CCAAT/Enhancer Binding Protein Control the Far Upstream Enhancer of the Rat alpha-Fetoprotein Gene. DNA and Cell. Biol. 1996;15:1063–1074. doi: 10.1089/dna.1996.15.1063. [DOI] [PubMed] [Google Scholar]
- 40.Bois-Joyeuz B, Chauvet C, Nacer-Cherif H, Bergeret W, Mazure N, Giguere V, Laudet V, et al. Modulation of the far-upstream enhancer of the rat α-fetoprotein gene by members of the RORα, Rev-erbα, and rev-erbβ groups of monomeric orphan nuclear receptors. DNA Cell Biol. 2000;19:589–599. doi: 10.1089/104454900750019344. [DOI] [PubMed] [Google Scholar]
- 41.Stanulovic VS, Kyrmizi I, Kruithof-de Julio M, Hoogenkamp M, Vermeulen JL, Ruijter JM, Talianidis I, et al. Hepatic HNF4alpha deficiency induces periportal expression of glutamine synthetase and other pericentral enzymes. Hepatology. 2007;45:433–444. doi: 10.1002/hep.21456. [DOI] [PubMed] [Google Scholar]
- 42.Colletti M, Cicchini C, Conigliaro A, Santangelo L, Alonzi T, Pasquini E, Tripodi M, et al. Convergence of Wnt signaling on the HNF4alpha-driven transcription in controlling liver zonation. Gastroenterology. 2009;137:660–672. doi: 10.1053/j.gastro.2009.05.038. [DOI] [PubMed] [Google Scholar]
- 43.Tan X, Yuan Y, Zeng G, Apte U, Thompson MD, Cieply B, Stolz DB, et al. Beta-catenin deletion in hepatoblasts disrupts hepatic morphogenesis and survival during mouse development. Hepatology. 2008;47:1667–1679. doi: 10.1002/hep.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng SY, Chen WJ, Lai PL, Jeng YM, Sheu JC, Hsu HC. High alpha-fetoprotein level correlates with high stage, early recurrence and poor prognosis of hepatocellular carcinoma: significance of hepatitis virus infection, age, p53 and beta-catenin mutations. Int J Cancer. 2004;112:44–50. doi: 10.1002/ijc.20279. [DOI] [PubMed] [Google Scholar]
- 45.Torbenson M, Kannangai R, Abraham S, Sahin F, Choti M, Wang J. Concurrent evaluation of p53, beta-catenin, and alpha-fetoprotein expression in human hepatocellular carcinoma. Am J Clin Pathol. 2004;122:377–382. doi: 10.1309/YH0H-3FKY-M4RM-U1JF. [DOI] [PubMed] [Google Scholar]
- 46.Gorog D, Regoly-Merei J, Paku S, Kopper L, Nagy P. Alpha-fetoprotein expression is a potential prognostic marker in hepatocellular carcinoma. World J Gastroenterol. 2005;11:5015–5018. doi: 10.3748/wjg.v11.i32.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loeppen S, Schneider D, Gaunitz F, Gebhardt R, Kurek R, Buchmann A, Schwarz M. Overexpression of glutamine synthetase is associated with beta-catenin-mutations in mouse liver tumors during promotion of hepatocarcinogenesis by phenobarbital. Cancer Res. 2002;62:5685–5688. [PubMed] [Google Scholar]
- 48.Emerson JA, Vacher J, Cirillo LA, Tilghman SM, Tyner AL. The zonal expression of α-fetoprotein transgenes in the livers of adult mice. Developmental Dynamics. 1992;195:55–66. doi: 10.1002/aja.1001950106. [DOI] [PubMed] [Google Scholar]