

Abstract
Neuromuscular diseases, which encompass disorders that affect muscle and its innervation, are highly heritable. Genetic diagnosis now frequently pinpoints the primary mutation responsible for a given neuromuscular disease. However, the results from genetic testing indicate that neuromuscular disease phenotypes may vary widely, even in individuals with the same primary disease-causing mutation. Clinical variability arises from both genetic and environmental factors. Genetic modifiers can now be identified using candidate gene as well as genomic approaches. The presence of genetic modifiers for neuromuscular disease helps define the clinical outcome and also highlights pathways of potential therapeutic utility. Herein, we will focus on single gene neuromuscular disorders, including muscular dystrophy, spinal muscular atrophy, and amyotrophic lateral sclerosis, and the methods that have been used to identify modifier genes. Animal models have been an invaluable resource for modifier gene discovery and subsequent mechanistic studies. Some modifiers, identified using animal models, have successfully translated to the human counterpart. Furthermore, in a few instances, modifier gene discovery has repetitively uncovered the same pathway, such as TGFβ signaling in muscular dystrophy, further emphasizing the relevance of that pathway. Knowledge of genetic factors that influence disease can have direct clinical applications for prognosis and predicted outcome.
Keywords: Genetic modifier, muscular dystrophy, amyotrophic lateral sclerosis, spinal muscular atrophy
Introduction
Genetic interactions were long hypothesized to be relevant for neuromuscular diseases (NMDs). With improvement in sequencing technology, the primary genetic mutation responsible for many monogenetic NMDs, especially the muscular dystrophies, is commonly determined. With knowledge of the primary gene defect, it is now apparent that the range of clinical presentation associated with mutation in a single gene is often broader than previously anticipated. Correspondingly, research efforts are now directed at unearthing genes that modify the effect of a primary disease causing mutation. Most monogenic diseases have phenotypes that vary widely, even in individuals with the same disease-causing mutation. This indicates the presence of additional factors, such as modifier genes, that alter disease outcome. Below we will discuss the current approaches used to discover modifier genes, different types of modifier genes that have been identified for NMDs, and the importance of modifier genes in revealing pathways involved in NMD pathogenesis.
Modifier genes
A modifier gene is a genetic locus that enhances or suppresses the outcome of the primary disease causing mutation. Modifier genes may affect different aspects of disease, such as age at onset, severity of disease, or duration of disease, and may act on some disease parameters but not others. For example, a modifier may regulate disease onset, but have no effect on disease duration. Genetic modifiers may act only on a specific subtype of NMD, while others may act on many NMDs. For example, the EPHA4 and SMN2 genes are modifiers of both amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) [1–4]. PGC1α has been found to modify ALS, Huntington's disease and Parkinson's disease [5, 6]. These findings emphasize the importance of collating findings across different NMDs, since pathways that modify more than one type of disease become excellent targets for therapy development.
Neuromuscular diseases and the significance of genetic modifiers
Inherited NMDs affect an estimated 1:3000 people worldwide, causing muscle weakness, chronic disability and even premature death [7]. NMDs create a significant financial burden, both on patients and families and the healthcare system; the total US cost of NMDs is estimated to be over $1 billion per year [8]. NMDs encompass the muscular dystrophies, motor neuron diseases, neuromuscular junction diseases, and others. Despite a good understanding of the primary genetic basis of many NMDs, there are few, if any, curative therapies. Considerable progress is being made in the areas of gene correction/restoration, cell based therapies, and supportive care, so that quality and quantity of life is improving with NMD. Despite progress, new approaches are needed, and determining the pathways that can alter the course of disease may reveal biological pathways useful for prognosis and therapy development.
Uncovering modifier genes can also have direct clinical application. For example, genetic markers that indicate an increased potential for cardiorespiratory complications can be used to institute earlier supportive therapy. Awareness of modifier genes can also be helpful when designing clinical trials. Clinical trials in rare diseases are complicated by having to recruit sufficient numbers of subjects. Identifying those subjects who are outliers can help stratify findings with strong scientific rationale. The ability to design a study equipped with the knowledge of disease modifying factors would make a clinical trial more efficient; one could stratify patients based on genotype at a modifier locus or use modifier loci as covariates in the analysis [9]. Reducing phenotypic variation permits smaller sample sizes that are more cost-effective, and also encourages the study of rare diseases where large sample sizes are often infeasible.
Approaches for Discovering Modifier Genes
The two major approaches for identifying genetic modifiers rely on examining candidate genes, based on biological knowledge of a gene or pathway, or unbiased approaches, which rely on genomewide scans or gene profiling. Most often, the search for modifiers relies on a blend of these methods. Candidate gene approaches can be augmented by gene expression profiling or other means, which can link a pathway to a given disease process. Limiting the number of genes to be tested avoids the burden of multiple testing, and the candidate gene approach has been successful in identifying many modifier genes including chondrolectin for SMA, CNTF for ALS, and osteopontin for Duchenne Muscular Dystrophy (DMD) [10–12].
Genomewide approaches to identify modifiers may employ quantitative trait locus (QTL) mapping or genomewide association studies (GWAS) to find statistical associations between genetic determinants across the genome and quantitative phenotypes, such as measurements of disease severity. GWAS necessitates a large sample size for sufficient power, and choosing the correct phenotypes for study requires a sound understanding of the disease process, the targeted cell or tissue type, as well as reproducible and reliable measures. There are some disadvantages to GWAS, such as the large sample size required and the high burden of multiple testing. Furthermore, ascribing the modifier effect to a gene within a GWAS locus is often biased toward genes that are already known to have a biological role related to the underlying disease. Genomewide approaches also entail whole exome, genome, transcriptome, and methylome analyses. These approaches have less bias, and thus the capacity to reveal surprising new pathways. For example, PLS3, a modifier of SMA in worms, flies, zebrafish, mice, and humans, was originally discovered by a transcriptome-wide differential expression analysis on a few individuals with highly discordant phenotypes [13–15].
Mapping modifiers in human NMD populations can be achieved using cohorts of related or unrelated individuals. The mapping cohort can be a large group of unrelated individuals who carry a primary mutation in the same gene. In this case, the primary gene mutations are likely to vary and analysis needs to account for heterogeneity that arises from having different primary gene mutations. An alternative approach relies on disease segregation in large families, where the genetic background may be less diverse and, importantly, individuals share the same primary gene mutation. For example, this technique was utilized in a study of the largest known Emery-Dreifuss muscular dystrophy (EDMD) family, consisting of 59 family members segregating an autosomal dominant form of EDMD due to a mutation in LMNA [16]. A strong linkage signal on chromosome 2 was associated with earlier disease onset, and this region includes the DES gene encoding desmin, an intermediate filament protein like LMNA [16].
Modifier gene discovery thus far mainly focused on identifying variants that affect the expression or sequence of genes that encode proteins. However non-coding RNAs, such as micro RNAs (miRNAs) and small nucleolar RNAs (snoRNAs), have been shown to play a role in human disease and are poised to act as modifiers as well [17]. miRNAs have been shown to play an important role in skeletal muscle homeostasis and function [18], and identifying variants that affect binding of these miRNAs could lead to novel therapeutic options for NMDs.
Animal models for mapping genetic modifiers
Animal models of NMD offer some advantages for QTL mapping. Using animal models for mapping reduces variability arising from nongenetic causes, especially those that derive from environmental differences. Since the environment can be better controlled for in animal models, as opposed to humans, the degree of phenotypic variability attributed to diet or other factors can be minimized. Additionally, population size is less problematic since sufficiently large mapping cohorts can be more easily developed using animal models. Like GWAS, the success or failure of QTL mapping is highly dependent on using reliable and representative measures of disease severity. The major disadvantage of using animal models is the modifier pathway may not translate to the human condition.
Genomewide RNAi screening has also been used to identify modifier genes of NMDs. For example, Dimitriadi et al. used a high-throughput RNAi screen for modifiers of SMA in C. elegans [14]. From this approach, four genes were identified that exacerbated SMN loss-of-function defects. Using functional conservation as a secondary screen, two of these four genes exerted a similar effect in a Drosophila model [14]. Conversely, an RNAi screen in C. elegans was used to evaluate 40 previously identified candidate modifier genes from a Drosophila SMA model [13]. This screen identified 12 genes that modified SMA models in a cross-species manner. This approach was also used to validate PLS3, initially identified as a modifier of human SMA, as a modifier of SMA in worms and flies.
For the muscular dystrophies, a genetic interaction screen in Drosophila was used to identify components that interact with the dystrogylcan-dystrophin complex [19]. Like model organisms, cell-based approaches can also be used to map modifiers. For example, Li et al. relied on a high throughput cell based screening approach used to identify modifiers of SMA [20]. An inducible model, where Smn was knocked down, produced a cell growth phenotype, which was ameliorated by both genetic and pharmacological factors. With increasing development of cell models for disease through IPSCs, this approach is likely to grow in the coming years.
Modifiers of Muscular Dystrophy
Osteopontin
Osteopontin, or secreted phosphoprotein 1 (SPP1) was identified as a modifier of DMD based on transcriptomic profiling [12]. SPP1 is upregulated in dystrophin deficient DMD patients and the mdx mouse model [21, 22]. SPP1 was found to be downregulated approximately 3 fold in mild patients versus severe DMD, indicating that downregulation of SPP1 may be protective in disease [12]. A single nucleotide variant referred to as −66T>G (and also known as rs28357094) in the promoter of SPP1 correlated with disease severity in two cohorts of human DMD patients. Disease severity was measured by age at loss of ambulation in the test cohort, and then verified in a validation cohort using grip strength as the measure. The rare G allele of SNP rs28357094 was associated with more severe disease [12]. The G allele has previously been shown to decrease promoter strength in luciferase gene reporter assays [23], which would seemingly contradict downregulation of SPP1 being protective. However, the SPP1 “G” allele may influence expression in a cell-type specific manner and/or be under the influence of steroids or hormones to account for its effect. Another study found no association between the same G allele in the SPP1 promoter and SPP1 mRNA or protein expression in DMD patients, although the sample size was small [24]. One possibility for these conflicting studies is complex transcriptional regulation at this locus. The SPP1 gene is known to be regulated by sex hormones, which may also influence disease [25]. It is also possible that another variant in the region of rs28357094 mediates the DMD disease modifying effect, as this region has strong linkage disequilibrium. Pegoraro et al. also identified two other genes, ACTN3 and CELSR2, as modifiers of DMD. However when corrected for multiple testing, these genes were not significantly associated with disease severity in the replication cohort [12].
Genetic variation in SPP1 has been implicated in a wide-variety of other diseases, including asthma, type 1 diabetes, rheumatoid arthritis, lupus, pseudoxanthoma elasticum (PXE), and multiple sclerosis (MS) [26–31]. SPP1 is a phosphorylated acidic glycoprotein with multiple functions, including bone-remodeling, cell-mediated immunity, and wound healing. In skeletal muscle, myoblasts are an important source of osteopontin when tissue integrity is compromised [32]. Osteopontin is expressed at low levels in normal mouse muscle, but is highly upregulated following damage and is associated with areas of regeneration in mdx mice [32, 33]. These data implicate osteopontin as an important modulator of matrix remodeling post-injury. Transforming growth factor β (TGFβ) is also critical to tissue injury and remodeling, and the SPP1/osteopontin and TGFβ pathways intersect. TGFβ binds and activates the SPP1 promoter [34]. Treatment of myoblast cultures with TGFβ1 results in increased osteopontin expression [32]. mdx mice deficient in SPP1 show a decrease in TGFβ1, which results in decreased fibrosis and increased muscle strength [21]. These results support a model where osteopontin expression leads to TGFβ activation and a feed forward mechanism where TGFβ also promotes osteopontin expression (Fig. 1). Interestingly, a SNP in TGFBR2, encoding the TGFβ receptor II protein, acts as a predictor of SPP1 mRNA levels in DMD, further establishing the interdependence of SPP1 and TGFβ [24]. There is evidence that SPP1 acts in a sex-specific manner, which should be kept in mind when designing studies involving both males and females. In healthy controls, the −66T>G variant of SPP1 is strongly associated with increased muscle volume in females, with individuals carrying the G allele showing 14–17% larger muscles [33]. In mice, Opn-null females had smaller muscles for five out of seven muscle groups examined, whereas males only displayed smaller muscle size for two of the muscle groups [33]. Genetic association studies of SPP1 in other diseases have also shown sex-specific effects [30, 35]. The SPP1 promoter is estrogen responsive [25], which may explain some of these sex-specific effects.
Fig. 1.
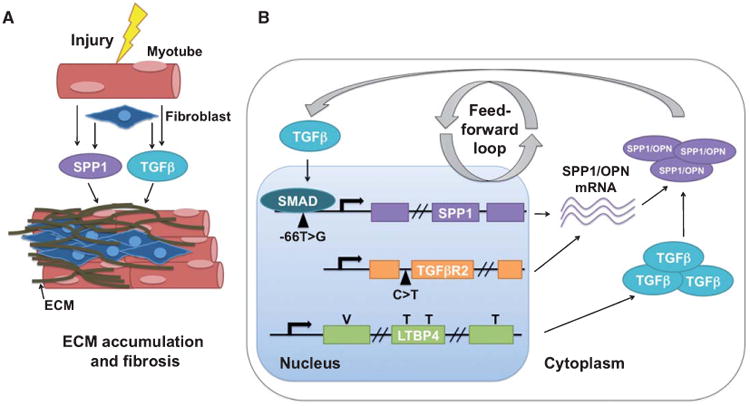
The TGFβ pathway in muscular dystrophy. SPP1, encoding osteopontin (OPN), and TGFβ are co-regulated. A) When myofibers are injured, osteopontin and TGFβ signaling pathways are activated, leading to ECM accumulation and fibrosis of the muscle. B) TGFβ activates the SPP1 promoter through SMAD signaling. A SNP in the SPP1 promoter is associated with a more severe form of DMD. A SNP in TGFBR2 results in increased SPP1 expression. The “VTTT” allele of LTBP4 leads to increased TGFβ signaling and is associated with a more severe form of DMD. Treatment of myoblasts with TGFβ results in increased osteopontin expression. The mdx mouse deficient for SPP1/osteopontin has reduced TGFβ signaling and reduced fibrosis.
Latent TGFβ binding protein 4 (LTBP4)
Ltbp4 was originally found in a genomewide screen for modifiers of muscular dystrophy using a mouse model of limb girdle muscular dystrophy (LGMD) [36]. Mice lacking the dystrophin associated protein γ-sarcoglycan (Sgcg) share a similar disease course as mdx mice. Sgcg mice in the DBA 2J strain have a more severe form of muscular dystrophy than Sgcg mice from the 129SV/J strain or C57Bl6/J backgrounds [37]. Interbreeding these two strains produces a more diverse phenotypic range, which is quantified by measuring Evans Blue dye within muscle. Evans blue dye gains entry into myofibers when the sarcolemma has been disrupted, as occurs more easily with dystrophin or sarcoglycan mutations [38]. Hydroxyproline content was also used a quantitative measure of fibrosis since this modified amino acid is found in collagen. Mice with a short in-frame deletion altering a proline rich hinge region of LTBP4 protein have increased membrane leak and fibrosis compared to mice carrying an insertion at this locus [36].
To test whether LTBP4 modifies human muscular dystrophy, Flanigan et al. genotyped nsSNPs in LTBP4 in DMD patients and correlated the genotypes at these SNPs with age at ambulatory loss [39]. The four nsSNPs in the human LTBP4 gene are distributed along the length of the protein, although one falls near the region implicated in binding TGFβ. Furthermore, the four nsSNPs are in linkage disequilibrium forming the “VTTT” or “IAAM” haplotypes, so named for the amino acid missense changes. DMD patients homozygous for the IAAM haplotype ambulated significantly longer than patients carrying LTBP4 SNPs constituting the VTTT haplotype [39]. The protective IAAM allele along with a history of steroid treatment had the largest advantage, having walked until age 12.5±3.3 years. In comparison, ambulatory loss occurred at age 10.7±2.1 years for those with the VTTT allele and steroid use. LTBP4 is an extracellular matrix protein that binds and sequesters TGFβ in the extracellular matrix and regulates its availability to the TGFβ receptor. Fibroblasts from humans carrying the risk allele of LTPB4 (VTTT) have increased TGFβ signaling compared to individuals carrying the protective allele (IAAM) [39].
TGFβ expression is increased in human DMD muscle and in the mdx mouse [40, 41]. Increased TGFβ expression is associated with excessive matrix accumulation in a wide-variety of diseases, such as liver cirrhosis and cardiac fibrosis [42]. LTBP4 SNPs from the VTTT haplotype, or SNPs in linkage disequilibrium with these, have been associated with deleterious phenotypes in other diseases, such as functional impairment in chronic obstructive pulmonary disease, more aggressive tumors in colorectal cancer, and the expansion of abdominal aortic aneurysm [43–45]. The fact that both LTBP4 and SPP1 modify multiple diseases suggests that these genes have broad effects and may modify other NMDs.
Replication of genetic modifiers of disease in independent cohorts can be helpful, but especially challenging when working with rare diseases. For example, the SPP1 SNP association in DMD was replicated in two cohorts by Pegoraro et al. but was not replicated in the cohort studied by Flanigan et al. [39]. However, the absence of replication may relate to altered population structure, small samples sizes, and the necessity of correcting for multiple testing.
Annexin A6
Anxa6, encoding the membrane associated annexin A6 protein, was identified recently using the same strategy that discovered Ltbp4. A similar intercross strategy, coupled with genomewide mapping, was applied to the Sgcg mouse model of LGMD [46]. QTL mapping revealed a locus on chromosome 11 that was associated with two traits, membrane damage in muscle and increased heart mass in mice with muscular dystrophy. Further assessment of the chromosome 11 locus, using RNA sequencing and whole genome analysis of the DBA 2J versus 129SV/J strains of mice, pointed to Anxa6 as a modifier of muscular dystrophy. Annexin A6 is a phospholipid binding protein and is the most abundant annexin in the heart [47]. Annexins have been implicated in a number of cellular processes including membrane repair. Annexin A1 and A2 interact with dysferlin, a protein responsible for LGMD 2B, another subtype of recessive muscular dystrophy [48]. Overexpression of annexin A1 and annexin A2 is associated with increased disease severity in dysferlinopathies [49]. Annexin A2 and A6 act as scaffolding proteins and become part of lipid rafts within plasma membranes in both smooth muscle and skeletal muscle [50, 51]. In an injury model in zebrafish, annexin A6 localized rapidly to the site of sarcolemmal disruption [52]. Using high resolution stimulated emission depletion microscopy studies in mammalian muscle, annexin A6 was shown to form a discreet protective cap over the site of muscle disruption [46].
Genetic variation in Anxa6 from the DBA 2J strain focused on two SNPs in exons 11 and 15 that produced low-level amounts of an Anxa6 splice variant. This splice variant encodes a truncated annexin A6 protein, which interfered with the normal translocation of annexin A6 to the site of sarcolemmal disruption [46]. Truncated annexin A6 protein acted in a dominant negative manner and was associated with a more severe form of muscular dystrophy. It remains to be seen if ANXA6 modifies human muscular dystrophy, but the identification of this pathway highlights other genes and proteins that may alter muscle membrane repair. Moreover, whether Anxa6 modifies other mouse models of muscular dystrophy has not yet been shown.
Modifiers of SMA
SMA is a progressive degenerative disorder affecting both lower and upper motor neurons. Loss and damage of motor neurons cause muscle weakness, atrophy, and eventual paralysis in SMA patients. Ninety-five percent of SMA cases are caused by homozygous deletion of the SMN1 gene, while the remaining 5% of cases are caused by missense mutations, splice site mutations, or small deletions in SMN1 [53–58]. The phenotype of SMA is variable in both severity and age of onset, which can happen as early as before birth to as late as adulthood. Clinically, patients are classified into subgroups: SMA type I(severe), type II (intermediate), type III (mild) and type IV (adult-onset) [59–61]. A summary of SMA modifiers is shown in Table 1.
Table 1. Cross-species modifiers of Spinal Muscular Atrophy.
| Human Gene | Ce Gene | Dm Gene | Modifies Ce SMA | Modifies Dm SMA | Modifies Human SMA | Function |
|---|---|---|---|---|---|---|
| PLS3 | plst-1 | Fim | x | x | x | Formation and stabilization of F-actin bundles |
| NCBP2L | ncbp-2 | Cbp20 | x | x | Nuclear export of mRNA, U snRNA transport, nonsense mediated decay, miRNA maturation | |
| NPVF | flp-4 | Fmrf | x | x | Activation of neuropeptide gated chloride channels and G-protein coupled receptors | |
| USO1 | uso-1 | p115 | x | x | Vesicle tethering during trans-Golgi transport | |
| PPARG | nhr-85 | Eip75B | x | x | Nuclear hormone receptor, regulation of circadian rhythms | |
| FGFR3 | egl-15 | Btl | x | x | FGF signaling, NMJ function and development | |
| ATF6 | atf-6 | CG3136 | x | x | Unfolded protein stress response | |
| PPP1R13 | ape-1 | CG18375 | x | x | Prevention of inappropriate apoptosis | |
| NEK2 | nekl-3 | Nek2 | x | x | Mitotic regulation | |
| ACTN | atn-1 | actinin | x | x | Actin-bundling | |
| STRN | cash-1 | CKA | x | x | Calveolin and calmodulin-binding | |
| DYNLL2 | dlc-1 | ctp | x | x | Intracellular trafficking, regulation of dynamin, F-actin assembly, transport of TGFβ | |
| RNF149 | kcnl-2 | SK | x | x | Potassium channel subunit | |
| BMPR2 | daf-4 | Wit | x | x | TGFβ receptor subunit, cell specification | |
| RXRA | nhr-25 | Usp | x | x | Ecdysone regulated molting, activation of Smad2 in muscles |
SMA: Spinal muscular atrophy. Dm: Drosophila melanogaster. Ce: Caenorhabditis elegans.
SMN2
The wide-range of clinical presentation associated with SMA and the genetic homogeneity underlying the primary genetic defect in SMA strongly supports the presence of genetic modifiers. SMN2 is a gene highly homologous to SMN1, arisen from gene duplication events of the SMN1 gene. Several independent studies have found SMN2 copy number to be a strong predictor of SMA severity [62–66]. The single C>T transition in SMN2 exon 7 results in exclusion of this exon in ∼85% of SMN2 transcripts [67]. The SMN2 transcripts missing exon 7 produce a less stable protein [67] with reduced oligomerization capacity [68]. SMN2 deletion on its own has no phenotype in humans, and 5–14% of normal individuals, with atleast two copies of SMN1, carry zero copies of SMN2 [54, 62]. In patients with SMA however, SMN2 copy number is inversely correlated with disease severity; SMA patients with one or two copies of SMN2 have a more severe form of disease than patients carrying 3 or 4 copies of SMN2 [62]. This inverse dose response was affirmed in a mouse model of SMA, where a human SMN2 transgene at high copy number rescued the motor neuron deficit of these mice [69]. Several small molecules have been shown to increase SMN2 expression and ameliorate disease phenotypes in mouse models, patient cells lines, and human pilot studies [70–75].
PLS3
Comparison of whole transcriptomes of lymphoblastoid cell lines from affected versus unaffected siblings with the same SMN1/2 status identified PLS3 as a protective modifier of SMA [15]. PLS3 encodes Plastin 3, also known as T-Plastin, a protein involved in the formation and stabilization of actin bundles [76]. PLS3 is a sex-specific modifier; unaffected SMN-deleted females expressed higher levels of PLS3 in blood, whereas males overexpressing PLS3 are not protected from SMA [15]. PLS3 is encoded on the X chromosome, and PLS3 expression in blood correlates inversely with disease severity, but only in post-pubertal female patients [77]. In a mouse model of SMA, PLS3 overexpression delays axonal pruning and rescues neuromuscular junction function by stabilizing F-actin dependent processes [78]. This modifying effect was also seen in zebrafish where overexpression of PLS3 was able to rescue neuromuscular defects in SMN deficient fish [15, 79]. The molecular basis of PLS3 overexpression in unaffected individuals is unknown.
ZPR1
ZPR1 encodes Zinc Finger Protein 1 and was identified as a protective modifier of SMA in a candidate gene study investigating the expression of SMN interacting proteins in lymphoblastoid cell lines from SMA patients versus controls [80]. ZPR1 protein levels are elevated in lymphoblastoid cell lines from mildly affected SMA patients and controls compared to severely affected patients [80]. ZPR1 facilitates SMN accumulation in nuclear bodies, and the ZPR1-SMN interaction is disrupted in SMA patients [81]. In a mouse model of SMA, reduction in the level of ZPR1 results in increased disease severity and decreased lifespan [82]. In cell lines from SMA patients, overexpression of ZPR1 results in increased SMN protein levels, without a change in SMN mRNA expression, suggesting that ZPR1's interaction with SMN stabilizes the SMN protein complex and prevents turnover [82]. As is the case with PLS3, the genetic basis of ZPR1 overexpression is unknown.
Modifiers of ALS
Similar to SMA, amyotrophic lateral sclerosis (ALS) is marked by progressive degeneration of motor neurons and muscle weakness. The genetic causes of ALS are far more heterogeneous than SMA, and only in 10% of ALS cases is there a known genetic cause. Approximately 20% of familial ALS cases are caused by mutation in the Cu/Zn superoxide dismutase 1 (SOD1) gene [83, 84]. Age of onset is typically later in life, on average around 50 years, and most patients die within 2–5 years of diagnosis. However, the clinical presentation of ALS is quite variable, both in age of onset and disease duration.
Mouse models
In a mouse model of ALS, mice that carry the same SOD1 mutation have considerable variation in disease severity depending on background strain [85, 86], indicating that genetic modifiers are playing a role. Transcriptomic analysis of mice from two of these strains revealed multiple potential modifier genes and pathways associated with slow or fast disease progression [87]. Candidate gene analysis has been widely tested by cross-breeding SOD1 mutant mice with genetically modified strains. These studies have identified many genes that, when perturbed, result in differences in disease onset or survival of these mice (reviewed in [2, 88]).
SMN
SMN, the gene responsible for SMA, has also been shown to modify ALS. A genomewide mapping screen for modifiers of the SOD1 mouse phenotype was conducted analyzing genetic loci for delayed onset of disease [86]. A strong linkage signal was found on chromosome 13, the locus containing Smn [86]. Follow up studies showed that cell lines expressing SOD1 mutant protein had less SMN protein expression than lines transfected with WT SOD1. Both SOD1 mutant mice and human patients with ALS have lower than normal levels of SMN in their spinal cords [2, 3]. Additionally, SOD1 mutant mice with a genetic reduction of SMN display a more severe ALS phenotype [3], whereas neuronal overexpression of SMN ameliorates the ALS phenotype [2]. Taken together, these data indicate that SMN acts as a protective modifier in ALS, just as SMN is protective in SMA.
EPHA4
Ephrin type A receptor 4 (EPHA4) was identified as a modifier of ALS in a genetic screen in zebrafish overexpressing mutant SOD1. A morpholino-based genetic screen identified 13 genes that, when reduced, rescued the mutant phenotype. The most protective morpholino was directed against zebrafish gene Rtk2, which is 67% identical to human EPHA4 [1]. Pharmacological inhibition of Epha4 using 2,5-dimethylpyrrolyl benzoic acid had the same rescue effect as the morpholino. EPHA4 is a receptor tyrosine kinase that interacts with ephrins—signaling molecules that are involved in axonal repulsion and regulation of synapse formation [89]. EPHA4 also acts as a modifier of ALS in mice, rats, and humans. In mice with SOD1 mutations, a 50% genetic reduction of Epha4 increases motor performance and survival [1]. In SOD1-mutant rats, treatment with an EPHA4-blocking peptide delayed disease onset and increased survival [1]. In humans, lower EPHA4 mRNA expression in blood from ALS patients is associated with later disease onset and prolonged survival. However, a genetic-association study of SNPs surrounding the EPHA4 locus did not find any associations between EPHA4 SNPs and ALS susceptibility, survival, or age at onset [1]. This negative result does not exclude EPH4A as a genetic modifier, since EPHA4 regulation may be regulated by trans-acting factors.
Conclusions
Neuromuscular disease has provided an excellent paradigm in which to demonstrate the role of genetic modifiers. The past decade has seen remarkable success in defining the complex genetic modifiers that influence neuromuscular disease outcome. For many of the modifier genes that have been identified, the precise genetic sequences that drive the modifier effect have not fully been explained. The observation that expression changes of many of these genes are sufficient to determine mild or severe disease is beneficial, and reveals essential information about new pathways involved in disease pathogenesis. Additional knowledge of the precise genetic changes that mediate these effects could augment genetic testing, prognosis, and treatment for individuals with different genetic makeup. As it stands now, those pathways by which expression levels can change the course of disease provide good drug targets.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- DMD
Duchenne Muscular Dystrophy
- EDMD
Emery Dreifuss Muscular Dystrophy
- EPHA4
Ephrin type A receptor 4
- GWAS
genomewide association study
- IPSC
induced pluripotent stem cell
- LTBP
Latent TGFβ binding protein
- LGMD
limb girdle muscular dystrophy
- PLS3
plastin 3
- TGFβ
transforming growth factor β
- TGFBR2
transforming growth factor β receptor II
- SMA
Spinal Muscular Atrophy
- SPP1
secreted phosphoprotein 1
- SMN
Survival of Motor Neuron
- QTL
quantitative trait locus
- ZPR1
Zinc finger protein 1
References
- 1.Van Hoecke A, Schoonaert L, Lemmens R, Timmers M, Staats KA, Laird AS, Peeters E, Philips T, Goris A, Dubois B, Andersen PM, Al-Chalabi A, Thijs V, Turnley AM, van Vught PW, Veldink JH, Hardiman O, Van Den Bosch L, Gonzalez-Perez P, Van Damme P, Brown RH, Jr, van den Berg LH, Robberecht W. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models in humans. Nat Med. 2012;18(9):1418–1422. doi: 10.1038/nm.2901. [DOI] [PubMed] [Google Scholar]
- 2.Turner BJ, Alfazema N, Sheean RK, Sleigh JN, Davies KE, Horne MK, Talbot K. Overexpression of survival motor neuron improves neuromuscular function and motor neuron survival in mutant SOD1 mice. Neurobiol Aging. 2014;35(4):906–915. doi: 10.1016/j.neurobiolaging.2013.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner BJ, Parkinson NJ, Davies KE, Talbot K. Survival motor neuron deficiency enhances progression in an amyotrophic lateral sclerosis mouse model. Neurobiology of Disease. 2009;34(3):511–517. doi: 10.1016/j.nbd.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Kariya S, Re DB, Jacquier A, Nelson K, Przedborski S, Monani UR. Mutant superoxide dismutase 1 (SOD1), a cause of amyotrophic lateral sclerosis, disrupts the recruitment of SMN, the spinal muscular atrophy protein to nuclear Cajal bodies. Hum Mol Genet. 2012;21(15):3421–3434. doi: 10.1093/hmg/dds174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eschbach J, Schwalenstocker B, Soyal SM, Bayer H, Wiesner D, Akimoto C, Nilsson AC, Birve A, Meyer T, Dupuis L, Danzer KM, Andersen PM, Witting A, Ludolph AC, Patsch W, Weydt P. PGC-1alpha is a male-specific disease modifier of human and experimental amyotrophic lateral sclerosis. Human Molecular Genetics. 2013;22(17):3477–3484. doi: 10.1093/hmg/ddt202. [DOI] [PubMed] [Google Scholar]
- 6.Weydt P, Soyal SM, Landwehrmeyer GB, Patsch W. A single nucleotide polymorphism in the coding region of PGC-1alpha is a male-specific modifier of Huntington disease age-at-onset in a large European cohort. BMC Neurol. 2014;14:1. doi: 10.1186/1471-2377-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emery AE. Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord. 1991;1(1):19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 8.Larkindale J, Yang W, Hogan PF, Simon CJ, Zhang Y, Jain A, Habeeb-Louks EM, Kennedy A, Cwik VA. Cost of illness for neuromuscular diseases in the United States. Muscle & Nerve. 2014;49(3):431–438. doi: 10.1002/mus.23942. [DOI] [PubMed] [Google Scholar]
- 9.Bello L, Piva L, Barp A, Taglia A, Picillo E, Vasco G, Pane M, Previtali SC, Torrente Y, Gazzerro E, Motta MC, Grieco GS, Napolitano S, Magri F, D'Amico A, Astrea G, Messina S, Sframeli M, Vita GL, Boffi P, Mongini T, Ferlini A, Gualandi F, Soraru G, Ermani M, Vita G, Battini R, Bertini E, Comi GP, Berardinelli A, Minetti C, Bruno C, Mercuri E, Politano L, Angelini C, Hoffman EP, Pegoraro E. Importance of SPP1 genotype as a covariate in clinical trials in Duchenne muscular dystrophy. Neurology. 2012;79(2):159–162. doi: 10.1212/WNL.0b013e31825f04ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sleigh JN, Barreiro-Iglesias A, Oliver PL, Biba A, Becker T, Davies KE, Becker CG, Talbot K. Chondrolectin affects cell survival and neuronal outgrowth in in vitro and in vivo models of spinal muscular atrophy. Hum Mol Genet. 2014;23(4):855–869. doi: 10.1093/hmg/ddt477. [DOI] [PubMed] [Google Scholar]
- 11.Giess R, Holtmann B, Braga M, Grimm T, Muller-Myhsok B, Toyka KV, Sendtner M. Early onset of severe familial amyotrophic lateral sclerosis with a SOD-1 mutation: Potential impact of CNTF as a candidate modifier gene. Am J Hum Genet. 2002;70(5):1277–1286. doi: 10.1086/340427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pegoraro E, Hoffman EP, Piva L, Gavassini BF, Cagnin S, Ermani M, Bello L, Soraru G, Pacchioni B, Bonifati MD, Lanfranchi G, Angelini C, Kesari A, Lee I, Gordish-Dressman H, Devaney JM, McDonald CM. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology. 2011;76(3):219–226. doi: 10.1212/WNL.0b013e318207afeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang HC, Dimlich DN, Yokokura T, Mukherjee A, Kankel MW, Sen A, Sridhar V, Fulga TA, Hart AC, Van Vactor D, Artavanis-Tsakonas S. Modeling spinal muscular atrophy in Drosophila. PLoS One. 2008;3(9):e3209. doi: 10.1371/journal.pone.0003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dimitriadi M, Sleigh JN, Walker A, Chang HC, Sen A, Kalloo G, Harris J, Barsby T, Walsh MB, Satterlee JS, Li C, Van Vactor D, Artavanis-Tsakonas S, Hart AC. Conserved genes act as modifiers of invertebrate SMN loss of function defects. PLoS Genet. 2010;6(10):e1001172. doi: 10.1371/journal.pgen.1001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oprea GE, Krober S, McWhorter ML, Rossoll W, Muller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320(5875):524–527. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Granger B, Gueneau L, Drouin-Garraud V, Pedergnana V, Gagnon F, Ben Yaou R, Tezenas du Montcel S, Bonne G. Modifier locus of the skeletal muscle involvement in Emery-Dreifuss muscular dystrophy. Hum Genet. 2011;129(2):149–159. doi: 10.1007/s00439-010-0909-1. [DOI] [PubMed] [Google Scholar]
- 17.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 18.Berardi E, Annibali D, Cassano M, Crippa S, Sampaolesi M. Molecular and cell-based therapies for muscle degenerations: A road under construction. Front Physiol. 2014;5:119. doi: 10.3389/fphys.2014.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kucherenko MM, Pantoja M, Yatsenko AS, Shcherbata HR, Fischer KA, Maksymiv DV, Chernyk YI, Ruohola-Baker H. Genetic modifier screens reveal new components that interact with the Drosophila dystroglycan-dystrophin complex. PLoS One. 2008;3(6):e2418. doi: 10.1371/journal.pone.0002418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li DK, Tisdale S, Espinoza-Derout J, Saieva L, Lotti F, Pellizzoni L. A cell system for phenotypic screening of modifiers of SMN2 gene expression and function. PLoS One. 2013;8(8):e71965. doi: 10.1371/journal.pone.0071965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vetrone SA, Montecino-Rodriguez E, Kudryashova E, Kramerova I, Hoffman EP, Liu SD, Miceli MC, Spencer MJ. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J Clin Invest. 2009;119(6):1583–1594. doi: 10.1172/JCI37662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turk R, Sterrenburg E, van der Wees CG, de Meijer EJ, de Menezes RX, Groh S, Campbell KP, Noguchi S, van Ommen GJ, den Dunnen JT, t Hoen PA. Common pathological mechanisms in mouse models for muscular dystrophies. FASEB J. 2006;20(1):127–129. doi: 10.1096/fj.05-4678fje. [DOI] [PubMed] [Google Scholar]
- 23.Giacopelli F, Marciano R, Pistorio A, Catarsi P, Canini S, Karsenty G, Ravazzolo R. Polymorphisms in the osteopontin promoter affect its transcriptional activity. Physiol Genomics. 2004;20(1):87–96. doi: 10.1152/physiolgenomics.00138.2004. [DOI] [PubMed] [Google Scholar]
- 24.Piva L, Gavassini BF, Bello L, Fanin M, Soraru G, Barp A, Ermani M, Angelini C, Hoffman EP, Pegoraro E. TGFBR2 but not SPP1 genotype modulates osteopontin expression in Duchenne muscular dystrophy muscle. J Pathol. 2012;228(2):251–259. doi: 10.1002/path.4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Craig AM, Denhardt DT. The murine gene encoding secreted phosphoprotein 1 (osteopontin): Promoter structure, activity, and induction in vivo by estrogen and progesterone. Gene. 1991;100:163–171. doi: 10.1016/0378-1119(91)90362-f. [DOI] [PubMed] [Google Scholar]
- 26.Arjomandi M, Galanter JM, Choudhry S, Eng C, Hu D, Beckman K, Chapela R, Rodriguez-Santana JR, Rodriguez-Cintron W, Ford J, Avila PC, Burchard EG. Polymorphism in osteopontin gene (SPP1) is associated with asthma and related phenotypes in a puerto rican population. Pediatr Allergy Immunol Pulmonol. 2011;24(4):207–214. doi: 10.1089/ped.2011.0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forton AC, Petri MA, Goldman D, Sullivan KE. An osteopontin (SPP1) polymorphism is associated with systemic lupus erythematosus. Hum Mutat. 2002;19(4):459. doi: 10.1002/humu.9025. [DOI] [PubMed] [Google Scholar]
- 28.Gazal S, Sacre K, Allanore Y, Teruel M, Goodall AH, Tohma S, Alfredsson L, Okada Y, Xie G, Constantin A, Balsa A, Kawasaki A, Nicaise P, Amos C, Rodriguez-Rodriguez L, Chioccia G, Boileau C, Zhang J, Vittecoq O, Barnetche T, Gonzalez Gay MA, Furukawa H, Cantagrel A, Le Loet X, Sumida T, Hurtado-Nedelec M, Richez C, Chollet-Martin S, Schaeverbeke T, Combe B, Khoryati L, Coustet B, El-Benna J, Siminovitch K, Plenge R, Padyukov L, Martin J, Tsuchiya N, Dieude P. Identification of secreted phosphoprotein 1 gene as a new rheumatoid arthritis susceptibility gene. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-204581. in press. [DOI] [PubMed] [Google Scholar]
- 29.Hendig D, Arndt M, Szliska C, Kleesiek K, Gotting C. SPP1 promoter polymorphisms: Identification of the first modifier gene for pseudoxanthoma elasticum. ClinChem. 2007;53(5):829–836. doi: 10.1373/clinchem.2006.083675. [DOI] [PubMed] [Google Scholar]
- 30.Marciano R, D'Annunzio G, Minuto N, Pasquali L, Santamaria A, Di Duca M, Ravazzolo R, Lorini R. Association of alleles at polymorphic sites in the Osteopontin encoding gene in young type 1 diabetic patients. Clin Immunol. 2009;131(1):84–91. doi: 10.1016/j.clim.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Niino M, Kikuchi S, Fukazawa T, Yabe I, Tashiro K. Genetic polymorphisms of osteopontin in association with multiple sclerosis in Japanese patients. J Neuroimmunol. 2003;136(1-2):125–129. doi: 10.1016/s0165-5728(03)00004-3. [DOI] [PubMed] [Google Scholar]
- 32.Uaesoontrachoon K, Yoo HJ, Tudor EM, Pike RN, Mackie EJ, Pagel CN. Osteopontin and skeletal muscle myoblasts: Association with muscle regeneration and regulation of myoblast function in vitro. Int J Biochem Cell Biol. 2008;40(10):2303–2314. doi: 10.1016/j.biocel.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 33.Hoffman EP, Gordish-Dressman H, McLane VD, Devaney JM, Thompson PD, Visich P, Gordon PM, Pescatello LS, Zoeller RF, Moyna NM, Angelopoulos TJ, Pegoraro E, Cox GA, Clarkson PM. Alterations in osteopontin modify muscle size in females in both humans and mice. Med Sci Sports Exerc. 2013;45(6):1060–1068. doi: 10.1249/MSS.0b013e31828093c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hullinger TG, Pan Q, Viswanathan HL, Somerman MJ. TGFbeta and BMP-2 activation of the OPN promoter: Roles of smad- and hox-binding elements. Exp Cell Res. 2001;262(1):69–74. doi: 10.1006/excr.2000.5074. [DOI] [PubMed] [Google Scholar]
- 35.Han S, Guthridge JM, Harley IT, Sestak AL, Kim-Howard X, Kaufman KM, Namjou B, Deshmukh H, Bruner G, Espinoza LR, Gilkeson GS, Harley JB, James JA, Nath SK. Osteopontin and systemic lupus erythematosus association: A probable gene-gender interaction. PLoS One. 2008;3(3):e0001757. doi: 10.1371/journal.pone.0001757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heydemann A, Ceco E, Lim JE, Hadhazy M, Ryder P, Moran JL, Beier DR, Palmer AA, McNally EM. Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J Clin Invest. 2009;119(12):3703–3712. doi: 10.1172/JCI39845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heydemann A, Huber JM, Demonbreun A, Hadhazy M, McNally EM. Genetic background influences muscular dystrophy. Neuromuscul Disord. 2005;15(9-10):601–609. doi: 10.1016/j.nmd.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139(2):375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flanigan KM, Ceco E, Lamar KM, Kaminoh Y, Dunn DM, Mendell JR, King WM, Pestronk A, Florence JM, Mathews KD, Finkel RS, Swoboda KJ, Gappmaier E, Howard MT, Day JW, McDonald C, McNally EM, Weiss RB. LTBP4 genotype predicts age of ambulatory loss in duchenne muscular dystrophy. Ann Neurol. 2013;73(4):481–488. doi: 10.1002/ana.23819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernasconi P, Di Blasi C, Mora M, Morandi L, Galbiati S, Confalonieri P, Cornelio F, Mantegazza R. Transforming growth factor-beta1 and fibrosis in congenital muscular dystrophies. Neuromuscul Disord. 1999;9(1):28–33. doi: 10.1016/s0960-8966(98)00093-5. [DOI] [PubMed] [Google Scholar]
- 41.Onofre-Oliveira PC, Santos AL, Martins PM, Ayub-Guerrieri D, Vainzof M. Differential expression of genes involved in the degeneration and regeneration pathways in mouse models for muscular dystrophies. Neuromolecular Med. 2012;14(1):74–83. doi: 10.1007/s12017-012-8172-3. [DOI] [PubMed] [Google Scholar]
- 42.Branton MH, Kopp JB. TGF-beta and fibrosis. Microbes Infect. 1999;1(15):1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 43.Hersh CP, Demeo DL, Lazarus R, Celedon JC, Raby BA, Benditt JO, Criner G, Make B, Martinez FJ, Scanlon PD, Sciurba FC, Utz JP, Reilly JJ, Silverman EK. Genetic association analysis of functional impairment in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(9):977–984. doi: 10.1164/rccm.200509-1452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forsti A, Li X, Wagner K, Tavelin B, Enquist K, Palmqvist R, Altieri A, Hallmans G, Hemminki K, Lenner P. Polymorphisms in the transforming growth factor beta 1 pathway in relation to colorectal cancer progression. Genes Chromosomes Cancer. 2010;49(3):270–281. doi: 10.1002/gcc.20738. [DOI] [PubMed] [Google Scholar]
- 45.Thompson AR, Cooper JA, Jones GT, Drenos F, van Bockxmeer FM, Biros E, Walker PJ, van Rij AM, Golledge J, Norman PE, Hafez H, Humphries SE. Assessment of the association between genetic polymorphisms in transforming growth factor beta, and its binding protein (LTBP), and the presence, and expansion, of Abdominal Aortic Aneurysm. Atherosclerosis. 2010;209(2):367–373. doi: 10.1016/j.atherosclerosis.2009.09.073. [DOI] [PubMed] [Google Scholar]
- 46.Swaggart KA, Demonbreun AR, Vo AH, Swanson KE, Kim EY, Fahrenbach JP, Holley-Cuthrell J, Eskin A, Chen Z, Squire K, Heydemann A, Palmer AA, Nelson SF, McNally EM. Annexin A6 modifies muscular dystrophy by mediating sarcolemmal repair. Proc Natl Acad Sci U S A. 2014;111(16):6004–6009. doi: 10.1073/pnas.1324242111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Camors E, Monceau V, Charlemagne D. Annexins and Ca2+ handling in the heart. Cardiovasc Res. 2005;65(4):793–802. doi: 10.1016/j.cardiores.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 48.Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH., Jr Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003;278(50):50466–50473. doi: 10.1074/jbc.M307247200. [DOI] [PubMed] [Google Scholar]
- 49.Cagliani R, Magri F, Toscano A, Merlini L, Fortunato F, Lamperti C, Rodolico C, Prelle A, Sironi M, Aguennouz M, Ciscato P, Uncini A, Moggio M, Bresolin N, Comi GP. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum Mutat. 2005;26(3):283. doi: 10.1002/humu.9364. [DOI] [PubMed] [Google Scholar]
- 50.Babiychuk EB, Draeger A. Annexins in Cell Membrane Dynamics: Ca2+-Regulated Association of Lipid Microdomains. The Journal of Cell Biology. 2000;150(5):1113–1124. doi: 10.1083/jcb.150.5.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Draeger A, Monastyrskaya K, Burkhard FC, Wobus AM, Moss SE, Babiychuk EB. Membrane segregation and downregulation of raft markers during sarcolemmal differentiation in skeletal muscle cells. Developmental Biology. 2003;262(2):324–334. doi: 10.1016/s0012-1606(03)00398-1. [DOI] [PubMed] [Google Scholar]
- 52.Roostalu U, Strahle U. In vivo imaging of molecular interactions at damaged sarcolemma. Dev Cell. 2012;22(3):515–529. doi: 10.1016/j.devcel.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 53.He J, Zhang QJ, Lin QF, Chen YF, Lin XZ, Lin MT, Murong SX, Wang N, Chen WJ. Molecular analysis of SMN1, SMN2, NAIP, GTF2H2, and H4F5 genes in 157 Chinese patients with spinal muscular atrophy. Gene. 2013;518(2):325–329. doi: 10.1016/j.gene.2012.12.109. [DOI] [PubMed] [Google Scholar]
- 54.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 55.Cobben JM, van der Steege G, Grootscholten P, deVisser M, Scheffer H, Buys CH. Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am J Hum Genet. 1995;57(4):805–808. [PMC free article] [PubMed] [Google Scholar]
- 56.Hahnen E, Forkert R, Marke C, Rudnik-Schoneborn S, Schonling J, Zerres K, Wirth B. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum Mol Genet. 1995;4(10):1927–1933. doi: 10.1093/hmg/4.10.1927. [DOI] [PubMed] [Google Scholar]
- 57.Bussaglia E, Clermont O, Tizzano E, Lefebvre S, Burglen L, Cruaud C, Urtizberea JA, Colomer J, Munnich A, Baiget M, et al. A frame-shift deletion in the survival motor neuron gene in Spanish spinal muscular atrophy patients. Nat Genet. 1995;11(3):335–337. doi: 10.1038/ng1195-335. [DOI] [PubMed] [Google Scholar]
- 58.Brahe C, Clermont O, Zappata S, Tiziano F, Melki J, Neri G. Frameshift mutation in the survival motor neuron gene in a severe case of SMA type I. Hum Mol Genet. 1996;5(12):1971–1976. doi: 10.1093/hmg/5.12.1971. [DOI] [PubMed] [Google Scholar]
- 59.Dubowitz V. Chaos in classification of the spinal muscular atrophies of childhood. Neuromuscular Disorders. 1991;1(2):77–80. doi: 10.1016/0960-8966(91)90051-s. [DOI] [PubMed] [Google Scholar]
- 60.Dubowitz V. Chaos in the classification of SMA: A possible resolution. Neuromuscular Disorders. 1995;5(1):3–5. doi: 10.1016/0960-8966(94)00075-k. [DOI] [PubMed] [Google Scholar]
- 61.Russman BS. Spinal muscular atrophy: Clinical classification and disease heterogeneity. J Child Neurol. 2007;22(8):946–951. doi: 10.1177/0883073807305673. [DOI] [PubMed] [Google Scholar]
- 62.Mailman MD, Heinz JW, Papp AC, Snyder PJ, Sedra MS, Wirth B, Burghes AH, Prior TW. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. 2002;4(1):20–26. doi: 10.1097/00125817-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 63.Burghes AHM. When is a deletion not a deletion? When it is converted. The American Journal of Human Genetics. 1997;61(1):9–15. doi: 10.1086/513913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harada Y, Sutomo R, Sadewa AH, Akutsu T, Takeshima Y, Wada H, Matsuo M, Nishio H. Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: Three SMN2 copies fail to rescue some patients from the disease severity. J Neurol. 2002;249(9):1211–1219. doi: 10.1007/s00415-002-0811-4. [DOI] [PubMed] [Google Scholar]
- 66.Wirth B, Brichta L, Schrank B, Lochmuller H, Blick S, Baasner A, Heller R. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet. 2006;119(4):422–428. doi: 10.1007/s00439-006-0156-7. [DOI] [PubMed] [Google Scholar]
- 67.Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9(2):259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 68.Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, Le T, Burghes AH, Androphy EJ. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19(1):63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 69.Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossoll W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9(3):333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 70.Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, Villanova M, Bertini E, Pini A, Neri G, Brahe C. Phenylbutyrate increases SMN expression in vitro: Relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12(1):59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- 71.Brahe C, Vitali T, Tiziano FD, Angelozzi C, Pinto AM, Borgo F, Moscato U, Bertini E, Mercuri E, Neri G. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur J Hum Genet. 2005;13(2):256–259. doi: 10.1038/sj.ejhg.5201320. [DOI] [PubMed] [Google Scholar]
- 72.Mercuri E, Bertini E, Messina S, Pelliccioni M, D'Amico A, Colitto F, Mirabella M, Tiziano FD, Vitali T, Angelozzi C, Kinali M, Main M, Brahe C. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscul Disord. 2004;14(2):130–135. doi: 10.1016/j.nmd.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 73.Brichta L, Holker I, Haug K, Klockgether T, Wirth B. In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann Neurol. 2006;59(6):970–975. doi: 10.1002/ana.20836. [DOI] [PubMed] [Google Scholar]
- 74.Swoboda KJ, Scott CB, Reyna SP, Prior TW, LaSalle B, Sorenson SL, Wood J, Acsadi G, Crawford TO, Kissel JT, Krosschell KJ, D'Anjou G, Bromberg MB, Schroth MK, Chan GM, Elsheikh B, Simard LR. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS One. 2009;4(5):e5268. doi: 10.1371/journal.pone.0005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Riessland M, Ackermann B, Forster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Human Molecular Genetics. 2010;19(8):1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- 76.Giganti A, Plastino J, Janji B, Van Troys M, Lentz D, Ampe C, Sykes C, Friederich E. Actin-filament cross-linking protein T-plastin increases Arp2/3-mediated actin-based movement. J Cell Sci. 2005;118(Pt 6):1255–1265. doi: 10.1242/jcs.01698. [DOI] [PubMed] [Google Scholar]
- 77.Stratigopoulos G, Lanzano P, Deng L, et al. ASsociation of plastin 3 expression with disease severity in spinal muscular atrophy only in postpubertal females. Archives of Neurology. 2010;67(10):1252–1256. doi: 10.1001/archneurol.2010.239. [DOI] [PubMed] [Google Scholar]
- 78.Ackermann B, Krober S, Torres-Benito L, Borgmann A, Peters M, Hosseini Barkooie SM, Tejero R, Jakubik M, Schreml J, Milbradt J, Wunderlich TF, Riessland M, Tabares L, Wirth B. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet. 2013;22(7):1328–1347. doi: 10.1093/hmg/dds540. [DOI] [PubMed] [Google Scholar]
- 79.Hao le T, Wolman M, Granato M, Beattie CE. Survival motor neuron affects plastin 3 protein levels leading to motor defects. J Neurosci. 2012;32(15):5074–5084. doi: 10.1523/JNEUROSCI.5808-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Helmken C, Hofmann Y, Schoenen F, Oprea G, Raschke H, Rudnik-Schoneborn S, Zerres K, Wirth B. Evidence for a modifying pathway in SMA discordant families: Reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Human Genetics. 2003;114(1):11–21. doi: 10.1007/s00439-003-1025-2. [DOI] [PubMed] [Google Scholar]
- 81.Gangwani L, Mikrut M, Theroux S, Sharma M, Davis RJ. Spinal muscular atrophy disrupts the interaction of ZPR1 with the SMN protein. Nat Cell Biol. 2001;3(4):376–383. doi: 10.1038/35070059. [DOI] [PubMed] [Google Scholar]
- 82.Ahmad S, Wang Y, Shaik GM, Burghes AH, Gangwani L. The zinc finger protein ZPR1 is a potential modifier of spinal muscular atrophy. Hum Mol Genet. 2012;21(12):2745–2758. doi: 10.1093/hmg/dds102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rosen DR, Sapp P, O'Regan J, McKenna-Yasek D, Schlumpf KS, Haines JL, Gusella JF, Horvitz HR, Brown RH., Jr Genetic linkage analysis of familial amyotrophic lateral sclerosis using human chromosome 21 microsatellite DNA markers. Am J Med Genet. 1994;51(1):61–69. doi: 10.1002/ajmg.1320510114. [DOI] [PubMed] [Google Scholar]
- 84.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261(5124):1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 85.Mancuso R, Olivan S, Mancera P, Pasten-Zamorano A, Manzano R, Casas C, Osta R, Navarro X. Effect of genetic background on onset and disease progression in the SOD1-G93A model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13(3):302–310. doi: 10.3109/17482968.2012.662688. [DOI] [PubMed] [Google Scholar]
- 86.Kunst CB, Messer L, Gordon J, Haines J, Patterson D. Genetic mapping of a mouse modifier gene that can prevent ALS onset. Genomics. 2000;70(2):181–189. doi: 10.1006/geno.2000.6379. [DOI] [PubMed] [Google Scholar]
- 87.Nardo G, Iennaco R, Fusi N, Heath PR, Marino M, Trolese MC, Ferraiuolo L, Lawrence N, Shaw PJ, Bendotti C. Transcriptomic indices of fast and slow disease progression in two mouse models of amyotrophic lateral sclerosis. Brain. 2013;136(Pt 11):3305–3332. doi: 10.1093/brain/awt250. [DOI] [PubMed] [Google Scholar]
- 88.Riboldi G, Nizzardo M, Simone C, Falcone M, Bresolin N, Comi GP, Corti S. ALS genetic modifiers that increase survival of SOD1 mice and are suitable for therapeutic development. Prog Neurobiol. 2011;95(2):133–148. doi: 10.1016/j.pneurobio.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 89.Klein R. Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat Neurosci. 2009;12(1):15–20. doi: 10.1038/nn.2231. [DOI] [PubMed] [Google Scholar]