

Abstract
Ciliopathies are an expanding group of rare conditions characterised by multiorgan involvement, that are caused by mutations in genes encoding for proteins of the primary cilium or its apparatus. Among these genes, CEP290 bears an intriguing allelic spectrum, being commonly mutated in Joubert syndrome and related disorders (JSRD), Meckel syndrome (MKS), Senior-Loken syndrome and isolated Leber congenital amaurosis (LCA). Although these conditions are recessively inherited, in a subset of patients only one CEP290 mutation could be detected.
To assess whether genomic rearrangements involving the CEP290 gene could represent a possible mutational mechanism in these cases, exon dosage analysis on genomic DNA was performed in two groups of CEP290 heterozygous patients, including five JSRD/MKS cases and four LCA, respectively. In one JSRD patient, we identified a large heterozygous deletion encompassing CEP290 C-terminus, that resulted in marked reduction of mRNA expression. No copy number alterations were identified in the remaining probands.
The present work expands the CEP290 genotypic spectrum to include multiexon deletions. Although this mechanism does not appear to be frequent, screening for genomic rearrangements should be considered in patients in whom a single CEP290 mutated allele was identified.
Keywords: Joubert syndrome and related disorders, Meckel syndrome, CEP290, genomic rearrangement
INTRODUCTION
Primary cilia are sensory “antenna-like” organelles found at the cell surface of several tissue types, such as the epithelium of renal tubules and bile ducts, retinal photoreceptors and neuronal cells in fetal and adult brain. These sophisticated microtubule-based organelles have been shown to sense multiple mechanical and chemical signals from the environment and to elicit specific cellular responses, which play crucial roles in embryonic development and homeostatic processes in adulthood [Singla and Reiter, 2006; Vogel, 2005; Eggenschwiler and Anderson, 2007]. Mutations in ciliary genes give rise to a multitude of human disorders that are collectively known as “ciliopathies”. This class of rare diseases can either involve a single organ or occur as multisystemic disorders, with variable and partly overlapping clinical features mainly involving the central nervous system, kidneys, eyes and liver. Typical ciliopathies include polycystic kidney diseases, isolated nephronophthisis, Bardet-Biedl (BBS), Alstrom, Meckel (MKS), Senior-Loken (SLS) and oro-facio-dygital type 1 (OFD1) syndromes [Badano et al., 2006].
In recent years, ciliopathies have been expanded to include Joubert syndrome and related disorders (JSRD), a group of autosomal recessive conditions characterized by a distinctive midbrain-hindbrain malformation (the “molar tooth sign” - MTS). In fact, protein products of all known JSRD genes (AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B and CC2D2A) have been found to localize and function in the primary cilium/basal body organelle [Valente et al., 2008; Cantagrel et al., 2008; Gorden et al., 2008]. Recent data indicate that JSRD and MKS are allelic at several of these loci (CEP290, TMEM67, RPGRIP1L and CC2D2A), and the MKS phenotype is now recognized as the most severe manifestation of the JSRD clinical spectrum [Baala et al., 2007;Baala et al., 2007; Delous et al., 2007; Gorden et al., 2008].
Mutation screenings of large cohorts of JSRD patients have allowed establishing solid correlates between these genes and specific clinical subgroups defined on the basis of multiorgan involvement [Valente et al., 2008; Zaki et al., 2008; Brancati et al., 2009]. In particular, we have recently demonstrated that mutations of the CEP290 gene are responsible for about 50% of the Cerebello-Oculo-Renal (COR) subgroup, characterized by neurological involvement, MTS, nephronophthisis and retinal dystrophy, while they are rarely detected in other JSRD phenotypes [Brancati et al., 2007].
The CEP290 gene has 54 exons and presents a wide allelic spectrum. Indeed, besides JSRD-COR and MKS, mutations in this gene can also cause SLS [Helou et al., 2007], and a recurrent intronic mutation leading to abnormal splicing has been found in a conspicuous proportion of cases with isolated Leber congenital amaurosis (LCA) [den Hollander et al., 2006; Perrault et al., 2007].
So far, nearly 100 distinct CEP290 mutations have been described, most of which were either nonsense, frameshift or splice-site mutations predicted to exert a loss-of-function effect on the resulting protein. In our screening of JSRD patients, we identified two CEP290 mutations in 16 probands, while in other five patients only a single mutated allele could be detected [Brancati et al., 2007]. Similarly, other studies have reported the occurrence of single heterozygous CEP290 mutations in patients with JSRD/MKS, SLS and isolated LCA [Helou et al., 2007; Baala et al., 2007; Perrault et al., 2007].
Here, we report on the identification of a large heterozygous genomic rearrangement leading to CEP290 partial deletion in one of these patients. This result broadens the genotypic spectrum of CEP290, with relevant implications in terms of its molecular analysis.
PATIENTS AND METHODS
Patients
Two groups of patients who carried a single CEP290 mutated allele were selected (Table I). The first group consisted of four JSRD probands with COR phenotype and one fetus with MKS, while the second group included four probands with isolated LCA [Brancati et al., 2007; Baala et al., 2007; Perrault et al., 2007]. Proband and parental DNA samples were available from all families, that had previously given written informed consent for CEP290 mutation screening. Family COR001 was re-consented in order to take fresh blood samples for mRNA analysis.
Table I.
Patients with a single CEP290 mutated allele who were included in the study.
| Patient | Allele 1 | Reference | |
|---|---|---|---|
|
| |||
| DNA Alteration | Effect | ||
| JSRD/MKS* | |||
| COR001 | c.5489_5493delA | p.A1832PfsX1 | [Brancati et al., 2007] |
| COR002a | c.6870delT | p.Q2291KfsX | [Brancati et al., 2007] |
| MTI125 | c.4393C>T | p.R1465X | [Brancati et al., 2007] |
| MTI286 | c.4393C>T | p.R1465X | [Brancati et al., 2007] |
| 05/158* | c.1984C>T | p.Q662X | [Baala et al., 2007] |
|
| |||
| Isolated LCA | |||
| 848,4 | c.4723A>T | p.L1575X | cousin of 848 [Perrault et al., 2007] |
| 196 | c.2991+1655A>G | p.C998X | [Perrault et al., 2007] |
| 317 | c.2991+1655A>G | p.C998X | [Perrault et al., 2007] |
| 446 | c.3934A>T | p.R1312X | unpublished |
Dosage analysis on genomic DNA
Genomic DNA was purified from peripheral blood leucocytes in patients and parents or frozen tissue in the fetal case, following standard methods. Dosage analysis of all exons of CEP290 and, subsequently, of the C12orf29 and C12orf50 genes (for family COR001) was performed by Quantitative Real-Time PCR (QRT-PCR), using Power SYBR Green I dye chemistry and an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, California, USA). Primer pairs for each exon of tested genes were designed by Primer Express 2.0 software (Applied Biosystems), and polymorphisms in the primer binding sites were excluded. CEP290 primers are listed in Table II (primers for C12orf29 and C12orf50 exons are available on request).
Table II.
CEP290 primers for genomic QRT-PCR
| Exons | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) |
|---|---|---|
| 5′-UTR | CGGTCGTGAACTGTAGGCTCT | GCTGCTAGGCGACACCATC |
| 2 | GGTGGAGCACAGTGAAAGAATTC | TCTGCCAGTTCTTCTTGACGG |
| 3 | GTTGCTTTTCTGTTTACTTAGGTGGA | AATTCTGAAAAGGTGTATCACATTTT |
| 4 | CTCAAGAAGTGGAGCTGGCTTT | TTTTTCCAAGGTGCTTACCAAATT |
| 5 | TTGTATTCCCTAGGATGTAATTGTCATT | TACAATCATCCTTATAATTTTTCCAGC |
| 6 | TTACGTAATGAAATTTGCCAACTTG | CTCCAACTCCTTTTCCATGTCC |
| 7 | GAGGAGGCAGAAAATGAAAACAG | AAGTACCTTTGTTGAACCACCACA |
| 8 | ACCGTTTTCCTGTCCTTTTCTG | AAATTCCCAAGATTTCACCACACT |
| 9 | AAGACAGTGACTACCGATCACAGTTG | GCCATTTTACCTGAATTTCATCAAG |
| 10 | TGAAAGCTATTGTGCATCAGACAG | TTCTTACTTGAAGTTGATAATGATCG |
| 11 | GTCTTTTAGGTGCAGGAGCTTACAG | TGCATTGACAGCTACCATAATTGG |
| 12 | AAAGATGATGAAATTATTGAGTATCAG | ATCAGCATCAAGCTGAGCATTC |
| 13 | TGCTCACCGAACAAGTAGAACAA | GGAGCTCATTTTTCAAATCTTCAATA |
| 14 | GCTTCAACCCTTTCTCAACAGAC | CTGTTCTCTCAGCCTCTTTAGTTTTC |
| 15 | TCGAGAGATTGAAATATTAACAAAGG | AGTGCCTCATTTTCATCAAGGAA |
| 16 | GCTTTCTATTGTAGGCCTTGAACC | TGCTGCTGTTTTAAGTGTTTGCTA |
| 17 | AAGAAGTGCAACTTCAGGTATACTCAG | TTTCATATCCAGACAACTCACTTATC |
| 18 | CACTGAGGACCTGAACCTAACTGA | TTGAGGCTCAATAAATCCAATTTTC |
| 19 | GAAAGGAGTAGGACAGTGATAGCCA | AAACAGAGAATGTGTTAACGCCCT |
| 20 | GCAACTTGAAGAAGGTATGAAAGAAAT | GATGTTTCTCCTCCTTTAACATCAGG |
| 21 | GCTAGGCTATAGAATCAAAGAATGCA | CGGTAAGCTGATCAACTTGGG |
| 22 | TCTGTCAATTTGTCTTTCTTTGGG | TTTCAAGATGGTCTATCTGGAAAAAA |
| 23 | TTTGCTGTAATTCGTCATCAACAA | ACTTTCACAATTAACAAGAATATACT |
| 24 | TAAAAGAGGAAAAGAGAAAACTTGAGG | TTATAGCATCTTGTTGGACTTGATCC |
| 25 | CAACCTTAGTAGAATTGGAGCGACA | CTTCAGCCTCCATTGACAACAA |
| 26 | ATGGCCATTTTCAAGATTGCA | TTTATTAGCCAGTTCTAGTTCAGACA |
| 27 | CCTTTATTCAGTGTGAAAACATCTCC | GTTTTTCCTTGGTAATCTCCAGTTCT |
| 28 | AATTAAATGAAAGGCAGCGGG | GTTCCTCCATTTGCTTTAACGAA |
| 29 | TCAATTTGGATGCACAGAAGGT | CAGCATCACTTACTGCCTTGCT |
| 30 | CACAACAACAATCTAGGGACAAGG | GAGCCAATACTGCACATACCTGAT |
| 31 | ATGGAGGCCTACAACTTGCG | TCTTCCCTCCAAACGAGCAT |
| 32 | ATCGGGAATTAGTCAAGGATAAAGAA | GACTGCTGATTGTACGTTCATATTCA |
| 33 | GAAGTTGACCTGGAACGCCA | CATTCATACCTTTTGTGCCGC |
| 34 | CACGGGCAACTTGCAAATC | GCCCCCCAAACATACCAAAT |
| 35 | ATGAACTGAGGCTTCGATTGC | TTGGTTCCATCTCTTTTCTGCC |
| 36 | TGTGAAGAAACATGAGGAAGACCTT | CCGTTTGTTTGAATTTATTTAGTGAA |
| 37 | AACAAGCATTTTATTCGTCTGGC | TTTGACCAAGAGTGAGGAAAGAGAG |
| 38 | AGAGGATTTAAAGTATCTTCTGGACCA | GCTTCTTTTTGAGCCTGAAGTTCA |
| 39 | CGGGCACTTTTAGAACTCCG | GGCCTCTTTTTGAGAAGTTGCA |
| 40 | TGCAAAAGAAACAAAAAGCCTATAATA | GCCTTTTCAGTTCATCATTCTCTTG |
| 41 | TAAACAAAGTCTAATTGAAGAACTCCA | TCTACTTCCTCCACCTTTCCCTCT |
| 42 | CTGGTAATTTTCCTACCTCCTTCG | CCACTTTTTACCTTCTTCCCACC |
| 43 | AAGAGAAACTTACTTTGCAGAGGAAAC | ACTCCAAAGCTCGTATTCCCAA |
| 44 | CAAGCTCTTCCTCGAGATTCTGTT | TTCTAAAGCATGAAGTTTTTCTTGGA |
| 45 | TGAAGATTTCAGGAATAGAGTCAGATG | CAGATGACAACTTCAAGTTTTCCTTC |
| 46 | TCAGAGATTTGAAGGAAATGTGTGA | CCCCTCTAACATGGCCAAGTT |
| 47 | GTGGAAAGACAATCCCAGAACTG | GTTCATTTTCTCTCTGGACTTTTTCA |
| 48 | ATCTTGGGCATCAGTTGAGCAT | GAAGCCTTTCATTTTCAGCAATAAT |
| 49 | TGAGAAGATGACAGTTCAACTAGAAG | GCACCTTCAAGCTGTGGACC |
| 50 | ACAATGAAGACCTTGAACAACAGG | GAACATCTTGCGATATTATGCATTAT |
| 51 | ATTTCCATGGCAAACCTTATCTTC | TCAGCACCTTCAGGAACATGTT |
| 52 | TCAGCTGGATAAAGAGAAAGCAGAA | GGTATGGTGCTTTCAGCTCCAC |
| 53 | ACCTTGAACTCATTCGTAGTGGG | TTTCCTTTAGTTGATCAGCATCTGTT |
| 54 | TCCCCATTTACTAAAGGTCACCTATAA | GTACAAGGTAGTGAGAAGGAAATAC |
To account for possible variations related to DNA input amounts or to the presence of PCR inhibitors, one exon of the reference gene TERT (telomerase reverse transcriptase) on chromosome 5 was simultaneously quantified. All experiments were performed in duplicate, and DNA samples of three healthy individuals were always included as normal controls. SYBR Green amplification mixture (25 μl) contained SYBR Green master mix 2x, 150 nM of each forward and reverse primer, and 20 ng of template DNA. After PCR amplification, a melting curve was generated for each PCR product to check the specificity of the reaction. Data analysis was performed using the ΔΔCt method [Marongiu et al., 2007]. A ratio of target exon/TERT exon between 0.8 and 1.2 was considered normal; ratios higher than 1.2 or lower than 0.6 were considered indicative of a heterozygous duplication or deletion of the target exon, respectively. Each result, outside the normal range, was subsequently confirmed with independent reaction.
CEP290 mRNA analysis
Total RNA from proband COR001 and her father was extracted from peripheral blood according to standard protocols and cDNA synthesis was performed using SuperScript II Reverse Transcriptase, RNase OUT Ribonuclease and random hexamers as primers (Invitrogen, Milan, Italy). The following primers were designed on CEP290 exon 12 to quantify mRNA expression: fw: 5′-GGGAGAAACTTAAGAATGCTCAGC-3′; rev: 5′-TGTCTCGTTCCTGTATACCCTGC-3′. After reverse transcription of mRNA, cDNAs from the patient, father and a control subject were tested by QRT-PCR using Glyceraldehyde phosphate deydrogenase (GAPDH) for normalization (GAPDH primers: fw: 5′-TCAATGGAAATCCCATCACCA-3′; rev: 5′-TGATTTTGGAGGGATCTCGCT-3′).
Characterization of the deletion breakpoints
In family COR001, the fragment containing the deletion breakpoints was amplified by PCR for direct sequencing. PCR primers adjacent to the presumably deleted region were generated within intron 3 of the C12orf50 gene and intron 41 of CEP290 gene, as follows: Intron3: 5′-CTTAAGCATCAGATCTGTCTC-3′; Intron41: 5′-CATCCAGTCTTCTTCCCTC-3′. PCR was cycled once at 94°C for 1 min, 35 times at 94°C for 45 sec, at 56°C for 30 sec, at 68°C for 45 sec, and once at 72°C for 10 min in 30 μl mixture using Platinum Taq DNA Polymerase High Fidelity (Invitrogen). After visualization on a 1.5% agarose gel, the purified PCR product was directly sequenced using BigDye™ terminator chemistry and an ABI 3100 Capillary Array Sequencer (Applied Biosystems).
Bioinformatic analysis
Sequences surrounding the identified breakpoints were analysed using the UCSC genome browser (http://genome.ucsc.edu/) and Repeat Masker (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker) for screening of interspersed repeats and low complexity DNA sequences. Their level of homology was then analyzed with BLAST (http://www.ncbi.nlm.nih.gov/BLAST).
RESULTS
Identification of CEP290 multiexon deletion in one patient
DNA samples from four JSRD-COR probands heterozygous for CEP290 mutations were available from a previous mutation screening. We further obtained DNA from one fetus with MKS and four patients with isolated LCA, also heterozygous for CEP290 point mutations (Table I). These samples were tested using QRT-PCR assay for dosage analysis of CEP290 54 exons and 5′-UTR. In one JSRD-COR proband (COR001), we obtained ratios consistently lower than 0.6 for exons 42 to 54 and normal values for all other exons (Fig. 1a). In the other probands, all exons generated normal ratios.
Fig. 1.
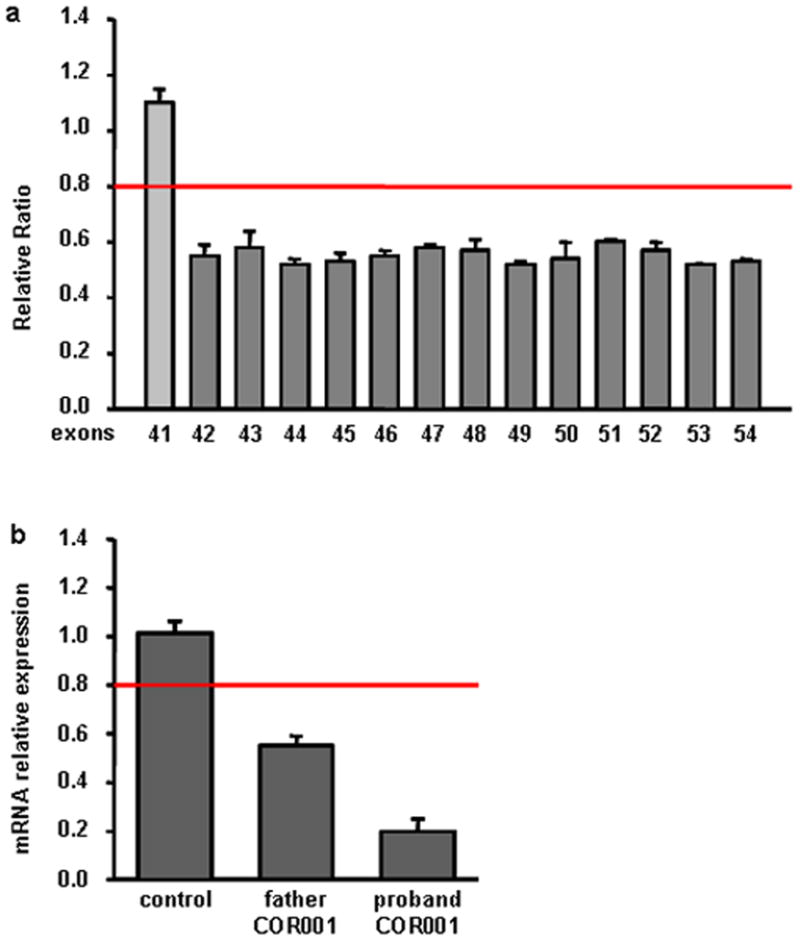
Characterization of the CEP290 gene deletion in COR001 family. a: CEP290 exon dosage in the proband showing the heterozygous deletion of exons 42 to 54. b: Measurement of CEP290 mRNA expression levels showing a reduction of about 50% in the father and 80% in the proband compared with those of normal control. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com].
Proband COR001 was known to have inherited the heterozygous mutation c.5493delA from her mother. In line with this finding, QRT-PCR in the father also showed ratios < 0.6 for exons 42 to 54, consistent with a paternally inherited heterozygous deletion of the C-terminal exons of CEP290. To analyze whether this heterozygous deletion could interfere with CEP290 expression, we performed cDNA QRT-PCR with primers designed outside the deleted region. In the proband’s father, we were able to show a reduction of CEP290 mRNA expression levels of about 50% compared with those of normal control, demonstrating a complete degradation of CEP290 mRNA transcribed from the deleted allele. Interestingly, the proband’s mRNA expression levels were reduced to about 20% of normal control (Fig. 1b). Because there is no evidence of preferential allele expression for CEP290, these data suggested that also an amount of mRNA produced by the c.5493delA mutant allele (resulting in the truncated protein p.A1832PfsX19) was being degraded.
Characterization of the deletion breakpoints in proband COR001
To define the deletion size, we extended dosage analysis of patient COR001 to all exons of genes C12ofr29 and C12orf50, that are located at 3′ of CEP290. The analysis showed a deletion of all exons of C12orf29 and of exons 1 to 3 of C12orf50, refining the deleted region to ~77kb interval between CEP290 intron 41 and C12orf50 intron 3 (Fig. 2a, b). Using a set of primers located adjacent to the supposed deletion breakpoints, we were able to amplify a ~700bp fragment in both the proband and her father, while no amplified fragment was obtained in a normal control (Fig. 2c). Direct sequencing of this fragment showed that the deleted region encompassed 76,844bp (CEP290:c.5709+2352_54_C12orf50:c.290-1375_77del). Since the newly generated junction fell within a stretch of six adenines, the two breakpoints cannot be exactly determined (Fig. 2d).
Fig. 2.
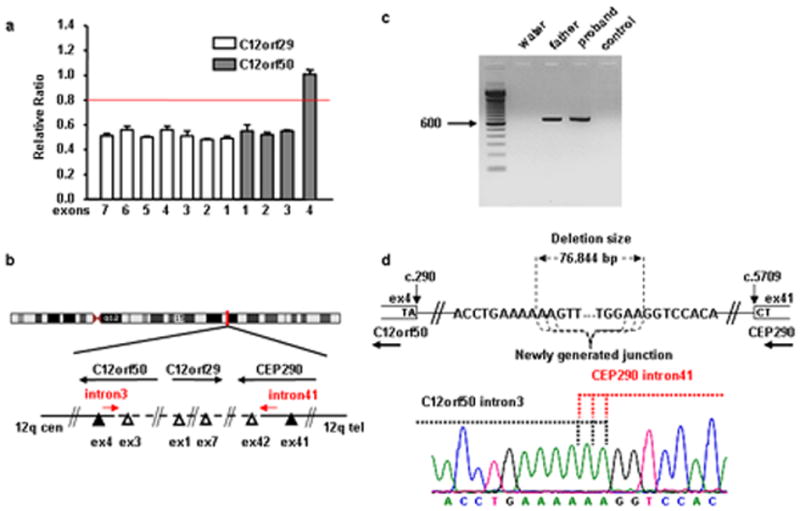
Characterization of the deletion breakpoints in COR001 family. a: Dosage analysis of CEP290 neighbouring genes showing the heterozygous deletion of all C12orf29 exons and of C12orf50 exons 1 to 3. b: schematic representation of the genomic region enclosing CEP290, C12orf29 and C12orf50 genes: open and solid triangles represent exons with ratio < 0.6 and between 0.8 and 1.2, respectively. c: agarose gel electrophoresis showing the ~700 bp fragment obtained with genomic primers located adjacent to the supposed deletion breakpoints. d: top schematic representation of the breakpoints. Bottom electropherogram of the fragment across the breakpoints, showing the newly generated junction between C12orf50 intron 3 and CEP290 intron 41 sequences. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com]
In silico analysis of the extended DNA sequences flanking the deletion indicated that the 5′ breakpoint within CEP290 intron 41 is located within a Long Terminal Repeat belonging to the ERV1 family, while the 3′ breakpoint within C12orf50 intron 3 does not contain any recognizable repeat motif.
DISCUSSION
JSRD, as well as many other ciliopathies, are known to be inherited in an autosomal recessive manner, yet mutation screenings of causative genes (including CEP290, MKS3, RPGRIP1L and CC2D2A) have failed to identify a second pathogenetic mutation in a subset of patients [Gorden et al., 2008; Brancati et al., 2007; Helou et al., 2007; Baala et al., 2007; den Hollander et al., 2006; Sayer et al., 2006; Baala et al., 2007; Wolf et al., 2007]. A first possibility to explain these findings is that these patients harbor mutation types that are undetectable with conventional screening techniques, such as heterozygous multiexon deletions or multiplications, or intronic mutations affecting exon splicing. In order to test for rearrangements involving one or more exons, we searched for genomic imbalances in two groups of probands with either JSRD/MKS or LCA phenotypes, heterozygous for CEP290 mutations, and identified a large multiexon deletion in one of five patients (20%) with the JSRD/MKS phenotype. As expected, no exon dosage imbalances were found in the four patients with LCA, a condition mostly associated with hypomorphic CEP290 mutations [den Hollander et al., 2006; Perrault et al., 2007].
The patient who carried the CEP290 deletion presented a typical COR phenotype with neurological signs of Joubert syndrome (hypotonia, ataxia, psychomotor delay, mental retardation and oculomotor apraxia), LCA with congenital blindness and nephronophthisis that evolved in renal failure at age nine years and requested kidney transplant one year later. The deletion was paternally inherited and spanned about 77Kb genomic DNA, encompassing the last 13 coding exons of CEP290, the entire C12orf29 gene and part of the C12orf50 gene. These two genes encode for uncharacterized proteins of 325 and 414 amino acids respectively. The heterozygous deletions of all or part of these genes does not seem to be associated with any clinical manifestation, as the proband’s phenotype can be fully explained by CEP290 loss of function, while the father is apparently healthy.
The molecular mechanism underlying the detected deletion is still unclear. Three major models have been proposed for genomic rearrangements in the human genome, including Non-Allelic Homologous Recombination, Non-Homologous End-Joining (NHEJ), and Fork Stalling and Template Swiching (FoSTeS) models [Gu et al., 2008]. In our case, in silico analysis of the sequences flanking the breakpoints junction identified several LINEs and Alu repetitive elements, but no significant homology between these sequences could be revealed. However, specific analysis of the breakpoint sequences demonstrated 2 bp micro-homologies, suggesting that microhomology-mediated end-joining could be involved in the origin of the deletion [Chan et al., 2007; Lee K and Lee SE, 2007]. In fact, this NHEJ-based mechanism requires only very short stretches of sequence identity (few bp) between the two ends of the junction. FoSTeS has also been associated with microhomology [Lee JA et al., 2007], yet it is unlikely to have been responsible for the deletion reported here, since so far it has only been implicated in the formation of duplications.
Although the overall number of tested patients is low, the identification of CEP290 exon dosage imbalances in one of five JSRD/MKS probands represents a potentially high yield, suggesting that this mutational mechanism may make a significant contribution to the burden of disease. In the four negative cases, other mutation types may have been missed. Of note, the most frequent CEP290 mutation identified in patients with isolated LCA is a substitution falling within intron 26 of the gene, that leads to the inclusion of a cryptic exon in CEP290 mRNA, with frameshift and premature introduction of a stop codon (p.C998X) [den Hollander et al., 2006; Perrault et al., 2007]. This mutation has been excluded by direct sequencing (or found only in the heterozygous state) in our patients, however similar mutations affecting cryptic splice sites would not have been detected by the employed screening techniques on genomic DNA. A second, intriguing possibility is that heterozygous mutations could represent “modifier” alleles, as already suggested for other ciliopathies. An obvious example is represented by BBS, a pleiotropic condition due to mutations in at least 12 genes. Some BBS patients have been found to harbour three mutations at two distinct loci, suggesting that single heterozygous mutations may contribute to the overall penetrance and expressivity of the disease [Katsanis, 2004; Badano et al., 2006]. A similar mechanism has been demonstrated also for NPHP genes in patients with isolated nephronophthisis [Hoefele et al., 2007]. Recently, heterozygous CEP290 and AHI1 mutations/rare variants have been identified in some JSRD probands homozygous for the NPHP1 deletion, possibly contributing to the phenotypic variability observed in these patients [Tory et al., 2007]. In our cohort of CEP290 heterozygotes, previous screenings have failed to identify mutations in several JBTS/MKS and LCA genes, however the possibility that additional mutations reside in still untested genes cannot be excluded at present. In this complex scenario, implicating several large genes, different mutational types and unconventional modes of inheritance variably contributing to the pathogenesis of these disorders, it appears that mRNA analysis would represent an optimal first-step strategy to perform comprehensive screenings of ciliary genes, allowing to detect not only mutations within exons and canonical splice junctions, but also exon dosage alterations and cryptic mutations affecting splicing. However, in our experience, mRNA from affected children is often difficult to obtain, since many patients live in remote locations or are unavailable for sampling, and some of them are no longer alive. In these cases, the genomic QRT-PCR-based strategy employed here, although time-consuming and labor-intensive, could be considered an alternative approach to search for exon dosage imbalances in patients found to carry single heterozygous mutations by conventional screening. In the near future, the mutation analysis of ciliary genes is expected to be greatly eased by large-scale adoption of innovative techniques such as SNP-arrays, capable of assessing copy number variations in the whole genome at high resolution, and high throughput re-sequencing, allowing rapid and simultaneous testing for point mutations in several large genes.
Acknowledgments
This work was supported by Fondazione Telethon Italy (grant nr. GGP08145 to EB/EMV), Italian Ministry of Health (Ricerca Finalizzata 2006 ex. articolo 56 to EMV, Ricerca Corrente 2009 to BD), the Mariani Foundation (to EMV), NIH (to JGG/EMV), NINDS, Burroughs Welcome Fund, the March of Dimes, and the Howard Hughes Medical Institute (to JGG), Agence Nationale pour la Recherche (ANR grant N° R06370KS to TAB).
Appendix
Members of the International JSRD Study Group are: A. Zankl (Brisbane, Australia); R. Leventer (Parkville, Australia); P. Grattan-Smith (Sydney, Australia); A. Janecke (Innsbruck, Austria); M. D’Hooghe (Brugge, Belgium); Y. Sznajer (Bruxelles, Belgium); R. Van Coster (Ghent, Belgium); L. Demerleir (Brussels, Belgium); K. Dias, C. Moco, A. Moreira (Porto Alegre, Brazil); C. Ae Kim (Sao Paulo, Brazil); G. Maegawa (Toronto, Canada); D. Petkovic (Zagreb, Croatia); G.M.H. Abdel-Salam, A. Abdel-Aleem, M.S. Zaki (Cairo, Egypt); I. Marti, S. Quijano-Roy (Garches, France); S. Sigaudy (Marseille, France); P. de Lonlay, S. Romano (Paris, France); R. Touraine (St. Etienne, France); M. Koenig, C. Lagier-Tourenne, J. Messer (Strasbourg, France); P. Collignon (Toulon, France); N. Wolf (Heidelberg, Germany); H. Philippi (Mainz, Germany); S. Kitsiou Tzeli (Athens, Greece); S. Halldorsson, J. Johannsdottir, P. Ludvigsson (Reykjavik, Iceland); S. R. Phadke (Lucknow, India); V. Udani (Mumbay, India); B. Stuart (Dublin, Ireland); A. Magee (Belfast, Northern Ireland); D. Lev, M. Michelson (Holon, Israel); B. Ben-Zeev (Ramat-Gan, Israel); R. Fischetto, (Bari, Italy); F. Benedicenti, F. Stanzial (Bolzano, Italy); R. Borgatti (Bosisio Parini, Italy); P. Accorsi, S. Battaglia, E. Fazzi, L. Giordano, L. Pinelli (Brescia, Italy); L. Boccone (Cagliari, Italy); S. Bigoni, A. Ferlini (Ferrara, Italy); M.A. Donati (Florence, Italy); G. Caridi, M.T. Divizia, F. Faravelli, G. Ghiggeri, A. Pessagno (Genoa, Italy); M. Briguglio, S. Briuglia, C.D. Salpietro, G. Tortorella (Messina, Italy); A. Adami, P. Castorina, F. Lalatta, G. Marra, D. Riva, B. Scelsa, L. Spaccini, G. Uziel (Milan, Italy); E. Del Giudice (Napoli, Italy); A.M. Laverda, K. Ludwig, A. Permunian, A. Suppiej (Padova, Italy); S. Signorini, C. Uggetti (Pavia, Italy); R. Battini (Pisa, Italy); M. Di Giacomo (Potenza, Italy); M.R. Cilio, M.L. Di Sabato, V. Leuzzi, P. Parisi (Rome, Italy); M. Pollazzon (Siena, Italy); M. Silengo (Torino, Italy); R. De Vescovi (Trieste, Italy); D. Greco, C. Romano (Troina, Italy); M. Cazzagon (Udine, Italy); A. Simonati (Verona, Italy); A.A. Al-Tawari, L. Bastaki, (Kuwait City, Kuwait); A. Mégarbané (Beirut, Lebanon); V. Sabolic Avramovska (Skopje, Macedonia); M.M. de Jong (Groningen, The Netherlands); P. Stromme (Oslo, Norway); R. Koul, A. Rajab (Muscat, Oman); M. Azam (Islamabad, Pakistan); C. Barbot (Oporto, Portugal); L. Martorell Sampol (Barcelona, Spain); B. Rodriguez (La Coruna, Spain); I. Pascual-Castroviejo (Madrid, Spain); S. Teber (Ankara, Turkey); B. Anlar, S. Comu, E. Karaca, H. Kayserili, A. Yüksel (Istanbul, Turkey); M. Akcakus (Kayseri, Turkey); L. Al Gazali, L. Sztriha (Al Ain, UAE); D. Nicholl (Birmingham, UK); C.G. Woods (Cambridge, UK); C. Bennett, J. Hurst, E. Sheridan (Leeds, UK); A. Barnicoat, R. Hennekam, M. Lees (London, UK); E. Blair (Oxford, UK); S. Bernes (Mesa, Arizona, US); H. Sanchez (Fremont, California, US); A.E. Clark (Laguna Niguel, California, US); E. DeMarco, C. Donahue, E. Sherr (San Francisco, California, US); J. Hahn, T.D. Sanger (Stanford California, US); T.E. Gallager (Manoa, Hawaii, US); W.B. Dobyns (Chicago, Illinois, US); C. Daugherty (Bangor, Maine, US); K.S. Krishnamoorthy, D. Sarco, C.A. Walsh (Boston, Massachusetts, US); T. McKanna (Grand Rapids, Michigan, US); J. Milisa (Albuquerque, New Mexico, US); W.K. Chung, D.C. De Vivo, H. Raynes, R. Schubert (New York, New York, US); A. Seward (Columbus, Ohio, US); D.G. Brooks (Philadephia, Pennsylvania, US); A. Goldstein (Pittsburg, Pennsylvania, US); J. Caldwell, E. Finsecke (Tulsa, Oklahoma, US); B.L. Maria (Charleston, South Carolina, US), K. Holden (Mt. Pleasant, South Carolina, US); R.P. Cruse (Houston, Texas, US); K.J. Swoboda, D. Viskochil (Salt Lake City, Utah, US).
References
- Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, Esculpavit C, Toutain A, Moraine C, Parent P, Marcorelles P, Dauge MC, Roume J, Le MM, Meiner V, Meir K, Menez F, Beaufrere AM, Francannet C, Tantau J, Sinico M, Dumez Y, MacDonald F, Munnich A, Lyonnet S, Gubler MC, Genin E, Johnson CA, Vekemans M, Encha-Razavi F, Attie-Bitach T. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler MC, Boddaert N, de LP, Johnson CA, Vekemans M, Antignac C, Attie-Bitach T. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badano JL, Leitch CC, Ansley SJ, May-Simera H, Lawson S, Lewis RA, Beales PL, Dietz HC, Fisher S, Katsanis N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439:326–330. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- Brancati F, Barrano G, Silhavy JL, Marsh SE, Travaglini L, Bielas SL, Amorini M, Zablocka D, Kayserili H, Al-Gazali L, Bertini E, Boltshauser E, D’Hooghe M, Fazzi E, Fenerci EY, Hennekam RC, Kiss A, Lees MM, Marco E, Phadke SR, Rigoli L, Romano S, Salpietro CD, Sherr EH, Signorini S, Stromme P, Stuart B, Sztriha L, Viskochil DH, Yuksel A, Dallapiccola B, Valente EM, Gleeson JG. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am J Hum Genet. 2007;81:104–113. doi: 10.1086/519026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancati F, Iannicelli M, Travaglini L, Mazzotta A, Bertini E, Boltshauser E, D’Arrigo S, Emma F, Fazzi E, Gallizzi R, Gentile M, Loncarevic D, Mejaski-Bosnjak V, Pantaleoni C, Rigoli L, Salpietro CD, Signorini S, Stringini GR, Verloes A, Zablocka D, Dallapiccola B, Gleeson JG, Valente EM The International JSRD Study Group. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum Mutat. 2009;30:E432–E442. doi: 10.1002/humu.20924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attie-Bitach T, Holden KR, Dobyns WB, Traver D, Al-Gazali L, Ali BR, Lindner TH, Caspary T, Otto EA, Hildebrandt F, Glass IA, Logan CV, Johnson CA, Bennett C, Brancati F, Valente EM, Woods CG, Gleeson JG. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CY, Kiechle M, Manivasakam P, Schiestl RH. Ionizing radiation and restriction enzymes induce microhomology-mediated illegitimate recombination in Saccharomyces cerevisiae. Nucleic Acids Res. 2007;35:5051–5059. doi: 10.1093/nar/gkm442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Berthélémé JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Rüther U, Schneider-Maunoury S, Attié-Bitach T, Saunier S. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, Hoyng CB, van den Born LI, Rohrschneider K, Cremers FP. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen D, Alswaid AF, Ozyurek H, Dibooglu S, Otto EA, Liu Y, Davis EE, Hutter CM, Bammler TK, Farin FM, Dorschner M, Topcu M, Zackai EH, Rosenthal P, Owens KN, Katsanis N, Vincent JB, Hildebrandt F, Rubel EW, Raible DW, Knoers NV, Chance PF, Roepman R, Moens CB, Glass IA, Doherty D. CC2D2A Is Mutated in Joubert Syndrome and Interacts with the Ciliopathy-Associated Basal Body Protein CEP290. Am J Hum Genet. 2008;83:559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helou J, Otto EA, Attanasio M, Allen SJ, Parisi M, Glass I, Utsch B, Hashmi S, Fazzi E, Omran H, O’ TJ, Sayer J, Hildebrandt F. Mutation analysis of NPHP6/CEP290 in patients with Joubert-Syndrome and Senior-Loken-Syndrome. J Med Genet. 2007;44:657–663. doi: 10.1136/jmg.2007.052027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefele J, Wolf MT, O’Toole JF, Otto EA, Schultheiss U, Deschenes G, Attanasio M, Utsch B, Antignac C, Hildebrandt F. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol. 2007;18:2789–2795. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- Katsanis N. The oligogenic properties of Bardet-Biedl syndrome. Hum Mol Genet. 2004;13(Spec No 1):R65–R71. doi: 10.1093/hmg/ddh092. [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Lee K, Lee SE. Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics. 2007;176:2003–2014. doi: 10.1534/genetics.107.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marongiu R, Brancati F, Antonini A, Ialongo T, Ceccarini C, Scarciolla O, Capalbo A, Benti R, Pezzoli G, Dallapiccola B, Goldwurm S, Valente EM. Whole gene deletion and splicing mutations expand the PINK1 genotypic spectrum. Hum Mutat. 2007;28:98. doi: 10.1002/humu.9472. [DOI] [PubMed] [Google Scholar]
- Perrault I, Delphin N, Hanein S, Gerber S, Dufier JL, Roche O, foort-Dhellemmes S, Dollfus H, Fazzi E, Munnich A, Kaplan J, Rozet JM. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2007;28:416. doi: 10.1002/humu.9485. [DOI] [PubMed] [Google Scholar]
- Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA, Glass IA, Petry M, Kispert A, Gloy J, Ganner A, Walz G, Zhu X, Goldman D, Nurnberg P, Swaroop A, Leroux MR, Hildebrandt F. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. 2006;313:629–633. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- Tory K, Lacoste T, Burglen L, Moriniere V, Boddaert N, Macher MA, Llanas B, Nivet H, Bensman A, Niaudet P, Antignac C, Salomon R, Saunier S. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18:1566–1575. doi: 10.1681/ASN.2006101164. [DOI] [PubMed] [Google Scholar]
- Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet. 2008;51:1–23. doi: 10.1016/j.ejmg.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Vogel G. News focus: Betting on cilia. Science. 2005;310:216–218. doi: 10.1126/science.310.5746.216. [DOI] [PubMed] [Google Scholar]
- Wolf MT, Saunier S, O’Toole JF, Wanner N, Groshong T, Attanasio M, Salomon R, Stallmach T, Sayer JA, Waldherr R, Griebel M, Oh J, Neuhaus TJ, Josefiak U, Antignac C, Otto EA, Hildebrandt F. Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int. 2007;72:1520–1526. doi: 10.1038/sj.ki.5002630. [DOI] [PubMed] [Google Scholar]
- Zaki MS, Abdel-Aleem A, Abdel-Salam G, Marsh SE, Silhavy JL, Barkovich AJ, Ross ME, Saleem SN, Dobyns WB, Gleeson JG. The molar tooth sign: a new Joubert syndrome and related cerebellar disorders classification system tested in Egyptian families. Neurology. 2008;70:556–565. doi: 10.1212/01.wnl.0000277644.12087.fd. [DOI] [PubMed] [Google Scholar]