

Abstract
Fluorescence spectroscopy is a sensitive technique for detecting protein-protein, protein-RNA and RNA-RNA interactions, requiring only nanomolar concentrations of labeled components. Fluorescence anisotropy provides information about the assembly of multi-subunit proteins, while molecular beacons provide a sensitive and quantitative reporter for base pairing between complementary RNAs. Here we present a detailed protocol for labeling Hfq protein with cyanine 3-maleimide and dansyl chloride to study the protein oligomerization and RNA binding by semi-native polyacrylamide gel electrophoresis (PAGE) and fluorescence anisotropy. We also present a detailed protocol for measuring the rate of annealing between a molecular beacon and a target RNA in the presence of Hfq using a stopped-flow spectrometer.
Keywords: Protein oligomerization, RNA chaperone, molecular beacon, fluorescence anisotropy, stopped-flow fluorescence, Hfq
1. Introduction
Hfq is a member of the Sm/Lsm protein superfamily abundant in most bacteria (1, 2). Hfq participates in multiple regulatory networks that mediate the response to stressors such as cold shock, low ion metabolism, oxidative stress, sugar metabolism, and quorum sensing (3, 4). Hfq acts in part by stabilizing regulatory small RNAs (sRNAs) and by facilitating base pairing between sRNAs and their target mRNA leader sequences. Hfq also interacts with RNase E and regulates the stability of mRNAs by either blocking or promoting RNA turnover by RNase E (5).
Bacterial Hfq is a homo-hexamer, and its highly conserved Sm core forms a ring-shaped structure with two RNA binding faces. The ‘proximal’ face of the ring binds single-stranded U-rich RNA sequences, while the ‘distal’ face binds single-stranded A-rich RNA sequences (6-8). Highly conserved arginine residues on the outer rim of Hfq form six active sites where sRNA-mRNA base pairing takes place (9, 10).
Structural methods such as X-ray crystallography and SAXS typically use 15 M to 1 mM protein concentrations. By contrast, the cellular concentration of Hfq is a few μM (11) and biochemical experiments typically use 50 nM to 1 μM Hfq. Therefore, sensitive methods are needed to measure the assembly and dynamics of multimeric proteins in solution at the lower protein concentrations typically used to measure Hfq function.
Conjugation of proteins with a fluorescent dye is a well-established method for measuring protein-protein and protein-ligand interactions (12). Labeling with extrinsic fluorophores increases the sensitivity of detection more than 100 times compared to Coomassie or silver staining. Although 35S labeling may be more sensitive than fluorescent labeling, it comes with significant hazards, and cannot be used for conformational studies by anisotropy or FRET.
Site-specific labeling with an extrinsic fluorescent dye can be achieved by treating proteins with maleimide esters, which form a covalent bond with the free sulfhydryl group of cysteine. To label a specific region of the protein, it may be necessary to mutate a surface-accessible serine or alanine to cysteine and replace natural cysteine residues with alanine (13, 14). Maleimide-linked cyanine (Cy) dyes are easily available, cost effective and widely used for fluorescence detection at nanomolar concentrations. A Typhoon scanner can detect as little as 10 fmol of fluorescent dye in a gel. Labeling of proteins with fluorescence dyes is typically more sensitive than the intrinsic fluorescence of tryptophan and tyrosine residues in the protein.
Native PAGE is capable of resolving different protein conformations without any structural destabilization (15). Unfortunately the pI of E. coli Hfq is 7.2, so the native protein will not electrophorese in a Laemmli buffer system. Here we describe a modified “semi-native” PAGE protocol for Hfq adapted from Updegrove and co-workers (16). The SDS concentration of the gel loading buffer is reduced to 0.5% (w/v) and the heat denaturing step is omitted, allowing the stable Hfq hexamer to remain intact. This technique is useful for determining the oligomeric states of stable proteins with a pI close to neutral.
Fluorescence anisotropy is directly correlated with the hydrodynamic volume and tumbling of the macromolecule (17). Since different oligomeric states of a protein will have different hydrodynamics, the transitions between them can be quantified by measuring the change in fluorescence anisotropy or polarization of the steady-state fluorescence emission. Dansyl chloride is a reagent that reacts with primary amino groups in both aliphatic and aromatic amines to produce stable fluorescent sulfonamide adducts (18, 19). It is a quick and inexpensive way to label proteins for anisotropy measurements, without making any mutations in the protein.
The base pairing between two complementary model RNAs can be visualized in real time by either FRET (20, 21) or molecular beacons (22). FRET is more expensive than molecular beacons, because all of the RNA strands must be labeled with a fluorescence donor or acceptor. By contrast, a strategy using molecular beacons requires only that beacon is fluorescently labeled; all other target RNAs are unlabelled. In addition, changes in fluorescence intensity for a molecular beacon experiment will be 3-5 times greater than the fluorescence intensity change in a typical FRET experiment. Finally, the increase in molecular beacon fluorescence intensity depends on base pairing between the beacon and target strands, while FRET reports co-localization of the labeled strands on Hfq whether or not they are base paired (23).
Short RNAs (12 to 25 nt) base pair with a complementary strand in 100 – 300 s at the typical concentrations used for our experiments (kon ~ 105 M−1s−1). Hfq protein increases the rate of RNA base pairing by 10 to 100 times (24), necessitating the use of a stopped-flow system for real time detection of the reaction. The large change in the fluorescence emission intensity of molecular beacons provides a strong signal highly suitable for use in a standard stopped-flow spectrometer (22). Therefore, the molecular beacon labeling strategy combined with stopped-flow spectroscopy offers an economical and effective technique for monitoring RNA annealing (or base pairing) kinetics in vitro.
Here we describe labeling of Hfq protein with maleimide-linked Cy3 or dansyl chloride, semi-native PAGE, and fluorescence anisotropy to determine the oligomeric state of the protein. We also describe how to use a molecular beacon and stopped-flow spectrometer to determine the rate of RNA annealing.
2. Materials
Standard precautions for avoiding contamination of samples with ribonucleases (RNases) should be followed. Experimenters should use high-quality pipet tips and tubes and always wear gloves when doing the experiments, purifying RNA or protein, and when handling any labware. Ultrapure, RNase-free deionized water (with a resistivity of ~18 MΩ at 25°C) should be used for making solutions.
2.1. Labeling Hfq with a fluorescent dye
50 μM E. coli Hfq:S65C protein (see Note 1).
10 mM Cy3–maleimide in anhydrous DMSO (see Note 2).
1 M tris(2-carboxyethyl)phosphine (TCEP) (see Note 3).
Wash buffer: 50 mM Na-HEPES, pH 7.5, 1 M NaCl, 2 M urea, 1 mM TCEP, 20 mM imidazole, 5% (v/v) glycerol.
Dialysis tubing or cassette (3,000 MWCO).
Elution buffer: 50 mM HEPES, pH 7.5, 1 M NaCl, 1 mM TCEP, 250 mM imidazole, 5% (v/v) glycerol.
15 mL centrifugal filter units (e.g., Amicon-Ultra 15 from Millipore) with 3,000 MWCO.
5X Hfq storage buffer A: 50 mM Tris-HCl pH 7.5, 250 mM NH4Cl, 1 mM TCEP, 1 mM EDTA, 10% (v/v) glycerol.
5 mL HiTrap Chelating Sepharose HP column (GE Healthcare).
100 mM cobalt sulfate in water.
2.2. Semi-native PAGE components
For best results use 4-20% acrylamide gradient pre-cast gels. Alternatively, materials for casting a 15% resolving gel are listed below.
10 μM Cy3–Hfq:S65C protein (see Note 4).
5X Hfq storage buffer B: 50 mM Tris-HCl pH 7.5, 250 mM NH4Cl, 1 mM EDTA, 10% (v/v) glycerol.
Running Buffer: 25 mM Tris, 250 mM glycine, 0.1% (w/v) SDS (see Note 5).
40% acrylamide (29:1 mono:bis) solution: mix 193.3 g acrylamide powder, 6.7 g bisacrylamide and bring the volume up to 500 ml with RNase-free water (see Note 6).
Resolving gel: 15% acrylamide-bisacrylamide (from 40% acryl-bisacryl stock solution), 375 mM Tris-HCl pH 8.8, 0.07% ammonium persulfate, 0.1% SDS, 2.5 μl TEMED.
Stacking gel: 3% acrylamide-bisacrylamide (from 40% acrylamide stock solution), 125 mM Tris-HCl, pH-6.8, 0.07% ammonium per sulfate, 0.1% SDS, 3 μl TEMED.
SDS-PAGE gel running system (e.g., Mini PROTEAN® tetra cell from Biorad).
3X gel loading buffer: 150 mM Tris-HCl pH 6.8, 1.5% SDS, 0.3% bromophenol blue, 30% (v/v) glycerol.
Typhoon gel scanner (GE Healthcare).
ImageQuant (Molecular Dynamics) or ImageJ software.
2.3. Dansyl chloride labeling components
250 μM Hfq (see Note 1).
250 mM potassium phosphate buffer pH 7.5.
50 mM dansyl chloride in acetone.
25 mg/ml dextran-coated charcoal in 50 mM potassium phosphate buffer pH 7.5.
5X Hfq storage buffer B (see section 2.2).
1 M NaCl.
Microvolume spectrophotometer (e.g., NanoDrop spectrometer from ThermoFisher).
2.4. RNA annealing kinetics experiment
10 μM molecular beacon oligoribonucleotide in TE buffer. The molecular beacon is labeled with a fluorophore (6-FAM) and quencher (DABCYL) (see Note 7).
20 μM target oligoribonucleotide in TE buffer (see Notes 7 and 8).
50 μM Hfq in 5X Hfq storage buffer B.
200 μM Hfq in 5X Hfq storage buffer B.
1X TNK buffer: 10 mM Tris–HCl pH 7.5, 50 mM NaCl, 50 mM KCl.
Fluorometer suitable for anisotropy measurements (e.g., L format Fluorolog-3 spectrophotometer from Horiba) (see Note 9).
500 μl quartz cuvettes.
Stopped-flow instrument (e.g., SX 18MV system from Applied Photophysics).
Data analysis and graphing software (e.g., Origin, Igor, or SigmaPlot).
3. Methods
3.1. Fluorescence labeling of Hfq
Mix 1 ml of 50 μM Hfq:S65C protein and 10 μl 1 M TCEP in a microcentrifuge tube. Mix gently and incubate at 37° C for 10 min (see Note 3).
Add 10 μl of 10 mM Cy3-maleimide in DMSO and cover the tube with aluminum foil. Mix gently and incubate 37° C for 2 h.
Dialyze the whole reaction overnight against 1 L of wash buffer at 4°C.
Charge the HiTrap Chelating Sepharose HP column with 2.5 ml 100 mM cobalt sulfate, then wash with 15 ml water and 15 ml wash buffer.
Load the dialyzed solution on the column and wash with 50 ml wash buffer.
Elute the labeled protein with 15 ml elution buffer.
Concentrate the eluted protein to 0.5 ml with a centrifugal filter unit (15 mL 3,000 MWCO).
Dialyze the concentrated protein overnight at 4°C against 1 L 5X Hfq storage buffer A.
Aliquot 50 μl labeled protein into 500-μl microcentrifuge tubes (see Notes 4 and 10). Snap freeze in a dry ice-ethanol bath and store at −80°C.
3.2. Semi-native PAGE
For best results use 4-20% gradient pre-cast gels (see Note 11). Alternatively, a 15% resolving gel will work well. Follow steps 1-2 below if not using a pre-cast gel.
Assemble two gel plates in a mini SDS PAGE gel casting apparatus. Make 5 ml 15% resolving gel solution by mixing all the ingredients listed in the subheading 2.2.6. Pour the resolving gel solution gently between two gel plates without making any air bubbles. Add 2 ml of acetone on the top of the resolving gel solution. Allow 10 min to polymerize the gel.
Remove all the acetone, wash with water, and dry the inner side of the gel plates with a piece of blotting paper. Make 2.5 ml stacking gel solution by mixing all the ingredients list in subheading 2.2.7. Pour the stacking gel solution on the top of the resolving gel. Insert a comb carefully without making any air bubbles. Allow another 10-15 min to polymerize the gel.
Remove the plates from the casting apparatus and mount it on a mini SDS PAGE gel running apparatus. Mount a plastic support on the other side of the gel holder if necessary. Add gel running buffer to the upper and lower reservoirs of the apparatus (see Note 6).
Beginning with the 10 μM solution of Cy3–Hfq:S65C from subheading 3.1.9, create serial dilutions of labeled Hfq with 5X HB buffer. It is better to use a wide range of dilutions to capture all possible conformations, for instance 0.1, 0.2, 1 and 5 μM Cy3-Hfq:S65C.
Place 10 μl of each dilution in a 1.5 ml microcentrifuge tube and add 5 μl 3X gel loading buffer. Immediately load 10 μl each sample into the wells of the SDS gel (see Note 12).
Run the gel at 20 V/cm until the bromophenol blue is at the bottom of the gel (about 30-40 min).
Separate the plates and pull the gel off the plates. Cover the gel carefully with Saran wrap (see Note 13).
Scan the gel with the Typhoon imager using preset settings for Cy3 imaging (see Note 14).
After scanning the gel, use ImageQuant or ImageJ software to quantify the intensity (pixel volume method) of monomeric, hexameric, and dodecameric Hfq bands (Fig. 1a,b). For E. coli Hfq, the molecular weights of these species are 11.2 kDa, 67 kDa and 134 kDa, respectively.
For each dilution of Hfq, calculate the percentage of each form of Hfq with the following formula: (Ii×100%)/(I1+I6+I12), in which Ii is the intensity of any band in a particular dilution and I1, I6 and I12 are the intensities of monomer, hexamer and dodecamer bands, respectively, from the same Hfq dilution. Plot the percentage of each form of Hfq against total protein concentration (Fig. 1c).
Figure 1.

Detection of Hfq oligomerization by semi-native PAGE. (a) Schematic representation of Hfq assembly; ribbon from pdb 3QHS. (b) Representative semi-native PAGE gel showing different oligomeric states of Hfq. Different dilutions of Cy3-Hfq were loaded (0.1 – 5 μM as shown above each lane) in a 15% polyacrylamide gel and scanned in the Cy3 channel of a Typhoon 9410 (GE Healthcare). (c) Concentration dependence of Hfq oligomerization. The fraction of each oligomeric state was calculated by dividing the counts of corresponding band by total counts in each lane (reprinted from Ref. (19) with permission from Elsevier)
3.3. Dansyl Chloride labeling of Hfq
Combine 200 μl 250 μM Hfq, 200 μl 250 mM potassium phosphate buffer pH 7.5, 10 μl 50 mM dansyl chloride in acetone and 590 μl H2O in a 1.5 ml microcentrifuge tube. Mix gently and incubate at room temperature for 1 h (see Notes 15 and 16).
Add 250 μl 25 mg/ml dextran-coated charcoal in 50 mM potassium phosphate pH 7.5 (see Note 16) and mix thoroughly to adsorb excess dansyl chloride. Incubate the mixture at 4° C for 30 min with slow continuous mixing.
Centrifuge the mixture in a microcentrifuge at 16,000 × g for 30 min to remove all the charcoal. Collect the supernatant and dialyze against 1 L of 5X Hfq storage buffer B at 4°C. Change the dialysis buffer twice.
Measure A280 of the solution using a microvolume spectrophotometer and calculate Hfq concentration using the absorption coefficient ε280 = 3840 M−1 cm−1.
3.4. Steady state anisotropy measurement
If using a Fluorolog-3 spectrophotometer (see Note 9), turn it on 30 min before the experiment. Choose the anisotropy option. Set up the parameters as follows: excitation wavelength 340 nm, emission wavelength 520 nm, excitation and emission slit widths 5 nm, temperature 30° C with 0.5° C tolerance. In the data display window, choose anisotropy, Ihh, Ihv, Ivh, Ivv. Use similar settings on other fluorometers.
Add 150 μl 50 μM dansyl-labeled Hfq from subsection 3.3, 100 μl 250 mM potassium phosphate buffer pH 7.5, 125 μl 1 M NaCl and 125 μl H2O in a microcentrifuge tube. Mix gently and transfer the mixture to a 500 μl quartz cuvette. In this example, the final protein concentration is 15 μM.
Put the cuvette in the fluorometer and let the temperature equilibrate at 30° C for 5 min. Measure all the parameters indicated in subheading 3.4.1. Repeat all the measurements five times. Use the anisotropy values directly from the display window or calculate the anisotropy from θ = (Ihh-Ihv)/(Ihh+2Ihv). Take the average of the measured anisotropy values.
Remove 100 μl sample from the cuvette and add 100 μl 50 mM potassium phosphate buffer pH 7.5 containing 250 mM NaCl. Mix gently, and allow the sample to equilibrate for 3 min. Measure all the parameters, repeat five times, and take the average value of the anisotropy as above.
Repeat the dilution in step 4 until the Hfq concentration reaches 100 nM.
Plot the anisotropy as a function of total Hfq concentration (Fig. 2) (see Note 17). If the protein goes through one or more a discrete disassembly transitions, one can fit the change in anisotropy to a two-stage cooperative binding isotherm, Δθ=Δθ1[Hfq]n1/([Hfq]n1+K1n1)+Δθ2[Hfq]n2/([Hfq]n2+K2n2)+θmin, in which Δθ1 and Δθ2 are the relative increase in anisotropy for each transition, K1 and K2 are the dissociation constants, n1 and n2 are the Hill co-efficients for forming hexamers and dodecamers, respectively. The increase in anisotropy can be alternatively fit to a statistical Ising model that describes each transition explicitly (19), Z = 1 + σ + σ2 + σ3 + σ4 + τσ5 + m(τσ5)2, in which σ = [Hfq]/κ is the dimensionless statistical weight for association of two Hfq monomers, τ represents cooperative hexamer assembly and m represents the association of two hexamers.
Figure 2.
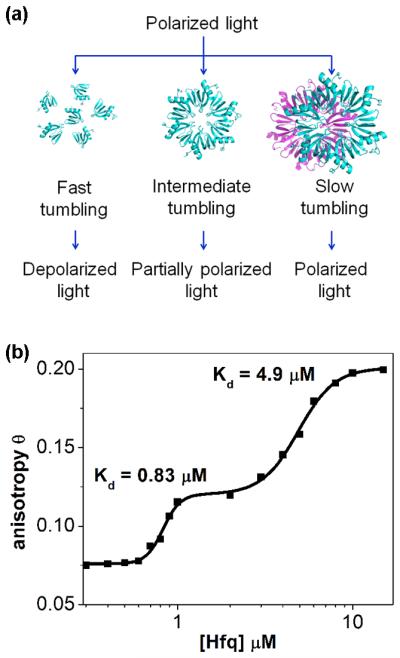
Detection of Hfq oligomerization states by fluorescence anisotropy. (a) Schematic illustration of how fluorescence anisotropy depends on the size of the macromolecule or macromolecular complex. The rotational diffusion or tumbling of the macromolecule in the solution is directly proportional to its hydrodynamic volume. Faster tumbling by small molecules results in increased average depolarization of the emission beam. (b) Representative plot showing fluorescence anisotropy of dansyl-labeled Hfq at different protein concentrations. The data are fit to two sequential cooperative binding transitions (reprinted from Ref. (19) with permission from Elsevier).
3.5. RNA annealing kinetics
The experiment rationale is depicted in Fig. 3a,b. The protocol below includes running/software instructions for the AP SX 18MV system, which should be adapted for other stopped flow instruments.
Figure 3.

RNA annealing kinetics using molecular beacons. (a) A diagram of stopped-flow spectrometer. (b) Experimental strategy for measuring the RNA annealing kinetics between a molecular beacon RNA and its target RNA (oligo C) in presence or absence of Hfq. The molecular beacon is labeled with a fluorophore (6-FAM) and quencher (DABCYL). The complementary region is 16 bp. Base pairing between oligo C and the molecular beacon increases the fluorescence emission intensity of the molecular beacon. (c) Representative kinetic traces showing increase in fluorescence intensity with time after mixing beacon and target RNA. Hfq concentrations were 0 μM (black) to 1.2 μM monomer (magenta). Hfq increases the rate of RNA annealing and the end point of the reaction. Each kinetic trace was fit with a double exponential rate equation to obtain the observed initial rate constant of RNA annealing. (d) Representative plot showing the dependence of the RNA annealing rate (kfast) on the Hfq concentration.
Thirty minutes before start of the experiment, turn on the lamp, controlling unit and computer of the stopped-flow system. Alternatively, the stopped-flow mixer can be attached to a standard fluorometer for kinetic measurements. Set the water bath at 30° C. Open the N2 regulator and set the output pressure to 125 psi.
In the instrument software, select fluorescence and set the excitation wavelength to 490 nm. A 515 nm cut off long pass filter for the emission signal can be installed in front of the photomultiplier tube instead of a monochromator to improve the signal-to-noise ratio.
Add 8.91 ml of 1X TNK and 90 μl of 20 μM target RNA in a 15-ml round bottom culture tube. Mix by gentle vortexing and subsequently spin down the solution.
Create a series of reactant solutions containing molecular beacon and varied amounts of Hfq protein (see Note 18). Add 10 μl 10 μM molecular beacon to each of 9 1.5-mL microcentrifuge tubes. Add 2, 4 and 8 μl 50 μM Hfq to tube numbers 2, 3 and 4, respectively. Add 3, 6, 9, 12 and 15 μl 200 μM Hfq to tube numbers 5–9, respectively. Bring the volume of each tube up to 1 ml with 1X TNK (see Note 19). Mix gently by pipetting the solution up and down few times.
Take a 2.5 ml syringe and draw a 1 ml RNA sample from step 3. Load this sample into the syringe B of the stopped-flow system (Fig. 3a). Move the syringes up and down three times to remove any trapped air bubbles from the syringes (see Note 20).
Use another 2.5 ml syringe to load 1 ml beacon-Hfq sample from tube 1 of step 4 on the syringe A of the stopped flow as above.
Wait for 5 min to equilibrate the samples to 30° C.
Advance (‘drive’) the syringes 3 times to remove any residual buffer from the optical path. Look at the live data display and press ‘auto PM’ after the signal stabilizes.
Set the acquisition time to 200 sec with 10, 000 total points. Press ‘acquire’ to start data acquisition. Wait until one cycle is complete. Repeat this step until the entire sample is consumed (see Note 21).
Repeat steps 5 to 9 for tube numbers 2 to 9 from step 4. Set the acquisition time to 200 sec, 100 sec and 60 sec for tube numbers 2, 3 and 4 respectively. For tube numbers 5– 9 use 20 sec total acquisition time.
Open each kinetic trace from ‘pro data viewer’ and ‘save the plot as’ ‘x.csv’ format.
Open the ‘x.csv’ file with Microsoft Office Excel or other spreadsheet program. Copy the X (time) and Y (emission) columns into a data analysis/graphing software.
Plot the relative fluorescence emission as a function of time (Fig. 3c), and fit the change in relative fluorescence to a single or double exponential rate equation, ΔF(t) = F0 – Afast exp(kfastt) – Aslow exp(kslowt).
Repeat each experiment three times. Take average value of observed rate constants (kobs = kfast) for a particular set of experiment and plot as a function of Hfq concentration (Fig. 3d).
Acknowledgements
The authors thank T. Soper, Y. Peng and A. Santiago-Frangos for helpful discussion. This work was supported by a grant from the NIH R01 GM46686.
Footnotes
Hfq hexamers used for fluorescence labeling were purified by passing E. coli cell lysate containing untagged Hfq over a Hi-trap HP Co+2 column and then re-purified by passing it over a cation exchange column (25). Protein purity was monitored by denaturing SDS-PAGE. The absence of nucleic acids in the protein preparation was judged from the relative absorption at 260 and 280 nm (A260/A280 < 0.7).
Anhydrous DMSO should be used to dissolve the lyophilized Cy3-maleimide activated dye. Exposure to air and light should be minimized throughout labeling process.
Most protein purification protocols use either DTT or BME, which will interfere with maleimide labeling. Before labeling with maleimide-linked Cy-dyes, all the DTT or BME should be removed from the protein solution through extensive dialysis. The dialysis buffer may contain 1 mM TCEP to reduce inter-protein disulfide bonds.
To reduce photobleaching, minimize exposure to light during purification/handling of fluorescently labeled protein.
It is better to use fresh running buffer to reduce heat generation during electrophoresis. If the gel apparatus becomes too warm, run the gel in the cold room.
Acrylamide is neurotoxic, so wear a mask, gloves, goggles and lab coat when handling powdered acrylamide, or purchase a pre-made acrylamide stock solution.
Use Mfold or another RNA structure prediction program to check the secondary structure of molecular beacon and target RNA. Molecular beacons should form a single stem-loop structure with typically 4-5 bp. The stem must be stable enough to prevent high background fluorescence in the absence of target, but weak enough to allow base pairing with the target RNA (26). The target RNA should be designed to have as little self-structure as possible. The ΔG of beacon-target annealing should be much lower than the secondary structure of the molecular beacon itself.
It is cost effective to gel purify oligoribonucleotides in the lab. Normally we order 200 nmol of synthetic RNA and purify the full-length RNA from a 12% preparative denaturing PAGE. Visualize the band by UV shadowing (taking care to minimize UV exposure), cut the band out of gel, and elute the RNA by soaking the gel in TEN (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 250 mM NaCl) buffer and rocking it in cold room for 16-24 hours. Recover RNA by ethanol precipitation from TEN buffer. Short oligoribonucleotides can also be purified by HPLC.
A sensitive spectro-fluorometer is advantageous when making polarization measurements on dilute protein samples. It is always advisable to take the average of three to five readings for a particular data point.
Hfq will lose activity after several freeze-thaw cycles. This can be easily avoided by making small aliquots of protein samples. This is also recommended for RNA samples.
A 4-20% gradient gel will resolve the Hfq hexamer (67,000 Da) and dodecamer (134,000 Da) better than a fixed percentage gel. Gradient gels are commercially available.
To maintain the semi-native condition, it is absolutely necessary to load the sample on the gel just after mixing the protein with the loading buffer. Heat denaturation should be avoided.
Cover the gel slices with saran wrap before placing them on the glass surface of the Typhoon scanner. Do not put glass plates directly on the scanner as they can scratch the imaging surface.
At first, choose the highest PMT voltage in the Typhoon scanner control window and do a quick scan by choosing a pixel size of 1000. Change the PMT voltage if more sensitivity is required. Once the proper PMT voltage is achieved, choose a pixel size of 200 for a higher resolution image.
Phosphate buffer pH 7.5 is ideal for dansyl chloride labeling of the protein. For best results, proteins in a different storage buffer can be dialyzed into the phosphate buffer pH 7.5, before treatment with dansyl chloride.
Use a freshly prepared solution of dansyl chloride for labeling and fresh suspension of dextran coated charcoal for purification.
To confirm that the decrease in anisotropy is due to dissociation of Hfq subunits, and not dilution of the fluorescence signal, perform a control experiment in which each 100 μl aliquot withdrawn from the cuvette is replaced by 100 μl of 15 μM unlabelled Hfq. The fluorescence anisotropy of the control should not change.
For a typical stopped-flow experiment, samples from syringe A and syringe B will be mixed equally in the observation cuvette, so the final concentrations of each component will be reduced by half.
Keep the experimental buffer at room temperature at least 30 min before preparing the samples. If all the samples are not at same temperature, an artifact may appear at the initial time points.
All the syringes and tubing in the stopped-flow system should be washed thoroughly with water before and after the experiment. Follow the instrument maker’s instructions for more stringent cleaning procedures.
One can usually obtain 6-8 kinetic traces from 1 ml sample A and 1 ml sample B. After the experiment, overlay all the traces to check for outliers. If 80% of the traces superimpose with each other, then it is a good data set. Use only the highly similar traces for the final processing.
References
- 1.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mura C, Randolph PS, Patterson J, Cozen AE. Archaeal and eukaryotic homologs of Hfq: A structural and evolutionary perspective on Sm function. RNA Biol. 2013;10(4):636–651. doi: 10.4161/rna.24538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaberdin VR, Blasi U. Translation initiation and the fate of bacterial mRNAs. FEMS Microbiol Rev. 2006;30:967–979. doi: 10.1111/j.1574-6976.2006.00043.x. [DOI] [PubMed] [Google Scholar]
- 4.Gottesman S, McCullen CA, Guillier M, Vanderpool CK, Majdalani N, Benhammou J, Thompson KM, FitzGerald PC, Sowa NA, FitzGerald DJ. Small RNA regulators and the bacterial response to stress. Cold Spring Harb Symp Quant Biol. 2006;71:1–11. doi: 10.1101/sqb.2006.71.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Göpel Y, Papenfort K, Reichenbach B, Vogel J, Görke B. Targeted decay of a regulatory small RNA by an adaptor protein for RNase E and counteraction by an anti-adaptor RNA. Genes Dev. 2013;27(5):552–564. doi: 10.1101/gad.210112.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schumacher MA, Pearson RF, Moller T, Valentin-Hansen P, Brennan RG. Structures of the pleiotropic translational regulator Hfq and an Hfq-RNA complex: a bacterial Sm-like protein. EMBO J. 2002;21:3546–3556. doi: 10.1093/emboj/cdf322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Link TM, Valentin-Hansen P, Brennan RG. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc Natl Acad Sci USA. 2009;106:19292–19297. doi: 10.1073/pnas.0908744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mikulecky PJ, Kaw MK, Brescia CC, Takach JC, Sledjeski DD, Feig AL. Escherichia coli Hfq has distinct interaction surfaces for DsrA, rpoS and poly(A) RNAs. Nat Struct Mol Biol. 2004;11:1206–1214. doi: 10.1038/nsmb858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sauer E, Schmidt S, Weichenrieder O. Small RNA binding to the lateral surface of Hfq hexamers and structural rearrangements upon mRNA target recognition. Proc Natl Acad Sci USA. 2012;109:9396–9401. doi: 10.1073/pnas.1202521109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panja S, Schu DJ, Woodson SA. Conserved arginines on the rim of Hfq catalyze base pair formation and exchange. Nucleic Acids Res. 2013;41(15):7536–7546. doi: 10.1093/nar/gkt521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol. 1999;181(20):6361–6370. doi: 10.1128/jb.181.20.6361-6370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu P, Brand L. Resonance energy transfer: methods and applications. Anal Biochem. 1994;218(1):1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 13.Hopkins JF, Panja S, Woodson SA. Rapid binding and release of Hfq from ternary complexes during RNA annealing. Nucleic Acids Res. 2011;39:5193–5202. doi: 10.1093/nar/gkr062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim H, Abeysirigunawardena SC, Chen K, Mayerle M, Ragunathan K, Luthey-Schulten Z, Ha T, Woodson SA. Protein-guided RNA dynamics during early ribosome assembly. Nature. 2014;506(7488):334–338. doi: 10.1038/nature13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wittig I, Schägger H. Advantages and limitations of clear-native PAGE. Proteomics. 2005;5(17):4338–4346. doi: 10.1002/pmic.200500081. [DOI] [PubMed] [Google Scholar]
- 16.Updegrove TB, Correia JJ, Chen Y, Terry C, Wartell RM. The stoichiometry of the Escherichia coli Hfq protein bound to RNA. RNA. 2011;17(3):489–500. doi: 10.1261/rna.2452111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3rd. Springer; 2006. Chapter 10-12. [Google Scholar]
- 18.Chen RF. Dansyl labeled proteins: Determination of extinction coefficient and number of bound residues with radioactive dansyl chloride. Anal Biochem. 1968;25:412–416. doi: 10.1016/0003-2697(68)90116-4. [DOI] [PubMed] [Google Scholar]
- 19.Panja S, Woodson SA. Hexamer to monomer equilibrium of E. coli Hfq in solution and its impact on RNA annealing. J Mol Biol. 2012;417:406–412. doi: 10.1016/j.jmb.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arluison V, Hohng S, Roy R, Pellegrini O, Regnier P, Ha T. Spectroscopic observation of RNA chaperone activities of Hfq in post-transcriptional regulation by a small non-coding RNA. Nucleic Acids Res. 2007;35:999–1006. doi: 10.1093/nar/gkl1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doetsch M, Stampfl S, Fürtig B, Beich-Frandsen M, Saxena K, Lybecker M, Schroeder R. Study of E. coli Hfq's RNA annealing acceleration and duplex destabilization activities using substrates with different GC-contents. Nucleic Acids Res. 2013 2013 Jan 7;41(1):487–97. doi: 10.1093/nar/gks942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 23.Rajkowitsch L, Schroeder R. Dissecting RNA chaperone activity. RNA. 2007;13:2053–2060. doi: 10.1261/rna.671807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopkins JF, Panja S, McNeil SA, Woodson SA. Effect of salt and RNA structure on annealing and strand displacement by Hfq. Nucleic Acids Res. 2009;37:6205–6213. doi: 10.1093/nar/gkp646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng Y, Soper TJ, Woodson SA. Positional effects of AAN motifs in rpoS regulation by sRNAs and Hfq. J Mol Biol. 2014;426(2):275–285. doi: 10.1016/j.jmb.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hopkins JF, Woodson SA. Molecular beacons as probes of RNA unfolding under native conditions. Nucleic Acids Res. 2005;33:5763–5770. doi: 10.1093/nar/gki877. [DOI] [PMC free article] [PubMed] [Google Scholar]