

Abstract
Cerebral ischemia was induced using photothrombosis 1 hour after intraperitoneal injection of the p38 mitogen-activated protein kinase (MAPK) inhibitor SB239063 into Swedish mutant amyloid precursor protein (APP/SWE) transgenic and non-transgenic mice. The number of surviving neurons in the penumbra was quantified using Nissl staining, and the activity of p38 MAPKs was measured by western blotting. The number of surviving neurons in the penumbra was significantly reduced in APP/SWE transgenic mice compared with non-transgenic controls 7 days after cerebral ischemia, but the activity of p38 MAPKs was significantly elevated compared with the non-ischemic hemisphere in the APP/SWE transgenic mice. SB239063 prevented these changes. The APP/SWE mutation exacerbated ischemic brain injury, and this could be alleviated by inhibiting p38 MAPK activity.
Keywords: cerebral ischemia, amyloid precursor protein, transgenic, Alzheimer's disease, p38 mitogen-activated protein kinase, SB239063, neural regeneration
Abbreviations
AD, Alzheimer's disease; Aβ, beta amyloid; MAPKs, mitogen activated protein kinases; APP/SWE, Swedish mutant amyloid precursor protein
INTRODUCTION
Many cases of familial Alzheimer's disease (AD) are caused by mutations in the amyloid precursor protein (APP)[1] gene. Increased brain beta amyloid (Aβ) levels were first reported in 1996 by Hsiao and colleagues in Swedish mutant amyloid precursor protein (APP/SWE) transgenic mice[2]. Aβ is overproduced in APP/SWE transgenic mice, which increases the vulnerability of neurons to cerebral ischemia via enhanced microglial activation, inflammation, disruption of endothelium-dependent vascular reactivity and inhibition of nitric oxide production[3,4]. Aβ also induces apoptosis via activation of mitogen activated protein kinases (MAPKs). MAPKs are a family of Ser/Thr kinases which transduce cell surface signals into changes in enzyme activity and gene expression. Stress, cytokines and ultraviolet radiation activate MAPKs, which play an important role in inflammation and cell death[5]. The p38 MAPKs are a MAPK subfamily, and are conserved from yeast to mammals. p38 MAPK signaling is linked to oxidative stress, not only in neonatal hippocampal slice cultures subjected to oxygen and glucose deprivation[6], but also in the rat brain after transient focal cerebral ischemia[7]. Accu Accumulating evidence indicates that p38 MAPKs may play roles in AD pathophysiology, including neuroinflammation, excitotoxicity, synaptic plasticity and tau phosphorylation[8,9]. SB239063, a new generation p38 MAPK inhibitor, has been studied in asthma, hepatic encephalopathy and cerebral ischemia due to its anti-inflammatory and anti-apoptotic properties[10,11,12]. However, only a few studies have focused on the effects of SB239063 on AD or AD concomitant with cerebral ischemia. In the present study, we induced cerebral ischemia in APP/SWE transgenic mice using photothrombosis to simulate mixed dementia, and investigated the effects of SB239063 on ischemic brain injury in APP/SWE transgenic mice.
RESULTS
Quantitative analysis of experimental animals
A total of 12 adult APP/SWE transgenic mice and 12 non-transgenic mice were selected and randomly assigned to two groups: vehicle (intraperitoneal injection of dimethyl sulphoxide; n = 6) and SB239063 (intraperitoneal injection of SB239063; n = 6). All mice were subjected to photothrombosis to induce cerebral ischemia 1 hour after administration. Nissl staining and western blot analyses were performed 7 days after ischemia. All 24 mice were included in the final analysis.
PCR results
PCR results revealed the presence of the expected bands of 0.4 kb for human APP and 0.7 kb for mouse prion protein (PrP) (Figure 1).
Figure 1.
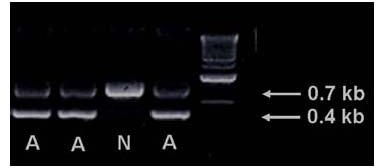
Identification of APP/SWE transgenic mice by PCR. A: APP/SWE transgenic mice; N: non-transgenic mice. The last lane contains the marker. APP/SWE: Swedish mutant amyloid precursor protein.
Effect of SB239063 on surviving neurons in the penumbra in APP/SWE transgenic mice
The ischemic index was determined by calculating the ratio of the amount of surviving neurons in the penumbra of the ischemic region in the hippocampus or in the cortex to that in the non-ischemic hemisphere. The ischemic index in the hippocampus of non-transgenic mice was significantly higher than in the APP/SWE transgenic mice (P < 0.05; Figure 2). Compared with the vehicle group, SB239063 administration significantly increased the ischemic index in the cortex and hippocampus of mice in both the non-transgenic and APP/SWE transgenic groups (P < 0.05; Figure 3).
Figure 2.
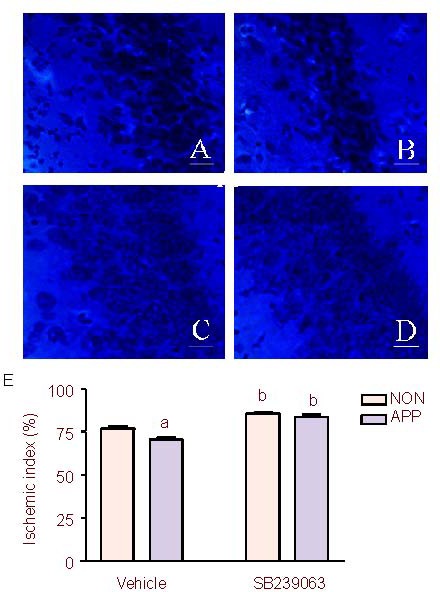
Effect of SB239063, a new generation p38 mitogen activated protein kinase inhibitor, on ischemic index in the hippocampus after photothrombosis in APP/SWE transgenic mice and non-transgenic mice (n = 6 mice per group).
(A–D) Nissl staining of the hippocampus: A, the non-transgenic mice in the vehicle group; B, the APP/SWE transgenic mice in the vehicle group; C, the non-transgenic mice in the SB239063 group; D, the APP/SWE transgenic mice in the SB239063 group. (E) Effect of SB239063 on ischemic index in the hippocampus after photothrombosis in APP/SWE transgenic and non-transgenic mice. Scale bars: 20 μm.
APP: APP/SWE transgenic mice group; NON: non-transgenic mice group. Ischemic index: ratio of amount of surviving neurons in the penumbra in the ischemic hemisphere to that in the non-ischemic hemisphere.
aP < 0.05, vs. non-transgenic mice group; bP < 0.05, vs. the vehicle group. Data were expressed as mean ± SEM (one-way analysis of variance followed by the Student-Newman-Keuls post hoc test). APP/SWE: Swedish mutant amyloid precursor protein.
Figure 3.
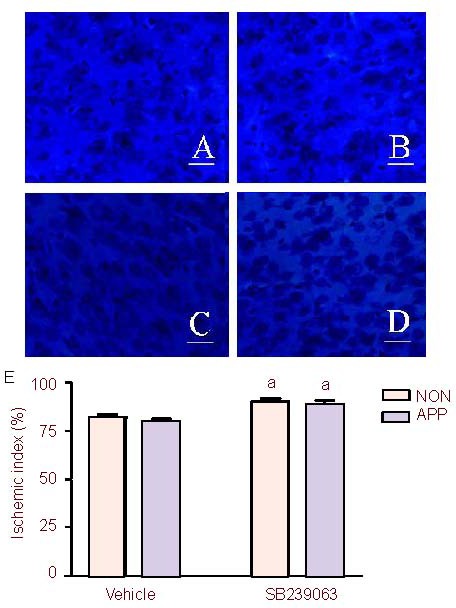
Effect of SB239063, a new generation p38 mitogen activated protein kinase inhibitor, on ischemic index in the cortex after photothrombosis in APP/SWE transgenic and non-transgenic mice (n = 6 mice per group).
(A-D) Nissl staining of the cortex: A; the non-transgenic mice in the vehicle group; B, the APP/SWE transgenic mice in the vehicle group; C, the non-transgenic mice in the SB239063 group; D, the APP/SWE transgenic mice in the SB239063 group. (E) Effect of SB239063 on ischemic index in the cortex after photothrombosis in APP/SWE transgenic and non-transgenic mice. Scale bars: 20 μm.
APP: APP/SWE transgenic mice group; NON: non-transgenic mice group. Ischemic index: ratio of amount of surviving neurons in the penumbra in the ischemic hemisphere to that in the non-ischemic hemisphere.
aP < 0.05, vs. vehicle group. Data were expressed as mean ± SEM (one-way analysis of variance followed by the Student-Newman-Keuls post hoc test). APP/SWE: Swedish mutant amyloid precursor protein.
Effect of SB239063 on p38 MAPK activity in APP/SWE transgenic mice
On day 7 after photothrombotic stroke, western blot analysis was used to determine the expression of p38 MAPKs. In non-transgenic mice, no p-p38 MAPK (active form of p38 MAPK) expression was detected in either the ischemic or non-ischemic hemisphere. In APP/SWE transgenic mice, the ratio of p-p38/p38 in the ischemic hemisphere was significantly higher than in the non-ischemic hemisphere (P < 0.05). Compared with the vehicle group, SB239063 administration significantly decreased p-38 MAPK activity in the ischemic hemisphere (P < 0.05), but not in the non-ischemic hemisphere (P > 0.05), indicating that there was no significant difference in p38 MAPK activity between the ischemic and non-ischemic hemispheres in APP/SWE transgenic mice (P > 0.05; Figure 4).
Figure 4.
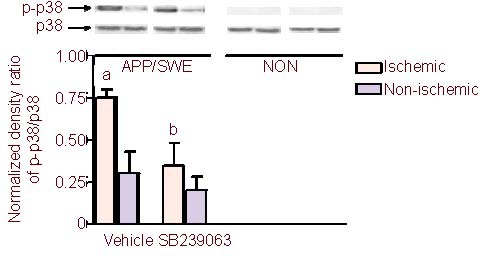
Effect of SB239063, a new generation p38 mitogen activated protein kinase inhibitor, on the activity of p38 MAPKs after photothrombosis in APP/SWE transgenic and non-transgenic mice (n = 6 mice per group).
Activity of p38MAPK is represented as the ratio of phosphorylated p38MAPK to p38MAPK. APP/SWE: APP/SWE transgenic mice; NON: non-transgenic mice.
aP < 0.05, vs. non-ischemic hemisphere; bP < 0.05, vs. vehicle group (one-way analysis of variance followed by the Student-Newman-Keuls post hoc test). APP/SWE: Swedish mutant amyloid precursor protein.
DISCUSSION
The APP/SWE transgenic mouse is the most widely used animal model of AD. Induced cerebral ischemia in this strain is used as an animal model of mixed dementia. Photothrombosis has been used to induce cerebral infarcts, which are highly reproducible in size and location in mice and rats[13], and this microvascular occlusion method is particular suitable for simulating ischemic brain damage in AD.
In the present study, cerebral ischemia was induced in APP/SWE transgenic mice after vehicle and SB239063 administration. Our results demonstrate that (1) APP/SWE mice treated with vehicle had greater neuronal loss in the penumbra in the hippocampus and higher p38 MAPK activity; and (2) SB239063 prevented neuronal loss in the hippocampus and cortex due to cerebral ischemia in both the APP/SWE transgenic and non-transgenic groups. Specifically, SB239063 rescued more neurons from photothrombosis in APP/SWE transgenic mice, and the ischemic index increased to the level of non-transgenic mice after treatment. Thus, the neuroprotective effect of SB239063 is likely due to its ability to reduce p-38 MAPK activity.
In our previous study, there was no significant difference in infarct volume between APP/SWE transgenic and non-transgenic mice on day 7 after photothrombotic stroke[14]. In the present study, the histological findings showed that neuronal loss in the penumbra of the hippocampus in APP/SWE mice was greater than in non-transgenic mice. Previous studies have demonstrated that susceptibility to ischemic brain damage caused by middle cerebral artery occlusion in transgenic mice overexpressing APP was increased, and the effect likely involved Aβ-induced disruption of endothelium-dependent vascular reactivity and enhanced microglial activation[3,4].
MAPKs are a family of Ser/Thr kinases which connect cell surface signals to changes in enzyme activity and gene expression. The signaling pathway of p38 MAPKs is responsible for tissue injury after ischemia/reperfusion and Aβ exposure[15,16]. In the present study, activities of p38 MAPKs after cerebral ischemia in non-transgenic and APP/SWE transgenic mice were measured. In non-transgenic mice, phosphorylated p38 was undetectable in the ischemic and non-ischemic hemispheres. Generally, expression of p38 MAPKs is very low in the absence of induction factors. AD, as an induction factor, activates p38 MAPK[17,18]. In the APP/SWE transgenic mice, p38 MAPKs were activated in the non-ischemic hemisphere, and further activation of p38 MAPKs was induced in the ischemic hemisphere on day 7 after stroke. Cerebral ischemia, also an induction factor, activates p38 MAPK. However, the activity of p38 MAPK declines to the normal level on day 7 after cerebral ischemia[19].
In the present study, APP/SWE prolonged the duration of p38 MAPK activation. On day 7 after cerebral ischemia, the activity of p38 MAPKs in the ischemic hemisphere was significantly higher than in the non-ischemic hemisphere. These results imply that both cerebral ischemia and APP/SWE induce the activation of p38 MAPKs; when cerebral ischemia and AD co-occurred, p38 MAPKs were further activated. Activated p38 MAPKs mediate inflammation and increase expression of nitric oxide synthetase, tumor necrosis factor and interleukin-1β, all of which induce neuronal apoptosis[20]. Therefore, APP/SWE aggravates ischemic neuronal damage after photothrombotic stroke by activating p38 MAPKs.
SB239063 is a new generation p38 MAPK inhibitor, which is selective for two isoforms of p38 MAP kinase, α and β. Ischemic conditions activate p38 MAPKs in the brain, and SB239063 has been shown to have a neuroprotective effect on cerebral ischemia. For example, SB239063 decreases the number of activated microglia, facilitates neurogenesis, reduces or abolishes ischemic factor stimulation of blood-brain barrier Na-K-Cl cotransporter activity and rescues astrocyte dysfunction[11,21,22]. In the present study, SB239063 prevented neuronal loss from cerebral ischemia in the hippocampus and cortex in both APP/SWE transgenic and non-transgenic mice. SB239063 inhibited p38 MAPK activity in the ischemic hemisphere, lowering it to a level similar to that in the non-ischemic hemisphere.
In summary, photothrombosis caused more severe damage in the APP/SWE transgenic mice than in non-transgenic mice. The potential mechanism includes increased p38 MAPK activity, which enhances neuronal apoptosis. Thus, SB239063 rescues neurons from photothrombosis-mediated ischemic injury by suppressing p38 MAPK activity.
MATERIALS AND METHODS
Design
A randomized, controlled, animal experiment.
Time and setting
The experiment was performed at the Medical Research Center, Second Clinical College, Jinan University, China, between May and October 2009.
Materials
APP/SWE transgenic mice were provided by the U.S. National Institutes of Health, which carry the human Swedish mutation. According to the study of Hsiao et al[2], the APP/SWE transgenic mice had normal learning and memory in spatial reference and alternation tasks at 3 months of age, but showed impairment by 9 to 10 months of age. Accompanying these behavioral deficits, numerous Aβ plaques were present in cortical and limbic structures, with elevated amounts of Aβ1-40 and Aβ1-42/43[2]. Three-month-old (25-30 g) male APP/SWE and male non-transgenic mice (C57BL/A2G; U.S. National Institutes of Health) were housed in a light-controlled environment at a standard temperature and allowed free access to food and water. All mice were genotyped to determine the presence of the transgenes.
Methods
Drug administration
Six APP/SWE transgenic mice and six non-transgenic mice received the vehicle dimethyl sulphoxide (3% in normal saline; 100 μL) intraperitoneally, followed by photothrombosis 1 hour later. The other six APP/SWE transgenic mice and six non-transgenic mice received SB239063 (Sigma-Aldrich, St. Louis, MO, USA; 92 μg dissolved in 100 μL of 3% dimethyl sulphoxide) intraperitoneally 1 hour prior to photothrombosis[23]. Mice were sacrificed on day 7 after photothrombosis for neuron quantification and western blotting.
Screening for APP/SWE double transgenic mice
A 1.0-cm length of mouse tail, cut from the tip, was dissected into 4-5 pieces and placed into a 1.5-mL microcentrifuge tube containing 0.5 mL of tail buffer and 20 μL of protease K (20 mg/mL), and incubated at 55-57°C overnight. On the second day, 0.5 mL of phenol was added into the tube and shaken vigorously for 1 minute. After centrifugation at 16 000 × g for 3 minutes, the upper phase was transferred to a new microcentrifuge tube, mixed with 0.5 mL of phenol/chloroform (1:1), shaken for 1 minute, and centrifuged at 16 000 × g for 3 minutes. The upper phase was transferred to a clear tube, mixed with 50 μL of sodium acetate (3 M, pH 6.0) and 0.5 mL of ethanol (100%). The tube was spun for 30 seconds and the supernatant was discarded, yielding the DNA pellet. After washing with 1 mL of 70% ethanol, the pellet was air-dried for 30 minutes, resuspended with 0.2 mL of Tris-EDTA (TE) buffer, heated at 55°C for 20 minutes, and stored at 4°C.
To screen for human APP DNA, a PCR reaction (10 μL) using the primers PrP-sense (5’ CCT CTT TGT GAC TAT GTG GAC TGA TGT CGG 3’), PrP-antisense (5’GTG GAT ACC CCC TCC CCC AGC GTA GAC C 3’) and APP (5’ CCG AGA TCT CTG AAG TGA AGA TGG ATG 3’) was thermocycled in a PTC-100™ Programmable Thermal Controller (MJ Research, Inc.) under the following conditions: 1 cycle of 95°C for 3 minutes; 30 cycles of 95°C for 1 minute, 59°C for 1 minute, 72°C for 1 minute and 40 seconds; and 1 cycle of 72°C for 10 minutes. The PCR product was mixed thoroughly with 6 × loading buffer (0.25% bromophenol blue, 0.025% xylene cyanol FF and 30% glycerol in water) at a ratio of 1:5, and loaded on a 1% agarose gel containing 0.25 μg/mL of ethidium bromide and run at 100 V for 30 minutes to reveal the expected bands of 0.4 kb for human APP and 0.7 kb for mouse PrP[2].
Establishment of the cerebral ischemia model
Mice were anesthetized with a mixture of ketamine 10% (at a dose of 150 mg/kg) and xylazine 2% at a ratio of 2:1. At the dorsal aspect of the head, the skull was exposed by a median incision of the skin, the periosteum was gently removed, and the bregma and lambda points were identified. A fiber optic bundle of a cold white light (Intralux 150 H; Volpi, Zurich, Switzerland) from a halogen bulb (15 V, 150 W Xenophot HLX64634 EFR; Osram, Munich, Germany) with a 1.5 mm aperture was centered using a micromanipulator at 2.0 mm posterior and 3.0 mm laterally from the bregma on the left side. According to the atlas by Paxinos et al[24], at this stereotactic position, the mouse sensorimotor cortex, caudate putamen and hippocampus were identified. The aperture of the cold light source was placed as close as possible to the skull to avoid scattering light that could cause variability. Rose Bengal (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in sterile saline at a concentration of 10 mg/mL, and 0.1 mL was injected intravenously 1 minute before illumination. The brains were then illuminated through the intact skull for 15 minutes. Subsequently, the skin was sutured and mice were allowed to regain consciousness. Mice tolerated the entire procedure and showed no visible neurological deficits[10].
Analysis of surviving neurons in the penumbra using Nissl staining
For the quantification of surviving neurons, 40-μm thick frozen brain sections were stained using the Nissl method. Briefly, the nuclei of neurons were stained with 0.1% cresyl violet solution (Sigma-Aldrich). The slices were rehydrated, stained in cresyl violet for 3-5 minutes, then dehydrated in 100% alcohol and cleared in xylene, and finally mounted with resin medium. Nissl bodies (neurons) were stained red-violet. The slices were then observed and captured under an AxioVision-4 Microscope Vision Imaging System (Carl Zeiss Vision, Germany). The neurons were quantified using an image analyzer (UTHSCSA ImageTool IT version 2.0; the University of Texas Health Science Center, San Antonio, USA). For frontal cortical neurons, all neurons with diameters greater than 6 μm were quantified in three horizontally consecutive areas near the infarction, using a 40 × objective lens and the image analyzer. CA1 hippocampal neurons with diameters greater than 8 μm were similarly quantified using a 40 × lens. In the non-ischemic hemisphere, symmetrical sites were selected to quantify neurons according to the above criteria. The mean neuronal number per mm2 was obtained from three different sections[25]. The ischemic index was determined by calculating the ratio of the amount of surviving neurons in the penumbra in the hippocampus or cortex in the ischemic hemisphere to that in the non-ischemic hemisphere.
Western blot for p38 MAPK activity
Mice were sacrificed, and two hemispheres were placed in a tissue homogenizer in 2 mL of lysis radioimmunoprecipitation buffer solution (phosphate buffered saline, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) with 10 mg/mL phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin and 1 μg/mL leupeptin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The samples were homogenized and centrifuged at 11 000 × g for 10 minutes, then the supernatant was quickly frozen at -70°C and stored for further analysis. The overall protein contents of the extracts were determined by Bio-Rad protein assay based on Bradford's method[26]. Equal amounts of total protein, 60 μg, were loaded on the lanes of sodium dodecyl sulfate-polyacrylamide gels and separated by electrophoresis (5% acrylamide stacking gel and 12% acrylamide separating gel). The separated proteins were transferred electrophoretically from the gel onto nitrocellulose membranes. After washing in buffer and blocking non-specific binding sites, the membranes were probed at 4°C overnight with rabbit anti-mouse p38 monoclonal antibody (1:1 000; Cat. 9212, Cell Signaling Technology, which detected three isoforms of p38 MAP kinase, α, β and γ) and mouse anti-phospho-p38 monoclonal antibody (1:500; Cat. 9216, Cell Signaling Technology, which detected three isoforms of p38 MAP kinase, α, β and γ only when activated by dual phosphorylation at Thr180 and Tyr182). After washes, the membranes were incubated with goat anti-rabbit or rabbit anti-mouse secondary antibodies conjugated to horseradish peroxidase (both at 1:2 000 dilution) for 1 hour at room temperature. The bands representing the proteins were visualized using an enhanced chemiluminescence detection system and X-ray film (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The intensity of bands in the autoradiograms was measured using a computer-assisted program (Image Processing and Analysis in Java, NIH Image). The activity of p38 MAPKs in each group was expressed as a percentage of phosphorylated p38 (p-p38) to p38[27].
Statistical analysis
All data were analyzed using SPSS 12.0 for Windows (SPSS, Chicago, IL, USA). Measurement data are expressed as mean ± SEM. The data were analyzed using one-way analysis of variance followed by the Student-Newman-Keuls post hoc test. A two-tailed P value less than 0.05 was used to indicate statistical significance.
Footnotes
Funding: The study was supported by the National Natural Science Foundation of China, No. 81171191; Shenzhen Bureau of Science Technology and Information, No. 201002013; Guangdong Province Medical Science Fund, No. A2008601; and Jinan University Scientific Research Foundation for Creation and Cultivation, No. 21609708.
Conflicts of interest: None declared.
Ethical approval: The experiment was conducted according to the institutional guidelines with the protocol approved by the Animal Ethics Committee of Jinan University, China.
(Edited by Zhu FQ, Huang G/Su LL/Song LP)
REFERENCES
- 1.Kamat CD, Gadal S, Mhatre M, et al. Antioxidants in central nervous system diseases: preclinical promise and translational challenges. J Alzheimers Dis. 2008;15:473–493. doi: 10.3233/jad-2008-15314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hsiao K, Chapman P, Nilsen S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 3.Koistinaho M, Koistinaho J. Interactions between Alzheimer's disease and cerebral ischemia--focus on inflammation. Brain Res Brain Res Rev. 2005;48:240–250. doi: 10.1016/j.brainresrev.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 4.Marchesi VT. Alzheimer's dementia begins as a disease of small blood vessels, damaged by oxidative-induced inflammation and dysregulated amyloid metabolism: implications for early detection and therapy. FASEB J. 2011;25:5–13. doi: 10.1096/fj.11-0102ufm. [DOI] [PubMed] [Google Scholar]
- 5.Wen J, Watanabe K, Ma M, et al. Edaravone inhibits JNK-c-Jun pathway and restores anti-oxidative defense after ischemia-reperfusion injury in aged rats. Biol Pharm Bull. 2006;29:713–718. doi: 10.1248/bpb.29.713. [DOI] [PubMed] [Google Scholar]
- 6.Lu Q, Rau TF, Harris V, et al. Increased p38 mitogen-activated protein kinase signaling is involved in the oxidative stress associated with oxygen and glucose deprivation in neonatal hippocampal slice cultures. Eur J Neurosci. 2011;34:1093–1101. doi: 10.1111/j.1460-9568.2011.07786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukerji SS, Rainey RN, Rhodes JL, et al. Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J Neurochem. 2009;111:1138–1148. doi: 10.1111/j.1471-4159.2009.06406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology. 2010;58:561–568. doi: 10.1016/j.neuropharm.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Munoz L, Ralay Ranaivo H, Roy SM, et al. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agusti A, Cauli O, Rodrigo R, et al. p38 MAP kinase is a therapeutic target for hepatic encephalopathy in rats with portacaval shunts. Gut. 2011;60:1572–1579. doi: 10.1136/gut.2010.236083. [DOI] [PubMed] [Google Scholar]
- 11.Strassburger M, Braun H, Reymann KG. Anti-inflammatory treatment with the p38 mitogen-activated protein kinase inhibitor SB239063 is neuroprotective, decreases the number of activated microglia and facilitates neurogenesis in oxygen-glucose-deprived hippocampal slice cultures. Eur J Pharmacol. 2008;592:55–61. doi: 10.1016/j.ejphar.2008.06.099. [DOI] [PubMed] [Google Scholar]
- 12.Duan W, Wong WS. Targeting mitogen-activated protein kinases for asthma. Curr Drug Targets. 2006;7:691–698. doi: 10.2174/138945006777435353. [DOI] [PubMed] [Google Scholar]
- 13.Schroeter M, Jander S, Stoll G. Non-invasive induction of focal cerebral ischemia in mice by photothrombosis of cortical microvessels: characterization of inflammatory responses. J Neurosci Methods. 2002;117:43–49. doi: 10.1016/s0165-0270(02)00072-9. [DOI] [PubMed] [Google Scholar]
- 14.Zou LY, Chu XF. Effect of Swedish amyloid precursor protein gene on cerebral ischemia induced by photothrombosis. Zhonghua Shenjing Yixue Zazhi. 2006;5:654–657. [Google Scholar]
- 15.Bartov O, Sultana R, Butterfield DA, et al. Low molecular weight thiol amides attenuate MAPK activity and protect primary neurons from Abeta(1-42) toxicity. Brain Res. 2006;1069:198–206. doi: 10.1016/j.brainres.2005.10.079. [DOI] [PubMed] [Google Scholar]
- 16.Yoo K, Choi JW, Choi MS, et al. Mitogen-activated protein kinases (MAPKs) mediate SIN-1/glucose deprivation-induced death in rat primary astrocytes. Arch Pharm Res. 2005;28:942–947. doi: 10.1007/BF02973881. [DOI] [PubMed] [Google Scholar]
- 17.Savage MJ, Lin YG, Ciallella JR, et al. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer's disease model is associated with amyloid deposition. J Neurosci. 2002;22:3376–3385. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology. 2010;58:561–568. doi: 10.1016/j.neuropharm.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Lennmyr F, Karlsson S, Gerwins P, et al. Activation of mitogen-activated protein kinases in experimental cerebral ischemia. Acta Neurol Scand. 2002;106:333–340. doi: 10.1034/j.1600-0404.2002.01313.x. [DOI] [PubMed] [Google Scholar]
- 20.Philpott KL, Facci L. MAP kinase pathways in neuronal cell death. CNS Neurol Disord Drug Targets. 2008;7:83–97. doi: 10.2174/187152708783885129. [DOI] [PubMed] [Google Scholar]
- 21.Wallace BK, Jelks KA, O’Donnell ME. Ischemia-induced stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransport involves p38 and JNK MAP kinases. Am J Physiol Cell Physiol. 2012;302:C505–517. doi: 10.1152/ajpcell.00261.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jayakumar AR, Panickar KS, Murthy ChR, et al. Oxidative stress and mitogen-activated protein kinase phosphorylation mediate ammonia-induced cell swelling and glutamate uptake inhibition in cultured astrocytes. J Neurosci. 2006;26:4774–4784. doi: 10.1523/JNEUROSCI.0120-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karunakaran S, Saeed U, Mishra M, et al. Selective activation of p38 mitogen-activated protein kinase in dopaminergic neurons of substantia nigra leads to nuclear translocation of p53 in 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine-treated mice. J Neurosci. 2008;28:12500–12509. doi: 10.1523/JNEUROSCI.4511-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paxinos G, Franklin K. 2nd ed. San Diego: Academic Press; 2001. The Mouse Brain in Stereotaxic Coordinates. [Google Scholar]
- 25.Chui DH, Tanahashi H, Ozawa K, et al. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- 26.Compton SJ, Jones CG. Mechanism of dye response and interference in the Bradford protein assay. Anal Biochem. 1985;151:369–374. doi: 10.1016/0003-2697(85)90190-3. [DOI] [PubMed] [Google Scholar]
- 27.Chen CH, Zhang DH, LaPorte JM, et al. Cyclic AMP activates p38 mitogen-activated protein kinase in Th2 cells: phosphorylation of GATA-3 and stimulation of Th2 cytokine gene expression. J Immunol. 2000;165:5597–5605. doi: 10.4049/jimmunol.165.10.5597. [DOI] [PubMed] [Google Scholar]